

Summary
Protein kinase C (PKC) is a family of kinases that plays diverse roles in many cellular functions, notably proliferation, differentiation, and cell survival. PKC is processed by phosphorylation and regulated by cofactor binding and subcellular localization. Extensive detail is available on the molecular mechanisms that regulate the maturation, activation, and signaling of PKC. However, less information is available on how signaling is terminated both from a global perspective and isozyme-specific differences. To target PKC therapeutically, various ATP-competitive inhibitors have been developed, but this method has problems with specificity. One possible new approach to developing novel, specific therapeutics for PKC would be to target the signaling termination pathways of the enzyme. This review focuses on the new developments in understanding how PKC signaling is terminated and how current drug therapies as well as information obtained from the recent elucidation of various PKC structures and down-regulation pathways could be used to develop novel and specific therapeutics for PKC.
Protein kinase C structure
PKC, a member of the larger superfamily of Ser/Thr kinases, the AGC kinases, consists of 10 isozymes divided into three subclasses based on their second messenger mode of regulation [1]: conventional (α, βI/βII, γ), novel (δ, ε, η, θ) and atypical (ι/λ, ζ),. Conventional PKCs respond to diacylglycerol and Ca2+; novel isozymes respond only to diacylglycerol; and atypical isozymes respond to neither. All PKC family members share a conserved domain architecture, consisting of a C-terminal kinase core and an N-terminal regulatory moiety (Fig. 1A) [2]. The regulatory moiety serves several functions: 1) it maintains the enzyme in an autoinhibited state in the absence of appropriate second messengers; 2) it targets the enzyme to specific cellular locations; and 3) it mediates protein-protein interactions [2]. Specifically, these functions are achieved by a pseudosubstrate peptide sequence and two membrane-targeting modules, the C1 and C2 domains.
Fig. (1).
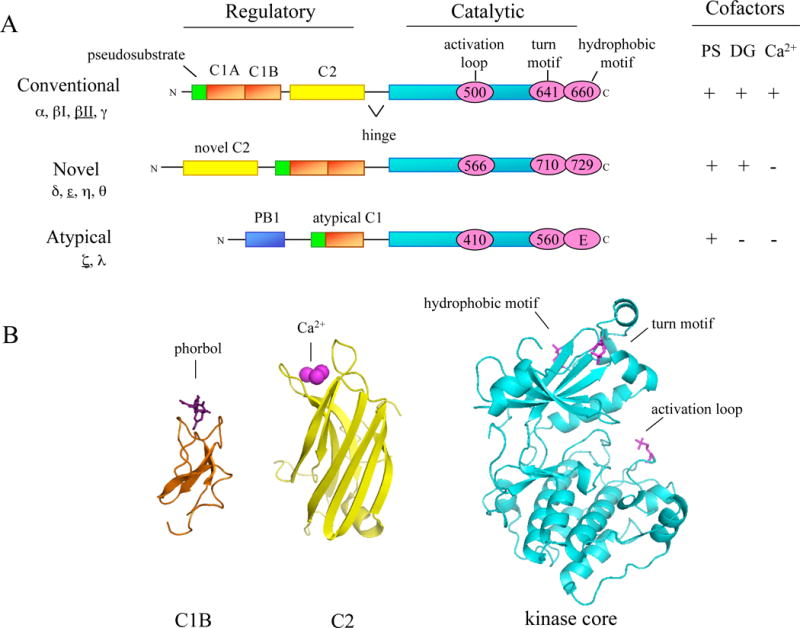
Domain composition of PKC family members. A, The primary structure of the three classes of PKC isozymes: conventional, novel, and atypical. The regulatory moiety contains the cofactor-binding modules: the pseudosubstrate (green); the C1A and C1B domains (orange) that bind phosphatidylserine for all PKCs and diacylglycerol/phorbol esters for conventional and novel PKCs; the C2 domain (yellow) that binds anionic lipids and Ca2+ for conventional PKCs; and a PB1 domain (blue) that serves as a protein:protein interaction module for atypical PKCs. The requirements for cofactor binding are shown to the right: PS, phosphatidylserine; DG, diacylglycerol; and Ca2+. The catalytic moiety contains the conserved kinase domain (cyan) with the activation loop phosphorylation site (pink circle) and a C-terminal tail that contains two conserved phosphorylation sites: the turn motif and the hydrophobic motif (pink circles; note that for atypical PKCs a glutamate occupies the hydrophobic motif). The numbering of the phosphorylation sites is representative of the PKC isozyme underlined to the left. B, Ribbon diagrams of the C1B domain of PKC δ (orange) with bound phorbol (purple), the C2 domain of PKC βII (yellow) with bound Ca2+ (pink), and the kinase domain of phosphorylated PKC βII (blue) with phosphate at the three conserved phosphorylation sites: activation loop (pink), turn motif (orange) and hydrophobic motif (green) [12, 20, 147]. (Adapted from [2, 23])
The pseudosubstrate of PKC lies N-terminal to the C1 domain. When PKC is inactive, this peptide sequence, resembling that of a substrate except for an Ala at the phospho-acceptor position, occupies the substrate-binding cavity of PKC [3]. In this closed, autoinhibited conformation, PKC is relatively resistant to proteolysis [4]. Upon activation of PKC, the pseudosubstrate is released from the kinase core, allowing the subsequent binding and phosphorylation of downstream substrates [5, 6]. The primary mechanism that drives release of the pseudosubstrate is the engagement of the membrane-targeting modules of PKC, the C1 and C2 (in the case of conventional isozymes) domains, to membranes.
The energy to release the pseudosubstrate from the substrate-binding cavity of PKC is provided by the high-affinity binding of the C1 and C2 domains to membrane lipids [7, 8]. The C1 domain is a small, Cys-rich globular structure that is present in all PKC isozymes [9, 10]. Both conventional and novel PKCs have a tandem C1 domain (C1A and C1B) that actively engage their ligands, diacylglycerol and their functional analogues, phorbol esters [11]. Atypical PKCs have a defective ligand binding pocket in their C1 domain and thus are unable to respond to either diacylglycerol or phorbol esters. When the C1 domain is bound to its ligand, a hydrophobic surface is created that allows for effective retention of the domain on membranes [12, 13] In addition to binding diacylglycerol and phorbol esters, the C1 domain also specifically binds the anionic phospholipid, phosphatidylserine [7]. In the case of conventional PKCs, the other membrane-targeting module, the C2 domain, also binds anionic phospholipids but in a Ca2+-dependent manner [7, 14]. Novel PKCs also contain a C2 domain but it lacks key residues required to bind Ca2+; it may play a role in protein:protein interactions [14]. Atypical PKCs lack a C2 domain entirely but have an additional N-terminal domain, a PB1 domain, which has been shown to serve an important role in mediating protein:protein interactions (Fig. 1A) [15].
The kinase core of PKCs and two AGC family members, protein kinase A (PKA) and protein kinase B (PKB)/Akt, are highly conserved with more than 40% sequence identity, primarily differing in the C-terminal tail. This tail is critical to the catalytic activity of the kinase because it contains important conserved regions that make key contacts with the kinase domain as first demonstrated in the hallmark structure of PKA [16]. Recent structural work has shown that the phosphorylated C-terminal tail of PKC (and other AGC kinases) positions a key regulatory helix, the C helix, for catalysis; it can also perform this stabilizing role in trans by binding the corresponding region in the upstream kinase phosphoinositide-dependent kinase 1, thus optimally positioning its C helix (PDK1) [17]. Even more insight into the structural mechanism of how PKC is activated is available with the recent elucidation of crystal structures for several PKC isoforms (ι, θ, & βII) bound to ATP-competitive inhibitors [18–20]. Like its relative PKA, the kinase domain of PKC is a bilobal structure with an N-terminal lobe that is primarily β-sheet and a C-terminal lobe that is primarily α helix; the ATP- and substrate-binding sites lie within a cleft between the two lobes (Fig. 1B) [21]. Unlike other PKC family members, PKC βII contains a novel α helix in the turn motif that associates with the N-terminal lobe of the kinase domain and may aid in stabilizing residues in the active site [20].
Signal propagation
In order for PKC to effectively transduce extracellular signals to downstream targets, PKC must be properly primed and positioned for optimal signaling. Perturbation of the phosphorylation state, conformation, or localization of PKC can disrupt these signaling events, leading to altered physiological states found in diseases such as cancer. These different levels of structural and spatial regulation of PKC allows for the design of more specific therapeutics.
Regulation by phosphorylation
1. Processing phosphorylation
Before PKC can respond to lipid second messengers, the enzyme must first be properly processed by three ordered phosphorylations: activation loop phosphorylation, turn motif phosphorylation, and hydrophobic motif phosphorylation [22, 23]. The first step in the maturation process of PKC is phosphorylation of the activation loop (Thr500 in PKC βII) by the upstream kinase, PDK1 (Fig. 2) [24–26]. PDK1 is not only responsible for phosphorylation of PKC but also other AGC kinases such as Akt [27, 28]. When PKC is newly synthesized, it is loosely tethered at the membrane in a conformation in which the pseudosubstrate is out of the active site, thus adopting an open conformation with the activation loop site exposed [4]. This open conformation is favorable for docking of PDK1 to the C-terminal tail of PKC and subsequent phosphorylation of the activation loop (Fig. 2; first species of PKC on left). Phosphorylation at the activation loop is critical for the maturation of PKC in that it allows for autophosphorylation at the C-terminus, properly positions residues necessary for catalysis, and reveals access to the substrate binding site [29–31]. However, once the activation loop is phosphorylated, phosphate at this site becomes dispensable for activity [23]. Thus, activation loop phosphorylation is merely a primer for the subsequent C-terminal phosphorylations at the turn and hydrophobic motif. These phosphorylations serve to stabilize mature PKC; unphosphorylated or dephosphorylated species of PKC are rapidly degraded. Thus, in cells deficient in PDK1, PKC levels are grossly reduced, attesting to the instability of the non-phosphorylated form [32].
Fig. (2).
