

Abstract
Peroxynitrite (−OON═O, PN) is a reactive nitrogen species (RNS) which can effect deleterious nitrative or oxidative (bio)chemistry. It may derive from reaction of superoxide anion (O2•−) with nitric oxide (·NO) and has been suggested to form an as-yet unobserved bound heme-iron-PN intermediate in the catalytic cycle of nitric oxide dioxygenase (NOD) enzymes, which facilitate a ·NO homeostatic process, i.e., its oxidation to the nitrate anion. Here, a discrete six-coordinate low-spin porphyrinate-FeIII complex [(PIm)-FeIII(−OON═O)] (3) (PIm; a porphyrin moiety with a covalently tethered imidazole axial “base” donor ligand) has been identified and characterized by various spectroscopies (UV–vis, NMR, EPR, XAS, resonance Raman) and DFT calculations, following its formation at −80 °C by addition of ·NO(g) to the heme-superoxo species, [(PIm)FeIII(O2•−)] (2). DFT calculations confirm that 3 is a six-coordinate low-spin species with the PN ligand coordinated to iron via its terminal peroxidic anionic O atom with the overall geometry being in a cis-configuration. Complex 3 thermally transforms to its isomeric low-spin nitrato form [(PIm)FeIII(NO3−)] (4a). While previous (bio)chemical studies show that phenolic substrates undergo nitration in the presence of PN or PN-metal complexes, in the present system, addition of 2,4-di-tert-butylphenol (2,4DTBP) to complex 3 does not lead to nitrated phenol; the nitrate complex 4a still forms. DFT calculations reveal that the phenolic H atom approaches the terminal PN O atom (farthest from the metal center and ring core), effecting O–O cleavage, giving nitrogen dioxide (·NO2) plus a ferryl compound [(PIm)FeIV═O] (7); this rebounds to give [(PIm)FeIII(NO3−)] (4a).The generation and characterization of the long sought after ferriheme peroxynitrite complex has been accomplished.
Graphical Abstract
INTRODUCTION
Nitric oxide (·NO; nitrogen monoxide) is an important regulatory molecule in mammalian biology functioning as an intracellular messenger which mediates multiple physiological processes, including regulation of blood pressure, neurotransmission, immune response, and platelet aggregation. It is generated enzymatically from L-arginine, NADPH, and O2 by nitric oxide synthases (NOS).1 Cellular overproduction of NO can lead to toxicological processes via the formation of reactive nitrogen species (RNS), including ·NO2 and peroxynitrite (ONOO−, PN) [oxoperoxonitrate(−1)] that are typically more reactive and toxic than NO. PN can be generated by the diffusion controlled (k = 3.9–19 × 109 M−1 s1−) reaction of superoxide (O2•−) and nitric oxide2 or by metal ion mediated chemistry (vide infra). The anionic form of peroxynitrite (O═NOO−) exists in equilibrium with its conjugate acid peroxynitrous acid form (O═NOOH), pKa = 6.8. PN and peroxynitrous acid decay rapidly by homolysis leading to the formation of ·NO2 and hydroxyl radicals (·OH) in ~30% yield or by isomerization to nitrate (NO3−).3
Nitrogen dioxide is a more moderate oxidant as well as a nitrating agent (R–H + 2 ·NO2 → R–NO2 + HNO2).4 Thus, PN and/or its “decay” radical products (e.g., ·NO2, ·OH) are capable of performing oxidation and/or nitration reactions, including reactions with biomolecules such as tyrosine, thiols, unsaturated fatty-acid-containing lipids, and DNA.4d,5 These reactions can contribute to a number of toxicological processes that include DNA damage, interference with critical cell–signaling functions, protein nitration leading to cell death, etc. Peroxynitrite is also implicated in a number of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Parkinson’s disease, and Alzheimer’s disease.5a Nitration of tyrosine residues in proteins can have significant effects that interfere with enzyme active site activity, protein folding, and consequently, function;6 evidence for the formation of 3–nitrotyrosine at stoichiometric levels has been associated with substantial loss of enzyme activity in the aging heart.7 Other metalloproteins such as manganese superoxide dismutase (MnSOD), and copper–zinc superoxide dismutase (CuZn-SOD) are known targets of tyrosine nitration.6c,d,8 It was shown that nitration/inactivation of MnSOD can lead to an O2•− enriched environment, and thus enhanced peroxynitrite formation, leading to further damage.6b,9,10
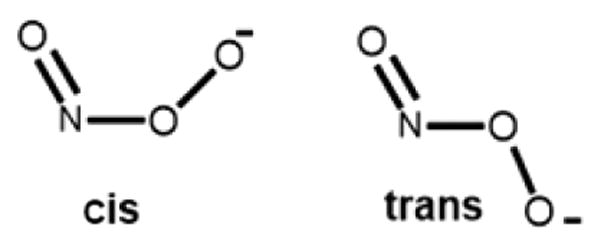
Because of the importance of PN (bio)chemistry, (H)OON═O has received the attention of many theoretical studies to understand its formation, structure, and reactivity. The peroxynitrite anion (O═NOO−) may exist in two geometric forms, the cis and the trans isomers (Chart 1).11 Tsai and co-workers12 employed a variety of computational methods and found that cis-(O═NOO−) is more stable than trans-(O═NOO−) by 3–4 kcal/mol. A theoretical study by Silaghi-Dumitrescu and co-workers13 on the nonheme models of active sites of superoxide reductase (SOR) and iron superoxide dismutase (FeSOD) investigated the formation and decomposition of possible peroxynitrite isomers in their ferrous and ferric oxidation states. Based on their results from DFT calculations, the iron-bound cis form of PN is more stable than the trans form. These researchers also found that all ferrous models of cis and trans Fe-(−OON═O) lead to the immediate heterolytic cleavage of the O–ONO bond to give nitrite (NO2−) and FeIV═O ferryl products, while on the other hand, the cis and trans ferric adducts homolytically cleaved to give nitrogen dioxide plus the ferryl product. Silaghi-Dumitrescu13b and has also carried out DFT calculations on peroxynitrite adducts of ferrous and ferric histidine-ligated hemes, and explored linkage isomerism and the effect of protonation in these adducts, providing insight into possible mechanisms employed by hemoproteins for scavenging peroxynitrite in vivo.
Chart 1.
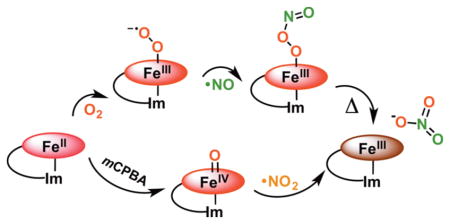
To avoid toxic ·NO(g) levels, microbial heme proteins known as nitric oxide dioxygenases (NODs) catalyze the dioxygenation of ·NO(g) to produce the biologically benign nitrate anion (NO3−).14 Oxygenated heme proteins are major targets/sinks of NO in mammalian biological systems, specifically hemoglobin (Hb) and myoglobin (Mb) which also have known NOD activity (Scheme 1).14a–c It is generally thought that this occurs via the formation of a coordinated ferric peroxynitrite intermediate FeIII(−OON═O). However, ambiguity remains regarding the lifetime of such intermediates and whether isomerization to nitrate is initiated by homolytic, heterolytic, or concerted O–O cleavage.
Scheme 1.
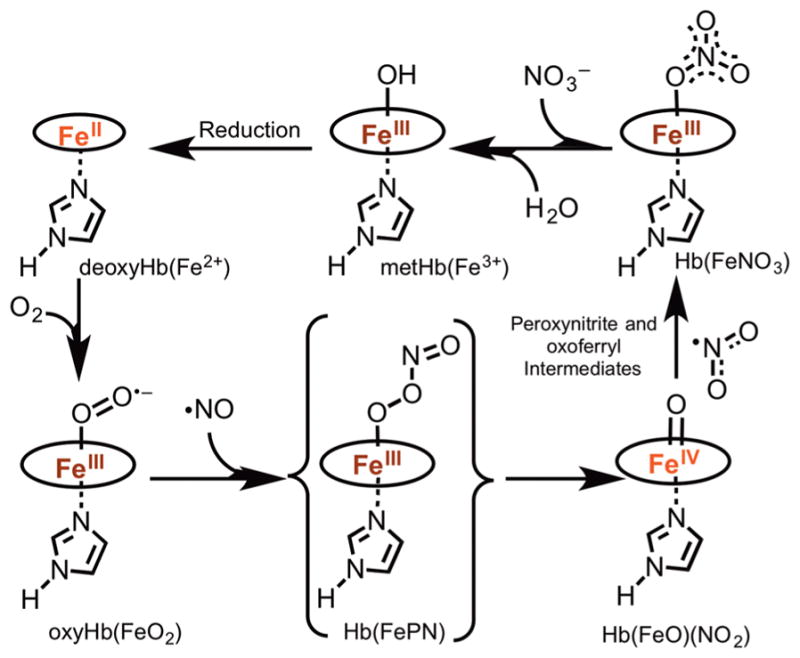
Proposed Catalytic Cycle of Hemoglobin (Hb) Acting as a Nitric Oxide Dioxygenase (NOD)
It is believed that the first stage of the reaction (oxy-heme + ·NO(g)) leads to the formation of a coordinated heme-peroxynitrite species, but at this time there is no strong experimental evidence supporting this assignment. Kinetics studies by Herold and co-workers using rapid scan UV–vis spectroscopy, in a reaction of NO(g) with oxy-myoglobin (MbO2) and oxy-hemoglobin (HbO2), resulted in the formation of transient species with millisecond lifetimes.15 Subsequently, Olson and co-workers14a used EPR to probe rapid freeze-quenched (RFQ) samples from the NO(g) reaction with HbO2, reported formation of a high-spin Fe(III) species on this same time scale. These intermediates were assigned as high-spin ferric heme peroxynitrite complexes on the basis of their electronic spectra. However, Goldstein and co-workers16 reported an intermediate with analogous spectral properties from the reaction of ferryl myoglobin with NO2(g) and concluded that it was the nitrato analog FeIII(ONO2). Similarly, using resonance Raman spectroscopy, Moënne-Loccoz and co-workers identified an intermediate formed in the reaction of NO(g) with MbO2 at 3 °C in alkaline solution and trapped by RFQ on the millisecond time-scale as the nitrato, not the peroxynitrite, complex.17 Ford and Kurtikyan also carried out the experiment to probe the feasibility of FeIII(−OON═O) intermediate using the six-coordinate oxyheme model complex (NH3)Fe(Por)(O2) and reacting it with NO(g) at low temperature via a sublimation and IR spectroscopy methodology. Surprisingly, even at 80–100 K, the six-coordinate nitrato complex was formed without spectral detection of any intermediate.18 However, using more kinetically stabilized d6 cobalt(III) complexes, Kurtikyan and co-workers showed that the oxy-coboglobin model compound (NH3)Co(Por)(O2)/(Py)Co(Por)(O2) also promotes the NOD reaction, and that they could unambiguously stabilize and characterize (by FT-IR spectroscopy and DFT calculations) the bound cobalt–peroxynitrite complex, also demonstrating that the reaction sequence paralleled the proposed scheme for hemeproteins (Scheme 1).19 However, so far clear experimental evidence confirming the formation of coordinated peroxynitrite at the initial stage of the NOD reaction has not been accomplished for heme-containing globins or synthetic iron-porphyrins.
