

Abstract
Ribavirin, a nucleic acid analog, has been employed as an antiviral agent against RNA and DNA viruses and has become the standard agent used for chronic hepatitis C in combination with interferon-α2a. Furthermore, the potential antitumor efficacy of ribavirin has attracted increasing interest. Recently, we demonstrated a dose-dependent antitumor effect of ribavirin for seven types of malignant glioma cell lines. However, the mechanism underlying the antitumor effect of ribavirin has not yet been fully elucidated. Therefore, the main aim of the present study was to provide further relevant data using two types of malignant glioma cell lines (U-87MG and U-138MG) with different expression of MGMT. Dotted accumulations of γH2AX were found in the nuclei and increased levels of ATM and phosphorylated ATM protein expression were also observed following ribavirin treatment (10 µM of ribavirin, clinical relevant concentration) in both the malignant glioma cells, indicating double-strand breaks as one possible mechanism underlying the antitumor effect of ribavirin. In addition, based on assessements using FACS, ribavirin treatment tended to increase the G0/G1 phase, with a time-lapse, indicating the induction of G0/G1-phase arrest. Furthermore, an increased phosphorylated p53 and p21 protein expression was confirmed in both glioma cells. Additionally, analysis by FACS indicated that apoptosis was induced following ribavirin treatment and caspase cascade, downstream of the p53 pathway, which indicated the activation of both exogenous and endogenous apoptosis in both malignant glioma cell lines. These findings may provide an experimental basis for the clinical treatment of glioblastomas with ribavirin.
Keywords: glioblastoma, ribavirin, MGMT
Introduction
The World Health Organization (WHO) classifies gliomas into four grades of malignancy according to their histopathology and clinical prognosis. Among these neoplasms, glioblastomas, classified as WHO grade IV, with the highest malignancy, account for 10.8% of all brain tumors and are the most common primary brain tumors in adults (1). Although current advancements in multimodality treatments including surgical resection, radiotherapy, and chemotherapy have become more widespread, the poor prognosis of glioblastomas has not improved for more than three decades. In 2005, Stupp et al (2) reported a phase III randomized controlled trial on concomitant and adjuvant temozolomide (TMZ), a second-generation alkylating agent, in addition to standard postoperative radiotherapy, as offering a first-line treatment for primary glioblastomas. They demonstrated that such therapy increased the median survival time of patients from 12.1 to 14.6 months (2). Furthermore, in 2009, they reported that these treatments increased the 5-year survival rate from 1.9 to 9.8% compared to radiotherapy alone (3). Subsequently, surgical resection and postoperative radiotherapy and chemotherapy including TMZ, have become the global standard as a first-line treatment for glioblastomas.
The underlying mechanism that may contribute to the effect of TMZ on tumors is considered to involve the adduction of the methyl base at the O6-position of guanine, forming O6-methylguanine in DNA, which mispairs with thymine instead of cytosine during the next cycle of DNA replication. As a result, futile cell cycles of the DNA-mismatch repair system lead to growth arrest and/or apoptosis induction. In contrast, O6-methylguanine-DNA methyltransferase (MGMT), a suicide DNA-repair enzyme, removes the methyl adduct formed by an alkylating (methylating) agent including TMZ and attenuates the effect of TMZ (4,5). The expression of MGMT, which is estimated to be 45–75% in glioblastomas, is closely correlated with clinical resistance to TMZ treatment (4,6–8).
Ribavirin (1-β-D-ribofuranosyl-1,2,4-triazole-3-carboxamide), which was first reported in 1972 by Sidwell et al (9) as an antiviral agent for the treatment of RNA and DNA viral infections, is a nucleic acid analog. To date, ribavirin has been used to treat respiratory syncytial virus as well as the Lassa virus and has become the standard agent for chronic hepatitis C in combination with interferon-α2a (10). The interest in the antitumor effect of ribavirin has been increasing due to its ability to inhibit inosine-5′-monophosphate dehydrogenase (IMPDH), eukaryotic translation initiation factor 4E (eIF4E) and histone methyltransferase enhancer of zeste homolog 2 (EZH2). Several studies have indicated an antitumor effect of ribavirin in breast cancer and acute myeloid leukemia (11–15). In addition, although there have been few studies on the antitumor effect of ribavirin against glioma, we demonstrated a dose-dependent antitumor effect of ribavirin for seven types of malignant glioma cell lines (16). Recently, Volpin et al (15) also demonstrated the antitumor effect of ribavirin on glioma cell lines and glioma stem-like cells. These findings clearly supported the antitumor effect of ribavirin, however the underlying mechanism has not yet been fully elucidated.
In the present study, we obtained further data, by examining the effects of ribavirin on the induction of apoptosis, the cell cycle, p53-pathway activation and DNA damage by employing the following two types of malignant glioma cell lines: the U-87MG cells with no MGMT expression and the U-138MG cells with MGMT expression. The findings may provide an experimental basis for the clinical therapy with ribavirin for glioblastomas.