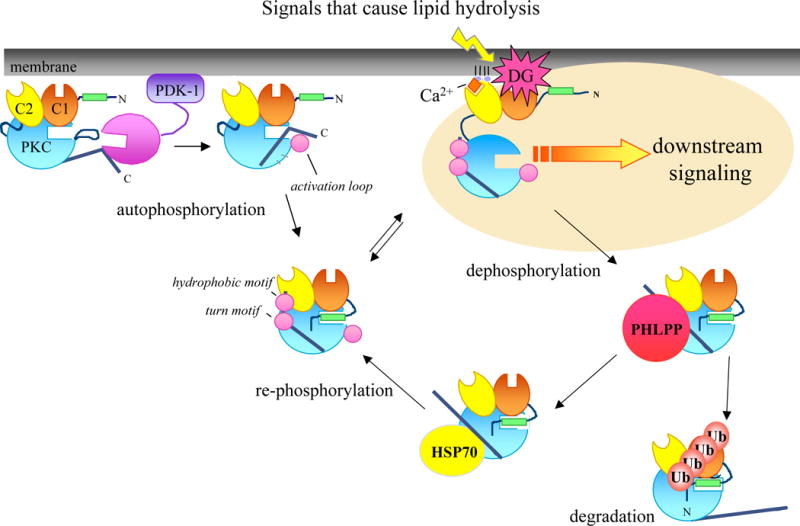
Model showing the life cycle of PKC. When PKC is newly synthesized, the enzyme is loosely tethered at the membrane in an open conformation with the pseudosubstrate (green) released from the substrate-binding cavity (rectangular indent in cyan circle), and the C-terminus is exposed to allow the upstream kinase, PDK-1, to bind (far left). PDK1 phosphorylates the activation loop and is released from the C-terminus. Once the C-terminus is free, PKC can autophosphorylate at the C-terminal phosphorylation sites by an intramolecular mechanism. Once PKC has been processed by phosphorylation, it is released into the cytosol and maintained in an inactive conformation with the pseudosubstrate lodged into the substrate-binding cavity (middle of the diagram). Signals that cause lipid hydrolysis and generate the second messengers, Ca2+ and diacylglycerol, cause the translocation of PKC from the cytosol to the membrane. Binding of Ca2+ and diacylglycerol to the C1 (orange) and C2 (yellow) domains, respectively, provides the energy to release the pseudosubstrate from the active site and allow downstream signaling. In this open, active conformation, PKC is susceptible to dephosphorylation by phosphatases such as PHLPP (red circle) and then targeted for ubiquitination and degradation. However, the molecular chaperone Hsp70 (yellow circle) can bind the dephosphorylated turn motif and stabilize PKC; this allows it to re-phosphorylate and re-enter the pool of competent, signaling enzyme.
Once PDK1 phosphorylates PKC at the activation loop, the enzyme undergoes a rapid phosphorylation at the turn motif site (Thr641 in PKC βII, Fig. 2; 3rd species of PKC from left) [23]. This site is conserved in all AGC kinases. In PKA, this phosphorylation serves to anchor the C-terminus at the upper lobe of the kinase domain by forming contacts with adjacent residues [2]. In the PKC βII structure, this phosphorylation forms ionic contacts with basic residues (Lys374 and Arg415) on opposing β strands, thus differing from PKA [20]. Novel isoforms do not have this Arg. Unlike phosphorylation of the activation loop site which is dispensable for activity, phosphorylation of the turn motif is absolutely required to maintain catalytic competence of the enzyme. Dephosphorylation abolishes activity [33, 34]. Phosphate on the turn motif locks PKC in a thermally stable conformation that, in the closed (inactive) state, is relatively resistant to phosphatases [23] .
The final step in the maturation and processing of PKC is autophosphorylation at the C-terminal hydrophobic motif, a Ser/Thr flanked by hydrophobic residues. This site is also conserved among AGC kinases. For PKC βII, this phosphorylation occurs through an intramolecular mechanism [22]. As discussed above, this phosphorylation aids in aligning the C helix for optimal catalytic activity; this site is absent in PKA [35]. Functionally, phosphorylation at the hydrophobic motif is not required; however, phosphorylation of this site affects the subcellular localization and stability of PKC [36]. In addition, the hydrophobic motif also provides a docking site for PDK1 [2].
Recent evidence suggests that the TORC2 complex (consisting of the mTOR kinase, rictor, mLST8, and Sin1) may be involved in regulation of the hydrophobic motif of PKC [37, 38]. In cells that lack components of this complex (either rictor or mLST8) PKC α is not phosphorylated at the hydrophobic motif and consequently is less stable [37]. Whether the mTORC2 complex controls events leading to the intramolecular autophosphorylation of this site remains to be established.
For conventional PKCs, phosphorylation at the C-terminal turn and hydrophobic motif sites is constitutive. Novel PKCs differ from their conventional counterparts in that their C-terminal phosphorylations are subject to modest regulation by outside stimuli; in addition, phosphorylation at this site for novel PKCs has been proposed to be catalyzed by a separate kinase [39]. In this regard, addition of phorbol esters or antigen-receptor stimulation of T cells results in an increase in hydrophobic motif phosphorylation of PKC θ that has been proposed to be independent of autophosphorylation [40]. The hydrophobic motif sites of both PKC δ and ε have been shown to be sensitive to rapamycin, an inhibitor the mTOR pathway [38]. Although kinase-dead mutants of novel isoforms are not phosphorylated at the hydrophobic motif, suggesting autophosphorylation, the possibility that these isoforms are controlled by a separate upstream kinase at this site remains to be unequivocally established [41].
2. Agonist-evoked autophosphorylation
In addition to the constitutive, processing autophosphorylations, PKC can autophosphorylate in response to agonist-evoked signaling. Novel autophosphorylation sites have been identified in PKC α, β, δ, η, and θ [42–45]. These additional autophosphorylation sites are isozyme-specific and mark activated PKC; a novel autophosphorylation site in the C2 domain of PKC α has been used as a dynamic marker in human cancer tissues [43]. In addition to marking activated PKC, these autophosphorylations regulate the cellular function of PKC. For example, autophosphorylation of PKC θ is required for T cell activation, proper localization, and cross-talk between other signaling pathways, such as with Akt [45]. In the invertebrate Aplysia, autophosphorylation of novel PKCs in the C2 domain is important for increased lipid binding and membrane translocation [46]. This role of autophosphorylation in the C2 domain of novel PKCs may be conserved through vertebrates; PKC η also has autophosphorylation sites that are thought to be important for lipid-regulation [47]. Autophosphorylation of PKC can fine-tune the differences and functional roles of each isozyme and provide an additional layer of regulation.
3. Tyrosine phosphorylation
Conventional, novel, and atypical PKC isozymes are phosphorylated on Tyr [48]. This phosphorylation is emerging as an additional mechanism to fine tuning PKC activity and has been most studied with the novel isoform, PKC δ. PKC δ is phosphorylated at Tyr311 and Tyr332 in response to H2O2 [49]. The Src family of kinases can phosphorylate PKC δ as a means to potentiate its activity and regulate activation loop phosphorylation [50–52]. The EGF receptor can also phosphorylate PKC δ in response to H2O2 [53]. Functionally, these tyrosine phosphorylations in PKC δ aid in its ability to induce apoptosis in response to etoposide [54]. Tyrosine phosphorylation may also serve an additional role in localization of PKC. Treatment of cells with tyrosine phosphatase inhibitors reverses the membrane translocation of PKC βII in phorbol ester-treated cells [55].
Regulation by lipid second messengers
The hallmark of PKC activation in cells is translocation to cellular membranes. Once PKC has been processed by phosphorylation, it is localized to the cytosol where it is inactive with the pseudosubstrate docked in the substrate binding cavity (Fig. 2; 3rd species of PKC from left) [4]. When extracellular signals cause hydrolysis of phosphatidylinositol-4,5-bisphosphate, diacylglycerol is generated and Ca2+ is released from intracellular stores. These second messengers, Ca2+ and diacylglycerol, initiate the membrane translocation and activation of PKC (Fig. 2; 4th species of PKC from left). In the case of conventional PKCs, Ca2+ binds the C2 domain and pretargets PKC to the membrane [23]. This initial binding to the membrane is of too low affinity to activate PKC; instead it allows the C1 domain to more effectively find its membrane-embedded ligand, diacylglycerol [56]. The coordinated engagement of both the C1 and C2 domain on membranes provides the energy to release the autoinhibitory pseudosubstrate [6]. Now in an open conformation, PKC can bind its substrates and initiate downstream signaling events (Fig. 2; 4th species of PKC from left). Note that the potent analogues of diacylglycerol, phorbol esters, cause translocation and activation of PKC in the absence of Ca2+ because the affinity of the C1 domain for phorbol ester-containing membranes is two orders of magnitude higher than that for diacylglycerol-containing membranes; thus, the binding energy of phorbol esters to the C1 domain of PKC is sufficiently high to allow pseudosubstrate removal [23].
Novel PKCs do not have a Ca2+-binding C2 domain and thus lack the membrane-pretargeting mechanism. Therefore, the novel isoforms compensate by having a C1 domain that binds diacylglycerol-containing membranes with an order of magnitude higher affinity than the C1 domain of conventional PKCs. Thus, whereas the C2 domain of the conventional isozyme PKC βII is the major determinant in driving membrane binding, it is the C1B domain of the novel isozyme PKC δ that is the major determinant for membrane binding [57]. The differential affinity of conventional vs novel C1 domains for diacylglycerol-containing membranes is tuned by a single residue on the C1B domain: when present as Trp, as it is in novel PKCs, it confers high-affinity membrane binding and when present as a Tyr, as it is in conventional PKCs, it confers low-affinity membrane binding [58].
Atypical PKCs respond to neither diacylglycerol nor calcium. The only mode of regulation that has been well-studied is phosphorylation by PDK1 [25, 26]. Some studies have shown that insulin and phosphatidylinositol-3,4,5-trisphosphate (PIP3) can increase PKC ζ activity through PDK1-dependent phosphorylation of the activation loop and autophosphorylation [59, 60]. Thus, unlike their other PKC counterparts, regulation of atypical PKCs depends on agonist stimulation.