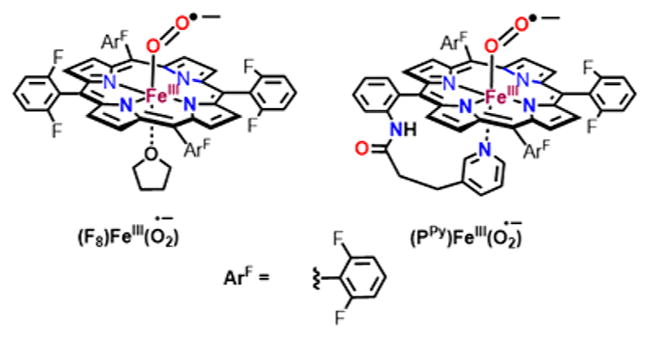
Previously, our laboratory carried out a spectroscopic study to probe the viability of a FeIII(−OON═O) intermediate in the reaction of oxy-heme model complexes of (F8)FeIII-(O2•−)20 and (PPy)FeIII-(O2•−)21 (Chart 2) with ·NO(g) at low temperature, both of which resulted in the formation of five coordinated nitrato complex without spectral detection of any intermediates. However, as the addition of a phenol resulted in its o-nitration, we inferred that a short-lived peroxynitrite or peroxynitrite-like species must have formed, but in the absence of the phenolic substrate isomerization to the nitrato complex was quite fast.
Chart 2.
In this work, we further probed for this most puzzling species expected to form upon heme-superoxo reaction with NO(g), i.e., a FeIII-(−OON═O) heme species, now employing the very robust five-coordinate reduced heme complex (PIm)FeII (1) (PIm = porphyrin with chelated axial imidazole base) as the starting material, which forms a stable six coordinate superoxo complex [(PIm)FeIII(O2•−)] (2) at low temperature (Scheme 2).22 In fact, reaction of 2 with NO(g) under cryogenic conditions resulted in the formation of a new species which was stable under these conditions, which we assign as the long sought after peroxynitrite ferriheme complex, [(PIm)-FeIII(−OON═O)] (3). This species was stable enough to perform spectroscopic analysis before decaying to the low-spin, six-coordinate nitrato compound [(PIm)FeIII(NO3)] (4a), as shown in Scheme 2 and further to the high-spin complex [(PIm)FeIII](NO3−) (4b) (see the Supporting Information (SI), Scheme S1). In this report, further details for these chemical transformations are provided by spectroscopic characterizations, and density functional theory (DFT) calculations offer insights into the structure of the peroxynitrite intermediate and the observed reactivity.
Scheme 2.
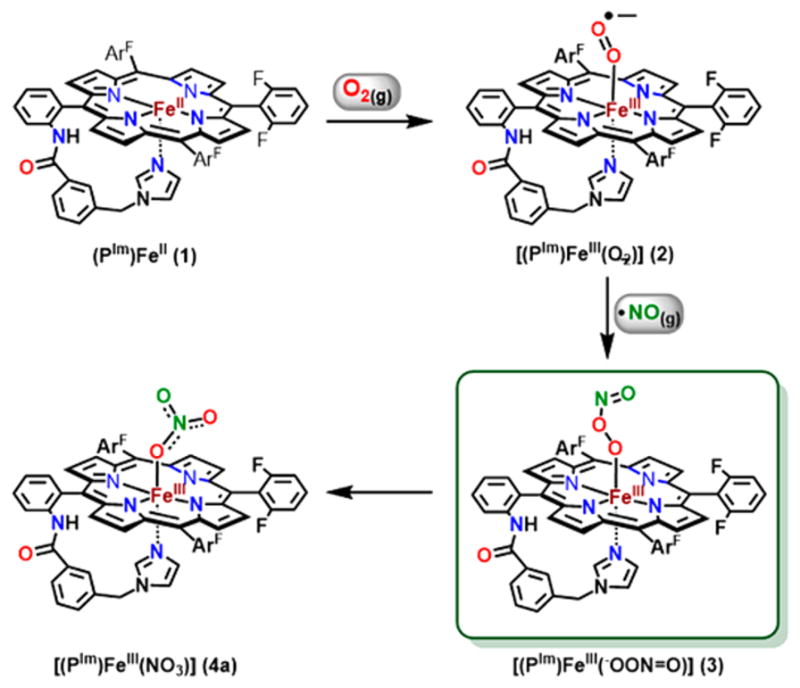
Reaction (Solvent: THF at −80 °C) Sequence Showing the Formation of Complex 2 by Bubbling O2(g) to a Solution of Complex 1 Followed by the Addition of ·NO(g) to Generate Complex 3 Which Thermally Decays to Give Complex 4a
EXPERIMENTAL SECTION
Materials and Methods
All reagents and solvents purchased and used were of commercially available analytical quality except as noted. Dioxygen was dried by passing through a short column of supported P4O10 (Aquasorb, Mallinckrodt). Nitrogen dioxide (·NO2) was obtained from Sigma-Aldrich (≥99.5%, 500 mL lecture bottle, ~14.33 psi @ 20 °C). Nitrogen monoxide (·NO) gas was obtained from Matheson Gases (High Purity grade, Full cylinder ~500 psi @ 20 °C) and purified as follows: it was first passed through multiple columns containing Ascarite II (Thomas Scientific) to remove higher nitrogen oxide impurities. Further purification by distillation was completed by warming frozen ·NO(g) (as crystalline N2O2) from 78 K in a liquid N2 cooled vacuum trap to 193 K through use of an acetone/ dry-ice (−80 °C) bath, and collection in a second liquid N2 cooled evacuated vacuum trap. This secondary flask was again warmed to −80 °C and the purified NO(g) was collected in an evacuated Schlenk flask (typically 50 mL) closed with a rubber septum secured tightly with copper wire. Using these procedures, as previously described in even more detail,20 ·NO(g) in the 50 mL Schlenk flask is sitting at pressures a bit above 1 atm. The ·NO2(g) was filled directly from the lecture bottle in a 10 mL Schlenk flask. Addition of ·NO(g), ·NO2(g), and O2(g) to metal complex solutions was done by transfer via a three-way long needle syringe connected to a Schlenk line. Preparation and handling of air-sensitive compounds were performed under an argon atmosphere using standard Schlenk techniques or in an MBraun Labmaster 130 inert atmosphere (<1 ppm of O2, <1 ppm of H2O) drybox filled with nitrogen gas. Solvents were purged with Ar prior to use. THF and pentane were distilled over Na/benzophenone ketyl or calcium hydride. 2,4-Di-tert-butylphenol (2,4DTBP) was purchased from Sigma-Aldrich and purified by multiple recrystallizations in toluene under Ar. meta-Chloroperbenzoic acid (mCPBA) was purchased from Sigma-Aldrich and purified by washing an ethereal solution with buffer (410 mL 0.1 M NaOH, 250 mL 0.2 M KH2PO4, diluted to 1 L, pH 7.5) and then dried over anhydrous MgSO4 before drying in vacuo. All other reagents were used as received.
Benchtop UV–vis measurements were carried out by using a Hewlett-Packard 8453 diode array spectrophotometer equipped with HP Chemstation software and a Unisoku thermostated cell holder for low temperature experiments. A 10 mm path quartz cell assembly (with extended glass tube with female 14/19 joint and stopcock) was used to perform all the experiments.