Materials and methods
Cell lines and cell culture
To elucidate the mechanisms of ribavirin sensitivity in malignant gliomas, we used two types of malignant glioma cell lines (U-87MG and U-138MG) which have different MGMT mRNA and MGMT protein expression.
The human malignant glioma U-87MG and U-138MG cell lines were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). These cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Nissui Pharmaceutical, Tokyo, Japan) containing 10% fetal calf serum (FCS; Life Technologies; Thermo Fischer Scientific, Grand Island, NY, USA) using plastic culture flasks (Corning, NY, USA) in a standard humidified incubator at 37°C with an atmosphere of CO2.
Growth inhibitory effect
We recently demonstrated the antitumor efficacy of ribavirin for malignant glioma cell lines (16). In this previous study, seven malignant glioma cell lines (A-172, AM-38, T98G, U-87MG, U-138MG, U-251MG and YH-13) were exposed to 0.1–1,000 µM of ribavirin and treated for 72 h and it was observed that ribavirin inhibited the growth of all malignant glioma cell lines in a dose-dependent manner (16). Based on these results on the growth inhibitory effect of ribavirin, the treatment concentration of ribavirin that was chosen for the present experiments was 10 µM, which also represents a clinically relevant concentration of ribavirin (17).
The growth inhibition of malignant glioma cells by ribavirin was evaluated by counting the cell numbers. Briefly, the cells were seeded at 1×104 cells/well in 24-well plates (Iwaki, Chiba, Japan) and cultured with medium for 24 h. Subsequently, the cells were washed twice with medium and further incubated with fresh medium (control) or medium containing 10 µM ribavirin for 96 h. After incubation, the cells were harvested with trypsin-EDTA solution (Invitrogen; Thermo Fisher Scientific, San Diego, CA, USA). The number of collected cells was assessed using a Coulter Counter (Coulter Counter Z1; Beckman Coulter, Fullerton, CA, USA). The experiments were repeated 6 times at each concentration. Student's t-tests were performed to compare pairs of groups. Data analyses were carried out using the statistical software IBM SPSS statistics version 21.0 (IBM Corporation, Armonk, NY, USA).
Cell cycle distribution analysis
Ribavirin-induced alterations of the cell cycle distribution were analyzed by flow cytometry. The cells were seeded in 6-well plates (Iwaki) at 1×105 cells/plate, incubated for 24 h and allowed to attach. Following 10 µM ribavirin treatment in the medium, the cells were harvested using trypsin-EDTA solution at 8 and 48 h and fixed in ice-cold 70% ethanol for 1 h. The fixed cells were treated with 500 µg/ml RNase A (Roche Diagnostics, Mannheim, Germany) for 1 h and stained with 12 µg/ml propidium iodide solution (PI; Miltenyi Biotech, Auburn, CA, USA) for 30 min at 4°C. The fluorescence was assessed with a FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) at a wavelength of 610 nm (FL3). The DNA histograms were analyzed using FlowJo software (BioLegend, San Diego, CA, USA). The experiments were repeated three times to confirm reproducibility.
Activation of apoptosis
Ribavirin-induced apoptosis was analyzed by flow cytometry, using dual staining with an Annexin V-FITC/PI Apoptosis Detection kit (BD Biosciences). The cells were seeded in 6-well plates (Iwaki) at 1×106 cells/well, incubated for 24 h and allowed to attach. The culture medium was then replenished with fresh medium containing 10 µM of ribavirin for 72 h. Subsequently, the cells were washed in phosphate-buffered saline (PBS) and harvested using trypsin-EDTA solution. After centrifugation and washing in PBS, the solution was agitated with 100 µl of binding buffer (Wako Pure Chemical Industries, Ltd., Tokyo, Japan), into which 5 µl of Annexin V Alexa Fluor 488 conjugate (Life Technologies; Thermo Fisher Scientific) and 10 µl of PI (Miltenyi Biotech) were added and incubated at room temperature for 10 min. An additional 400 µl of binding buffer was added in order to reach a total sample volume of 500 µl. The fluorescence was assessed with a FACSCalibur flow cytometer (BD Biosciences). The apoptotic cells were analyzed using FlowJo software (BioLegend). The experiments were repeated three times to confirm reproducibility.
Western blot analysis
Soluble protein lysates of sub-confluent glioma cells were obtained using lysate buffer (Medical and Biological Laboratories, Woburn, MA, USA) for 20 min on ice. The proteins (50 µg proteins) were loaded and separated by 12% polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes (GE Healthcare, Tokyo, Japan) for 30 min at 10 V with a Bio-Rad Trans Blot (Bio-Rad Laboratories, Franklin Lakes, NJ, USA). Non-specific binding was blocked with a washing buffer (PBS/0.05% containing 1% skimmed milk) for 60 min at room temperature. The primary antibody employed for the immunoblotting was β-actin mouse mAb (cat. no. 013-24553; 1:2,000; Wako Pure Chemical Industries) which was used as a loading control. The secondary antibodies employed were anti-mouse IgG (whole molecule) peroxidase conjugate (cat. no. A4416; 1:5,000; Sigma-Aldrich, St. Louis, MO, USA) for 60 min at room temperature. The immune complex was visualized using an ECL detection system (GE Healthcare) and ImageQuant Las4000 (GE Healthcare), and then analyzed using ImageJ (National Institutes of Health, Bethesda, MD, USA). The same experiments were repeated three times to confirm reproducibility.
