

Abstract
Salivary gland cancers are an incredibly heterogeneous group of tumors that include 24 histologically distinct tumor types. The use of new genetic methods has paved the way for promising advancements in our understanding of the molecular biology underlying each type of tumor. The objective of this review was to highlight common oncogenes, tumor suppressor genes, and cytogenetic and epigenetic changes associated with the most common tumor types: mucoepidermoid carcinoma, adenoid cystic carcinoma, salivary duct carcinoma, mammary analogue secretory carcinoma, hyalinizing clear cell carcinoma, carcinoma ex pleomorphic adenoma, and acinic cell carcinoma. Recent insights into the pathogenesis of each cancer subtype have helped better define and classify these tumors. Further research in salivary gland cancers should focus on determining the key genes involved in the tumorigenesis of each distinct malignancy and identifying individualized chemotherapies directed at these targets.
Keywords: acinic cell carcinoma, adenoid cystic carcinoma, cytogenetics, epigenetics, genetics, hyalinizing clear cell carcinoma, mammary analogue secretory carcinoma, mucoepidermoid carcinoma, salivary duct carcinoma, salivary gland cancer
Introduction
Salivary gland malignancies are a heterogeneous group of tumors that currently involves 24 histologically distinct cancer subtypes.1 Thus there is also significant heterogeneity in the aberrant genetic and molecular pathways that contribute to the development of each specific tumor. Systemic therapies for salivary gland cancers are reserved for advanced disease and have only achieved a modest response, with cisplatin-based regimens the most frequently studied.2 However, the advent and the now widespread use of new genetic methods have paved the way for promising advancements in our understanding of the molecular biology underlying each type of tumor.
The objective of this review was to present a broad current understanding of the genetic landscape that defines this heterogeneous group of tumors. We highlight the common cytogenetic and epigenetic changes associated with each type of salivary gland cancer as well as any oncogenes and tumor-suppressor genes (TSGs) that may play an important role in the pathogenesis of disease (Fig. 1). We hope that the information provided here will offer a solid foundation for the future exploration of more specific diagnostic tools and individualized, genetically targeted therapies.
Figure 1.
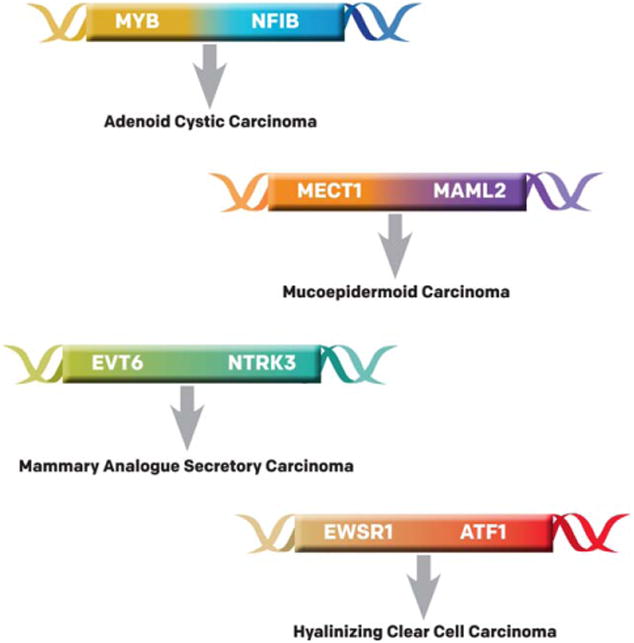
Known translocations identified in salivary gland cancers are illustrated. ATF1 indicates activating transcription factor 1; EVT6, ets variant 6; EWSR1, Ewing sarcoma binding protein 1; MAML2, mastermind-like transcriptional coactivator 2; MECT1, mucoepidermoid carcinoma translocated-1; MYB, v-myb avian myeloblastosis viral oncogene homolog; NFIB, nuclear factor I/B; NTRK3, neurotrophic tyrosine kinase, receptor, type 3.
Mucoepidermoid Carcinoma
Mucoepidermoid carcinoma (MEC) is the most common salivary gland malignancy, accounting for 30% to 35% of all malignant neoplasms of the major and minor salivary glands.3-5 Histologically, it is mainly composed of mucous, epidermoid, and intermediate cell types that form cysts.6,7 Current grading systems are based on the extent of cyst formation, differentiation of the 3 main cell types, and cytomorphologic changes.8-11 However, prognosis and treatment strategies based on these traditional grading systems can be controversial, and promising genetic and molecular markers can offer new outlooks in diagnosis and treatment.8,12
Mucoepidermoid Carcinoma Translocated-1–Mastermind-Like Gene 2 Translocation
The most common genetic alteration in MECs is a unique translocation t(11;19)(q21;p13) producing a fusion transcript of exon 1 of the mucoepidermoid carcinoma translocated-1 gene (MECT1 [aka CRTC1) ]) at 19p13 with exons 2 through 5 of a mastermind-like gene (MAML2) at 11q21.13 The fusion transcript can activate transcription of targets in the Notch pathway and can be present in 38% to 81% of MECs of the salivary gland.14-18 Specifically, it has been demonstrated that dysregulated Notch signaling underlies the pathogenesis of other malignancies, and the MECT1-MAML2 transcript can activate the Notch target gene Hes-1 (hes family basic helix-loop-helix transcription factor 1) in the absence of Notch ligand.19 The MECT1-MAML2 transcript can also be recruited to 3′,5′-cyclic adenosine monophosphate (cAMP)-responsive element-binding protein (CREB) sites to activate CREB-inducible genes that regulate cell proliferation and differentiation.13,19 This biologic effect of deregulated cAMP signaling may be essential for tumor cell growth.20
Presence of the fusion transcript can be identified in low-grade and high-grade MECs but is associated with a significantly better prognosis (less distant metastasis, higher disease-specific and overall survival, reduced local recurrence).15,18,21,22 This translocation is so specific for MEC that further studies have suggested that translocation-negative, high-grade MEC may be more appropriately categorized as a different tumor type alto-gether.15,21 In fact, multiple studies have suggested that most MECT1-MAML2 fusion-positive MECs are of the low-grade and intermediate-grade types and that high-grade MECs are a distinctive group of tumors.23
Other Genetic Considerations in MEC
In MECs, the overexpression of epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) has been explored. EGFR and HER2 overexpression was present in a minority of MECs and was associated with higher rate of metastasis and worse overall survival.24,25 Other evidence indicates that HER2 and EGFR copy number variations can be observed in high-grade MEC, irrespective of MECT1-MAML2 fusion status, offering another “hit” in the progression to malignancy and dedifferentiation.23 Overall, fusion-positive tumors have lower copy number variations compared with fusion-negative tumors, with the most frequent copy number variations detected in a variety of TSGs, such as deleted in colorectal carcinoma (DCC), SMAD family member 4 (SMAD4), cyclin-dependent kinase inhibitor 2A/2B (CDKN2A/B), and oncogenes, such as LYN proto-oncogene SRC family tyrosine kinase (LYN), v-mos Moloney murine sarcoma viral oncogene homolog (MOS), and pleomorphic adenoma 1 (PLAG1).26
Another specific genetic alteration that has been investigated in conjunction with the MECT1-MAML2 fusion is deletion in the CDKN2A/p16 gene. Fusion-positive MEC cases with CDKN2A deletion or hyper-methylation are associated with metastases and death, and patients who have MEC with better survival do not have this deletion.27 Fusion-negative cases with poor survival also have a CDKN2A deletion of methylation, suggesting that the p16 pathway may be responsible for additional events in the pathogenesis of MEC.