UV–Vis Samples Preparation
In a standard UV–vis experiment, in the glovebox, 3.0 mL of a complex (PIm)FeII (1) (range: 0.1–0.015 mM) solution in THF was placed in the cuvette assembly, a stir bar was added, and the assembly capped with a rubber septum. After transfer to the benchtop and spectrometer with cryostat, the sample was cooled to −80 °C. Dioxygen gas was bubbled through the solution using a gastight microsyring, until full formation of the superoxo [(PIm)FeIII(O2•−)] (2) complex occurred. Similarly, complex [(PIm)-FeIII(−OON═O)] (3) was generated by addition of NO(g) to complex 2. Excess O2(g)/·NO(g) was removed by several vacuum/Ar cycles.
Synthesis
(PIm)FeII/d8-(PIm)FeII (1), [(PIm)FeIII(OH)] (5), [(PIm)FeIII](SbF6) (6) and [(PIm)FeIV=O] (7) were synthesized as previously described.22,23 The oxy-heme complex [(PIm)FeIII(O2•−)] (2) was generated by bubbling dioxygen to a solution of complex 1. Similarly, complex [(PIm)FeIII(−OON═O)] (3) was generated by removing excess of O2(g) from complex 2 by several cycles of vacuum/Ar purge, followed by addition of ·NO(g) via gastight syringe under Ar. (For the synthesis of authentic [(PIm)FeIII](NO3−) (4b), see the SI.)
Resonance Raman Spectroscopy
Samples were excited at a variety of wavelengths, using either a Coherent I90C-K Kr+ion laser, Liconix HeCd laser, a Coherent 25/7 Sabre Ar+ ion laser, or a Lighthouse Photonics Sprout-D pumped M-Squared SolsTiS Ti:Saph laser while the sample was immersed in a liquid nitrogen cooled (77 K) EPR finger dewar (Wilmad), or cooled at 110 K through contacted with the liquid nitrogen cooled sample holder. The laser power (<2 mW) and sample spinning conditions were set such that the RR spectra obtain before and after extended data acquisition showed no detectable differences. Resonance Raman sample preparation: In a typical experiment, 0.650 mL of a complex (PIm)FeII (1) (2 mM) solution in THF was placed in a 9 mm economy rubber septum capped NMR tube. After cooling the NMR tube to −80 °C (acetone/N2(liq) bath), O2(g) was bubbled through the solution mixture to form the complex [(PIm)FeIII(O2•−)] (2). Similar to the procedure used for UV–vis experiments, complex [(PIm)FeIII(−OON═O)] (3) was prepared by removing excess of dioxygen by vacuum/Ar cycles from 2 and careful addition of NO(g) was carried out using three-way gastight syringe. Complex [(PIm)FeIII](NO3−)] (4b) was prepared by thermal decay of complex 3. After generation of all complexes, tubes were frozen in N2(liq) and flame-sealed. Isotopically labeled (18O2/15N18O) samples were prepared in analogous manner.
X-ray Absorption Spectroscopy
Fe K-edge X-ray absorption spectroscopy (XAS) was conducted at the Stanford Synchrotron Radiation Lightsource (SSRL) on the unfocused 20-pole, 2.0 T wiggler beamline 7-3 under storage ring parameters of 3 GeV and ~500 mA. A Rh-coated premonochromator mirror was used for harmonic rejection and vertical collimation. A Si(220) double-crystal monochromator was used for energy selection. The samples were loaded into delrin XAS cells with 38 μm Kapton windows. The samples were maintained at a constant temperature of ~10 K during data collection using an Oxford Instruments CF 1208 continuous flow liquid helium cryostat. A Canberra solid-state Ge 30-element array detector was used to collect Fe Kα fluorescence data to k = 16 Å−1. Internal energy calibration was accomplished by simultaneous measurement of the transmission of a Fe foil placed between two ionization chambers situated after the sample. The first inflection point of the foil spectrum was assigned to 7111.2 eV. For edge and pre-edge analysis, an average of the first scans collected from physically separated measurement spots on the sample was used for complex [(PIm)FeIII(O2•−)] (2), [(PIm)FeIII(−OON═O)] (3), and [(PIm)FeIII(NO3)] (4a), and the first three-scan average was used for complex [(PIm)FeIII(OH)] (5) (see the SI for the further method of XAS analysis).
NMR Spectroscopy
All NMR spectra were recorded in 7 in., 5 mm o.d. NMR tubes. Low–temperature 2H NMR (Bruker 300 MHz (46.05 MHz for 2H NMR) spectroscopy was carried out using the spectrometer equipped with a tunable deuterium probe to enhance deuterium detection); measurements were performed at −80 °C under a N2 atmosphere. The 2H chemical shifts were calibrated to natural abundance deuterium solvent peaks. 2H NMR samples preparation: Same as rRaman experiment, 0.50 mL of a complex d8-(PIm)FeII (1) (2 mM) solution in THF was placed in a 5 mm rubber septum capped NMR tube. After cooling to −80 °C (acetone/N2(liq) bath), dioxygen was bubbled through the solution mixture to form the complex [(PIm)FeIII(O2•−)] (2). The NMR tube was transferred rapidly into the NMR instrument which was precooled to −85 °C. Complexes [(PIm)FeIII(−OON═O)] (3) and [(PIm)FeIII(NO3−)] (4a) were prepared in a similar manner.
Electron Paramagnetic Resonance (EPR) Spectroscopy
Electron paramagnetic resonance (EPR) spectra for all frozen samples were recorded on a Bruker EMX spectrometer equipped with a Bruker ER 041 X G microwave bridge and a continuous-flow liquid helium cryostat (ESR900) coupled to an Oxford Instruments TC503 temperature controller. Spectra were obtained at 14 K under nonsaturating microwave power conditions (ν = 9.4108 GHz, microwave power = 0.201 mW, modulation amplitude = 10 G, microwave frequency = 100 kHz, receiver gain = 5.02 × 103). EPR samples preparation: All samples were prepared in a similar way as NMR and rRaman samples.
DFT Calculations
Density functional theory calculations were performed using Gaussian 09, revision D.01. All geometry optimizations and energies were obtained using the spin-unrestricted B3LYP functional and ultrafine integration grid. The basis sets used were as follows: 6-311g* for Fe, peroxynitrite (N and O), and all N atoms immediately bound to Fe; 6-31g for all other atoms in the molecule (C, F, H, and remaining N and O). All calculations were performed using a THF solvent PCM.
RESULTS AND DISCUSSION
Interaction of (PIm)FeII (1) with O2(g)/·NO(g)
UV–Vis Spectroscopy
In THF solvent, ferrous heme complex 1 (0.015 mM) is a six-coordinate low-spin complex (at all temperatures)22 (THF as sixth ligand) [λmax = 417 nm (Soret), ε = 258 600 M−1 cm−1, 525 nm, ε = 28 650 M−1 cm−1, 554 nm (sh), ε = 7 250 M−1 cm−1]. Bubbling with O2(g) generates superoxo species [(PIm)FeIII(O2•−)] (2) [λmax = 423 nm, ε = 247 440 M−1 cm−1, 533 nm, ε = 23 037 M−1 cm−1].22 After removing the excess of O2(g) using vacuum/Ar cycles, addition of 250 μL from our standard freshly purified NO(g) flask (see Experimental Section) to the solution of complex 2 causes an immediate change in the absorption spectrum, with a blue shift in the Soret band from 423 to 417 nm and concomitant Q-band red shift from 533 to 541 nm, leading to the formation of the putative peroxynitrite species [(PIm)FeIII(−OON═O)] (3) (λmax = 417 nm, ε = 220 700 M−1 cm−1, 541 nm, ε = 15 287 M−1 cm−1) (Figure 1, Scheme 2). This is stable at −80 °C for minutes before slowly transforming to complex 4a [λmax = 414 (Soret), ε = 220 500 M−1 cm−1, 544 nm, ε = 15 000 M−1 cm−1], which is a six-coordinate nitrato species [(PIm)- FeIII(NO3)] 4a (Scheme 2).
Figure 1.
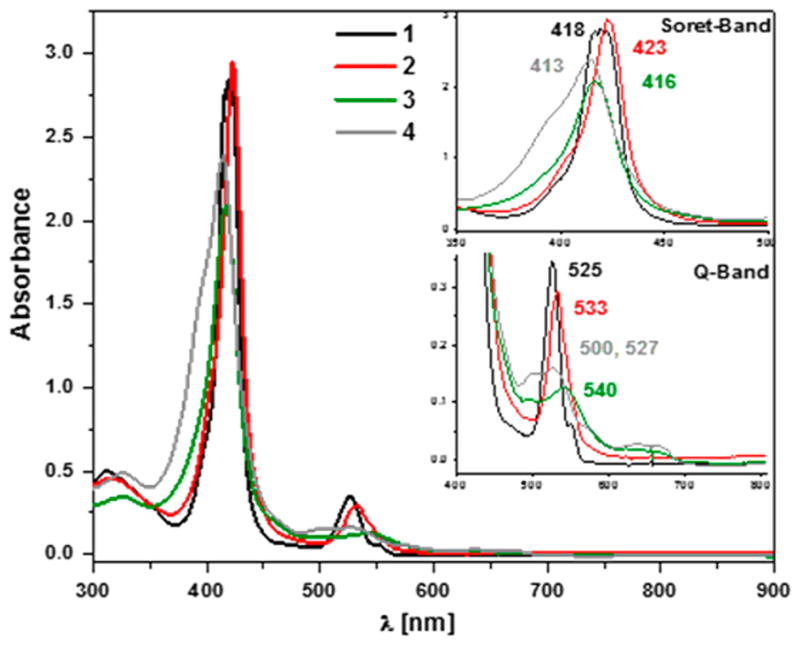
UV–vis spectroscopy showing generation of superoxo complex 2 (red spectrum) by bubbling O2(g) through the THF solution of (PIm)FeII (1) (black) at −80 °C, followed by addition of ·NO(g) to generate peroxynitrite complex 3 (green) with the final decay product (warming to RT) being nitrate complex 4b (gray) (inset: Soret band and Q-band for 1, 2, 3, and 4b).