Regulation by scaffolding proteins
Specificity in PKC signaling is achieved by proper spatial localization [61]. Targeted kinase activity reporters have shown that PKC signals in all regions of the cell: plasma membrane, nucleus, Golgi apparatus, mitochondria, and cytosol [62]. One must assume that in order for PKC to signal in these distinct cellular compartments, precise targeting mechanisms must be in place. One such mechanism is targeting by scaffolding proteins. Scaffolding proteins position PKC near its activators and substrates or at a particular cellular compartment, which allow for isozyme-specific signaling. PKC mediates protein:protein interactions through its regulatory C1 and C2 domains as well as through the C-terminal tail. In some cases, PKC binding proteins interact with a particular conformation (unphosphorylated, phosphorylated but inactive, phosphorylated and active) of PKC, adding an additional layer of complexity to the regulation by scaffolding proteins [23]. This form of regulation is unique in that therapeutics can be developed in order to disrupt or enhance PKC signaling in various regions of the cell.
When PKC is activated, it translocates from the cytosol to the plasma membrane (or other cellular membranes). This localization of activated PKC isozymes is achieved by protein:protein interactions between PKC and specific anchoring proteins. The proteins responsible for binding to activated PKC and thus regulating its activity are RACKS (receptors for activated C-kinase); this interaction is mediated by the C2 domain (reviewed in [63]). Different PKC isozymes bind to specific RACKs to target them to their proper location for cellular function. Mutation of the RACK binding site alters PKC activation in cells [63]. RACKS not only bind PKC but also other signaling molecules such as PLCγ, Src, and integrins [64]. By serving as adaptors for other proteins, RACKS facilitate signaling by bringing enzymes in close proximity with their substrates or their activators. Understanding how the interaction between PKC and RACK is mediated has been informative in the design of activators and inhibitors of PKC that have therapeutic potential in cardiac disease (which will be discussed later).
Another important protein:protein interaction module is the C1 domain. A yeast two-hybrid screen using the N-terminal region of PKC βII as bait has identified some novel binding partners for PKC. PKC βII localizes to the centrosome, an organelle involved in spindle formation and cytokinesis, via the interaction of its C1A domain with pericentrin, a scaffolding protein. If this interaction is disrupted, PKC βII is released from the centrosome and cell division is inhibited [65]. Another protein that interacts with the C1A domain of PKC βII is a novel E3 ligase, RINCK (RING finger protein that interacts with C kinase). Overexpression of RINCK results in ubiquitination and degradation of PKC [66]. By binding the C1 domain of PKC, these various proteins can alter the subcellular localization and activity of PKC.
One of the most promising regions for development of isozyme-specific therapeutics is the C-terminal tail of PKC. For example, PKC α has been shown to play a role in synaptic plasticity by regulating the trafficking of cellular receptors that underlie the processes of memory and learning [67–69]. PKC α can induce cerebellar long-term synaptic depression (LTD) through protein interactions mediated by its type I PDZ (PSD-95, dishevelled, ZO1) ligand, QSAV, at its C-terminus [67]. Deletion of this motif results in prevention of LTD and insertion of this motif into another conventional isoform that lacks the PDZ motif can induce LTD [67]. PKC α has been shown to interact with PICK1, another PDZ-containing protein, through its PDZ ligand in order to facilitate this process; thus, the PDZ ligand of PKC positions it with other signaling proteins in order to regulate important cellular functions [70].
Signal termination
Extensive studies have detailed the molecular mechanisms of the maturation and activation of PKC (reviewed in [2, 23]). We also now know more about where PKC signals in the cell and what are the downstream targets of PKC signaling. What is less understood are the molecular mechanisms that dictate the signal termination and “down-regulation” of PKC. It is in this part of PKC’s life cycle where there is a great opportunity for targeting specific PKC isoforms and designing novel therapeutics to regulate specific PKC signaling pathways.
Reverse translocation
PKC’s translocation to the membrane is a regulated process controlled by the generation of lipid second messengers and the subsequent allosteric activation of PKC by binding these cofactors. Just as translocation to the membrane initiates activation, translocation from the membrane initiates the termination process, and event which is also regulated. Acute activation of PKC by activation of Gq-protein coupled receptors results in translocation to the membrane that is rapidly reversed; stimulation by phorbol esters prolongs interaction with the membrane [71]. This reversal of PKC translocation under physiological agonist stimulation is coincident with desensitization of those receptors [71]. Interestingly, PKC can still translocate to the membrane in response to phorbol esters after receptor stimulation indicating that acute activation of PKC is not enough to induce desensitization of PKC itself [71]. Further studies investigating the molecular mechanism of this reverse translocation show that the C1 and C2 domains drive the membrane translocation and the catalytic activity of PKC is required for this process; it has been proposed that PKC must maintain its priming autophosphorylations at the C-terminus in order to disengage from the membrane [72, 73]. In phorbol ester-treated cells, tyrosine phosphorylation may also serve a role in facilitating the reverse translocation process [55]. Thus, PKC is responsible for regulating its own dynamic membrane trafficking with autophosphorylation serving as a priming step to initiate the signal termination process.
Removal of second messengers
Signaling pathways that activate phospholipase C (PLC) enzymes result in hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2), giving the products diacylglycerol and inositol-1,4,5-trisphosphate (IP3). When IP3 binds to receptors on the intracellular stores, Ca2+ is released. PKC is activated by binding of these second messengers, diacylglycerol and Ca2+, to its regulatory modules, C1 and C2 domains, respectively.
Increasing the intracellular concentrations of Ca2+ and diacylglycerol activates PKC; conversely, decreasing these ligands to basal levels inactivates PKC. PKC is acutely regulated by the presence of its activating cofactors [23]. Studies using a membrane-tethered FRET-based activity reporter CKAR (C kinase activity reporter) show that PKC substrate phosphorylation oscillates with spiking levels of Ca2+, reflecting activation of conventional PKC isoforms [74]. This correlation of increasing substrate phosphorylation (CKAR) with increasing Ca2+ levels can be independent of diacylglycerol as seen in HeLa cells or coincident with diacylglycerol oscillations as seen in MDCK cells [74]. These temporal dynamics in PKC signaling reflect a tight coupling between PKC activation and the influence of PKC and phosphatases on substrate phosphorylation. Indeed, targeting CKARs to various regions of the cell (plasma membrane, Golgi, cytosol, etc.) reveal a biphasic nature of PKC activity in response to agonist-induced signaling. Studies using these various reporters have shown that PKC has an early peak in activity followed by a late, sustained plateau [62]. These differential activity profiles track with second messenger responses: Ca2+ is responsible for the early peak in activity at the plasma membrane while diacylglycerol is responsible for the second, sustained phase at Golgi [62]. The levels of diacylglycerol vary at these different regions in the cell (plasma membrane, Golgi), possibly reflecting differences in activity of different PLC isoforms [75].
One way to deplete levels of diacylglycerol in the cell is through metabolism by diacylglycerol kinases (DGKs). DGK phosphorylates diacylglycerol, converting it into phosphatidic acid [76]. Thus, DGKs, by removing the activating cofactor, serve an opposing role to PKCs in signaling pathways. In DGKδ knockout mice, there are increased levels of diacylglycerol and subsequently increased PKC activity that leads to aberrant phosphorylation of downstream targets [77]. In cardiac remodeling pathways, DGK inhibits activation of Gq-coupled signaling that leads to the activation of PKC [78]. DGKs and PKCs can also physically interact, which allows for spatio-temporal control of localized diacylglycerol in the cell. PKC has been shown to catalyze an activating phosphorylation of DGK γ leading to a negative feedback mechanism [79]. Conversely, phosphorylation of DGK ζ by PKC is inhibitory [80]. Activation of PKC can also cause translocation of DGK to where PKC is localized initiating another method of negative feedback [81]. Thus, the location of PKCs and DGKs are precisely controlled in order to allow for rapid and efficient signaling by diacylglycerol. When diacylglycerol is produced, PKC is activated and that signal is terminated by the action of DGKs.
Activation-induced down-regulation
Twenty-five years ago, Nishizuka and coworkers reported that protein kinase C was the receptor for the potent tumor promoting phorbol esters; it was later shown that phorbol esters trigger the rapid redistribution of PKC from the cytosol to the membrane fraction of cells, the hallmark of PKC activation [82–84]. Blumberg and coworkers showed that phorbol esters, such as phorbol-12,13-dibutyrate (PDBu) and phorbol 12-myristate 13-acetate (PMA), elicit their actions by binding to the C1 domain, specifically competing with diacylglycerol [85–87]. Conventional and novel PKCs could therefore bind to phorbol esters while atypical PKCs remain immune to their effects. Thus, PKC became known as the “receptor” for the tumor-promoting phorbol esters [88].
Chronic activation of PKC that occurs with treatment of cells with phorbol esters and bryostatins (another C1 domain binding compound) leads to the loss in activity and disappearance of PKC protein: ultimately, the down-regulation of PKC. The mechanism by which phorbol esters down-regulate PKC was first shown to be via an increased rate in proteolysis (degradation) [89]. Although phorbol esters are the classical reagent used in down-regulation studies, they are not physiologically relevant agonists. The challenge in this field has been to demonstrate that natural agonists (such as hormones and growth factors) can initiate the down-regulation response. Studies have shown that the neuropeptide bombesin, platelet-derived growth factor (PDGF), or exogenous diacylglycerol can initiate the down-regulation of PKC isoforms in Swiss 3T3 fibroblasts [90]. In gonadotrope cell lines, gonadotropin-releasing hormone (GnRH) can activate several PKC isozymes via a PLC-mediated pathway and initiate their down-regulation [91]. Indeed, the detailed molecular mechanism of how phorbol esters and other agonists promote the down-regulation of PKC still remains elusive. However, the factors that are important for the signal propagation of PKC (activity, conformation, phosphorylation state, localization) also contribute to the regulation of the signal termination of PKC.