Taken together, Jee et al proposed that MECs should be thought of as 3 distinct groups of tumors: 1) a low-grade, fusion-positive MEC with few genomic imbalances and a good prognosis (fewer recurrences, metastases, and disease-related deaths); 2) a high-grade, fusion-positive MEC with other genomic imbalances and an unfavorable prognosis; and 3) a high-grade, fusion-negative adenocarcinoma (based on molecular characterization) with multiple genomic imbalances and an unfavorable prognosis.26
Other less frequently detected MEC-related fusions involve other members of the MECT/CRTC family. A CRTC3-MAML2 fusion has been discovered in 6% of MECs and appears to be mutually exclusive with the CRTC1-MAML2 fusion transcript. However, it is not associated with significant differences in disease-free survival compared with the CRTC1-MAML2 fusion.28
The potential role of high-risk human papilloma virus (HPV) in MEC remains controversial. Because high-risk HPV has been identified as a major prognostic bio-marker for oropharyngeal squamous cell carcinoma (SCC), there is great interest in exploring the role that HPV may play in other types of head and neck cancer. An initial study reported that E6/7 oncogene transcripts of HPV type 16 (HPV16) and HPV18 were identified in 30% and 13% of MECs, respectively, using reverse transcriptase-polymerase chain reaction.29 However, in subsequent studies, no detectable high-risk HPV was detected by in situ hybridization in MECs, and there was no significant correlation between HPV status and MECT1-MAML2 status.30,31
Adenoid Cystic Carcinoma
Adenoid cystic carcinoma (ACC) is a rare salivary gland malignancy with a yearly incidence of from 3 to 4.5 cases per million, but it is the most common malignant tumor to arise from the minor salivary glands.32,33 Clinically, ACC has a rather unusual and indolent course characterized by slow growth but late distant recurrence. Although most patients with ACC are alive at 5 years, a majority of patients die from their disease. Even from 5 to 20 years after diagnosis, the number of deaths is equal between ACC and other competing causes.34,35
Histologically, ACC is characterized by a mixture of myoepithelial and luminal cells that grow in a cribriform, tubular, or solid pattern. Solid tumors are considered the highest grade of ACC and are associated with a worse prognosis, specifically advanced stage and development of distant metastasis. Tumor cells are small, cuboidal, and deeply basophilic with little cytoplasm.36 Perineural invasion is also an unfavorable prognostic factor and is associated with distant metastasis.33 The mechanisms of ACC for both tumorigenesis and invasion are poorly understood, and current research in ACC has a strong focus on the potential genetic alterations that lead to these processes.
Myeloblastosis and the Myeloblastosis-Nuclear Factor I/B Fusion
Similar to MECs, translocations have been studied in ACCs for the purpose of both molecular diagnostic tools and target-specific therapies. An important discovery was made when Persson et al observed that a recurrent t(6;9)(q22-23;p23-24) translocation in ACC tumors consistently resulted in a fusion of the myeloblastosis (MYB) oncogene to the transcription factor nuclear factor I/B (NFIB).37 The MYB-NFIB fusion oncogene can be identified in approximately 1/3 to 1/2 of salivary ACCs and is not observed in other salivary gland tumors.38,39 MYB has an important role in cell proliferation, apoptosis, and differentiation; and downstream targets of MYB include mast/stem cell growth factor receptor (proto-oncogene c-Kit or tyrosine protein kinase Kit [c-kit]), cytochrome c oxidase subunit II (cox-2), and B-cell chronic lymphocytic lymphoma/leukemia 2 (bcl2).40
Studies have demonstrated that MYB is overexpressed in the majority of ACCs that have MYB-NFIB fusions, but is also overexpressed in fusion-negative ACCs, suggesting that MYB may be crucial in the pathogenesis of ACC.38 It is suspected that the mechanism for MYB up-regulation is caused by loss of its the 30-untranslated region, which includes regulatory micro-RNA target sites. Thus, translocation of other sequences to the flanking sites of the MYB gene can also lead to MYB overexpression.41 The methylation status of the MYB promoter has also been investigated. Cytosineguanine (CpG) islands in the MYB promoter did not exhibit differential methylation between ACC and normal salivary gland samples, suggesting that promoter hypomethylation does not contribute to the differential expression of MYB in ACC.42
The clinical significance of MYB overexpression in ACC remains a mystery. One study indicated that high MYB expression was associated with worse overall patient survival.41 However, more recent studies determined that MYB-NFIB tumor status and individual MYB expression were not significantly associated with disease-free or overall survival.40,43 More investigations are needed to better understand the exact role that MYB plays in the development and progression of ACC. Currently, there are no drugs available to target either the MYB-NFIB fusion gene or the MYB pathway.44
Cell Surface Receptors and Ligands Gain-of-function mutations in cell surface growth factors are a common feature of many malignancies and thus have been explored extensively in ACC. In general, luminal cells in ACC express high amounts of c-kit (aka cluster of differentiation 117 [CD117], a tyrosine kinase receptor), and myoepithelial cells express large amounts of EGFR.45,46
The c-kit transmembrane tyrosine kinase receptor is structurally related to platelet-derived growth factor and colony-stimulating factor-1 receptors. Expression of c-kit is often identified in ACCs and MECs, but common genetic alterations in c-kit observed in other types of cancers (ie, alterations in exons 11 and 17) are not present in salivary gland cancers.47 Nonetheless, c-kit expression in ACC does correlate with tumor grade, suggesting that there may be an alternative, unknown mechanism for c-kit activation in ACC.48 Investigations into this mechanism have demonstrated copy number gains of c-kit in a few ACCs.49 More recently, Tang et al identified a c-kit– associated zinc-finger transcription factor (Slug) that may act as a mediator of epithelial-mesenchymal transitions50 and could be associated with worse TNM stage, perineural invasion, locoregional recurrence, and distant metastases.51 Studies into c-kit have promising potential for targeted therapies, because the popular drug imatinib can inhibit c-kit receptors. Unfortunately, clinical trials involving imatinib in patients with ACC have not yielded positive results.52
EGFR and HER2 can be significantly overexpressed in all types of salivary gland malignancies, and EGFR/HER2 gene status correlates with tumor stage, tumor grade, disease-specific survival, and loss of phosphatase and tensin homolog (PTEN).24,53 In ACCs specifically, it has been observed that EGFR is overexpressed, whereas HER2 expression status remains controversial.54,55 Over-expression of epiregulin, an EGFR ligand, can promote in vitro migration and invasion in an ACC cell line. These effects may be mediated through activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), protein kinase C (Akt), and Cox-2.56 EGFR expression levels are correlated with worse histologic grade but not disease-free or overall survival.57 EGFR-targeted therapies like erlotinib have not yet been studied in depth among patients with ACC. The exact role of EGFR in ACC pathogenesis remains in question.
Important Epigenetic Changes in ACC Epigenetic alterations like DNA methylation and histone acetylation are important factors in human carcinogenesis. Methylation of cytosines in CpG islands in gene promoters can lead to transcriptional inactivation by blocking RNA polymerase binding sites and thus inactivate TSGs. Similarly, hypomethylation of these sites may lead to relative overexpression of oncogenes. The advent of quantitative methylation-specific polymerase chain reaction has allowed for researchers to take a closer look at the epigenetic differences between cancer and normal salivary glands.