Confirmation that 4a is a nitrate compound (nitrogen-containing product derived from ·NO(g)) was obtained using QUNTAFIX nitrate/nitrite test paper (semiquantitative) and by ion chromatography (see SI).24 The test was performed after warming 4a to RT, where the UV–vis spectrum is that of a typical five-coordinate high-spin complex 4b (Scheme S1, Figure 1). Thus, we postulate that the nitrato ligand is not bound but rather serves as a counteranion at RT, i.e., with [(PIm)FeIII](NO3) (4b) as the formulation [λmax = 413, 501, 527, 572 (sh), 634 (sh) nm], as is known for a well-characterized analog compound, [(PIm)FeIII](SbF6) (6).23,25 Further, because it is relevant for studies described below, we note that superoxo compound 2 does thermally decay and at RT transforms to the high-spin hydroxy complex [(PIm)-FeIII(OH)] (5).22 In our UV–vis experiment we did not observe any ferric or ferrous nitrosyl species (see Figure S9).
Resonance Raman Spectroscopy
Further properties of the products of dioxygen and NO(g) (Scheme 2) were interrogated using resonance Raman (rR) spectroscopy. The high-frequency rR spectra of 1, 2, and 3 obtained with a 406.7 nm excitation show oxidation state marker band (ν4) at 1359, 1368, and 1371 cm−1, and spin-state marker band (ν2) at 1567, 1569, and 1571 cm−1, respectively. DFT calculations (vide infra) support the slight increase in ν4 and ν2 frequencies (~1 cm−1 calcd) for 3 relative to 2, which derives from the more π acidic peroxynitrite ligand causing a decrease in backbonding from Fe into the porphyrin eg orbitals (resulting in higher energy ν4 and ν2 core modes in 3).26 Residual signals from 1 are clearly observed in the rR spectra of 2 and 3, presumably due to some loss of the superoxo complex during the degassing of excess O2(g) prior to exposure to NO, but spectra obtained with 406.7, 413.1 nm and lower energy excitations like 441.6 and 457.9 nm (Figures 2 and S4) identify 2 and 3 as distinct six–coordinate low-spin Fe(III) complexes (Figure 2). Two 16O2/18O2 isotopic sensitive shifts were observed for the superoxo complex [(PIm)-FeIII(O2•−)] (2), at 1180 cm−1 [Δ(18O2), −56 cm−1] and 575 cm−1 [Δ(18O2), −23 cm−1] corresponding to the O–O and Fe–O stretches, respectively (Figure S4), as previously observed.22 As a control, we collected rRaman data on the ferrous-nitrosyl complex, which has a ν(Fe–NO) stretch of 570 (Δ15N = −20) cm−1 (Figure S4); this mode is not observed in the data for complex 3. Complex [(PIm)FeIII(−OON═O)] (3) was also excited at several wavelengths ranging from 406.7 to 900 nm, but no enhancement of an isotope-sensitive mode (15N or 18O) was observed.
Figure 2.
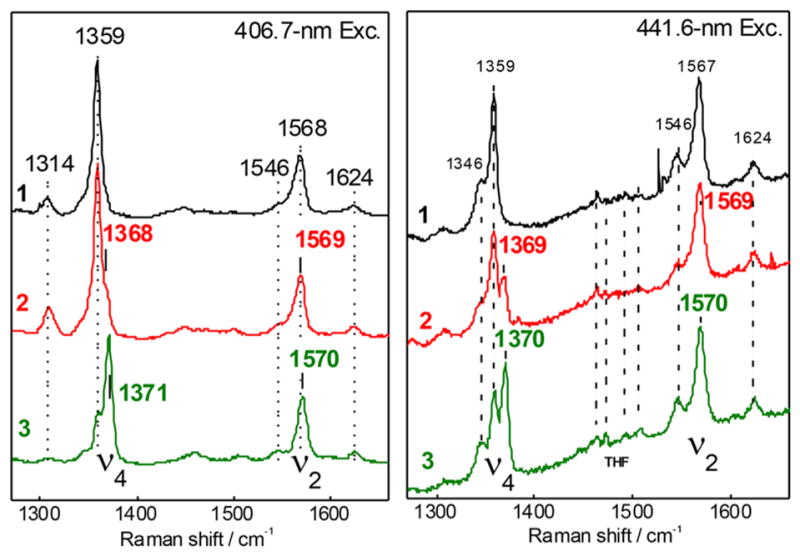
rRaman spectroscopy collected at 406.7 nm (left) and 441.6 (right) nm excitation for complexes [(PIm)FeII] (1, black), [(PIm)FeIII- (O2•−)] (2, red), and [(PIm)FeIII-(−OON═O)] (3, green), showing the oxidation state marker band (ν4) and spin state marker band (ν2). Note that apparent peak maxima can vary with excitation due to differences in relative enhancement of contributing species.
X-ray Absorption Spectroscopy
Figure 3 shows the Fe Kedge XAS spectra and the enlarged pre-edge region of 2, 3, 4a, and 5. In comparing the data obtained for 2 and 3, the edge and pre-edge features show notable changes. In addition, the spectral features at 7130–7140 eV are different between 3 and 4a. These indicate that 3 has distinctive properties compared to the superoxo complex 2 and the decay product 4a. Further information about the spin state and the coordination number can be obtained from the pre-edge region and the extended X-ray absorption fine structure (EXAFS) data. The Fe K pre-edge feature at ~7112 eV originates from a 1s → 3d transition. This transition is electric dipole forbidden, but gains weak intensity through a quadrupole mechanism. For noncentrosymmetric complexes, the intensity of this pre-edge feature increases due to metal 4p mixing into the 3d orbitals. A systematic study using various Fe model complexes showed that this feature is sensitive to oxidation state, spin state, and site symmetry.27 The second derivative of the pre-edge of 3 exhibits three features centered at 7111.3, 7112.7, and 7113.9 eV with intensities of 1.0, 3.6, and 1.2 units, respectively, for a total intensity of 5.8 units (Figure 3 and Table S1). The shoulder at lower energy (~7111 eV), which was not observed in the high-spin species of 5, is a key feature for low-spin Fe(III) model complexes including heme complexes.27,28 The total intensity of 5.8 units is typical for six-coordinate Fe(III) synthetic complexes. The EXAFS data show that 3 is a six-coordinate species with the first shell fit best using six Fe–N/O contributions at 1.99 Å (Figures S10, S11 and Table S2). Taken together, XAS suggests that peroxynitrite complex 3 is a six-coordinate low-spin Fe(III) species.
Figure 3.

Fe K-edge XAS spectra (top) and enlarged pre-edge region (bottom) for complex 2 (red), 3 (green), 4a (gray), and 5 (blue). Inset shows smoothed second derivatives of the pre-edge region.
NMR Spectroscopy
More structural information was obtained for these complexes from low temperature 2H NMR spectroscopy (see Experimental Section). We previously reported on complex d8-[(PIm)FeIII(O2•−)] (2)22 (δ 10.0 ppm, low-spin, Fe(III), d5, diamagnetic) obtained by bubbling molecular oxygen into a solution of complex 1 (δ 9.5 ppm, low-spin, Fe(II), d6), shown in Figure 4 (top). Addition of NO(g) to complex 2 resulted in the formation of complex [(PIm)- FeIII(−OON═O)] (3) with pyrrolic-proton resonances occurring at three different frequencies (δ −1.0, 8.94, and 18.10 ppm) in a ratio of 1:2:1. Splitting in deuterated pyrrolic-protons indicates the presence of low symmetry in the compound, which arises from the covalently tethered imidazole group, as well as coordination of the peroxynitrite. The small upfield shift of pyrrole-proton peak, here found at −1.0 ppm, is common for low-spin S = 1/2 iron(III) six-coordinate compounds.29 With thermal decay of peroxynitrite complex 3, a nitrato product forms, [(PIm)FeIII](NO3) (4b), as presented in Schemes 2 and S1, and this exhibits typical (for high spin) downfield shifts in the pyrrole hydrogen resonances, at 52.0, 42.0, and 34.0 ppm. Therefore, the 2H NMR spectroscopic data agree with our analysis of the UV–vis and rR spectropsopic experiments, in further support of our formulation of 3 as a peroxynitrite complex [(PIm)FeIII(−OON═O)].
Figure 4.

2H NMR (top) in THF at −80 °C for complexes (1, 2, 3, and 4b). Asterisk (*) is for solvent THF; (bottom) EPR of peroxynitrite complex 3 (green) in THF at 14 K. Decay of complex 3 leads to the formation of [(PIm)FeIII](NO3) (4b) (gray); note that only partial formation of 4b (and increase in the g = 5.96 signal intensity) is shown. See text for details.