MGMT
To confirm the protein expression of MGMT in the U-87MG and U-138MG cells, anti-MGMT mouse mAb (cat. no. MT 3.1; 1:500; Thermo Fisher Scientific) was employed as the primary antibody for western blotting.
p53, phosphorylated p53 and p53 related gene products
The protein expression of p53, phosphorylated p53 (p-p53) and important factors of the p53 pathway, p21, Bax, Fas, caspase-8, caspase-9 and caspase-3, were analyzed at 0, 4, 8, 24 and 48 h after being treated with 10 µM ribavirin. As primary antibodies, anti-p53 mouse mAb (cat. no. sc-126; 1:500), anti-p-p53 mouse mAb (cat. no. sc-101762; 1:500), anti-p21 mouse mAb (cat. no. sc-6246; 1:500), anti-Bax mouse mAb (cat. no. sc-20067; 1:500), anti-Fas mouse mAb (cat. no. sc-8009; 1:500), anti-caspase-3 mouse mAb (cat. no. sc-7272; 1:500) (all from Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-caspase-8 mouse mAb (cat. no. 1C12; 1:500) and caspase-9 mouse mAb (cat. no. C9; 1:500) (both from Cell Signaling Technology, Tokyo, Japan) were employed.
Ataxia telangiectasia mutated (ATM) and phosphorylated ATM
ATM detects double-strand breaks (DSBs), a type of DNA damage and activates p53 (18,19). Therefore, we analyzed the changes in ATM and phosphorylated ATM (p-ATM) at 0, 4, 8, 24 and 48 h after treatment with 10 µM ribavirin. As primary antibodies, anti-ATM mouse mAb (cat. no. sc-23921; 1:500) and anti-p-ATM mouse mAb (cat. no. sc-47739; 1:500) (both from Santa Cruz Biotechnology, Inc.) were employed.
Fluorescence microscopy
To ascertain DNA damage, especially DSBs caused by ribavirin, the expression of phosphorylated histone H2AX (γH2AX) was investigated using the fluorescence antibody technique at 4 h after treatment. The cells were seeded in a collagen-coated glass bottom dish (Matsunami Glass Ind., Ltd., Osaka, Japan) at a concentration of 2×105 cells and allowed to proliferate for 24 h. Subsequently, the cells were treated with DMEM containing ribavirin and FBS for 4 h at 37°C. After washing with PBS, the cells were fixed in 95% ethanol and 5% acetic acid for 10 min at room temperature, and then fixed using PBS containing 1% formaldehyde and 0.25% Triton X-100 for 5 min at room temperature. Following fixation, the cells were blocked at room temperature for 30 min using PBS containing 5% FBS and stained for 1 h at room temperature using anti-phospho-Histone H2A.X (Ser139) clone JBW301, FITC conjugate (Merck Millipore, Billerica, MA, USA). After washing with PBS, the cells were observed under a fluorescence microscope (Olympus IV70; Olympus, Tokyo, Japan). The experiments were repeated three times to confirm reproducibility.
Results
MGMT protein expression
One important mechanism of resistance to methylating agents such as TMZ is DNA repair mediated by MGMT. We observed that the absolute value of MGMT mRNA, obtained using real-time quantitative RT-PCR, in the U-138MG cells was 6.3×103 copies/mg RNA. In contrast, such expression was not detected in the U-87MG cell line (20). Furthermore, in the present study, western blot analysis revealed an MGMT expression at the protein level in the U-138MG cells, but an absence of the MGMT expression in the U-87MG cells (Fig. 1). Thus, the U-138MG cell line was MGMT-proficient, whereas the U-87MG cell line was MGMT-deficient.
Figure 1.
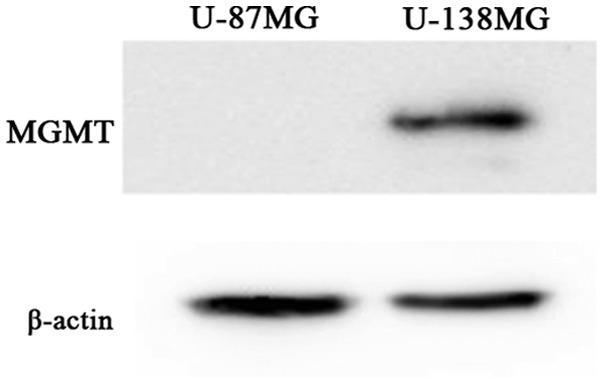
MGMT protein expression of the U-87MG and U-138MG cells as observed by western blot analysis. MGMT protein was not expressed in the U-87MG cells, however it was expressed in the U-138 MG cells.