Quantitative methylation-specific polymerase chain reaction has demonstrated that salivary gland cancers have increased methylation in several TSGs, including adenomatous polyposis coli (APC); amyloid β precursor protein-binding family A, member 1 (Mint 1); P-glycoprotein 9.5 (PGP9.5); retinoic acid receptor β (RAR-β); and TIMP metallopeptidase inhibitor 3 (Timp3).58 Similar methods have also demonstrated that several cyclin-dependent kinase inhibitors (CKIs) are frequently methylated in ACCs.59 CKIs are cell cycle regulators and tumor suppressors in various cancers. Hypermethylation of 1 specific CKI, p27, may lead to its down-regulation and contribute to disruption of the cell cycle in ACC tumorigenesis.59 Large-scale microarrays comparing ACC with normal salivary tissues have reported hypermethylation at up to 32 CpG islands; these islands were associated with genes involved in developmental, apoptotic, and other fundamental cellular pathways. One in particular, the engrailed homeobox 1 (EN1) gene, was correlated with histologic tumor grade and patient survival and has some potential as a possible biomarker.60
A genome-wide screen for epigenetic oncogene candidates also identified aquaporin-1 (AQP1) and suprabasin (SBSN) as promising candidates.61 Aquaporin is a small transmembrane protein responsible for water transport across membranes. The AQP1 promoter is significantly hypomethylated in ACC tumors compared with normal salivary gland tissues; this promoter hypomethylation is associated with increased AQP1 expression. In addition, AQP1 promoter hypermethylation is associated with improved overall survival.62 SBSN plays an important role in epidermal differentiation and maintaining anchorage-independent and anchorage-dependent cell proliferation in ACC. SBSN is significantly hypomethylated and up-regulated in ACC tumors compared with normal salivary glands, and this hypomethylation is associated with an increased risk of regional recurrence.63
Other Genetic Considerations in ACC
The potential role of epigenetics alterations in ACC is not at all surprising, considering the finding that 2 separate, large, whole-exome sequencing studies of ACC tumors have revealed a large number of variants in chromatin remodeling genes.64,65 These genes, which are responsible for histone modification and epigenetic regulation, are also often mutated in breast ACC.66 Stephens et al reported aberrations in chromatin remodeling genes in half of all salivary ACC samples sequenced, including some chromatin regulator genes that had not previously been implicated in cancer (SW/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 2 [SMARCA2]; chromodomain helicase DNA-binding protein 2 [CHD2]; bromodomain containing 2 [BRD2]; AT-rich interaction domain containing 5B [ARID5B]; and lysine-specific demethylase 5A [KDM5A]).64 Another study similarly reported chromatin regulator mutations in 35% of ACC tumors tested.65
Using whole-exome sequencing, Ho et al also identified recurrent mutations in the fibroblast growth factor– insulin-like growth factor–phosphatidylinositol 3-kinase (FGF-IGF-PI3K) pathway in 30% of tumors, establishing a potential targetable pathway for future therapy. However, relatively few consistent, individual mutations were identified in either study, confirming that ACC maintains a stable genome aside from the frequently identified translocation.
In addition to MYB-NFIB translocations, other cytogenetic changes have also been explored in ACC. Because ACCs do not display the degree of genomic instability associated with many other cancer types,67 many believe that chromosome regions displaying loss of heterozygosity are not simply nonspecific changes but, rather, are important clues to the pathogenesis of ACC. ACC tumors have exhibited copy number losses at chromosome 12q12-q13 and 1p32-36 and gains at 22q12-q13, 8, 16p, 17q, and 18.60 Rutherford et al interpret the high incidence of chromosome 12 deletions as consistent with the presence of specific TSGs on this chromosome.68 Genomic deletions in chromosome 6 can also occur in up to 57% of ACCs, and the hotspot of deletion (6q24.1-q25.1) contains candidate TSGs, including pleomorphic adenoma of the salivary gland gene like 1 (PLAG1) and large tumor suppressor 1 (LATS1).68
The scarcity of knowledge about the pathogenesis of ACC has also prompted investigators to explore novel methods in the detection of genetic abnormalities. Whole mitochondrial sequencing has demonstrated that ACC tumors contain a high incidence of mitochondrial mutations.69 The clinical and molecular implications of these mutations are not yet known. A comprehensive summary of the genetic alterations involved in ACC as well as all the other tumor types featured in this review is provided in Table 1.72
Table 1. Important Genetic Alterations in Salivary Gland Cancers.
| Cancer Type/Genetic Alteration | Gene(s) | Role in Tumor Pathogenesis |
|---|---|---|
| Mucoepidermoid carcinoma (MEC) Translocation, fusion transcript | MECT1-MAML2 (O'Neill 200913) | Activate the Notch target gene Hes-1 in the absence of Notch ligand (Kaye 200619); activate CREB inducible genes that regulate cell proliferation and differentiation (O'Neill 2009,13 Kaye 200619) Exact function unknown |
| Copy number variation | CRTC3-MAML2 (Nakayama 200928) HER2, EGFR (Ettl 201224) | Growth factor receptors, overexpression associated with poor rates of metastases and survival (Ettl 201224) |
| DCC, SMAD4 (Jee 201326) | Various tumor suppressor genes, exact role in MEC unknown (Jee 201326) | |
| Epigenetic | LYN, MOS, PLAG6 (Jee 201326) CDKN2A/p16 (Anzick 201027) | Various oncogenes, exact role in MEC unknown (Jee 201326) Tumor suppressor gene, cell cycle regulator, deletion/hypermethylation associated with poor survival (Anzick 201027) |
| Adenoid cystic carcinoma (ACC) Translocation, fusion transcript | MYB-NFIB (Persson 200937) | MYB is an oncogene involved in cell proliferation, apoptosis, and differentiation; downstream targets include c-kit, cox-2, and bcl2 (Bell 201140); controversial association with patient survival (Mitani 2011,41 Rettig 201543) |
| Epigenetic changes | APC, Mint1, PGP9, RAR-β, Timp3 (Durr 201058) p27 (Daa 200859) | Various tumor suppressor genes, hypermethylated in ACC, exact role unknown (Durr 201058) Cell cycle regulator hypermethylated in ACC, leads to down-regulation and disruption of cell cycle (Daa 200859), role in prognosis unknown |
| EN1 (Liu 201260) | Homeobox gene hypermethylated in ACC, correlates with histologic tumor grade and patient survival (Liu 201260) | |
| AQ1 (Shao 2011,61 Tan 201462) | Transmembrane protein for water transport, hypomethylated in ACC, hypermethylation is associated with better survival (Shao 2011,61 Tan 201462) | |
| SBSN (Shao 201263) | Involved in epidermal differentiation, anchorage-independent growth, hypomethylated in ACC and associated with increased risk of recurrence (Shao 201263) | |
| Copy number losses/deletion | 12q12-q13, 1p32-36, and gains at 22q12-q13, 8, 16p, 17q, 18 (Liu 201260) | High presence of tumor suppressor genes at these loci, exact role in ACC unknown (Liu 201260) |
| Hotspot 6q24.1-q25.1 including PLAG1, LATS1 (Rutherford 200668) | Tumor suppressor genes, exact role in ACC unknown (Rutherford 200668) | |
| Copy number gains/up-regulation | c-kit (Holst 1999,48 Freier 200549) | Transmembrane tyrosine kinase receptor, overexpression associated with worse tumor grade (Holst 1999,48 Freier 200549) |
| EGFR (Monteiro 200957) | Growth factor receptor, overexpression associated with worse histologic grade (Monteiro 200957) | |