EPR Spectroscopy
EPR experiments also reveal that complex 3 exhibits a low-spin, rhombic signature that is typical for six-coordinated ferri-hemes (features at gx = 2.70, gy = 2.25 and gz = 1.72, Figure 4, bottom).29a,c There is also a signal at g = 5.96 that represents a rather small amount (5–10%) of a high-spin Fe(III) species arising from decomposition; see Figures S5 and S6 with text. The EPR spectroscopic experiments also demonstrate that putative peroxynitrite compound [(PIm)FeIII(−OON═O)] (3) is not a nitrato species, as thermal decay leads to the formation of product 4b, associated with the spin state change to high-spin (S = 5/2) (control experiments involving nitrate addition to [(PIm)FeIII]+ showed that the low-spin nitrato species 4a could only be observed in the presence of a large excess of NO3− and low temperatures, saturating at ~50% 4a formation). Furthermore, the signal for decay product 4b matches the authentically synthesized nitrato complex (see the SI, Figures S1 and S5–S7).30
Thus, the overall chemistry sequence shown in Scheme 2 parallels that found in the enzymes, NOD activity, where ·NO(g) is oxidized to the nitrate anion in the Fe–O2 + ·NO(g) reaction. We note that there is no evidence that a nitrito complex [(PIm)FeIII(NO2)] ever forms; we also independently generated this species and it possesses markedly differing UV–vis and EPR spectroscopic properties, see the SI, Figure S7. Under these conditions, we also do not observe formation of any ferrous/ferric nitrosyl complex. As we have shown earlier, the ferrous nitrosyl31 complex has characteristic UV–vis and EPR spectroscopic features, shown in Figure S9.
Reactivity Studies
To gain additional insight into the properties of [(PIm)FeIII(−OON═O)] (3), we investigated the addition of 2,4-di-tert-butylphenol (2,4DTBP) to in situ generated 3. Such reactions are known to give o-nitration of phenol for some heme containing systems20,21 and for complexes of other transition metals.32 However, addition of 2,4DTBP to 3 yielded no immediate spectral changes (via UV–vis and rR), and formation of complex 4a was still observed after minutes at −80 °C (Scheme 3, top). Warming to RT yielded a positive test for nitrate ion and only unreacted 2,4DTBP. This result suggests that isomerization of peroxyni-trite (−OON═O) complex 3 to nitrate complex 4a is faster than phenol nitration. As depicted in Scheme 1, isomerization of heme-peroxynitrite (−OON═O) to heme-nitrate (O-NO2) involves homolysis of the O–O bond to give a ferryl-oxo species plus ·NO2, whereupon rebound of nitrogen dioxide and formation of a new O–N bond gives nitrate as product. However, under the present reaction conditions, monitoring either by UV–vis, rRaman, NMR, or EPR spectroscopies, we do not detect any high-valent compound-II-like (FeIV═O) species.
Scheme 3.
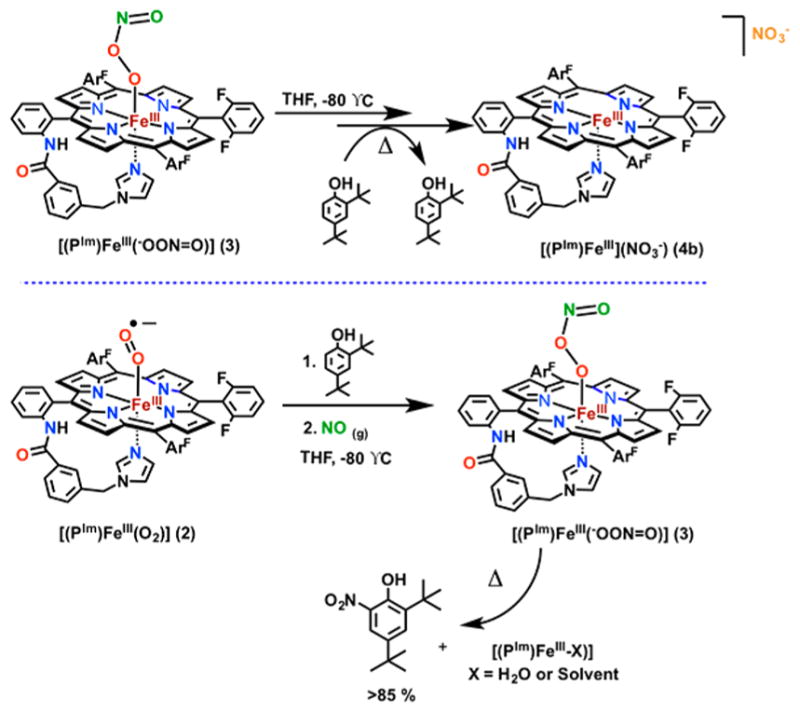
Reactivity of Complex 3 toward 2,4-Di-tert-butyl Phenol (2,4DTBP): (Top) 2,4DTPB Added to Complex 3; (Bottom) 2,4DTBP Added to 2 Prior to Complex 3 Formation, Which Results in Nitration of Phenol
To probe the plausibility of this fast isomerization reaction, we first generated the compound-II-like [(PIm)FeIV=O] (7) complex23 by adding 1.5 equiv of mCPBA (meta-chloroperoxybenzoic acid) to complex 1 at −80 °C in THF and monitoring the reaction by UV–vis spectroscopy (Figure 5, bottom left). Following the formation of complex 7, addition of ·NO2(g) (see Experimental Section) resulted in a very short-lived intermediate observed in a single spectrum at −80 °C using our benchtop UV–vis spectrometer, which immediately converted into the final Fe(III)-nitrato product 4b, confirmed by a positive nitrate test as well as characteristic UV–vis and EPR spectroscopic features (Figure 5, bottom right; see also Figure S8 with explanations).
Figure 5.
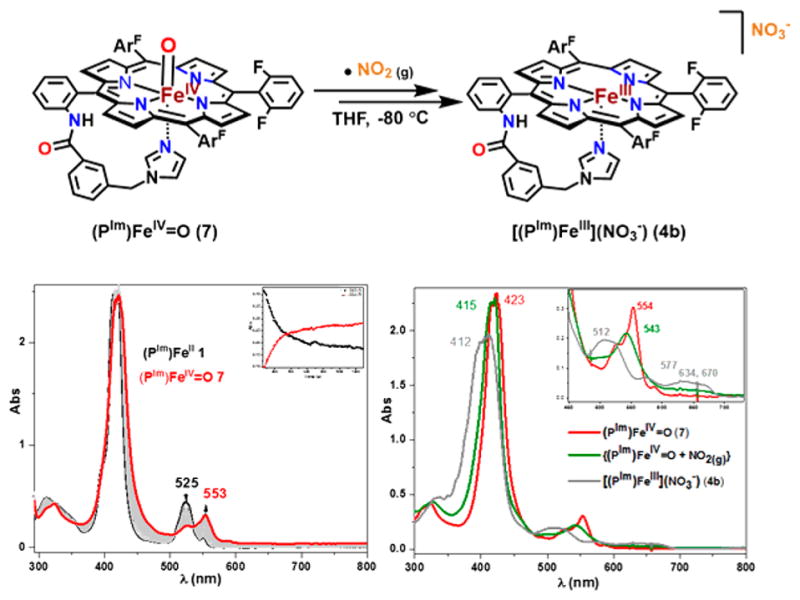
(top) Reaction of compound II with ·NO2(g) leads to ferric nitrate complex 4b (see the text and SI for detailed discussion). (Bottom left) UV–vis spectroscopy of in situ generated ferryl complex 7 by reacting 1 with mCPBA; (bottom right) the UV–vis of in situ generated complex 4b by addition of ·NO2(g) to 7.
Interestingly, we do observe effective nitration chemistry when 2,4DTBP (1.1 equiv) is added to 2 prior to ·NO(g) at −80 °C (Scheme 3, bottom). In this case, workup of the reaction solution (requiring warming to RT) reveals that the ferric product [(PIm)FeIII(OH2)] forms along with high yields (>85%) of 2,4-di-tert-butyl-6-nitrophenol (2-NO2-DTBP) (GC analysis; see the SI), as well as a negative test for NO3−/NO2− ions (see the SI).33 The nitrated phenol product is only observed after stirring the reaction mixture for hours at RT; however, analysis of the solution immediately after warming yields only 10–15% of unreacted 2,4DTBT, indicating that the nitration reaction occurs prior to stirring at RT. Interestingly, the species formed upon ·NO(g) addition at −80°C (i.e., 2 + 2,4DTBP + ·NO(g)) is spectrally indistinguishable (via UV–vis, rR, EPR, and NMR) from that of 3 generated without 2,4DTBP present (as in Scheme 2), suggesting that a product comparable to 3 has formed. Furthermore, the decay after several minutes at −80 °C is likewise indistinguishable from the decay of 3 to 4a (although the UV–vis features are slightly broader for the reaction where 2,4DTBP is added before ·NO(g)).34 We are as of yet unable to fully explain the observed difference in reactivity based on our current data and DFT calculations (vide infra) (also see ref 33).
DFT Calculations
To better understand the nature of the peroxynitrite complex [(PIm)FeIII(−OON═O)] (3), and explore possible effects of phenol addition both before and after formation of 3, we turned to DFT calculations. For peroxynitrite binding to a metal ion, several possible binding modes and conformations must be considered, including N atom11 vs O atom ligation, cis vs trans isomers (Figure 6a and b, respectively), and even O,O′-chelation.31 Peroxynitrite N-binding to a heme-Fe(III) has been proposed based on theoretical calculations,13 but peroxo anion O atom binding is observed experimentally (and supported by DFT calculations) for several porphyrinate cobalt(III)-peroxynitrite complexes.19a
Figure 6.