Anticancer effect and cell sensitivity to ribavirin in malignant glioma cell lines
Growth inhibitory effect
To assess the antitumor effect of ribavirin in malignant glioma cells, we treated the U-87MG and U-138MG malignant glioma cell lines, with 10 µM of ribavirin for 96 h and determined the number of viable cells. As depicted in Fig. 2, a cell growth inhibitory effect of 10 µM ribavirin was observed in both the U-87MG and U-138MG cell lines. Although the U-138MG cells exhibited a significant suppression of cell proliferation, the inhibitory effect of ribavirin was less pronounced in comparison to that in the U-87MG cells. These findings were consistent with those that we have previously reported (16).
Figure 2.
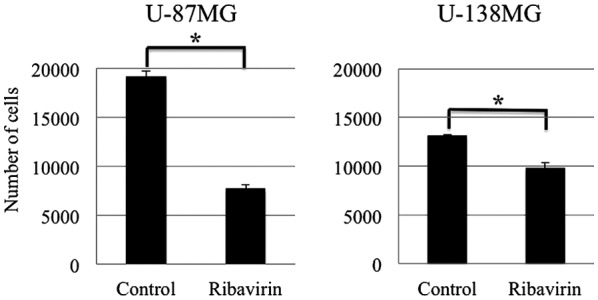
Antitumor effect of ribavirin in malignant glioma cell lines. Ribavirin exerted a cell growth inhibitory effect in both the U-87MG and U-138MG cells. Although the U-138MG cells exhibited a significant suppression of cell proliferation, the effect of ribavirin on cell proliferation was less pronounced in comparison to that in the U-87MG cells. The results are expressed as the mean ± SE. *P<0.05 (Student's t-test).
Cell cycle analysis
We performed DNA flow cytometric analysis to investigate whether alterations in the cell cycle distribution were induced in malignant glioma cells following 10 µM of ribavirin treatment for 8 or 48 h. The proportion of cells in each cell cycle phase are presented in Fig. 3. We observed that this amount of ribavirin increased the G0/G1 phase at 8 and 48 h following treatment, with a time-lapse, in both the U-87MG and U-138MG cells, indicating a G0/G1-phase arrest. These findings were consistent with those that we have previously reported and not contradictory with those previously reported by Volpin et al (15) as well as by Ogino et al (16).
Figure 3.
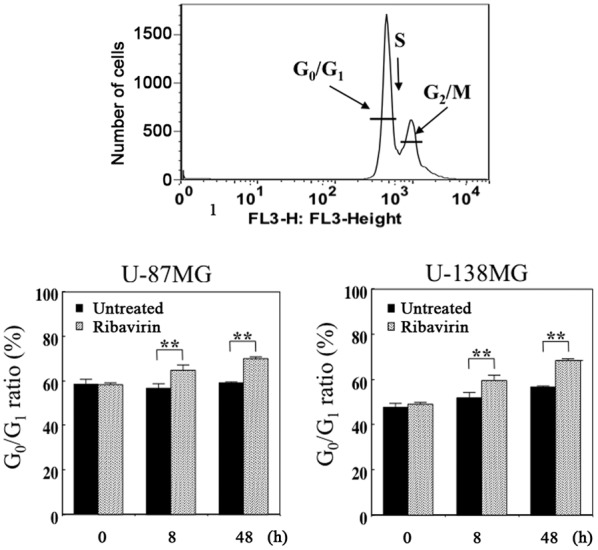
Cell cycle distribution analysis of the U-87MG and U-138MG cells which underwent treatment with ribavirin (untreated cells serving as the control) for 8 or 48 h. The histogram depicting the FACS data demonstrates an increase in the population of cells in the G0/G1 phase following ribavirin treatment, with a time-lapse, indicating that the antitumor efficacy of ribavirin resulted from the accumulation of cells in the G0/G1 phase. The results are expressed as the mean ± SE. **P<0.01 (Student's t-test).
We observed the protein expression involved in the cell cycle mediated by p53 using western blot analysis at 0, 4, 8, 24 and 48 h following 10 µM of ribavirin treatment in malignant glioma cells. The p-p53 and p21 protein expression was increased after 4 h of ribavirin treatment in the U-87MG and U-138MG malignant glioma cells (Figs. 4 and 5).
Figure 4.
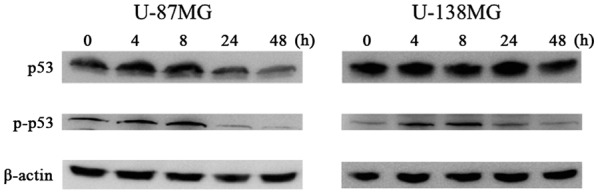
p53 and phosphorylated p53 protein expression following ribavirin treatment. The p-p53 protein expression was increased following 4 h of ribavirin treatment in U-87MG and U-138MG malignant glioma cells.
Figure 5.