| Salivary duct carcinoma (SDC) Aneuploidy/up-regulation | HER2 (Zhu 20156) | Growth factor receptor, overexpression in SDC associated with worse recurrence rates, metastases, and survival (Zhu 20156) |
| p53 (Zhu 20156) | Plays a role in genome stability, overexpression in SDC associated with worse recurrence rates, metastases, and survival (Zhu 20156) | |
| AR (Simpson 201470) | Androgen receptor (AR)-negative tumors are more aggressive than AR-positive tumors (Simpson 201470) | |
| Mammary analogue secretory carcinoma (MASC) Translocation | ETV6-NTRK3 (Stenman 201471) | Fusion product is a chimeric tyrosine kinase with the ability to activate the Ras-MAP pathway and PI3K-Akt pathway (Stenman 201471), role in prognosis unknown |
| Hyalinizing clear cell carcinoma (HCCC) Translocation | EWSR1-ATF (Stenman 201372) | Exact function unknown (Stenman 201372) |
| Carcinoma ex pleomorphic adenoma Rearrangements | PLAG1, HMGA2 | Transcription factors that can undergo a variety of rearrangements; consequences of rearrangements unknown (Stenman 201372) |
Abbreviations: Akt, protein kinase B; APC, adenomatosis polyposis coli; AQ1, aquaporin 1; ATF, activating transcription factor; bcl2, B-cell chronic lymphocytic lymphoma/leukemia 2; CDKN2A/p16, cyclin-dependent kinase inhibitor 2A; c-kit, mast/stem cell growth factor receptor (proto-oncogene c-Kit or tyrosine protein kinase Kit); cox-2, cytochrome c oxidase subunit II; CREB, cyclic AMP responsive binding protein; CRTC3, CREB regulated transcription coactivator 3; DCC, deleted in colorectal carcinoma; EGFR, epidermal growth factor receptor; EN1, engrailed homeobox 1; ETV6, ets variant 6; EWSR1, Ewing sarcoma RNA binding protein 1; HER2, human epidermal growth factor receptor 2; Hes-1, hes family basic helix-loop-helix transcription factor 1; HMGA2, high-mobility group AT-hook 2; LATS1, large tumor suppressor kinase 1; LYN, LYN proto-oncogene, Src family tyrosine kinase; MAP, mitogen-activated protein; MAML2, mastermind-like transcriptional coactivator 2; MECT1, mucoepidermoid carcinoma translocated-1; Mint1, amyloid β precursor protein-binding family A, member 1 (APBA1) adaptor protein; MOS, v-mos Moloney murine sarcoma viral oncogene homolog; MYB, v-myb avian myeloblastosis viral oncogene homolog; NFIB, nuclear factor I/B; NTRK3, neurotrophic tyrosine kinase, receptor, type 3; p16, CDKN2A multiple tumor suppressor; p27, cyclin-dependent kinase inhibitor 1B; p53, tumor protein 53; PGP9, P-glycoprotein 9; PI3K, phosphoinositide 3-kinase; PLAG1, pleomorphic adenoma 1; PLAG4, pleomorphic adenoma 4; RAR-β, retinoic acid receptor β; Ras, rat sarcoma; SBSN, suprabasin; SMAD4, SMAD family member 4; Timp3, TIMP metallopeptidase inhibitor 3.
Salivary Duct Carcinoma
Salivary duct carcinoma (SDC) can account for 5% to 10% of salivary carcinomas, often occurs in men aged >50 years, and is one of the most aggressive cancers that can arise in the salivary glands.70,71 It is classified as an aggressive adenocarcinoma that histologically resembles high-grade breast ductal carcinoma and can arise as a malignant component of carcinoma ex pleomorphic adenoma.70 On immunohistochemistry, cells are usually positive for cytokeratin 7 (CK7), CK20, and HER2 but negative for S100. Unlike ductal carcinoma of the breast, the majority of SDCs do not stain positive for the estrogen receptor.70 Instead, from 67% to 83% of SDCs express the androgen receptor (AR).70 Clinically, SDC has an aggressive course, and patients commonly develop regional and distant metastases. Most die from the disease within 3 to 4 years of diagnosis.71
Investigations into the pathogenesis of SDC have revealed overexpression of HER2 in greater than 1/3 of patients with SDC, similar to its breast counterpart.71 Overexpression of p53 and high frequency of DNA aneuploidy are also common.73 Both HER2 and p53 overexpression in SDCs are correlated with early local disease recurrence, distant disease metastasis, and survival rates.6,74 Conversely, expression of AR is associated with greater disease-free survival.75 Other mutations described in SDC include v-Raf murine sarcoma viral oncogene homolog B (BRAF); phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit α (PIK3CA); receptor tyrosineprotein kinase erbB-2 (ERBB2); PTEN; and EGFR.76-80
On the basis of the differential expression and prognostic value of HER2 and AR in SDCs, some investigators have proposed that SDC should be classified based on the presence or absence of AR and HER2.70 This not only would allow for prognostic stratification but also would make way for new therapeutic strategies directly targeted toward these molecules. The HER2 inhibitor trastuzumab has exhibited great success in the treatment of breast cancer. In patients with progressive SDC, trastuzumab may improve disease-free and overall survival.71 Androgen-deprivation therapies (ADTs) have also been explored in small studies and have produced promising results.70 One case series of 10 patients with recurrent or metastatic SDC who received ADT had a median progression-free survival of 12 months.81 Other reports suggest extending ADT beyond patients with advanced disease, using a combination of ADT and radiation as definitive therapy in poor surgical candidates.82
Mammaryanalogue Secretory Carcinoma
Mammary analogue secretory carcinoma (MASC) is a relatively new salivary gland tumor first recognized in 2010 that histologically resembles secretory carcinoma of the breast.83 It is an infiltrative tumor that grows in microcystic and tubular patterns, with PAS-positive/diastase-resistant secretory material within the luminal spaces,84 and can often be misclassified as acinic cell carcinoma.85 Since its definition, many tumors that were previously labeled as acinic cell carcinomas have now been reclassified as MASCs.70 On immunohistochemistry, MASC can be distinguished from other salivary gland tumors by its strong positivity for S100, mammaglobin, cytokeratin, and vimentin.86 Clinically, MASC can occur at any age and has a slight male predominance.70 Although few long-term follow-up studies have been conducted, it is considered a low-grade neoplasm with recurrence and metastases rates <20%. However, MASC usually has a more aggressive clinical course compared with its breast counterpart, and it has the capacity for high-grade transformation.86
The genetic hallmark of this tumor, similar to its counterpart in the breast, is a translocation between the ets variant 6 (ETV6) and neurotrophic tyrosine kinase, receptor, type 3 (NTRK3) genes. This t(12;15)(p13;q25) translocation produces a fusion product that contains the transcriptional regulator ETV6 fused with the membrane receptor kinase NTRK3, which activates cell proliferation and survival.83 Although the exact function of this fusion product is not yet known, it may be related to the high expression of signal transducer and activator of transcription molecule 5a (STAT5α) in MASCs.83 The fusion product, a chimeric tyrosine kinase, has the ability to activate both the rat sarcoma (Ras) mitogen-activated protein (MAP) kinase (Ras-MAP) pathway and the PI3K-Akt pathway.71 One case series of 14 Japanese patients with MASC patients indicated that, although all had an ETV6 split, only 43% had the fusion transcript, suggesting that EVT6 may also occasionally fuse with non-NTRK3 genes.87
The significance of this ETV6-NTRK3 fusion for future treatment is not well understood either. The transcript target has promising pharmaceutical potential, because some ETV6-NTRK3–positive leukemias have responded well to tyrosine inhibitors.88 More research is needed to characterize the exact role that this fusion product plays in prognosis and management.