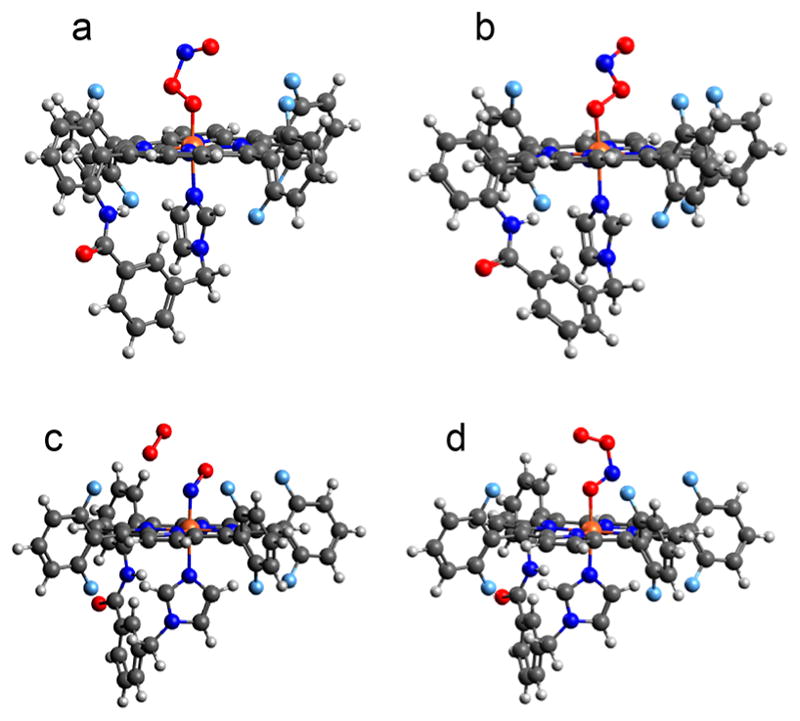
DFT optimized structures for possible orientations of intermediate 3, as discussed in the text.
Indeed, our calculations predict that the lowest energy structure of 3 is cis-peroxynitrite bound to Fe(III) through the terminal peroxy O (Figure 6a), which is 3.7 kcal/mol (all energies given herein are ΔG at −80 °C unless otherwise noted) more stable than the trans isomer (Figure 6b), in agreement with computational results reported for other metal–peroxynitrite systems.11–13 We also investigated the possibility of a cis-peroxynitrite binding via the O atom of the N═O moiety (Figure 6d), but this is found to be much higher in energy (~20 kcal/mol) than the peroxo O-bound isomers shown in Figure 6a,b. Geometry optimization of an N-bound peroxynitrite (Figure 6c and Table S3), which could form by attack of dioxygen on an FeIII-(−NO) complex, results in cleavage of the interior N–O bond and dissociation of molecular O2. It is interesting to note that a comparison of {FeIII(−NO) + O2 and {FeIII(O2•−) + NO}, where the metal complex and gas molecule are noninteracting species, shows the {FeIII(−NO) + O2} to be 7.0 kcal/mol lower in energy. However, the cis-PN structure given for 3 is 8.5 kcal/mol lower than the separated {FeIII(O2•−) + NO} (15.2 kcal/mol lower in ΔH, but this neglects the entropic cost of ·NO(g) association), and therefore, formation of FeIII(-OONO) is thermodynamically favored. This is in agreement with our experimental data demonstrating that 3 is in fact distinguishable from FeIII(NO)31,35 and FeIII(O2•−).22
Relative to FeIII(−OONO) (Figure 6a), the geometry optimized structure of FeIII(NO3−) is lower in energy by 31.5 kcal/mol for the LS (S = 1/2) and 28.5 kcal/mol for the HS (S = 5/2) surfaces. The preference for the LS state is likely due to the 6C structure, in which NO3− ion remains bound. Nevertheless, our calculations support that the nitrato complex is the decay product of 3.
Taking the optimized structures for 3 and 4a, we evaluated a reaction coordinate between them, and found that O–O cleavage was the lowest energy barrier (ΔG‡ = 9.3 kcal/mol). Elongating the O–O bond past the barrier, an intermediate is found that is 4.1 kcal/mol lower energy than 3, and consists of an Fe(IV)═O and ·NO2, where the ·NO2 molecule has reoriented such that the N atom is pointed toward the Fe-bound O atom (Figure 7a). The ·NO2 can then rebound onto the Fe(IV) ═O moiety with a negligible barrier (ΔG‡ = 2.5 kcal/mol) to form the N–O bond and yield the nitrate product shown in Figure 7b, which is 31.5 kcal/mol below the bound peroxynitrite structure (vide supra).
Figure 7.
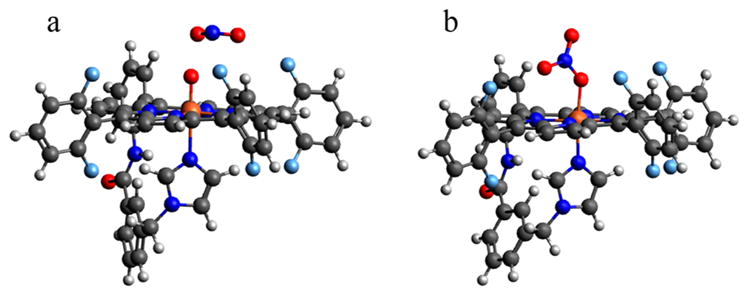
DFT optimized structures for the (a) FeIV═O + ·NO2 intermediate and (b) FeIII(NO3−) (4a) product following decay of the FeIII(−OONO) 3 complex.
We next sought to understand the difference in reactivity observed for phenol addition before vs after ·NO(g), where only the latter produces effective phenol nitration. Since our data indicate that a peroxynitrite species forms initially in both cases (vide supra), we first considered the interaction of phenol with a fully formed peroxynitrite to understand how it might impact the decay process. Exploring several approach vectors of phenol onto the optimized cis-FeIII(−OONO) (Figure 6a), it was found that phenol binding to the O atom directly bound to Fe(O(1)) is most favorable, while the next-lowest energy binding mode (which is on the O(2) atom) is 2.6 kcal/mol higher. Relative to the fully separated species, the O(1)-bound adduct has an energy of ΔE = −7.3 kcal/mol and ΔG = +0.5 kcal/mol, indicating an essentially thermoneutral process. Phenol (bound to O(1)) is calculated to have a minimal impact on the barrier to O–O cleavage (ΔG‡ = 9.4 kcal/mol), and yields similar thermodynamics for formation of the {FeIV═O + ·NO2} intermediate (ΔG = −4.6 kcal/mol), where phenol ends up H-bonded to the ferryl-oxo (Figure 8b). Note that O–O cleavage is again the more favorable process, as Fe–O cleavage gives a much higher barrier. As was the case without phenol present, rebound of NO2 to form nitrate has a very low barrier (ΔG‡ = 3.2 kcal/mol) and proceeds to form the FeIII(NO3−) product 4a (ΔG = −25.9 kcal/mol) shown in Figure 8c. In contrast, we note that H atom abstraction by the FeIV═O (Figure 8b) to yield {FeIII–OH + PhO• + ·NO2} is thermodynamically uphill by 4.1 kcal/mol, greater even than the barrier for isomerization to nitrate. Taken together, these results are in good agreement with our observation that addition of 2,4DTBP to 3 still yields decay to the nitrate product 4a (at a rate comparable to that observed without 2,4DTBP), along with unreacted 2,4DTBP.
Figure 8.
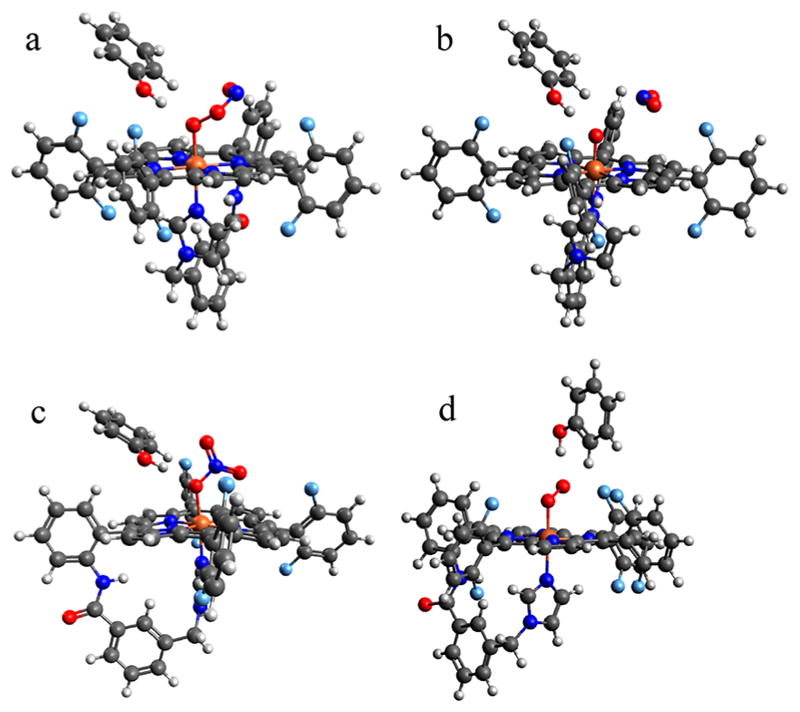
DFT-optimized structures for phenol-bound (a) (PIm)-FeIII(−OONO) (3), (b) {(PIm)FeIV=O + ·NO2}, (c) (PIm)FeIII(NO3−) (4a), and (d) (PIm)FeIII(O2•−) (2).
