

Abstract
The HBS1L-MYB intergenic region (chr6q23) regulates erythroid cell proliferation, maturation, and fetal hemoglobin (HbF) expression. An enhancer element within this locus, highlighted by a 3-bp deletion polymorphism (rs66650371), is known to interact with the promoter of the neighboring gene, MYB, to increase its expression, thereby regulating HbF production. RNA polymerase II binding and a 50-bp transcript from this enhancer region reported in ENCODE datasets suggested the presence of a long noncoding RNA (lncRNA). We characterized a novel 1283 bp transcript (HMI-LNCRNA; chr6:135,096,362–135,097,644; hg38) that was transcribed from the enhancer region of MYB. Within erythroid cells, HMI-LNCRNA was almost exclusively present in nucleus, and was much less abundant than the mRNA for MYB. HMI-LNCRNA expression was significantly higher in erythroblasts derived from cultured adult peripheral blood CD34+ cells which expressed more HBB, compared to erythroblasts from cultured cord blood CD34+ cells which expressed much more HBG. Down-regulation of HMI-LNCRNA in HUDEP-2 cells, which expressed mostly HBB, significantly upregulated HBG expression both at the mRNA (200-fold) and protein levels, and promoted erythroid maturation. No change was found in the expression of BCL11A and other key transcription factors known to modulate HBG expression. HMI-LNCRNA plays an important role in regulating HBG expression, and its downregulation can result in a significant increase in HbF. HMI-LNCRNA might be a potential therapeutic target for HbF induction treatment in sickle cell disease and β-thalassemia.
Keywords: Long noncoding RNA, HbF quantitative trait loci, Regulation of HbF expression
INTRODUCTION
Sickle cell disease (SCD) and β-thalassemia major are the most prevalent hemoglobinopathies worldwide. Once found primarily in regions of the world where malaria was and may still be endemic, these diseases are now widespread due to human migration and are increasingly important to global health. Fetal hemoglobin (HbF; α2γ2) can inhibit polymerization of deoxy-sickle hemoglobin, and can also compensate for the lack of adult hemoglobin (HbA; α2β2) in β-thalassemia major. Therefore, HbF is the major modifier of disease severity for both SCD and β-thalassemia major [1,2].
Genome-wide association studies (GWAS) have found multiple single nucleotide polymorphisms (SNPs) marking three major quantitative trait loci (QTL) associated with HbF levels — chr11p15 (HBB gene cluster), chr2p16 (BCL11A) and chr6q23 (HBS1L-MYB intergenic polymorphisms or HMIP) [3]. Together they account for 20–45% of HbF variance in different populations. In addition, other cis-acting elements such as the HBD-HBBP1 intergenic region and transcription factors including MYB, KLF1, BCL11A, ZBTB7A, CHD4, NR2C1/NR2C2 and KDM1α, play important roles in regulating HBG expression [4–7]. Nevertheless, significant gaps of knowledge on the regulation of HBG still remain.
The 126 kb HBS1L-MYB intergenic region on chr6q23 is between the genes HBS1L which is a member of the GTP-binding elongation factor family with no known association with erythroid-specific traits, and MYB which encodes for the transcription factor c-MYB. c-MYB regulates proliferation and maturation of erythroid cells, and modulates gene expression within the HBB gene cluster [8,9]. A distal enhancer located at ~84 kb upstream of MYB has been shown by GWAS, insertional mutagenesis, long-range interaction demonstrable by chromosome conformation capture (3C) analysis, and gene editing with Cas9 nucleases [10–13]. This enhancer encompasses a 3-bp deletion polymorphism (rs66650371), which is surrounded by binding sites for erythroid-specific transcription factors TAL1/E47, GATA, RUNX1, LDB1 and KLF1, and is likely the functional motif to account for most of the effect upon HbF level by this QTL [10,12,13]. Alteration of this enhancer by polymorphisms such as rs66650371 reduced its interaction with the MYB promoter, which led to downregulation of MYB and upregulation of HBG expression. Furthermore, ENCODE datasets annotated RNA polymerase II occupancy and a 50-bp RNA transcript adjacent to rs66650371. This led us to hypothesize that this transcript is part of a long noncoding RNA (lncRNA) [10]. LncRNAs are usually greater than 200 nucleotides long, are transcribed throughout the genome, and have broad functionality.
We now report the characterization of a novel 1283 bp lncRNA, herein named the HBS1L-MYB intergenic long noncoding RNA (HMI-LNCRNA). HMI-LNCRNA is transcribed from the enhancer for MYB, and its downregulation significantly increased HBG expression at both the mRNA and protein levels in human adult-like erythroid cells. These observations suggest that HMI-LNCRNA has an important role in silencing HBG expression in adults, and could become a therapeutic target for increasing HbF in patients with SCD and β-thalassemia major.
MATERIALS AND METHODS
K562 cells
K562 cells were cultured at 37°C in RPMI medium containing 10% FBS and 2% penicillin/streptomycin.
RNA extraction
Total RNA was extracted using RNeasy Mini Kit (Qiagen), treated with DNase (RNase-Free DNase Set, Qiagen), followed by RNA cleanup using RNeasy Mini Kit. For tissue-specificity experiment, multiple human organ RNA panels (Invitrogen and Clontech) were also treated with DNase, followed by RNA cleanup.