Hyalinizing Clear Cell Carcinoma
Hyalinizing clear cell carcinoma (HCCC) is an epithelial malignancy of the salivary glands composed of clear cells that form nests and cords. Because many other salivary gland tumors can have a clear cell component, clear cell carcinoma is a diagnosis of exclusion.86 Histologically, the clear cells grow in a pattern of hyalinization with focal mucinous differentiation. HCCC is a very rare, low-grade tumor with a very good prognosis and rare metastases.71
Recently, a disease-defining translocation in the Ewing sarcoma RNA-binding protein 1 (EWSR1) gene was identified in >80% of HCCCs and may be used to distinguish it from its many mimics.70 The most common fusion, a t(12;22)(q13;q12) translocation, produces a fusion transcript consisting of the genes EWSR1 and activating transcription factor 1 (ATF1).72 Although the translocation appears to be very specific to HCCC, its exact function is not well known. It is noteworthy that this specific translocation is also present in a majority of odonogenic clear cell carcinomas, suggesting a biologic link between the 2 malignancies.86
Carcinoma Ex Pleomorphic Adenoma
Carcinoma ex pleomorphic adenoma is a carcinoma that arises from a benign pleomorphic adenoma and can account for 10% to 15% of salivary gland cancers.8 The carcinomatous component of this tumor is usually an aggressive, high-grade adenocarcinoma, but it also can comprise other types of carcinomas.8 Because of the tremendous diversity in its histologic appearance, recent molecular studies have attempted to identify the unifying genetic changes that define this tumor. After all, pleomorphic adenoma, the benign counterpart to carcinoma ex pleomorphic adenoma, is characterized by a very specific pattern of translocations involving the transcription factors PLAG1 and high-mobility group AT-hook 2 (HMGA2) and various fusion partner genes.89 Small studies have demonstrated that carcinoma ex pleomorphic adenomas also harbor common rearrangements and copy number variations in the pleomorphic adenoma-specific genes PLAG1 and HMGA2.72 The consequences of these rearrangements are not well understood.
Salivary Gland SCC
Although SCC is the most common form of head and neck cancer, primary SCC is rare in the salivary glands, and only 7% of parotid cancers are identified as primary SCC.90 Recent work in mouse models has demonstrated that mice with increased Wnt/β-catenin signaling rapidly develop salivary gland SCC.91 When cancer stem cells were isolated from these SCC tumors, investigators were able to demonstrated that the β-catenin pathway drove tumor proliferation through histone modifications at the promoters of stem cell-associated genes.91 It is noteworthy that mutations in components of the Wnt/β-catenin pathway have also recently been implicated in ACC—another type of salivary gland malignancy that displays a high prevalence of epigenetic aberrations.60
Acinic Cell Carcinoma
Acinic cell carcinoma is traditionally considered another low-grade tumor with a good prognosis; however, like many other salivary gland cancers, late recurrence and metastatic disease can occur.8 Very little is known about the genetic alterations that contribute to this disease. Since the emergence of MASCs as a distinct tumor category, the defining characteristics of acinic cell carcinoma have come under question. New evidence suggests that it may be a far more aggressive tumor than originally reported.92 The mammalian target of rapamycin (mTOR) pathway has been explored in the pathogenesis of acinic cell carcinoma, and some immunohistochemical evidence supports the overactivation of this pathway.71 Further evidence implicating the mTOR pathway in acinic cell carcinoma may lead to new drug trials involving the many mTOR pathway inhibitors (ie, rapamycin).
Conclusion
Salivary gland cancers are a histologically and molecularly heterogeneous group of tumors that have distinct genetic origins. Currently, the mainstay of treatment for salivary gland cancer is surgical resection and postoperative radiotherapy. At this point, chemotherapy and targeted therapy play only a limited role. In fact, the most recent National Comprehensive Cancer Network guidelines only recommend systemic therapy for unresectable, persistent, or recurrent disease. Platinum-based agents have been the most well studied systemic therapies, but the evidence behind this practice is solely based on a mild response observed in nonrandomized studies.93
Recent discoveries in the genetic alterations of each tumor type have helped us better define and categorize these salivary malignancies. However, although many candidate oncogenes and TSGs have been discovered, the diagnostic and therapeutic potential of these genetic anomalies remains to be seen. Although the genetic makeup of each tumor type appears to be unique, there are common pathways and themes shared among the various types of salivary gland tumors—abnormalities in epigenetic regulation, oncogenic fusion products, and overexpression of cell surface receptors. Therefore, effective drug development should focus on targeting the key players upon which multiple oncogenic pathways converge. Some targeted therapy clinical trials have begun to make progress, but several promising aforementioned leads require more evidence to impact clinical practice. Future systemic therapy for salivary gland cancers hopefully will use a combination of whole-exome tumor sequencing and specific molecular inhibitors to form individualized treatment regimens for every patient afflicted with this disease.
Acknowledgments
Funding Support: This study was supported by a grant from the National Institutes of Health/National Institute of Dental and Craniofacial Research (R01-023227).
Footnotes
Conflict of Interest Disclosures: The authors made no disclosures.