In order for PKC to be down-regulated, PKC must have its intrinsic catalytic activity. Mutation of the ATP binding site in PKC renders it insensitive to phorbol ester-mediated degradation [92]. However such kinase-inactive constructs of PKC still translocate to the membrane in response to phorbol ester treatment indicating that the membrane translocation of PKC is independent of its activity [92]. This finding that catalytically-inactive PKC constructs are not sensitive to phorbol ester-dependent down-regulation was the first clue that autophosphorylation of PKC might be a prerequisite for the initiation of the down-regulation pathway. However, there was initial conflict over whether this was the mechanism because other studies had shown that a kinase-dead PKC could down-regulate through the activity of other endogenous PKCs (transphosphorylation) [93–95]. Indeed, isozyme differences exist in down-regulation profiles. Studies performed in Schizosaccharomyces pombe indicated that kinase activity was required for specific isozymes (PKC δ) but those that could not down-regulate themselves (PKC ε) could be affected by trans PKC activity [96]. However, later studies using PKC inhibitors confirmed the hypothesis that the catalytic activity of PKC was required for its down-regulation [91, 97]. What remains unclear is whether the increased proteolysis observed with phorbol ester activation of PKC is a consequence of a conformational change, a secondary effect of autophosphorylation, or whether a specific protease is activated.
When PKC is activated by its lipid cofactors, phosphatidylserine and diacylglycerol, PKC translocates to the membrane and the autoinhibitory pseudosubstrate is pulled out of the active site, leaving PKC in an open conformation [4, 5]. In the case of diacylglycerol, this response is transient as diacylglycerol is rapidly metabolized [98]. Conversely, with the higher affinity binding of phorbol esters, translocation of PKC to the membrane is prolonged, leaving PKC in an open conformation and susceptible to the activity of proteases. Not only does activation of PKC expose the pseudosubstrate, which can be cleaved, but also the proteolytically-labile hinge region between the regulatory and catalytic domains. The Ca2+-dependent neutral proteases (m-calpain and μ-calpain) can cleave PKC in this hinge; however, mutation of the calpain cleavage sites failed to alter the down-regulation in response to phorbol esters indicating that these proteases were not involved in the process [99]. Later studies using calpain protease inhibitors failed to inhibit down-regulation confirming that they were not primarily responsible for the degradation [91]. Therefore, having PKC in an open conformation is important for mediating activation-induced down-regulation but not by the action of calpain proteases.
Dephosphorylation
As mentioned earlier, activation of PKC by its lipid cofactors allosterically alters the conformation of PKC by removing the pseudosubstrate out of the substrate-binding cavity. Not only is activated PKC susceptible to cleavage by proteases but it also has a markedly increased sensitivity to dephosphorylation by phosphatases. Phosphorylation of PKC is critical for maintenance of catalytic competence. Dephosphorylation of the three processing sites (activation loop, turn motif, hydrophobic motif) of PKC inactivates the kinase. Studies show that chronic activation of PKC results in a fully dephosphorylated, inactive kinase, which precedes its degradation [97, 100, 101]. This dephosphorylated form accumulates in a cytoskeletal, detergent-insoluble fraction of cells. This mechanism is the classical model of PKC down-regulation. Thus, discovery of the phosphatases that might control dephosphorylation of PKC under agonist-evoked activation would be important for understanding how PKC is desensitized. Upon activation, PKC translocates to the membrane compartment. The heterotrimeric type 2A phosphatase (PP2A) is localized to the membrane and dephosphorylates PKC upon stimulation with phorbol esters [102]. Addition of okadaic acid, a PP2A phosphatase inhibitor, slows this process and potentially protects PKC from down-regulation [102].
The newly-discovered protein phosphatase 2C (PP2C) family of phosphatases, the PHLPP family (for PH domain Leucine-rich repeat Protein Phosphatase), has recently been shown to regulate the phosphorylation state, and thus levels, of PKC [103, 104]. There are three isoforms of PHLPP: the alternatively spliced PHLPP1α/β and PHLPP2. PHLPP was first characterized as the phosphatase responsible for dephosphorylating the hydrophobic motif of Akt [105]. Since hydrophobic motif phosphorylation is primarily responsible for Akt’s intrinsic catalyticactivity, dephosphorylation by PHLPP ultimately inactivates the kinase [105, 106]. In addition, depletion of PHLPP by siRNA increases the level and duration of agonist-evoked Akt signaling, causing apoptosis and decreased cell proliferation [105, 107]. In contrast, phosphorylation of the hydrophobic motif in PKC primarily controls the stability of PKC. Overexpression of PHLPP leads to dephosphorylation of PKC at the hydrophobic motif which ultimately shunts it to the detergent-insoluble fraction of cells for degradation (Fig. 2; species of PKC on bottom right) [104]. Conversely, depletion of PHLPP results in an up-regulation of PKC levels. Thus, PHLPP plays an important role in the regulation of PKC levels and stability.
Rescue by heat shock proteins
Once PKC has been activated by membrane binding, it is in an open conformation rendering it more sensitive to phosphatases, proteolysis, and ultimately, degradation. However, dephosphorylated PKC can be rescued with the help of heat shock proteins. When PKC is dephosphorylated at the turn motif (one of the three processing sites), Hsp70 can bind and stabilize it, allowing it to re-phosphorylate and enter back into the pool of signaling PKC (Fig. 2) [108]. Disruption of this interaction causes PKC to accumulate in the detergent-insoluble fraction of cells, targeting it for down-regulation [108]. Specifically, an invariant Leu that precedes the turn motif phosphorylation site is responsible for mediating the interaction between Hsp70 and PKC; mutation of this site subsequently increases the dephosphorylation and ubiquitination of PKC [109]. Thus, Hsp70 prolongs the lifetime of PKC. Specifically, Hsp70 binds the dephosphorylated form of PKC; this differs from other binding partners such as PDK1 which binds to the C-terminus of newly synthesized PKC that has never been phosphorylated [108, 110]. Thus, the C-terminus of PKC serves an important role in modulating the signaling lifetime of PKC by mediating important protein:protein interactions. Through regulation of the phosphorylation state of the C-terminus, PKC can interact with specific binding partners to target it to different signaling pathways.
Degradation / down-regulation pathways
1. Degradation by proteasomal pathways
A ubiquitous mechanism for protein degradation in the cell is the ubiquitin-proteasome system (UPS) (reviewed in [111]). By labeling proteins with a “tag” (ubiquitin), they are targeted to a multi-enzymatic complex, the proteasome, where they are proteolyzed and degraded [112]. Ubiquitin is an 8.5 kDa polypeptide that is conjugated to proteins which serves as a molecular marker for degradation. Degradation by the proteasome involves a two successive steps: 1) priming the protein for degradation by addition of ubiquitin and 2) proteolysis of the protein via the proteasome machinery [112]. In the first step, ubiquitin is activated and conjugated to the protein by the action of the three enzymes: 1) E1, ubiquitin-activating enzyme, 2) E2, ubiquitin-conjugating enzyme and 3) E3, ubiquitin ligase [112]. These enzymes work in concert to add the molecular tag that will target them to the proteasome for degradation. The second step is the actual degradation of the tagged protein by the proteasome to produce smaller peptides and free ubiquitin [112]. Typically, ubiquitin is conjugated to proteins in the long chains which serve not only to target them to the proteasome, as described above, but also can also lead to other cellular functions [111]. Many cellular proteins utilize the UPS for degradation.
PKC was first shown to be degraded by the ubiquitin-proteasome pathway with treatment of bryostatins; like phorbol esters, they are also strong activators of PKC and lead to the rapid dephosphorylation and down-regulation of PKC [101, 113]. Treatment of cells with bryostatin results in an accumulation of higher molecular weight species that are labeled with ubiquitin antibody, an accumulation that is inhibited by proteasome inhibitors [113]. Later studies in human fibroblasts indicated that the UPS system is primarily responsible for degradation of PKC α and PKC ε isozymes upon treatment of bryostatin and PMA; addition of proteasome inhibitors inhibited down-regulation whereas inhibitors of calpain proteases, lysosomal enzymes, and vesicle trafficking had no effect [114].
What targets PKC to be degraded by the proteasome? Debate currently exists as to whether or not the phosphorylation state of PKC dictates whether it will be ubiquitinated. Several studies have suggested that dephosphorylation of the three priming sites of PKC (activation loop, turn motif, hydrophobic motif) and inactivation precedes the degradation of the enzyme [97, 113, 114]. However, this phenomenon may be isozyme- and cell-type specific. Although the conventional isoforms PKC α and PKC β and the novel isoform PKC ε have been shown to be dephosphorylated prior to degradation, the novel isoform PKC δ is hyper-phosphorylated in response to PMA-induced degradation and phosphatase inhibitors such as calyculin A promote its down-regulation [97, 114, 115]. In studies using a rat intestinal epithelial cell line, fully phosphorylated, active PKC is ubiquitinated at the plasma membrane and degraded by the proteasome in response to PMA; phosphatase inhibitors accelerate this process [116]. Differences in cell-type, localization, type and duration of stimuli all could account for the differences in down-regulation mechanisms. However, fewer studies have shown ubiquitination of the fully phosphorylated enzyme [117]. Additionally, other phosphorylations could play a role in targeting PKC for degradation. All PKC isozymes contain PEST sequences which are proline, glutamate, serine, and threonine residues that are thought to predispose a protein for degradation [114, 118]. Phosphorylation at these sequences in proteins such as Iκα and cyclins triggers ubiquitination and degradation by the proteasome [118]. Certainly phosphorylation of PEST sequences could serve as a common mechanism for targeting proteins for degradation.
In summary, activation of PKC triggers its own down-regulation. Not only does chronic activation that occurs with phorbol esters promotes the ubiquitination and degradation of PKC, but also natural agonists such as diacylglycerol, bombesin, and hormones all induce ubiquitination of PKC [90, 91, 119]. Down-regulation that occurs via the ubiquitin-proteasome pathway requires the catalytic activity of PKC; addition of kinase inhibitors inhibit this process and a kinase-dead PKC cannot be degraded [119, 120]. Essentially, PKC initiates its own suicide mechanism.