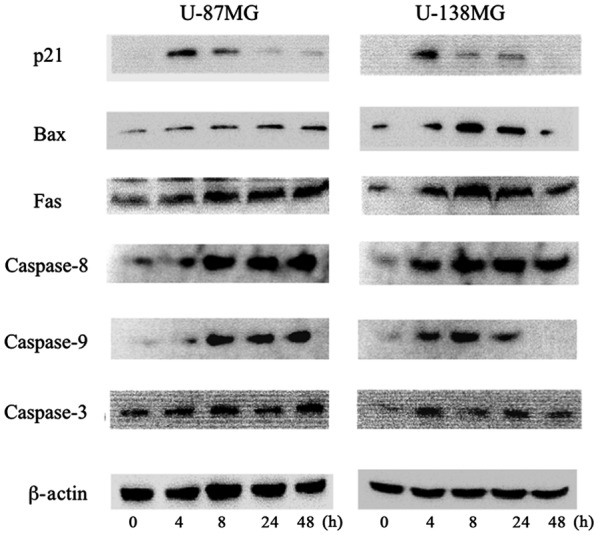
Apoptotic pathway induction by ribavirin. Western blot analysis demonstrated that the protein expression of p21, Bax, Fas, caspase-8, caspase-9 and caspase-3 was increased following 4 h of ribavirin treatment in both U-87MG and U-138MG malignant glioma cells.
Activation of apoptosis
The induction of apoptosis by ribavirin in malignant glioma cells was investigated by Annexin V/PI double staining and assessed using flow cytometry. After 72 h of 10 µM ribavirin treatment, the proportion of living and apoptotic cells was compared with the control in both the U-87MG and U-138MG cell lines. The distribution of apoptotic cells (Annexin V-positive: early-stage apoptosis; Annexin V/PI-positive: late-stage apoptosis) are displayed in Fig. 6. The results revealed that the apoptotic cells were increased in both cell lines. These findings were not contradictory with the results previously reported by Volpin et al (15).
Figure 6.
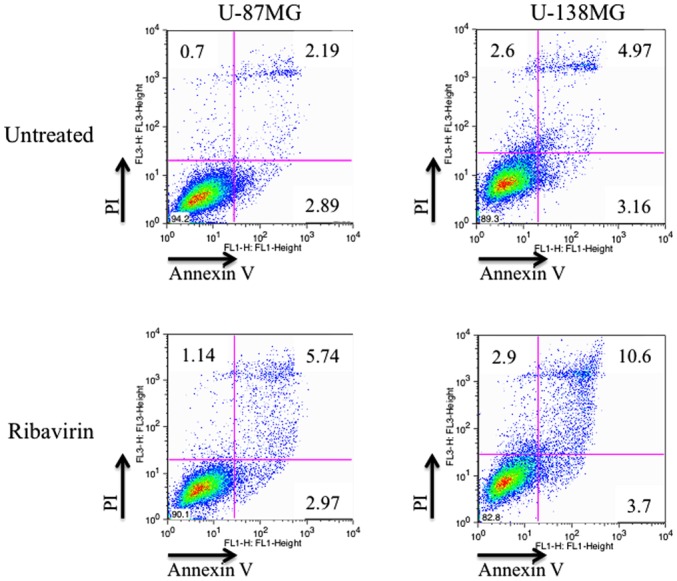
Induction of apoptosis by ribavirin. In the U-87MG and U-138MG cells, the ratio of detection of Annexin V-positive and Annexin V/PI-positive cells, indicating early-stage apoptosis and late-phase apoptosis, respectively, as determined by FACS, increased after 72 h of ribavirin treatment.
The underlying mechanisms of the apoptotic effect of ribavirin were examined by western blot analysis. The intrinsic mitochondrial pathway associated with apoptosis, involving Bax, caspase-9 and caspase-3, was analyzed. In addition, the extrinsic apoptotic pathway mediated by Fas, caspase-8 and caspase-3 was investigated in the malignant glioma cells. In both the U-87MG and U-138MG cell lines, after 4 h of 10 µM ribavirin treatment, the protein expression of Bax, Fas, caspase-8, caspase-9 and caspase-3 was increased (Fig. 5). Thus, ribavirin induced apoptosis in the glioma cells through both the intrinsic and extrinsic apoptotic pathways.
DNA damage
To ascertain the DNA damage caused by ribavirin in the malignant glioma cells, investigations of γH2AX using the fluorescence antibody technique and ATM and p-ATM protein expression by western blot analysis were performed. The accumulation of γH2AX in the cell nuclei was confirmed by fluorescence microscopy at 4 h following 10 µM of ribavirin treatment in both the U-87MG and U-138MG cell lines (Fig. 7). Furthermore, in each of these cell lines, the expression of p-ATM was increased after ribavirin treatment (Fig. 8).
Figure 7.