References
- 1.Gillespie MB, Albergotti WG, Eisele DW. Recurrent salivary gland cancer. Curr Treat Options Oncol. 2012;13:58–70. doi: 10.1007/s11864-011-0174-0. [DOI] [PubMed] [Google Scholar]
- 2.Adelstein DJ, Koyfman SA, El-Naggar AK, Hanna EY. Biology and management of salivary gland cancers. Semin Radiat Oncol. 2012;22:245–253. doi: 10.1016/j.semradonc.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Coca-Pelaz A, Rodrigo JP, Triantafyllou A, et al. Salivary mucoepidermoid carcinoma revisited. Eur Arch Otorhinolaryngol. 2015;272:799–819. doi: 10.1007/s00405-014-3053-z. [DOI] [PubMed] [Google Scholar]
- 4.Luna MA. Salivary mucoepidermoid carcinoma: revisited. Adv Anat Pathol. 2006;13:293–307. doi: 10.1097/01.pap.0000213058.74509.d3. [DOI] [PubMed] [Google Scholar]
- 5.Speight PM, Barrett AW. Salivary gland tumours. Oral Dis. 2002;8:229–240. doi: 10.1034/j.1601-0825.2002.02870.x. [DOI] [PubMed] [Google Scholar]
- 6.Zhu S, Schuerch C, Hunt J. Review and updates of immunohistochemistry in selected salivary gland and head and neck tumors. Arch Pathol Lab Med. 2015;139:55–66. doi: 10.5858/arpa.2014-0167-RA. [DOI] [PubMed] [Google Scholar]
- 7.Adams A, Warner K, Nor JE. Salivary gland cancer stem cells. Oral Oncol. 2013;49:845–853. doi: 10.1016/j.oraloncology.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seethala RR. Histologic grading and prognostic biomarkers in salivary gland carcinomas. Adv Anat Pathol. 2011;18:29–45. doi: 10.1097/PAP.0b013e318202645a. [DOI] [PubMed] [Google Scholar]
- 9.Batsakis JG, Luna MA. Histopathologic grading of salivary gland neoplasms: I. Mucoepidermoid carcinomas. Ann Otol Rhinol Laryngol. 1990;99(10 pt 1):835–838. doi: 10.1177/000348949009901015. [DOI] [PubMed] [Google Scholar]
- 10.Goode RK, Auclair PL, Ellis GL. Mucoepidermoid carcinoma of the major salivary glands: clinical and histopathologic analysis of 234 cases with evaluation of grading criteria. Cancer. 1998;82:1217–1224. doi: 10.1002/(sici)1097-0142(19980401)82:7<1217::aid-cncr2>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 11.Brandwein MS, Ferlito A, Bradley PJ, Hille JJ, Rinaldo A. Diagnosis and classification of salivary neoplasms: pathologic challenges and relevance to clinical outcomes. Acta Otolaryngol. 2002;122:758–764. [PubMed] [Google Scholar]
- 12.Katabi N, Ghossein R, Ali S, Dogan S, Klimstra D, Ganly I. Prognostic features in mucoepidermoid carcinoma of major salivary glands with emphasis on tumour histologic grading. Histopathology. 2014;65:793–804. doi: 10.1111/his.12488. [DOI] [PubMed] [Google Scholar]
- 13.O'Neill ID. t(11;19) translocation and CRTC1-MAML2 fusion oncogene in mucoepidermoid carcinoma. Oral Oncol. 2009;45:2–9. doi: 10.1016/j.oraloncology.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 14.Verdorfer I, Fehr A, Bullerdiek J, et al. Chromosomal imbalances, 11q21 rearrangement and MECT1-MAML2 fusion transcript in mucoepidermoid carcinomas of the salivary gland. Oncol Rep. 2009;22:305–311. [PubMed] [Google Scholar]
- 15.Tirado Y, Williams MD, Hanna EY, Kaye FJ, Batsakis JG, El-Naggar AK. CRTC1/MAML2 fusion transcript in high grade mucoepidermoid carcinomas of salivary and thyroid glands and War-thin's tumors: implications for histogenesis and biologic behavior. Genes Chromosomes Cancer. 2007;46:708–715. doi: 10.1002/gcc.20458. [DOI] [PubMed] [Google Scholar]
- 16.Tonon G, Modi S, Wu L, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet. 2003;33:208–213. doi: 10.1038/ng1083. [DOI] [PubMed] [Google Scholar]
- 17.Martins C, Cavaco B, Tonon G, Kaye FJ, Soares J, Fonseca I. A study of MECT1-MAML2 in mucoepidermoid carcinoma and War-thin's tumor of salivary glands. J Mol Diagn. 2004;6:205–210. doi: 10.1016/S1525-1578(10)60511-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Behboudi A, Enlund F, Winnes M, et al. Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer. 2006;45:470–481. doi: 10.1002/gcc.20306. [DOI] [PubMed] [Google Scholar]
- 19.Kaye FJ. Emerging biology of malignant salivary gland tumors offers new insights into the classification and treatment of mucoepidermoid cancer. Clin Cancer Res. 2006;12:3878–3881. doi: 10.1158/1078-0432.CCR-06-0791. [DOI] [PubMed] [Google Scholar]
- 20.Komiya T, Park Y, Modi S, Coxon AB, Oh H, Kaye FJ. Sustained expression of Mect1-Maml2 is essential for tumor cell growth in salivary gland cancers carrying the t(11;19) translocation. Oncogene. 2006;25:6128–6132. doi: 10.1038/sj.onc.1209627. [DOI] [PubMed] [Google Scholar]
- 21.Seethala RR, Dacic S, Cieply K, Kelly LM, Nikiforova MN. A reappraisal of the MECT1/MAML2 translocation in salivary mucoepidermoid carcinomas. Am J Surg Pathol. 2010;34:1106–1121. doi: 10.1097/PAS.0b013e3181de3021. [DOI] [PubMed] [Google Scholar]
- 22.Okabe M, Miyabe S, Nagatsuka H, et al. MECT1-MAML2 fusion transcript defines a favorable subset of mucoepidermoid carcinoma. Clin Cancer Res. 2006;12:3902–3907. doi: 10.1158/1078-0432.CCR-05-2376. [DOI] [PubMed] [Google Scholar]
- 23.Nakano T, Yamamoto H, Hashimoto K, et al. HER2 and EGFR gene copy number alterations are predominant in high-grade salivary mucoepidermoid carcinoma irrespective of MAML2 fusion status. Histopathology. 2013;63:378–392. doi: 10.1111/his.12183. [DOI] [PubMed] [Google Scholar]
- 24.Ettl T, Stiegler C, Zeitler K, et al. EGFR, HER2, survivin, and loss of pSTAT3 characterize high-grade malignancy in salivary gland cancer with impact on prognosis. Hum Pathol. 2012;43:921–931. doi: 10.1016/j.humpath.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 25.Ettl T, Schwarz S, Kleinsasser N, Hartmann A, Reichert TE, Driemel O. Overexpression of EGFR and absence of C-KIT expression correlate with poor prognosis in salivary gland carcinomas. Histopathology. 2008;53:567–577. doi: 10.1111/j.1365-2559.2008.03159.x. [DOI] [PubMed] [Google Scholar]
- 26.Jee KJ, Persson M, Heikinheimo K, et al. Genomic profiles and CRTC1-MAML2 fusion distinguish different subtypes of mucoepidermoid carcinoma. Mod Pathol. 2013;26:213–222. doi: 10.1038/modpathol.2012.154. [DOI] [PubMed] [Google Scholar]
- 27.Anzick SL, Chen WD, Park Y, et al. Unfavorable prognosis of CRTC1-MAML2 positive mucoepidermoid tumors with CDKN2A deletions. Genes Chromosomes Cancer. 2010;49:59–69. doi: 10.1002/gcc.20719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakayama T, Miyabe S, Okabe M, et al. Clinicopathological significance of the CRTC3-MAML2 fusion transcript in mucoepidermoid carcinoma. Mod Pathol. 2009;22:1575–1581. doi: 10.1038/modpathol.2009.126. [DOI] [PubMed] [Google Scholar]