Finally, we examined the case in which phenol is added prior to ·NO(g) to consider whether the observed change in nitration reactivity could be attributable to a different peroxynitrite structure forming initially, caused by the interaction with phenol. Phenol binding to the Fe-superoxo species 2 is thermodynamically favorable by ΔE = −9.5 kcal/mol and ΔG = −1.2 kcal/mol, depicted in Figure 8d. With the phenol bound to the terminal O, reaction with NO initially yields a cis-peroxynitrite structure where the phenol is H-bonded to O(2) that is downhill to form by 3.0 kcal/mol (see the SI, Figure S12). However, this can easily isomerize (with a barrier lower than that of intraperoxynitrite cleavage) to the O(1)-bound structure shown in Figure 8a, lowering the energy an additional 2.6 kcal/mol and yielding the same peroxynitrite-phenol adduct as in the case of phenol addition to an already-formed [(PIm)FeIII(−OONO)] (3). This supports our data suggesting that FeIII-bound peroxynitrite forms independent of the sequence of ·NO(g)/ 2,4DTBP addition.
Thus, our calculations modeling the formation and subsequent decay (via O–O cleavage) of [(PIm)-FeIII(−OONO)] (3) with and without phenol present indicate that phenol would have a minimal impact on either process. However, our current computations do not adequately explain the observation of nitrated phenol upon warming to RT. A simple calculation of {(PIm)FeIII(NO3−) + PhOH} to {(PIm)-FeIII(OH−) + 2-NO2PhOH} shows that this process is favorable by 15 kcal/mol in ΔG at 298 K, but given that observation of 2-NO2PhOH requires warming to room temperature, the nature of the active nitrating species remains uncertain.33
CONCLUSION
In summary, for the first time, we were able to observe an intermediate in a reaction of heme-superoxo complex with ·NO(g) using a synthetically designed heme (PIm)FeII complex and were able to characterize the long sought after peroxynitrite ferriheme species [(PIm)FeIII(−OON═O)] (3), using a number of spectroscopic techniques. The results led to the formulation of this complex as a low–spin, six-coordinate FeIII–PN species. Employing DFT calculations, we determined that the cis conformation of complex 3 is more stable than the trans. The addition of phenol to 3 leads to homolysis of the O–O bond resulting in fast isomerization of PN to nitrate, rather than the nitration of phenol that has been observed for other peroxynitrite systems. The data presented here concerning synthetic heme/O2/·NO reactions offer important insights into the chemistry associated with heme–peroxynitrite species. From the present research along with our published Cu/O2/ ·NO studies,36 it is clear that heme and possibly other metal/O2/·NO systems can be associated with oxidation and/or nitration of exogenous substrates. Thus, peroxynitrite-derived chemistry may not only derive from the superoxide (O2•−) plus nitrogen monoxide (·NO) reactivity as mainly discussed in the biological/biochemical literature, but also metal/O2 + ·NO or metal/· NO + O2 reactivity.32c,f,g,36
Supplementary Material
Acknowledgments
For K.D.K., this research was supported by the USA National Institutes of Health (GM60353). H.L. was supported by an Abbott Laboratories Stanford Graduate Fellowship. For E.I.S., this research is supported by the USA National Institutes of Health (GM040392). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393). For P.M-L., this research was supported by the USA National Institutes of Health (GM074785). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b08468.
Experimental details, synthesis, further spectroscopic characterization (UV–vis, EPR, NMR), control experiments, and DFT calculations (PDF)
References
- 1.(a) Ignarro LJ. Annu Rev Pharmacol Toxicol. 1990;30:535–560. doi: 10.1146/annurev.pa.30.040190.002535. [DOI] [PubMed] [Google Scholar]; (b) Tennyson AG, Lippard SJ. Chem Biol. 2011;18:1211–1220. doi: 10.1016/j.chembiol.2011.09.009. [DOI] [PubMed] [Google Scholar]; (c) Toledo JC, Jr, Augusto O. Chem Res Toxicol. 2012;25:975–989. doi: 10.1021/tx300042g. [DOI] [PubMed] [Google Scholar]
- 2.(a) Blough NV, Zafiriou OC. Inorg Chem. 1985;24:3502–3504. [Google Scholar]; (b) Nauser T, Koppenol WH. J Phys Chem A. 2002;106:4084–4086. [Google Scholar]
- 3.(a) Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Chem Res Toxicol. 1992;5:834–842. doi: 10.1021/tx00030a017. [DOI] [PubMed] [Google Scholar]; (c) Szabo C, Ischiropoulos H, Radi R. Nat Rev Drug Discovery. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]; (d) Augusto O, Gatti RM, Radi R. Arch Biochem Biophys. 1994;310:118–125. doi: 10.1006/abbi.1994.1147. [DOI] [PubMed] [Google Scholar]; (e) Goldstein S, Czapski G. Inorg Chem. 1995;34:4041–4048. [Google Scholar]; (f) Gerasimov OV, Lymar SV. Inorg Chem. 1999;38:4317–4321. [Google Scholar]
- 4.(a) Radi R. Acc Chem Res. 2013;46:550–559. doi: 10.1021/ar300234c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Campolo N, Bartesaghi S, Radi R. Redox Rep. 2014;19:221–231. doi: 10.1179/1351000214Y.0000000099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bartesaghi S, Peluffo G, Zhang H, Joseph J, Kalyanaraman B, Radi R. Methods Enzymol. 2008;441:217–236. doi: 10.1016/S0076-6879(08)01212-3. Nitric Oxide, Part G. [DOI] [PubMed] [Google Scholar]; (d) Radi R. Proc Natl Acad Sci U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Pacher P, Beckman JS, Liaudet L. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schopfer FJ, Baker PRS, Freeman BA. Trends Biochem Sci. 2003;28:646–654. doi: 10.1016/j.tibs.2003.10.006. [DOI] [PubMed] [Google Scholar]; (c) Dedon PC, Tannenbaum SR. Arch Biochem Biophys. 2004;423:12–22. doi: 10.1016/j.abb.2003.12.017. [DOI] [PubMed] [Google Scholar]; (d) Beckman JS, Koppenol WH. Am J Physiol Cell Physiol. 1996;271:C1424. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 6.(a) Batthyány C, Souza JM, Durán R, Cassina A, Cerveñansky C, Radi R. Biochemistry. 2005;44:8038–8046. doi: 10.1021/bi0474620. [DOI] [PubMed] [Google Scholar]; (b) Demicheli V, Quijano C, Alvarez B, Radi R. Free Radical Biol Med. 2007;42:1359–1368. doi: 10.1016/j.freeradbiomed.2007.01.034. [DOI] [PubMed] [Google Scholar]; (c) Reynolds MR, Berry RW, Binder LI. Biochemistry. 2007;46:7325–7336. doi: 10.1021/bi700430y. [DOI] [PubMed] [Google Scholar]; (d) Quint P, Reutzel R, Mikulski R, McKenna R, Silverman DN. Free Radical Biol Med. 2006;40:453–458. doi: 10.1016/j.freeradbiomed.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 7.Knyushko TV, Sharov VS, Williams TD, Schöneich C, Bigelow DJ. Biochemistry. 2005;44:13071–13081. doi: 10.1021/bi051226n. [DOI] [PubMed] [Google Scholar]
- 8.(a) MacMillan-Crow LA, Crow JP, Thompson JA. Biochemistry. 1998;37:1613–1622. doi: 10.1021/bi971894b. [DOI] [PubMed] [Google Scholar]; (b) MacMillan-Crow LA, Crow JP, Kerby JD, Beckman JS, Thompson JA. Proc Natl Acad Sci U S A. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Radi R. Proc Natl Acad Sci U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.As pointed out by one reviewer, there is some disagreement among researchers as to whether peroxynitrite (PN) is directly responsible for (a) damaging consequences to biomolecules or (b) disease states. Some in the community suggest that the reaction of ·NO(g) with superoxide detoxifies the latter since formation of peroxynitrite will lead to its isomerization to innocuous nitrate ion. However, chemical observations (e.g., (i) superoxide itself is very unreactive and not damaging, (ii) it reacts faster with nitric oxide than it undergoes the Haber–Weiss reaction), biochemical findings, (e.g., (i) ·NO(g) itself is not so reactive toward biomolecules, (ii) PN diffuses over longer distances than superoxide or hydroxyl radical, and (iii) it has been directly detected in activated macrophages), and logic in a chemical perspective very strongly implicate peroxynitrite in the roles described here in this Introduction. See also relevant refs 5a, 5d, 10, and 33a.