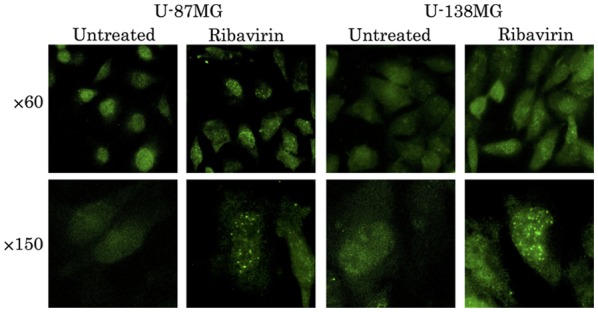
Observation of DSBs by γH2AX fluorescence microscopy. The accumulation of γH2AX in the cell nuclei was confirmed by fluorescence microscopy at 4 h following ribavirin treatment in the U-87MG and U-139MG cell lines.
Figure 8.
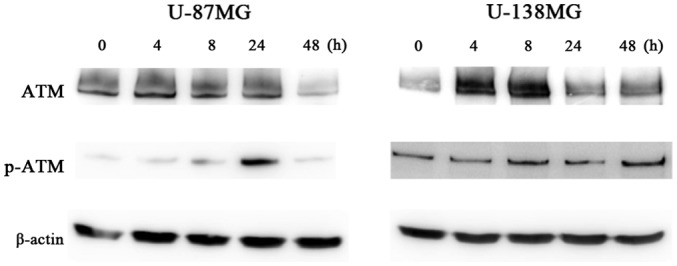
DSBs detected from ATM and p-ATM protein expression by western blot analysis. The expression of p-ATM was increased in both the U-87MG and U-138MG cell lines after the ribavirin treatment.
Discussion
The interest in the antitumor effect of ribavirin for tumor treatment has been increasing due to its ability to inhibit IMPDH, eIF4E and EZH2. It has been observed that ribavirin exhibits an antitumor effect in breast cancer and chronic myeloid leukemia (11–15). Recently, we demonstrated a dose-dependent antitumor effect of ribavirin on seven types of malignant glioma cell lines (16). In clinical practice for brain tumors, one of the most important problems is whether ribavirin crosses the blood-brain barrier. It has been observed that when administered at a dose of 800 mg/day as a therapeutic agent for chronic hepatitis C, the blood concentration of ribavirin was 13 µM and the cerebrospinal transitivity of ribavirin, with a low molecular weight of 244.2, was 70% (17). In addition, in the present study, a satisfactory cell proliferation inhibitory effect on both the U-87MG and U-138MG cells was observed when the concentration of ribavirin was 10 µM, indicating that ribavirin could represent a new therapeutic agent for glioblastomas. Recently, Volpin et al (15) demonstrated that 30 µM of ribavirin inhibited the proliferation and migration and increased the cell arrest and cell death of glioma cells, potentially through the modulation of elF4E, EZH2 and extracellular regulated protein kinase (ERK) pathways. However, the mechanism underlying the antitumor effect of ribavirin on malignant glioma cells has not yet been fully elucidated. Therefore in the present study, we further investigated the processes involved in this effect.
When DSBs occur for various reasons, H2AX is phosphorylated (then referred to as γH2AX) and accumulates at the site of the DNA damage (21). The DSB is recognized by ATM, and subsequently induces autophosphorylation of ATM and then p-ATM activates p53 (22). On the other hand, recent evidence revealed that γH2AX does not always indicate the presence of DSB (23). Tu et al (23) revealed that an increased level of γH2AX occurred in the cell cycle-dependent phosphorylation of H2AX when the G2/M arrest was induced by ionizing radiation and demonstrated that DNA-dependent protein kinase catalytic subunit and cell cycle checkpoint protein 2, but not ATM, were two important kinases involved in this process. In the present study, dotted accumulations of γH2AX in the nuclei were observed at 4 h after ribavirin treatment in the U-87MG and the U-138MG cells. The cell cycle distribution analysis revealed an increase in the population of cells in the G0/G1 phase after ribavirin treatment. Furthermore, the p-ATM, p-p53 and p21 protein expression, as investigated by western blot analysis, was increased after the ribavirin treatment. It is known that p21, known as cyclin-dependent kinase inhibitor 1, is activated by p53 and induces cell cycle arrest in the G0/G1 phase (24). Therefore, these findings indicated that ribavirin treatment may increase the G0/G1 arrest, but not the G2/M arrest and DSBs could represent one of the mechanisms underlying the antitumor effect of ribavirin on malignant glioma cell lines.
There are two major DSB repair pathways in human cells (25): one is homologous recombination (HR) and the other is non-homologous end joining (NHEJ). Repair by NHEJ is possible throughout the cell cycle, whereas HR occurs only in the S-phase to the G2-phase when sister chromatids are present. This indicates that ribavirin-induced DSB may activate the NHEJ repair pathway, with low restoration accuracy, rather than the HR pathway.