- 29.Isayeva T, Said-Al-Naief N, Ren Z, Li R, Gnepp D, Brandwein-Gensler M. Salivary mucoepidermoid carcinoma: demonstration of transcriptionally active human papillomavirus 16/18. Head Neck Pathol. 2013;7:135–148. doi: 10.1007/s12105-012-0411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bishop JA, Yonescu R, Batista D, Yemelyanova A, Ha PK, Westra WH. Mucoepidermoid carcinoma does not harbor transcriptionally active high risk human papillomavirus even in the absence of the MAML2 translocation. Head Neck Pathol. 2014;8:298–302. doi: 10.1007/s12105-014-0541-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jour G, West K, Ghali V, Shank D, Ephrem G, Wenig BM. Differential expression of p16(INK4A) and cyclin D1 in benign and malignant salivary gland tumors: a study of 44 cases. Head Neck Pathol. 2013;7:224–231. doi: 10.1007/s12105-012-0417-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bjorndal K, Krogdahl A, Therkildsen MH, et al. Salivary gland carcinoma in Denmark 1990–2005: a national study of incidence, site and histology. Results of the Danish Head and Neck Cancer Group (DAHANCA) Oral Oncol. 2011;47:677–682. doi: 10.1016/j.oraloncology.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 33.Coca-Pelaz A, Rodrigo JP, Bradley PJ, et al. Adenoid cystic carcinoma of the head and neck—an update. Oral Oncol. 2015;51:652–661. doi: 10.1016/j.oraloncology.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 34.Lloyd S, Yu JB, Wilson LD, Decker RH. Determinants and patterns of survival in adenoid cystic carcinoma of the head and neck, including an analysis of adjuvant radiation therapy. Am J Clin Oncol. 2011;34:76–81. doi: 10.1097/COC.0b013e3181d26d45. [DOI] [PubMed] [Google Scholar]
- 35.Spiro RH. Distant metastasis in adenoid cystic carcinoma of salivary origin. Am J Surg. 1997;174:495–498. doi: 10.1016/s0002-9610(97)00153-0. [DOI] [PubMed] [Google Scholar]
- 36.Gondivkar SM, Gadbail AR, Chole R, Parikh RV. Adenoid cystic carcinoma: a rare clinical entity and literature review. Oral Oncol. 2011;47:231–236. doi: 10.1016/j.oraloncology.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 37.Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A. 2009;106:18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitani Y, Li J, Rao PH, et al. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin Cancer Res. 2010;16:4722–4731. doi: 10.1158/1078-0432.CCR-10-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.West RB, Kong C, Clarke N, et al. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol. 2011;35:92–99. doi: 10.1097/PAS.0b013e3182002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell D, Roberts D, Karpowicz M, Hanna EY, Weber RS, El-Naggar AK. Clinical significance of Myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer Biol Ther. 2011;12:569–573. doi: 10.4161/cbt.12.7.17008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitani Y, Rao PH, Futreal PA, et al. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: association with MYB-NFIB chimeric fusion, MYB expression, and clinical outcome. Clin Cancer Res. 2011;17:7003–7014. doi: 10.1158/1078-0432.CCR-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shao C, Bai W, Junn JC, et al. Evaluation of MYB promoter methylation in salivary adenoid cystic carcinoma. Oral Oncol. 2011;47:251–255. doi: 10.1016/j.oraloncology.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rettig EM, Tan M, Ling S, et al. MYB rearrangement and clinico-pathologic characteristics in head and neck adenoid cystic carcinoma. Laryngoscope. 2015;125:E292–E299. doi: 10.1002/lary.25356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chae YK, Chung SY, Davis AA, et al. Adenoid cystic carcinoma: current therapy and potential therapeutic advances based on genomic profiling. Oncotarget. 2015;6:37117–37134. doi: 10.18632/oncotarget.5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheuk W, Chan JK. Advances in salivary gland pathology. Histopathology. 2007;51:1–20. doi: 10.1111/j.1365-2559.2007.02719.x. [DOI] [PubMed] [Google Scholar]
- 46.Bell D, Hanna EY. Head and neck adenoid cystic carcinoma: what is new in biological markers and treatment? Curr Opin Otolaryngol Head Neck Surg. 2013;21:124–129. doi: 10.1097/MOO.0b013e32835c05fd. [DOI] [PubMed] [Google Scholar]
- 47.Jeng YM, Lin CY, Hsu HC. Expression of the c-kit protein is associated with certain subtypes of salivary gland carcinoma. Cancer Lett. 2000;154:107–111. doi: 10.1016/s0304-3835(00)00387-6. [DOI] [PubMed] [Google Scholar]
- 48.Holst VA, Marshall CE, Moskaluk CA, Frierson HF., Jr KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod Pathol. 1999;12:956–960. [PubMed] [Google Scholar]
- 49.Freier K, Flechtenmacher C, Walch A, et al. Differential KIT expression in histological subtypes of adenoid cystic carcinoma (ACC) of the salivary gland. Oral Oncol. 2005;41:934–939. doi: 10.1016/j.oraloncology.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 50.Tang YL, Fan YL, Jiang J, et al. C-kit induces epithelial-mesenchymal transition and contributes to salivary adenoid cystic cancer progression. Oncotarget. 2014;5:1491–1501. doi: 10.18632/oncotarget.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang Y, Liang X, Zheng M, et al. Expression of c-kit and Slug correlates with invasion and metastasis of salivary adenoid cystic carcinoma. Oral Oncol. 2010;46:311–316. doi: 10.1016/j.oraloncology.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 52.Hotte SJ, Winquist EW, Lamont E, et al. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: a Princess Margaret Hospital phase II consortium study. J Clin Oncol. 2005;23:585–590. doi: 10.1200/JCO.2005.06.125. [DOI] [PubMed] [Google Scholar]
- 53.Ettl T, Baader K, Stiegler C, et al. Loss of PTEN is associated with elevated EGFR and HER2 expression and worse prognosis in salivary gland cancer. Br J Cancer. 2012;106:719–726. doi: 10.1038/bjc.2011.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dori S, Vered M, David R, Buchner A. HER2/neu expression in adenoid cystic carcinoma of salivary gland origin: an immunohistochemical study. J Oral Pathol Med. 2002;31:463–467. doi: 10.1034/j.1600-0714.2002.00017.x. [DOI] [PubMed] [Google Scholar]
- 55.Vered M, Braunstein E, Buchner A. Immunohistochemical study of epidermal growth factor receptor in adenoid cystic carcinoma of salivary gland origin. Head Neck. 2002;24:632–636. doi: 10.1002/hed.10104. [DOI] [PubMed] [Google Scholar]
- 56.Hu K, Li SL, Gan YH, Wang CY, Yu GY. Epiregulin promotes migration and invasion of salivary adenoid cystic carcinoma cell line SACC-83 through activation of ERK and Akt. Oral Oncol. 2009;45:156–163. doi: 10.1016/j.oraloncology.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 57.Monteiro LS, Bento MJ, Palmeira C, Lopes C. Epidermal growth factor receptor immunoexpression evaluation in malignant salivary gland tumours. J Oral Pathol Med. 2009;38:508–513. doi: 10.1111/j.1600-0714.2009.00770.x. [DOI] [PubMed] [Google Scholar]
- 58.Durr ML, Mydlarz WK, Shao C, et al. Quantitative methylation profiles for multiple tumor suppressor gene promoters in salivary gland tumors [serial online] PLoS One. 2010;5:e10828. doi: 10.1371/journal.pone.0010828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Daa T, Kashima K, Kondo Y, Yada N, Suzuki M, Yokoyama S. Aberrant methylation in promoter regions of cyclin-dependent kinase inhibitor genes in adenoid cystic carcinoma of the salivary gland. APMIS. 2008;116:21–26. doi: 10.1111/j.1600-0463.2008.00773.x. [DOI] [PubMed] [Google Scholar]