Since PKC is ubiquitinated and degraded, now the question arises as to what molecular machinery is responsible for this process. The prime target would be to identify the E3 ligase that tags PKC with ubiquitin. Several E3 ligases have been identified that are responsible for ubiquitination of PKC. As mentioned earlier, RINCK is an E3 ligase that ubiquitinates PKC [66]. However, RINCK is the not the E3 ligase responsible for the activation-induced down-regulation pathways [66]. RINCK controls the amplitude of the PKC signal by regulating the basal levels of PKC in the cell; depletion of RINCK with siRNA increases PKC protein levels by a mechanisms independent of the activation or phosphorylation state of PKC [66]. Recently, an ubiquitin complex called LUBAC (linear ubiquitin assembly complex) comprised of HOIL-1, an E3 ligase, and a binding protein, HOIP, was shown to bind and ubiquitinate activated PKC α and βII [121]. The von Hippel-Lindau tumor suppressor protein (pVHL), another E3 ligase, targets atypical PKCs; pVHL ubiquitinates activated PKC λ [122]. As there are ten isozymes of PKCs with a wide range of function, it is certainly possible that there could be a specific E3 ligase for not only each PKC, but perhaps in particular cellular functions and processes. Understanding the mechanisms that recruit E3 ligases to target PKCs for degradation would provide another means of therapeutic regulation of PKC.
2. Down-regulation by internalization / trafficking pathways
The down-regulation of PKC is not passive process; activation of PKC is the first step in triggering this mechanism and a catalytically-active enzyme is required. Studies have shown that the phorbol ester-induced down-regulation of PKC correlates with an increase in endocytic, membrane transport processes [96, 123, 124]. The desensitization of receptors occurs through internalization and targeting to endosomes [125]. Active PKC is associated with membranes and thus it is highly possible that PKC is involved with this increased membrane trafficking. Here, localization and scaffolding proteins can dictate the fate of PKC. Now the question arises: how does PKC disengage from the membrane and begin the desensitization process?
Upon stimulation with PMA, PKC accumulates at a perinuclear compartment [97, 124]. This perinuclear accumulation is a temperature-sensitive process, suggesting that vesicular trafficking is involved [97]. Decreasing the temperature also inhibits the dephosphorylation of PKC upon PMA stimulation suggesting that the dephosphorylation step coincides with membrane trafficking events [97]. Indeed, the down-regulation of PKC shares similar characteristics to receptor desensitization processes. Upon stimulation with phorbol esters, PKC trafficks to an endosomal compartment as an active kinase and then is subsequently transported to a perinuclear region where it is dephosphorylated and degraded [123]. This trafficking is mediated by a caveolae-dependent process [123]. Interestingly, it has been shown that phorbol ester treatment disrupts caveolae; however, most likely in the time frame of treatment, the caveolae are internalized as part of this membrane trafficking event [123, 126]. A similar mechanism of internalization and dephosphorylation of PKC has been shown in rat intestinal epithelial cells upon bryostatin treatment [116].
Other studies have characterized PKC’s activation-induced translocation to this perinuclear/juxtanuclear compartment as a subset of recycling endosomes that colocalize with the centrosome [127]. Here, under long-term stimulation with phorbol esters, PKC remains as a fully active kinase that is not degraded; PKC colocalizes with markers of recycling endosomes and not cellular compartments involved in degradation such as the proteasome and lysosome [127]. Instead, PKC is actively involved in the sequestration of proteins such as transferrin, a marker for membrane recycling, to this juxtanuclear, centrosomal compartment (independent of the Golgi), deemed the “pericentrion,” and inhibitors of clathrin-dependent endocytosis can prevent this process [127, 128]. Interestingly, the caveolae-dependent, clathrin-independent trafficking of PKC as seen with other studies can occur through this clathrin-dependent process and thus reconcile the differences seen between these two pathways [123, 128, 129]. However, these membrane trafficking/recycling events are observed following 60 minutes of phorbol ester treatment; it is certainly possible that the degradative events for PKC occur beyond that time frame either in that pericentrion compartment or somewhere else in the cell. The temporal and spatial dynamics of PKC down-regulation still remain to be fully elucidated.
PKC in disease
PKC isozymes are involved in a wide variety of cellular processes. Since PKC isozymes are so diverse in their function, disruption of PKC signaling can have multiple cellular effects. Diseases that are affected by aberrant PKC signaling pathways include metabolic disorders such as diabetes, cardiovascular and pulmonary disorders, central nervous system dysfunction, and neuronal degeneration [130]. PKC undergoes a series of processing phosphorylations that controls its stability and ultimately sets the amplitude for agonist-induced signaling in the cell. Dysregulation of PKC that alters the protein levels in the cell affect the magnitude and duration of downstream signaling; these altered levels of PKC are associated with a variety of pathologies, most notably cancer [131–133]. Indeed, identification of PKC as the receptor for the tumor-promoting phorbol esters provided the first substantial link that this enzyme may be involved in carcinogenesis [131].
The role of PKC in cancer has been reviewed quite extensively over the past few years [130–133]. Almost all of the ten isozymes have been implicated in some form of cancer. The expression levels of PKCs as well as the specific cellular pathways that are affected (i.e. proliferation, apoptosis, angiogenesis) vary based on specific isozyme and tissue type. However, the genetic factors that control PKC’s role in cancer are less well understood.
Advances have been made towards discovering the genes involved in carcinogenesis. With the availability of the entire human genome sequence, it is now possible to identify genes important in pathological pathways. Recently, 210 human cancer genomes were sequenced, specifically looking at the 518 kinases, in order to identify putative “passenger” and “driver” mutations that might predispose one to cancer [134]. The study identified mutations predicted to be driver mutations based on the following: if a kinase possesses a higher ratio of nonsynonymous mutations compared to synonymous mutations, and this frequency is greater than the frequency expected by chance, then the given kinase is likely to possess a driver mutation, which is hypothesized to play a role in the process of tumorigenesis. Driver mutations can confer a growth advantage to the cell, inhibit apoptosis, inhibit migration, and essentially prevent the cell from being able to regulate properly; passenger mutations do not and are therefore not selected [134]. Out of the 518 kinases in the human genome, approximately 120 kinases carry at least one putative driver mutation, PKC included [134]. In fact, of the 10 PKC isozymes, the conventional isozymes PKC α and PKC β had the highest probability of carrying driver mutations; these data are consistent with proposed roles of conventional PKCs in cancer [132–134]. For these two isozymes, mutations were identified in both the regulatory and kinase domains [134]. Additionally, mutations have been characterized in the C1B domain and C-terminus of PKC γ which contribute to the development of spinocerebellar ataxia [135]. Understanding how these mutations alter PKC function, stability, and localization could be key in designing therapeutics to target PKCs involved in various cancers and diseases.
PKC as a drug target
Since disruption of PKC regulation has been implicated in tumorigenesis and drug resistance, PKC has become a prime candidate for the design of therapeutics, specifically as cancer therapies [132, 133, 136]. However, the involvement of PKC isozymes in cancer is very complex due to the number of different PKC isozymes and the various roles they play in different cancer types. PKC isozymes can directly oppose each other in function: PKC δ is typically a pro-apoptotic and anti-proliferative whereas PKC ε is anti-apoptotic and proliferative [131]. Within individual isozymes, differences can vary based on the cancer type: PKC βII is up-regulated in B-cell lymphomas and colon cancer and down-regulated in bladder cancer; similarly, PKC ε is up-regulated in breast cancer and down-regulated in colon cancer [131]. Thus, designing specific therapeutics for specific PKC isozymes is of great importance to prevent unwanted side effects (i.e., down-regulating PKC δ if trying to specifically target PKC ε). PKC drugs have been targeted against two regions of the kinase: the catalytic domain and the regulatory domain.
Catalytic domain
Currently there are a number of PKC inhibitors undergoing clinical trials. Two of these inhibitors, enzastaurin (LY-317615) and ruboxistaurin (Arxxant, LY-333531), which are specific for PKC β, are ATP-binding competitive inhibitors and are in Phase III of clinical trials for cancer drugs [132]. Enzastaurin has shown promise in the treatment of colon and lung cancers, which have increased levels of PKC β, by inducing apoptosis, reducing proliferation, and suppressing angiogenesis, primarily mediated through the Akt/PI3K and VEGF (vascular endothelial growth factor) pathways [137, 138]. Ruboxistaurin has been developed as a treatment for diabetic retinopathy, which has hyperactivated PKC β [139, 140].
With the recent elucidation of various crystal structures of the catalytic domain of PKC, optimization of PKC inhibitors targeting the catalytic domain can be achieved. For example, the PKC θ structure has been used in structure-based design of inhibitors to target PKC θ in the treatment of asthma and autoimmune diseases [141]. The goal would be to use the structure to design specific inhibitors to test in cell-based assays and ultimately take to clinical trials. The catalytic domain of various isozymes could be compared and then tuned for selectivity for that specific isozyme [141].
Regulatory domain
Naturally-occurring agonists of PKC such as bryostatin have been also tested in clinical trials but have shown less success [132]. These compounds bind to the C1 domain and result in the acute activation of conventional and novel PKCs, similarly to phorbol esters; however, unlike phorbol esters, bryostatins have shown anti-tumor effects such as inhibiting cell growth and promoting apoptosis by differential regulation of PKC isozymes [132, 136]. As mentioned earlier, bryostatin inhibits PKC activity by down-regulating the enzyme through the ubiquitin-proteasome pathway [113, 114, 136, 142]. However, since bryostatin does target several PKC isozymes, it is hard to measure the anticancer effects.