We evaluated the apoptosis rate by flow cytometric analysis and the key regulators of apoptosis, the caspase cascade, downstream of the p53 pathway, by western blot analysis. Flow cytometry revealed an increased proportion of Annexin V-positive cells and Annexin V/PI-positive cells, which indicated early-stage apoptosis and late-phase apoptosis respectively, after 72 h of ribavirin treatment in both the U-87MG and U-138MG cells. The induction of apoptosis initiated the signaling pathway called the caspase cascade. Caspases can be broadly divided into initiator caspases involved in the relatively early stage of apoptosis and effector caspases involved in the actual execution of apoptosis. Apoptosis is broadly divided into exogenous apoptosis occurring through the cell membrane receptors (via death receptors; the extrinsic pathway) and endogenous apoptosis via the mitochondrial intrinsic pathway (26). The present study revealed that ribavirin activated caspase-3 (an effector caspase) and increased the expression of Fas (a death receptor) and caspase-8, which confirmed induction of exogenous apoptosis. In addition, increases in Bax and caspase-9, inducing endogenous apoptosis, were also observed in both the U-87MG and U-138MG cells following ribavirin treatment. Thus, ribavirin induces apoptosis in malignant glioma cells by activating both exogenous and endogenous apoptosis.
Finally, previous research associated with the present study will be briefly discussed. Surgical resection and concomitant radiotherapy with TMZ followed by adjuvant TMZ chemotherapy have become the current standard treatment for glioblastomas. However, the prognosis is still poor and a more effective TMZ treatment regimen needs to be established. Among the factors that may contribute to TMZ resistance, MGMT is thought to be involved in its principal mechanisms (3,27,28). In addition, it has been indicated that MGMT methylation status has not only a predictive but also a prognostic value in glioblastomas (4). In the present study, a cell growth inhibitory effect of ribavirin was observed in both cell lines. Specifically, the U-138MG cell line was MGMT-proficient, whereas the U-87MG cell line was MGMT-deficient. The antitumor effect of ribavirin may therefore not be dependent on the expression of MGMT. On the other hand, Volpin et al (15), for the first time in 2017, reported the efficacy of ribavirin in combination with radio/chemotherapy as an anti-glioma agent. They demonstrated that ribavirin (30 µM) in combination with TMZ (100 µM) and irradiation (5 Gy) potentially enhanced the efficacy of the antitumor response in glioma cells and glioma stem-like cells and that the median survival of animals (rats; intracranial implantation of 9L gliosarcoma) treated with a combination of ribavirin (daily i.p. injection of 10 mg/kg) and irradiation (one session, 10 Gy) and TMZ (50 mg/kg for 5 days) was significantly increased compared with animals treated with irradiation and TMZ (15). However, further studies are warranted to assess whether ribavirin is effective against MGMT. In addition, it is important to conduct more studies to evaluate whether ribavirin exhibits a synergistic effect with irradiation and TMZ.
In conclusion, the present study indicated that ribavirin exerted an antitumor effect on malignant glioma cells due to the induction of DSBs and the cell cycle arrest in the G0/G1 phase, both in exogenous and endogenous apoptosis. In addition, such effects may not be dependent on the expression of MGMT.
Acknowledgements
The present study was supported in part by the Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (grant no. 16K10772) and in part by a grant from the Health Sciences Research Institute, Inc. (Yokohama, Japan) for the Division of Companion Diagnostics, Department of Pathology and Microbiology, Nihon University School of Medicine. The authors are grateful to Hiroyuki Satake and Nobuo Miyazaki, Toray Industries Inc. (Tokyo, Japan) for their invaluable discussions. Some parts of this study have been submitted within a Japanese-language thesis for Yushi Ochiai's Ph.D. degree at Nihon University School of Medicine.
References
- 1.Committee of Brain Tumor Registry of Japan, corp-author. Report of Brain Tumor Registry of Japan (2001–2004). Vol. 13. Neurol Med Chir (Tokyo) 2014;54:1–102. [Google Scholar]
- 2.Stupp R, Mason WP, Van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Hegi ME, Mason WP, Van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups. National Cancer Institute of Canada Clinical Trials Group Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 4.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 5.Fukushima T, Takeshima H, Kataoka H. Anti-glioma therapy with temozolomide and status of the DNA-repair gene MGMT. Anticancer Res. 2009;29:4845–4854. [PubMed] [Google Scholar]