- 60.Liu J, Shao C, Tan ML, Mu D, Ferris RL, Ha PK. Molecular biology of adenoid cystic carcinoma. Head Neck. 2012;34:1665–1677. doi: 10.1002/hed.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shao C, Sun W, Tan M, et al. Integrated, genome-wide screening for hypomethylated oncogenes in salivary gland adenoid cystic carcinoma. Clin Cancer Res. 2011;17:4320–4330. doi: 10.1158/1078-0432.CCR-10-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan M, Shao C, Bishop JA, et al. Aquaporin-1 promoter hypermethylation is associated with improved prognosis in salivary gland adenoid cystic carcinoma. Otolaryngol Head Neck Surg. 2014;150:801–807. doi: 10.1177/0194599814521569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shao C, Tan M, Bishop JA, et al. Suprabasin is hypomethylated and associated with metastasis in salivary adenoid cystic carcinoma [serial online] PLoS One. 2012;7:e48582. doi: 10.1371/journal.pone.0048582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stephens PJ, Davies HR, Mitani Y, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965–2968. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martelotto LG, De Filippo MR, Ng CK, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol. doi: 10.1002/path.4573. published online ahead of print June 12, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu Y, Baras AS, Shirasuna K, Frierson HF, Jr, Moskaluk CA. Concurrent loss of heterozygosity and copy number analysis in adenoid cystic carcinoma by SNP genotyping arrays. Lab Invest. 2007;87:430–439. doi: 10.1038/labinvest.3700536. [DOI] [PubMed] [Google Scholar]
- 68.Rutherford S, Yu Y, Rumpel CA, Frierson HF, Jr, Moskaluk CA. Chromosome 6 deletion and candidate tumor suppressor genes in adenoid cystic carcinoma. Cancer Lett. 2006;236:309–317. doi: 10.1016/j.canlet.2005.05.049. [DOI] [PubMed] [Google Scholar]
- 69.Mithani SK, Shao C, Tan M, et al. Mitochondrial mutations in adenoid cystic carcinoma of the salivary glands [serial online] PLoS One. 2009;4:e8493. doi: 10.1371/journal.pone.0008493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Simpson RH, Skalova A, Di Palma S, Leivo I. Recent advances in the diagnostic pathology of salivary carcinomas. Virchows Arch. 2014;465:371–384. doi: 10.1007/s00428-014-1639-x. [DOI] [PubMed] [Google Scholar]
- 71.Stenman G, Persson F, Andersson MK. Diagnostic and therapeutic implications of new molecular biomarkers in salivary gland cancers. Oral Oncol. 2014;50:683–690. doi: 10.1016/j.oraloncology.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 72.Stenman G. Fusion oncogenes in salivary gland tumors: molecular and clinical consequences. Head Neck Pathol. 2013;7(suppl 1):S12–S19. doi: 10.1007/s12105-013-0462-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoang MP, Callender DL, Sola Gallego JJ, et al. Molecular and biomarker analyses of salivary duct carcinomas: comparison with mammary duct carcinoma. Int J Oncol. 2001;19:865–871. doi: 10.3892/ijo.19.4.865. [DOI] [PubMed] [Google Scholar]
- 74.Jaehne M, Roeser K, Jaekel T, Schepers JD, Albert N, Loning T. Clinical and immunohistologic typing of salivary duct carcinoma: a report of 50 cases. Cancer. 2005;103:2526–2533. doi: 10.1002/cncr.21116. [DOI] [PubMed] [Google Scholar]
- 75.Masubuchi T, Tada Y, Maruya S, et al. Clinicopathological significance of androgen receptor, HER2, Ki-67 and EGFR expressions in salivary duct carcinoma. Int J Clin Oncol. 2015;20:35–44. doi: 10.1007/s10147-014-0674-6. [DOI] [PubMed] [Google Scholar]
- 76.Nardi V, Sadow PM, Juric D, et al. Detection of novel actionable genetic changes in salivary duct carcinoma helps direct patient treatment. Clin Cancer Res. 2013;19:480–490. doi: 10.1158/1078-0432.CCR-12-1842. [DOI] [PubMed] [Google Scholar]
- 77.Williams MD, Roberts DB, Kies MS, Mao L, Weber RS, El-Naggar AK. Genetic and expression analysis of HER-2 and EGFR genes in salivary duct carcinoma: empirical and therapeutic significance. Clin Cancer Res. 2010;16:2266–2274. doi: 10.1158/1078-0432.CCR-09-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ku BM, Jung HA, Sun JM, et al. High-throughput profiling identifies clinically actionable mutations in salivary duct carcinoma [serial online] J Transl Med. 2014;12:299. doi: 10.1186/s12967-014-0299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qiu W, Tong GX, Turk AT, Close LG, Caruana SM, Su GH. Oncogenic PIK3CA mutation and dysregulation in human salivary duct carcinoma [serial online] Biomed Res Int. 2014;2014:810487. doi: 10.1155/2014/810487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Griffith CC, Seethala RR, Luvison A, Miller M, Chiosea SI. PIK3CA mutations and PTEN loss in salivary duct carcinomas. Am J Surg Pathol. 2013;37:1201–1207. doi: 10.1097/PAS.0b013e3182880d5a. [DOI] [PubMed] [Google Scholar]
- 81.Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29:e473–e476. doi: 10.1200/JCO.2010.32.8351. [DOI] [PubMed] [Google Scholar]
- 82.Soper MS, Iganej S, Thompson LD. Definitive treatment of androgen receptor-positive salivary duct carcinoma with androgen deprivation therapy and external beam radiotherapy. Head Neck. 2014;36:E4–E7. doi: 10.1002/hed.23383. [DOI] [PubMed] [Google Scholar]
- 83.Skalova A, Vanecek T, Sima R, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol. 2010;34:599–608. doi: 10.1097/PAS.0b013e3181d9efcc. [DOI] [PubMed] [Google Scholar]
- 84.Hunt JL. An update on molecular diagnostics of squamous and salivary gland tumors of the head and neck. Arch Pathol Lab Med. 2011;135:602–609. doi: 10.5858/2010-0655-RAIR.1. [DOI] [PubMed] [Google Scholar]
- 85.Urano M, Nagao T, Miyabe S, Ishibashi K, Higuchi K, Kuroda M. Characterization of mammary analogue secretory carcinoma of the salivary gland: discrimination from its mimics by the presence of the ETV6-NTRK3 translocation and novel surrogate markers. Hum Pathol. 2015;46:94–103. doi: 10.1016/j.humpath.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 86.Fonseca FP, Sena Filho M, Altemani A, Speight PM, Vargas PA. Molecular signature of salivary gland tumors: potential use as diagnostic and prognostic marker. J Oral Pathol Med. doi: 10.1111/jop.12329. published online ahead of print May 20, 2015. doi:10,1111/jop.12329. [DOI] [PubMed] [Google Scholar]
- 87.Ito Y, Ishibashi K, Masaki A, et al. Mammary analogue secretory carcinoma of salivary glands: a clinicopathologic and molecular study including 2 cases harboring ETV6-X fusion. Am J Surg Pathol. 2015;39:602–610. doi: 10.1097/PAS.0000000000000392. [DOI] [PubMed] [Google Scholar]
- 88.Bishop JA. Unmasking MASC: bringing to light the unique morphologic, immunohistochemical and genetic features of the newly recognized mammary analogue secretory carcinoma of salivary glands. Head Neck Pathol. 2013;7:35–39. doi: 10.1007/s12105-013-0429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Persson F, Andren Y, Winnes M, et al. High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement, and fusion of HMGA2. Genes Chromosomes Cancer. 2009;48:69–82. doi: 10.1002/gcc.20619. [DOI] [PubMed] [Google Scholar]
- 90.Ying YL, Johnson JT, Myers EN. Squamous cell carcinoma of the parotid gland. Head Neck. 2006;28:626–632. doi: 10.1002/hed.20360. [DOI] [PubMed] [Google Scholar]
- 91.Wend P, Fang L, Zhu Q, et al. Wnt/beta-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J. 2013;32:1977–1989. doi: 10.1038/emboj.2013.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chiosea SI, Griffith C, Assaad A, Seethala RR. The profile of acinic cell carcinoma after recognition of mammary analog secretory carcinoma. Am J Surg Pathol. 2012;36:343–350. doi: 10.1097/PAS.0b013e318242a5b0. [DOI] [PubMed] [Google Scholar]
- 93.Licitra L, Marchini S, Spinazze S, et al. in advanced salivary gland carcinoma. A phase II study of 25 patients. Cancer. 1991;68:1874–1877. doi: 10.1002/1097-0142(19911101)68:9<1874::aid-cncr2820680904>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]