The C2 domain is critical for determining the subcellular localization of PKC [63]. Peptides have been derived from the C2 domain to serve as isozyme-specific activators and inhibitors by regulating PKC’s interaction with its RACK [63]. In PKC ε, for example, short peptides have been developed from its C2 domain that confer cardiac protective effects against ischemic injury [143]. These peptides are being investigated as potential leads to treat various cardiac conditions and have entered clinical trials [144]. The benefit of this type of rational drug design is that it allows more selectivity and specificity for PKC isozymes since greater variation exists among the isozymes in their C2 domain. Indeed, targeting protein:protein interaction modules is becoming a more plausible way to target specific PKC isozymes. PKC ι-mediated oncogenic signaling pathways can be altered by disrupting PKC ι’s interaction with PAR6, mediated through its PB1 domain [145]. Since PKC ι mediates its oncogenic effects through its PB1 domain, disrupting that interaction has shown promise in treating lung cancer, where PKC ι signals through its complex with PAR6 [145].
Finally, another potential region that can be used to develop specific PKC therapeutics is the C-terminal tail of PKC, which is quite variable among the isoforms. As mentioned earlier, PKC α and PKC ζ have unique PDZ ligands at their C-termini to mediate important protein:protein interactions; these can be disrupted with therapeutics to modulate PKC’s activity and their downstream signaling effects. Additionally, the C-terminal tail is important for mediating intramolecular interactions between the C2 domain and catalytic core, thus indicating that it would be a prime target for designing isozyme-specific drugs [63].
Targeting protein regulators of PKC activity
Another possibility for the development of therapeutics to target PKC would be to look at upstream regulators of PKC activity, i.e., phosphatases, E3 ligases. Specifically, the focus should look at the various down-regulation pathways of PKCs which seem to differ among the isozymes. The PHLPP phosphatase, which directly dephosphorylates PKC and regulates its cellular levels, is down-regulated in colon cancers which have high levels of PKCs [103]. Perhaps designing drugs to target PHLPP in colon cancer would be a better way to regulate PKC’s effects in this type of cancer. Additionally, the E3 ligase RINCK lies on a chromosomal position that is frequently deleted in non-small cell lung carcinoma, which also has high levels of PKCs [63, 146]. Since the PKC isozymes share a highly conserved catalytic core, other avenues for designing novel and specific PKC therapeutics must be considered. Targeting subcellular localization and binding partners, which vary among the isozymes, is a likely candidate.
Conclusion
The PKC family represents a gold-mine for the development of novel, specific therapeutics. However, the existence of multiple highly-related isozymes with conserved catalytic and substrate-recognition mechanisms has proven a challenge for the development of specific inhibitors. Our increasing understanding of the spatio-temporal dynamics of signaling by these family members holds promise for the design of novel therapeutics that will disrupt the localization and hence function of specific isoforms.
References
- 1.Mellor H, Parker PJ. Biochem J. 1998;332(Pt 2):281–92. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newton AC. Biochem J. 2003;370:361–71. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.House C, Kemp BE. Science. 1987;238:1726–8. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 4.Dutil EM, Newton AC. J Biol Chem. 2000;275:10697–701. doi: 10.1074/jbc.275.14.10697. [DOI] [PubMed] [Google Scholar]
- 5.Orr JW, Newton AC. J Biol Chem. 1994;269:8383–7. [PubMed] [Google Scholar]
- 6.Orr JW, Keranen LM, Newton AC. J Biol Chem. 1992;267:15263–6. [PubMed] [Google Scholar]
- 7.Johnson JE, Giorgione J, Newton AC. Biochemistry. 2000;39:11360–9. doi: 10.1021/bi000902c. [DOI] [PubMed] [Google Scholar]
- 8.Cho W. J Biol Chem. 2001;276:32407–10. doi: 10.1074/jbc.R100007200. [DOI] [PubMed] [Google Scholar]
- 9.Colon-Gonzalez F, Kazanietz MG. Biochim Biophys Acta. 2006;1761:827–37. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Hurley JH. Biochim Biophys Acta. 2006;1761:805–11. doi: 10.1016/j.bbalip.2006.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurley JH, Newton AC, Parker PJ, Blumberg PM, Nishizuka Y. Protein Sci. 1997;6:477–80. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Cell. 1995;81:917–24. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 13.Dries DR, Newton AC. J Biol Chem. 2008 doi: 10.1074/jbc.M709943200. [DOI] [PubMed] [Google Scholar]
- 14.Cho W, Stahelin RV. Biochim Biophys Acta. 2006;1761:838–49. doi: 10.1016/j.bbalip.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 15.Hirano Y, Yoshinaga S, Ogura K, Yokochi M, Noda Y, Sumimoto H, Inagaki F. J Biol Chem. 2004;279:31883–90. doi: 10.1074/jbc.M403092200. [DOI] [PubMed] [Google Scholar]
- 16.Knighton DR, Zheng JH, Ten Eyck LF, Xuong NH, Taylor SS, Sowadski JM. Science. 1991;253:414–20. doi: 10.1126/science.1862343. [DOI] [PubMed] [Google Scholar]
- 17.Kannan N, Haste N, Taylor SS, Neuwald AF. Proc Natl Acad Sci U S A. 2007;104:1272–7. doi: 10.1073/pnas.0610251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Messerschmidt A, Macieira S, Velarde M, Badeker M, Benda C, Jestel A, Brandstetter H, Neuefeind T, Blaesse M. J Mol Biol. 2005;352:918–31. doi: 10.1016/j.jmb.2005.07.060. [DOI] [PubMed] [Google Scholar]
- 19.Xu ZB, Chaudhary D, Olland S, Wolfrom S, Czerwinski R, Malakian K, Lin L, Stahl ML, Joseph-McCarthy D, Benander C, Fitz L, Greco R, Somers WS, Mosyak L. J Biol Chem. 2004;279:50401–9. doi: 10.1074/jbc.M409216200. [DOI] [PubMed] [Google Scholar]
- 20.Grodsky N, Li Y, Bouzida D, Love R, Jensen J, Nodes B, Nonomiya J, Grant S. Biochemistry. 2006;45:13970–81. doi: 10.1021/bi061128h. [DOI] [PubMed] [Google Scholar]
- 21.Johnson LN, Lewis RJ. Chem Rev. 2001;101:2209–42. doi: 10.1021/cr000225s. [DOI] [PubMed] [Google Scholar]
- 22.Keranen LM, Dutil EM, Newton AC. Curr Biol. 1995;5:1394–1403. doi: 10.1016/s0960-9822(95)00277-6. [DOI] [PubMed] [Google Scholar]
- 23.Newton AC. Chem Rev. 2001;101:2353–64. doi: 10.1021/cr0002801. [DOI] [PubMed] [Google Scholar]
- 24.Dutil EM, Toker A, Newton AC. Curr Biol. 1998;8:1366–75. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 25.Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Science. 1998;281:2042–5. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- 26.Chou MM, Hou W, Johnson J, Graham LK, Lee MH, Chen CS, Newton AC, Schaffhausen BS, Toker A. Curr Biol. 1998;8:1069–77. doi: 10.1016/s0960-9822(98)70444-0. [DOI] [PubMed] [Google Scholar]
- 27.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Curr Biol. 1997;7:261–9. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 28.Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR. Biochem J. 1998;331(Pt 1):299–308. doi: 10.1042/bj3310299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nolen B, Taylor S, Ghosh G. Mol Cell. 2004;15:661–75. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 30.Dutil EM, Keranen LM, DePaoli-Roach AA, Newton AC. J Biol Chem. 1994;269:29359–62. [PubMed] [Google Scholar]
- 31.Orr JW, Newton AC. J Biol Chem. 1994;269:27715–8. [PubMed] [Google Scholar]
- 32.Balendran A, Hare GR, Kieloch A, Williams MR, Alessi DR. FEBS Lett. 2000;484:217–23. doi: 10.1016/s0014-5793(00)02162-1. [DOI] [PubMed] [Google Scholar]
- 33.Edwards AS, Faux MC, Scott JD, Newton AC. J Biol Chem. 1999;274:6461–8. doi: 10.1074/jbc.274.10.6461. [DOI] [PubMed] [Google Scholar]
- 34.Bornancin F, Parker PJ. Curr Biol. 1996;6:1114–23. doi: 10.1016/s0960-9822(02)70678-7. [DOI] [PubMed] [Google Scholar]
- 35.Frodin M, Antal TL, Dummler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM. Embo J. 2002;21:5396–407. doi: 10.1093/emboj/cdf551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards AS, Newton AC. J Biol Chem. 1997;272:18382–90. doi: 10.1074/jbc.272.29.18382. [DOI] [PubMed] [Google Scholar]