- 6.Bello MJ, Alonso ME, Amiñoso C, Anselmo NP, Arjona D, Gonzalez-Gomez P, Lopez-Marin I, de Campos JM, Gutierrez M, Isla A, et al. Hypermethylation of the DNA repair gene MGMT: association with TP53 G:C to A:T transitions in a series of 469 nervous system tumors. Mutat Res. 2004;554:23–32. doi: 10.1016/j.mrfmmm.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Kamiryo T, Tada K, Shiraishi S, Shinojima N, Kochi M, Ushio Y. Correlation between promoter hypermethylation of the O6-methylguanine-deoxyribonucleic acid methyltransferase gene and prognosis in patients with high-grade astrocytic tumors treated with surgery, radiotherapy, and 1-(4-amino-2-methyl-5-pyrimidinyl)methyl-3-(2-chloroethyl)-3-nitrosourea-based chemotherapy. Neurosurgery. 2004;54:349–357, discussion 357. doi: 10.1227/01.NEU.0000103422.51382.99. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura M, Watanabe T, Yonekawa Y, Kleihues P, Ohgaki H. Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C -> A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis. 2001;22:1715–1719. doi: 10.1093/carcin/22.10.1715. [DOI] [PubMed] [Google Scholar]
- 9.Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT, Robins RK. Broad-spectrum antiviral activity of Virazole: 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science. 1972;177:705–706. doi: 10.1126/science.177.4050.705. [DOI] [PubMed] [Google Scholar]
- 10.Kohli A, Shaffer A, Sherman A, Kottilil S. Treatment of hepatitis C: A systematic review. JAMA. 2014;312:631–640. doi: 10.1001/jama.2014.14905. [DOI] [PubMed] [Google Scholar]
- 11.Kentsis A, Topisirovic I, Culjkovic B, Shao L, Borden KL. Ribavirin suppresses eIF4E-mediated oncogenic transformation by physical mimicry of the 7-methyl guanosine mRNA cap; Proc Natl Acad Sci USA; 2004; pp. 18105–18110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borden KL, Culjkovic-Kraljacic B. Ribavirin as an anti-cancer therapy: Acute myeloid leukemia and beyond? Leuk Lymphoma. 2010;51:1805–1815. doi: 10.3109/10428194.2010.496506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Assouline S, Culjkovic B, Cocolakis E, Rousseau C, Beslu N, Amri A, Caplan S, Leber B, Roy DC, Miller WH, Jr, et al. Molecular targeting of the oncogene eIF4E in acute myeloid leukemia (AML): A proof-of-principle clinical trial with ribavirin. Blood. 2009;114:257–260. doi: 10.1182/blood-2009-02-205153. [DOI] [PubMed] [Google Scholar]
- 14.De la Cruz-Hernandez E, Medina-Franco JL, Trujillo J, Chavez-Blanco A, Dominguez-Gomez G, Perez-Cardenas E, Gonzalez-Fierro A, Taja-Chayeb L, Dueñas-Gonzalez A. Ribavirin as a tri-targeted antitumor repositioned drug. Oncol Rep. 2015;33:2384–2392. doi: 10.3892/or.2015.3816. [DOI] [PubMed] [Google Scholar]
- 15.Volpin F, Casaos J, Sesen J, Mangraviti A, Choi J, Gorelick N, Frikeche J, Lott T, Felder R, Scotland SJ, et al. Use of an anti-viral drug, Ribavirin, as an anti-glioblastoma therapeutic. Oncogene. 2017;36:3037–3047. doi: 10.1038/onc.2016.457. [DOI] [PubMed] [Google Scholar]
- 16.Ogino A, Sano E, Ochiai Y, Yamamuro S, Tashiro S, Yachi K, Ohta T, Fukushima T, Okamoto Y, Tsumoto K, et al. Efficacy of ribavirin against malignant glioma cell lines. Oncol Lett. 2014;8:2469–2474. doi: 10.3892/ol.2014.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naik GS, Tyagi MG. A pharmacological profile of ribavirin and monitoring of its plasma concentration in chronic hepatitis C infection. J Clin Exp Hepatol. 2012;2:42–54. doi: 10.1016/S0973-6883(12)60090-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: Variations on a theme? Curr Opin Cell Biol. 2001;13:225–231. doi: 10.1016/S0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 19.Kurz EU, Lees-Miller SP. DNA damage-induced activation of ATM and ATM-dependent signaling pathways. DNA Repair (Amst) 2004;3:889–900. doi: 10.1016/j.dnarep.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 20.Yoshino A, Ogino A, Yachi K, Ohta T, Fukushima T, Watanabe T, Katayama Y, Okamoto Y, Naruse N, Sano E. Effect of IFN-beta on human glioma cell lines with temozolomide resistance. Int J Oncol. 2009;35:139–148. doi: 10.3892/ijo_00000322. [DOI] [PubMed] [Google Scholar]
- 21.Löbrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. gammaH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle. 2010;9:662–669. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 22.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 23.Tu WZ, Li B, Huang B, Wang Y, Liu XD, Guan H, Zhang SM, Tang Y, Rang WQ, Zhou PK. γH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013;587:3437–3443. doi: 10.1016/j.febslet.2013.08.028. [DOI] [PubMed] [Google Scholar]
- 24.Harris SL, Levine AJ. The p53 pathway: Positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 25.Goodarzi AA, Jeggo PA. The repair and signaling responses to DNA double-strand breaks. Adv Genet. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 26.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: Regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50:6119–6129. [PubMed] [Google Scholar]
- 28.Yoshino A, Tashiro S, Ogino A, Yachi K, Ohta T, Fukushima T, Watanabe T, Katayama Y, Okamoto Y, Sano E, et al. Gene expression profiles predicting the response to IFN-β and a combination of temozolomide and IFN-β in malignant gliomas. Int J Oncol. 2011;39:529–542. doi: 10.3892/ijo.2011.1068. [DOI] [PubMed] [Google Scholar]