- 10.(a) Ross AB, Bielski BHJ, Buxton GV, Cabelli DE, Greenstock CL, Helman WP, Huie RE, Grodkowski J, Neta P. NDRL-NIST Solution Kinetics Database, ver. 2. National Institute of Standards and Technology; Gaithersburg, MD: 1994. [Google Scholar]; (b) Koppenol WH. Peroxynitrite Detection in Biological Media: Challenges and Advances. Ch 1. The Royal Society of Chemistry; Cambridge, UK: 2016. Peroxynitrite: The Basics; pp. 1–11. [Google Scholar]; (c) Prolo C, Álvarez MN, Radi R. BioFactors. 2014;40:215–225. doi: 10.1002/biof.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagy PI. J Phys Chem A. 2002;106:2659–2670. [Google Scholar]
- 12.Tsai HH, Hamilton TP, Tsai JHM, van der Woerd M, Harrison JG, Jablonsky MJ, Beckman JS, Koppenol WH. J Phys Chem. 1996;100:15087–15095. [Google Scholar]
- 13.(a) Attia AA, Silaghi-Dumitrescu R. Int J Quantum Chem. 2014;114:652–665. [Google Scholar]; (b) Silaghi-Dumitrescu R. J Mol Struct: THEOCHEM. 2005;722:233–237. [Google Scholar]
- 14.(a) Olson JS, Foley EW, Rogge C, Tsai AL, Doyle MP, Lemon DD. Free Radical Biol Med. 2004;36:685–697. doi: 10.1016/j.freeradbiomed.2003.11.030. [DOI] [PubMed] [Google Scholar]; (b) Gardner PR, Gardner AM, Brashear WT, Suzuki T, Hvitved AN, Setchell KD, Olson JS. J Inorg Biochem. 2006;100:542–550. doi: 10.1016/j.jinorgbio.2005.12.012. [DOI] [PubMed] [Google Scholar]; (c) Gardner PR, Gardner AM, Martin LA, Salzman AL. Proc Natl Acad Sci U S A. 1998;95:10378–10383. doi: 10.1073/pnas.95.18.10378. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ouellet H, Ouellet Y, Richard C, Labarre M, Wittenberg B, Wittenberg J, Guertin M. Proc Natl Acad Sci U S A. 2002;99:5902–5907. doi: 10.1073/pnas.092017799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Herold S, Matsui T, Watanabe Y. J Am Chem Soc. 2001;123:4085–4086. doi: 10.1021/ja010111d. [DOI] [PubMed] [Google Scholar]; (b) Herold S, Exner M, Nauser T. Biochemistry. 2001;40:3385–3395. doi: 10.1021/bi002407m. [DOI] [PubMed] [Google Scholar]; (c) Herold S. FEBS Lett. 1999;443:81–84. doi: 10.1016/s0014-5793(98)81345-8. [DOI] [PubMed] [Google Scholar]; (d) Herold S. FEBS Lett. 1998;439:85–88. doi: 10.1016/s0014-5793(98)01345-3. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein S, Merenyi G, Samuni A. J Am Chem Soc. 2004;126:15694–15701. doi: 10.1021/ja046186+. [DOI] [PubMed] [Google Scholar]
- 17.Yukl ET, de Vries S, Moenne-Loccoz P. J Am Chem Soc. 2009;131:7234–7235. doi: 10.1021/ja9026924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurtikyan TS, Ford PC. Chem Commun. 2010;46:8570–8572. doi: 10.1039/c0cc02665d. [DOI] [PubMed] [Google Scholar]
- 19.(a) Kurtikyan TS, Eksuzyan SR, Hayrapetyan VA, Martirosyan GG, Hovhannisyan GS, Goodwin JA. J Am Chem Soc. 2012;134:13861–13870. doi: 10.1021/ja305774v. [DOI] [PubMed] [Google Scholar]; (b) Kurtikyan TS, Eksuzyan SR, Goodwin JA, Hovhannisyan GS. Inorg Chem. 2013;52:12046–12056. doi: 10.1021/ic4018689. [DOI] [PubMed] [Google Scholar]
- 20.Schopfer MP, Mondal B, Lee DH, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2009;131:11304–11305. doi: 10.1021/ja904832j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma SK, Rogler PJ, Karlin KD. J Porphyrins Phthalocyanines. 2015;19:352–360. doi: 10.1142/S108842461550025X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Sharma SK, Karlin KD. Polyhedron. 2013;58:190–196. doi: 10.1016/j.poly.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-Bosch I, Sharma SK, Karlin KD. J Am Chem Soc. 2013;135:16248–16251. doi: 10.1021/ja405739m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Jones WR, Jandik P. J Chromatogr A. 1992;608:385–393. [Google Scholar]; (b) Melanson JE, Lucy CA. J Chromatogr A. 2000;884:311–316. doi: 10.1016/s0021-9673(99)01340-0. [DOI] [PubMed] [Google Scholar]
- 25.In other experiments, we showed that nitrate binding to 6 is weak and does not occur at RT, whereas addition of excess nitrate ion, and lowering of the temperature leads to a low-spin complex, thus where nitrate ion is bound, in [(PIm)FeIII(NO3)] 4a; see the SI.
- 26.Spiro TG, Strekas TC. J Am Chem Soc. 1974;96:338–345. doi: 10.1021/ja00809a004. [DOI] [PubMed] [Google Scholar]
- 27.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 28.Wilson SA, Green E, Mathews II, Benfatto M, Hodgson KO, Hedman B, Sarangi R. Proc Natl Acad Sci U S A. 2013;110:16333–16338. doi: 10.1073/pnas.1315734110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Walker FA. Coord Chem Rev. 1999;185–186:471–534. [Google Scholar]; (b) Nakamura M, Ikeue T, Neya S, Funasaki N, Nakamura N. Inorg Chem. 1996;35:3731–3732. doi: 10.1021/ic9514962. [DOI] [PubMed] [Google Scholar]; (c) Walker FA. NMR and EPR spectroscopy of paramagnetic metalloporphyrins and heme proteins. World Scientific Publishing Co. Pte. Ltd; Singapore: 2010. pp. 1–337. [Google Scholar]
- 30.Nitrato complex 4 can exist in two forms, with nitrate bound in a six-coordinated complex, or in a form where nitrate is outside of the coodination sphere; what forms depends on temperature and/or if excess nitrate anion is present. Details of these aspects are given in the SI; the key observation is that both forms are distinguishable from peroxynitrite complex 3.
- 31.Sharma SK, Kim H, Rogler PJA, Siegler M, Karlin KD. JBIC, J Biol Inorg Chem. 2016;21:729–743. doi: 10.1007/s00775-016-1369-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Yokoyama A, Han JE, Cho J, Kubo M, Ogura T, Siegler MA, Karlin KD, Nam W. J Am Chem Soc. 2012;134:15269–15272. doi: 10.1021/ja307384e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yokoyama A, Cho KB, Karlin KD, Nam W. J Am Chem Soc. 2013;135:14900–14903. doi: 10.1021/ja405891n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kumar P, Lee YM, Park YJ, Siegler MA, Karlin KD, Nam W. J Am Chem Soc. 2015;137:4284–4287. doi: 10.1021/ja513234b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tran NG, Kalyvas H, Skodje KM, Hayashi T, Moënne-Loccoz P, Callan PE, Shearer J, Kirschenbaum LJ, Kim E. J Am Chem Soc. 2011;133:1184–1187. doi: 10.1021/ja108313u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Skodje KM, Williard PG, Kim E. Dalton Trans. 2012;41:7849–7851. doi: 10.1039/c2dt30443k. [DOI] [PubMed] [Google Scholar]; (f) Kalita A, Kumar P, Mondal B. Chem Commun. 2012;48:4636–4638. doi: 10.1039/c2cc31117h. [DOI] [PubMed] [Google Scholar]; (g) Kalita A, Deka RC, Mondal B. Inorg Chem. 2013;52:10897–10903. doi: 10.1021/ic400890f. [DOI] [PubMed] [Google Scholar]
- 33.(a) Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]; (b) Beckman JS, Ischiropoulos H, Zhu L, Van der Woerd M, Smith C, Chen J, Harrison J, Martin JC, Tsai M. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]; (c) Gunaydin H, Houk KN. Chem Res Toxicol. 2009;22:894–898. doi: 10.1021/tx800463y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Beckman JS. Chem Res Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]; (e) O’Donnell VB, Eiserich JP, Chumley PH, Jablonsky MJ, Krishna NR, Kirk M, Barnes S, Darley-Usmar VM, Freeman BA. Chem Res Toxicol. 1999;12:83–92. doi: 10.1021/tx980207u. [DOI] [PubMed] [Google Scholar]; (f) Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, Challis GL. Nat Chem Biol. 2012;8:814–816. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Note: addition of phenol to the superoxo complex resulted in no change in UV–vis/EPR/rRaman spectroscopies, in agreement with the nearly thermoneutral binding from DFT. (PIm)FeIII-(O2−) (2) is not a good proton abstracter (even 10 equiv of phenol shows no reaction); also, proton transfer from phenol to the Fe-superoxo moiety is calculated from DFT to be a highly endergonic process. Therefore, we do not believe we are generating HO2.
- 35.Hunt AP, Lehnert N. Acc Chem Res. 2015;48:2117–2125. doi: 10.1021/acs.accounts.5b00167. [DOI] [PubMed] [Google Scholar]
- 36.(a) Park GY, Deepalatha S, Puiu SC, Lee D-H, Mondal B, Narducci Sarjeant AA, del Rio D, Pau MYM, Solomon EI, Karlin KD. JBIC, J Biol Inorg Chem. 2009;14:1301–1311. doi: 10.1007/s00775-009-0575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maiti D, Lee D-H, Narducci Sarjeant AA, Pau MYM, Solomon EI, Gaoutchenova K, Sundermeyer J, Karlin KD. J Am Chem Soc. 2008;130:6700–6701. doi: 10.1021/ja801540e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.