- 37.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Dev Cell. 2006;11:859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Parekh D, Ziegler W, Yonezawa K, Hara K, Parker PJ. J Biol Chem. 1999;274:34758–64. doi: 10.1074/jbc.274.49.34758. [DOI] [PubMed] [Google Scholar]
- 39.Cameron AJ, De Rycker M, Calleja V, Alcor D, Kjaer S, Kostelecky B, Saurin A, Faisal A, Laguerre M, Hemmings BA, McDonald N, Larijani B, Parker PJ. Biochem Soc Trans. 2007;35:1013–7. doi: 10.1042/BST0351013. [DOI] [PubMed] [Google Scholar]
- 40.Freeley M, Volkov Y, Kelleher D, Long A. Biochem Biophys Res Commun. 2005;334:619–30. doi: 10.1016/j.bbrc.2005.06.136. [DOI] [PubMed] [Google Scholar]
- 41.Cenni V, Doppler H, Sonnenburg ED, Maraldi N, Newton AC, Toker A. Biochem J. 2002;363:537–45. doi: 10.1042/0264-6021:3630537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durgan J, Michael N, Totty N, Parker PJ. FEBS Lett. 2007;581:3377–81. doi: 10.1016/j.febslet.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 43.Ng T, Squire A, Hansra G, Bornancin F, Prevostel C, Hanby A, Harris W, Barnes D, Schmidt S, Mellor H, Bastiaens PI, Parker PJ. Science. 1999;283:2085–9. doi: 10.1126/science.283.5410.2085. [DOI] [PubMed] [Google Scholar]
- 44.Flint AJ, Paladini RD, Koshland DE., Jr Science. 1990;249:408–11. doi: 10.1126/science.2377895. [DOI] [PubMed] [Google Scholar]
- 45.Thuille N, Heit I, Fresser F, Krumbock N, Bauer B, Leuthaeusser S, Dammeier S, Graham C, Copeland TD, Shaw S, Baier G. Embo J. 2005;24:3869–80. doi: 10.1038/sj.emboj.7600856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pepio AM, Sossin WS. J Biol Chem. 2001;276:3846–55. doi: 10.1074/jbc.M006339200. [DOI] [PubMed] [Google Scholar]
- 47.Littler DR, Walker JR, She YM, Finerty PJ, Jr, Newman EM, Dhe-Paganon S. Biochem Biophys Res Commun. 2006;349:1182–9. doi: 10.1016/j.bbrc.2006.08.160. [DOI] [PubMed] [Google Scholar]
- 48.Konishi H, Tanaka M, Takemura Y, Matsuzaki H, Ono Y, Kikkawa U, Nishizuka Y. Proc Natl Acad Sci U S A. 1997;94:11233–7. doi: 10.1073/pnas.94.21.11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinberg SF. Biochem J. 2004;384:449–59. doi: 10.1042/BJ20040704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joseloff E, Cataisson C, Aamodt H, Ocheni H, Blumberg P, Kraker AJ, Yuspa SH. J Biol Chem. 2002;277:12318–23. doi: 10.1074/jbc.M111618200. [DOI] [PubMed] [Google Scholar]
- 51.Hall KJ, Jones ML, Poole AW. Biochem J. 2007;406:501–9. doi: 10.1042/BJ20070244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rybin VO, Guo J, Gertsberg Z, Elouardighi H, Steinberg SF. J Biol Chem. 2007;282:23631–8. doi: 10.1074/jbc.M701676200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morita M, Matsuzaki H, Yamamoto T, Fukami Y, Kikkawa U. J Biochem (Tokyo) 2007 doi: 10.1093/jb/mvm190. [DOI] [PubMed] [Google Scholar]
- 54.Blass M, Kronfeld I, Kazimirsky G, Blumberg PM, Brodie C. Mol Cell Biol. 2002;22:182–95. doi: 10.1128/MCB.22.1.182-195.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takahashi H, Suzuki K, Namiki H. Cell Struct Funct. 2003;28:123–30. doi: 10.1247/csf.28.123. [DOI] [PubMed] [Google Scholar]
- 56.Nalefski EA, Newton AC. Biochemistry. 2001;40:13216–29. doi: 10.1021/bi010761u. [DOI] [PubMed] [Google Scholar]
- 57.Giorgione JR, Lin JH, McCammon JA, Newton AC. J Biol Chem. 2006;281:1660–9. doi: 10.1074/jbc.M510251200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dries DR, Gallegos LL, Newton AC. J Biol Chem. 2007;282:826–30. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 59.Standaert ML, Bandyopadhyay G, Kanoh Y, Sajan MP, Farese RV. Biochemistry. 2001;40:249–55. doi: 10.1021/bi0018234. [DOI] [PubMed] [Google Scholar]
- 60.Standaert ML, Bandyopadhyay G, Perez L, Price D, Galloway L, Poklepovic A, Sajan MP, Cenni V, Sirri A, Moscat J, Toker A, Farese RV. J Biol Chem. 1999;274:25308–16. doi: 10.1074/jbc.274.36.25308. [DOI] [PubMed] [Google Scholar]
- 61.Mochly-Rosen D. Science. 1995;268:247–51. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- 62.Gallegos LL, Kunkel MT, Newton AC. J Biol Chem. 2006;281:30947–56. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- 63.Kheifets V, Mochly-Rosen D. Pharmacol Res. 2007;55:467–76. doi: 10.1016/j.phrs.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schechtman D, Mochly-Rosen D. Oncogene. 2001;20:6339–47. doi: 10.1038/sj.onc.1204778. [DOI] [PubMed] [Google Scholar]
- 65.Chen D, Purohit A, Halilovic E, Doxsey SJ, Newton AC. J Biol Chem. 2004;279:4829–39. doi: 10.1074/jbc.M311196200. [DOI] [PubMed] [Google Scholar]
- 66.Chen D, Gould C, Garza R, Gao T, Hampton RY, Newton AC. J Biol Chem. 2007 doi: 10.1074/jbc.M703320200. [DOI] [PubMed] [Google Scholar]
- 67.Leitges M, Kovac J, Plomann M, Linden DJ. Neuron. 2004;44:585–94. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 68.Perez JL, Khatri L, Chang C, Srivastava S, Osten P, Ziff EB. J Neurosci. 2001;21:5417–28. doi: 10.1523/JNEUROSCI.21-15-05417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hanley JG, Henley JM. Neuron. 2006;49:778–80. doi: 10.1016/j.neuron.2006.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Staudinger J, Lu J, Olson EN. J Biol Chem. 1997;272:32019–24. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- 71.Feng X, Zhang J, Barak LS, Meyer T, Caron MG, Hannun YA. J Biol Chem. 1998;273:10755–62. doi: 10.1074/jbc.273.17.10755. [DOI] [PubMed] [Google Scholar]
- 72.Feng X, Hannun YA. J Biol Chem. 1998;273:26870–4. doi: 10.1074/jbc.273.41.26870. [DOI] [PubMed] [Google Scholar]
- 73.Feng X, Becker KP, Stribling SD, Peters KG, Hannun YA. J Biol Chem. 2000;275:17024–34. doi: 10.1074/jbc.275.22.17024. [DOI] [PubMed] [Google Scholar]
- 74.Violin JD, Zhang J, Tsien RY, Newton AC. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJ. J Biol Chem. 2006;281:2639–48. doi: 10.1074/jbc.M507681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luo B, Regier DS, Prescott SM, Topham MK. Cell Signal. 2004;16:983–9. doi: 10.1016/j.cellsig.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 77.Crotty T, Cai J, Sakane F, Taketomi A, Prescott SM, Topham MK. Proc Natl Acad Sci U S A. 2006;103:15485–90. doi: 10.1073/pnas.0604104103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arimoto T, Takeishi Y, Takahashi H, Shishido T, Niizeki T, Koyama Y, Shiga R, Nozaki N, Nakajima O, Nishimaru K, Abe J, Endoh M, Walsh RA, Goto K, Kubota I. Circulation. 2006;113:60–6. doi: 10.1161/CIRCULATIONAHA.105.560771. [DOI] [PubMed] [Google Scholar]
- 79.Yamaguchi Y, Shirai Y, Matsubara T, Sanse K, Kuriyama M, Oshiro N, Yoshino K, Yonezawa K, Ono Y, Saito N. J Biol Chem. 2006;281:31627–37. doi: 10.1074/jbc.M606992200. [DOI] [PubMed] [Google Scholar]
- 80.Luo B, Prescott SM, Topham MK. J Cell Biol. 2003;160:929–37. doi: 10.1083/jcb.200208120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.van Baal J, de Widt J, Divecha N, van Blitterswijk WJ. J Biol Chem. 2005;280:9870–8. doi: 10.1074/jbc.M409301200. [DOI] [PubMed] [Google Scholar]
- 82.Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. J Biol Chem. 1982;257:7847–51. [PubMed] [Google Scholar]
- 83.Kraft AS, Anderson WB. Nature. 1983;301:621–3. doi: 10.1038/301621a0. [DOI] [PubMed] [Google Scholar]
- 84.Newton AC. Trends Pharmacol Sci. 2004;25:175–7. doi: 10.1016/j.tips.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 85.Konig B, DiNitto PA, Blumberg PM. J Cell Biochem. 1985;29:37–44. doi: 10.1002/jcb.240290105. [DOI] [PubMed] [Google Scholar]
- 86.Blumberg PM, Sharkey NA, Konig B, Jaken S, Leach KL, Jeng AY. Princess Takamatsu Symp. 1983;14:75–87. [PubMed] [Google Scholar]
- 87.Sharkey NA, Leach KL, Blumberg PM. Proc Natl Acad Sci U S A. 1984;81:607–10. doi: 10.1073/pnas.81.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nishizuka Y. Nature. 1984;308:693–8. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- 89.Young S, Parker PJ, Ullrich A, Stabel S. Biochem J. 1987;244:775–9. doi: 10.1042/bj2440775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Olivier AR, Parker PJ. J Biol Chem. 1994;269:2758–63. [PubMed] [Google Scholar]
- 91.Junoy B, Maccario H, Mas JL, Enjalbert A, Drouva SV. Endocrinology. 2002;143:1386–403. doi: 10.1210/endo.143.4.8752. [DOI] [PubMed] [Google Scholar]
- 92.Ohno S, Konno Y, Akita Y, Yano A, Suzuki K. J Biol Chem. 1990;265:6296–300. [PubMed] [Google Scholar]
- 93.Pears C, Parker PJ. FEBS Lett. 1991;284:120–2. doi: 10.1016/0014-5793(91)80776-y. [DOI] [PubMed] [Google Scholar]
- 94.Lindner D, Gschwendt M, Marks F. Biochem Biophys Res Commun. 1991;176:1227–31. doi: 10.1016/0006-291x(91)90416-5. [DOI] [PubMed] [Google Scholar]
- 95.Freisewinkel I, Riethmacher D, Stabel S. FEBS Lett. 1991;280:262–6. doi: 10.1016/0014-5793(91)80307-o. [DOI] [PubMed] [Google Scholar]
- 96.Goode NT, Hajibagheri MA, Parker PJ. J Biol Chem. 1995;270:2669–73. doi: 10.1074/jbc.270.6.2669. [DOI] [PubMed] [Google Scholar]
- 97.Hansra G, Garcia-Paramio P, Prevostel C, Whelan RD, Bornancin F, Parker PJ. Biochem J. 1999;342(Pt 2):337–44. [PMC free article] [PubMed] [Google Scholar]
- 98.Carrasco S, Merida I. Trends Biochem Sci. 2007;32:27–36. doi: 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 99.Junco M, Webster C, Crawford C, Bosca L, Parker PJ. Eur J Biochem. 1994;223:259–63. doi: 10.1111/j.1432-1033.1994.tb18990.x. [DOI] [PubMed] [Google Scholar]
- 100.Borner C, Filipuzzi I, Wartmann M, Eppenberger U, Fabbro D. J Biol Chem. 1989;264:13902–9. [PubMed] [Google Scholar]
- 101.Lee HW, Smith L, Pettit GR, Bingham Smith J. Am J Physiol. 1996;271:C304–11. doi: 10.1152/ajpcell.1996.271.1.C304. [DOI] [PubMed] [Google Scholar]
- 102.Hansra G, Bornancin F, Whelan R, Hemmings BA, Parker PJ. J Biol Chem. 1996;271:32785–8. doi: 10.1074/jbc.271.51.32785. [DOI] [PubMed] [Google Scholar]
- 103.Brognard J, Newton AC. Trends Endocrinol Metab (in press) [Google Scholar]
- 104.Gao T, Brognard J, Newton AC. J Biol Chem. 2008 doi: 10.1074/jbc.M707319200. in press. [DOI] [PubMed] [Google Scholar]
- 105.Gao T, Furnari F, Newton AC. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 106.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Embo J. 1996;15:6541–51. [PMC free article] [PubMed] [Google Scholar]
- 107.Brognard J, Sierecki E, Gao T, Newton AC. Mol Cell. 2007;25:917–31. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 108.Gao T, Newton AC. J Biol Chem. 2002;277:31585–92. doi: 10.1074/jbc.M204335200. [DOI] [PubMed] [Google Scholar]
- 109.Gao T, Newton AC. J Biol Chem. 2006;281:32461–8. doi: 10.1074/jbc.M604076200. [DOI] [PubMed] [Google Scholar]
- 110.Gao T, Toker A, Newton AC. J Biol Chem. 2001;276:19588–96. doi: 10.1074/jbc.M101357200. [DOI] [PubMed] [Google Scholar]
- 111.Herrmann J, Lerman LO, Lerman A. Circ Res. 2007;100:1276–91. doi: 10.1161/01.RES.0000264500.11888.f0. [DOI] [PubMed] [Google Scholar]
- 112.Reinstein E, Ciechanover A. Ann Intern Med. 2006;145:676–84. doi: 10.7326/0003-4819-145-9-200611070-00010. [DOI] [PubMed] [Google Scholar]
- 113.Lee HW, Smith L, Pettit GR, Vinitsky A, Smith JB. J Biol Chem. 1996;271:20973–6. [PubMed] [Google Scholar]
- 114.Lee HW, Smith L, Pettit GR, Smith JB. Mol Pharmacol. 1997;51:439–47. [PubMed] [Google Scholar]
- 115.Srivastava J, Procyk KJ, Iturrioz X, Parker PJ. Biochem J. 2002;368:349–55. doi: 10.1042/BJ20020737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leontieva OV, Black JD. J Biol Chem. 2004;279:5788–801. doi: 10.1074/jbc.M308375200. [DOI] [PubMed] [Google Scholar]
- 117.Carmena D, Sardini A. Biochem Soc Trans. 2007;35:1043–5. doi: 10.1042/BST0351043. [DOI] [PubMed] [Google Scholar]
- 118.Rechsteiner M, Rogers SW. Trends Biochem Sci. 1996;21:267–71. [PubMed] [Google Scholar]
- 119.Lu Z, Liu D, Hornia A, Devonish W, Pagano M, Foster DA. Mol Cell Biol. 1998;18:839–45. doi: 10.1128/mcb.18.2.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kang BS, French OG, Sando JJ, Hahn CS. Oncogene. 2000;19:4263–72. doi: 10.1038/sj.onc.1203779. [DOI] [PubMed] [Google Scholar]
- 121.Nakamura M, Tokunaga F, Sakata S, Iwai K. Biochem Biophys Res Commun. 2006;351:340–7. doi: 10.1016/j.bbrc.2006.09.163. [DOI] [PubMed] [Google Scholar]
- 122.Okuda H, Saitoh K, Hirai S, Iwai K, Takaki Y, Baba M, Minato N, Ohno S, Shuin T. J Biol Chem. 2001;276:43611–7. doi: 10.1074/jbc.M107880200. [DOI] [PubMed] [Google Scholar]
- 123.Prevostel C, Alice V, Joubert D, Parker PJ. J Cell Sci. 2000;113(Pt 14):2575–84. doi: 10.1242/jcs.113.14.2575. [DOI] [PubMed] [Google Scholar]
- 124.Becker KP, Hannun YA. J Biol Chem. 2004;279:28251–6. doi: 10.1074/jbc.M400770200. [DOI] [PubMed] [Google Scholar]
- 125.von Zastrow M. Life Sci. 2003;74:217–24. doi: 10.1016/j.lfs.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 126.Smart EJ, Ying YS, Anderson RG. J Cell Biol. 1995;131:929–38. doi: 10.1083/jcb.131.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Becker KP, Hannun YA. J Biol Chem. 2003;278:52747–54. doi: 10.1074/jbc.M305228200. [DOI] [PubMed] [Google Scholar]
- 128.Idkowiak-Baldys J, Becker KP, Kitatani K, Hannun YA. J Biol Chem. 2006;281:22321–31. doi: 10.1074/jbc.M512540200. [DOI] [PubMed] [Google Scholar]
- 129.Alvi F, Idkowiak-Baldys J, Baldys A, Raymond JR, Hannun YA. Cell Mol Life Sci. 2007;64:263–70. doi: 10.1007/s00018-006-6363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fields AP, Gustafson WC. Methods Mol Biol. 2003;233:519–37. doi: 10.1385/1-59259-397-6:519. [DOI] [PubMed] [Google Scholar]
- 131.Griner EM, Kazanietz MG. Nat Rev Cancer. 2007;7:281–94. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 132.Martiny-Baron G, Fabbro D. Pharmacol Res. 2007;55:477–86. doi: 10.1016/j.phrs.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 133.Koivunen J, Aaltonen V, Peltonen J. Cancer Lett. 2006;235:1–10. doi: 10.1016/j.canlet.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 134.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, Menzies A, Mironenko T, Perry J, Raine K, Richardson D, Shepherd R, Small A, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Cahill DP, Louis DN, Goldstraw P, Nicholson AG, Brasseur F, Looijenga L, Weber BL, Chiew YE, DeFazio A, Greaves MF, Green AR, Campbell P, Birney E, Easton DF, Chenevix-Trench G, Tan MH, Khoo SK, Teh BT, Yuen ST, Leung SY, Wooster R, Futreal PA, Stratton MR. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Seki T, Adachi N, Ono Y, Mochizuki H, Hiramoto K, Amano T, Matsubayashi H, Matsumoto M, Kawakami H, Saito N, Sakai N. J Biol Chem. 2005;280:29096–106. doi: 10.1074/jbc.M501716200. [DOI] [PubMed] [Google Scholar]
- 136.Mackay HJ, Twelves CJ. Nat Rev Cancer. 2007;7:554–62. doi: 10.1038/nrc2168. [DOI] [PubMed] [Google Scholar]
- 137.Graff JR, McNulty AM, Hanna KR, Konicek BW, Lynch RL, Bailey SN, Banks C, Capen A, Goode R, Lewis JE, Sams L, Huss KL, Campbell RM, Iversen PW, Neubauer BL, Brown TJ, Musib L, Geeganage S, Thornton D. Cancer Res. 2005;65:7462–9. doi: 10.1158/0008-5472.CAN-05-0071. [DOI] [PubMed] [Google Scholar]
- 138.Herbst RS, Oh Y, Wagle A, Lahn M. Clin Cancer Res. 2007;13:s4641–6. doi: 10.1158/1078-0432.CCR-07-0538. [DOI] [PubMed] [Google Scholar]
- 139.Ryan GJ. Am J Health Syst Pharm. 2007;64:S15–21. doi: 10.2146/ajhp070332. [DOI] [PubMed] [Google Scholar]
- 140.Joy SV, Scates AC, Bearelly S, Dar M, Taulien CA, Goebel JA, Cooney MJ. Ann Pharmacother. 2005;39:1693–9. doi: 10.1345/aph.1E572. [DOI] [PubMed] [Google Scholar]
- 141.Mosyak L, Xu Z, Joseph-McCarthy D, Brooijmans N, Somers W, Chaudhary D. Biochem Soc Trans. 2007;35:1027–31. doi: 10.1042/BST0351027. [DOI] [PubMed] [Google Scholar]
- 142.Szallasi Z, Smith CB, Pettit GR, Blumberg PM. J Biol Chem. 1994;269:2118–24. [PubMed] [Google Scholar]
- 143.Brandman R, Disatnik MH, Churchill E, Mochly-Rosen D. J Biol Chem. 2007;282:4113–23. doi: 10.1074/jbc.M608521200. [DOI] [PubMed] [Google Scholar]
- 144.Churchill E, Budas G, Vallentin A, Koyanagi T, Mochly-Rosen D. Annu Rev Pharmacol Toxicol. 2007 doi: 10.1146/annurev.pharmtox.48.121806.154902. [DOI] [PubMed] [Google Scholar]
- 145.Fields AP, Frederick LA, Regala RP. Biochem Soc Trans. 2007;35:996–1000. doi: 10.1042/BST0350996. [DOI] [PubMed] [Google Scholar]
- 146.Lahn M, Su C, Li S, Chedid M, Hanna KR, Graff JR, Sandusky GE, Ma D, Niyikiza C, Sundell KL, John WJ, Giordano TJ, Beer DG, Paterson BM, Su EW, Bumol TF. Clin Lung Cancer. 2004;6:184–9. doi: 10.3816/clc.2004.n.032. [DOI] [PubMed] [Google Scholar]
- 147.Sutton RB, Sprang SR. Structure. 1998;6:1395–405. doi: 10.1016/s0969-2126(98)00139-7. [DOI] [PubMed] [Google Scholar]