Reverse transcription polymerase chain reaction RT-PCR
cDNA was synthesized from total RNA using SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen). PCR reactions were done using the Multiplex PCR kit (Qiagen). The following primers were used to amplify the 1180 bp product: 5′-ATCGCTCATGAGAAATGTGG-3′ (forward) and 5′-GGAACCGCCCTGATAACATT-3′ (reverse).
Rapid amplification of cDNA ends (RACE)
5′- and 3′-RACE were done using the FirstChoice RLM-RACE Kit (Ambion), following the manufacturer’s instructions, using SuperTaq Plus Polymerase (Life Technologies) for PCR reactions. The following gene-specific primers were used: 5′-GTCTAATGGTGTGGCTCACAAA-3′ (5′-outer), 5′-CCCCAGCTTCCTTATCTGTAAA-3′ (5′-inner), 5′-TTCACTCTGGACAGCAGATGTT-3′ (3′-outer) and 5′-CGGTTCCCTCAGAAGACACTTA-3′ (3′-inner). RACE PCR products were ligated to pCRII vector using TA Cloning Dual Promoter Kit (Invitrogen), transformed into One Shot INVαF chemically competent E. coli (Invitrogen), and grown on LB plates containing ampicillin and X-Gal. Insert-positive white colonies were picked and grown for DNA extraction. PCR to amplify insert (Forward: 5′-TGTGGAATTGTGAGCGGA TA-3′ and Reverse: 5′-GTTTTCCCAGTCACGACGTT-3′), and DNA sequencing were done to determine the 5′- and 3′-ends.
DNA sequencing
PCR products were purified using AccuPrep PCR Purification Kit, and prepared for sequencing using ABI Big Dye Terminator v3.1 Cycle Sequencing Kit. Sequence data was analyzed on FinchTV version 1.5.0. NCBI BLAST was used to determine length and location of sequence.
Human Umbilical Cord Blood-Derived Erythroid Progenitor (HUDEP) cells
HUDEP cells are an immortalized erythroid cell line derived from cord blood CD34+ mononuclear cells [14]. HUDEP-1 and HUDEP-2 cells were maintained in expansion medium—StemSpan SFEM medium (StemCell Technologies) supplemented with SCF (50 ng/ml, Invitrogen), EPO (3 U/ml, Invitrogen), dexamethasone (1 μM, Sigma), doxycycline (1 μg/ml, Clontech), L-glutamine (1%, Life Technologies) and penicillin/streptomycin (2%, Life Technologies). For erythroid maturation, cells were cultured in differentiation medium—IMDM medium (Invitrogen) supplemented with heat inactivated human serum from human male AB plasma (5%, Sigma), EPO (3 U/ml, Invitrogen), insulin (10 μg/ml, Sigma), doxycycline (1 μg/ml, Clontech), holo-transferrin (500 μg/ml, Sigma), heparin (3 U/ml, Sigma), SCF (100 ng/ml, Invitrogen), L-glutamine (1%, Life Technologies) and penicillin/streptomycin (2%, Life Technologies)—for 5 days. For further erythroid maturation, doxycycline was removed and cells were cultured for 2 more days.
Primary CD34+ mononuclear cells
Primary CD34+ mononuclear cells derived from cord blood and peripheral blood (StemCell Technologies) were expanded for up to 7 days in StemSpan SFEM II medium (StemCell Technologies) supplemented with StemSpan CC100 (1×, StemCell Technologies) and penicillin/streptomycin (2%, Life Technologies). To induce erythroid differentiation, cells were cultured for up to 10 days in StemSpan SFEM II medium (StemCell Technologies) supplemented with SCF (10 ng/ml, Invitrogen), EPO (5 U/ml, Invitrogen), IL-6 (10 ng/ml, Sigma) and penicillin/streptomycin (2%, Life Technologies).
Nuclear and cytoplasmic fractionation
Nuclear/Cytosol Fractionation Kit (BioVision) was used following manufacturer’s instructions.
Quantitative PCR (qPCR)
RNA was used for qPCR using TaqMan RNA-to-CT 1-Step Kit (Applied Biosystems) and the following TaqMan gene expression assays (Applied Biosystems): HBG1/2 (Hs00361131_g1), HBB (Hs00758889_s1), MYB (Hs00920556_m1), HBS1L (Hs04188641_g1), HMI-LNCRNA (custom TaqMan assay designed by Applied Biosystems to target genome position chr6: 135,096,354–135,097,644, hg38; assay ID number AJI1MTQ), BCL11A (Hs01093197_m1), CHD4 (Hs00172349_m1), KLF1 (Hs00610592_m1), ZBTB7A (Hs00252415_s1), NR2C1 (Hs00915957_m1), NR2C2 (Hs00991824_m1), KDM1a (Hs01002741_m1) and ACTB (Hs01060665_g1). QPCR reactions were run on a StepOne Plus qPCR machine (Applied Biosystems). ACTB was used as the endogenous control.
Western blot analysis
Cell pellets were suspended in Roche lysis buffer (protease inhibitor, 0.3% NP40, 10% glycerine, 2 mM EDTA, 246 mM NaCl, 10% phosphatase inhibitor, PBS and water), placed on ice for 1 hour and centrifuged at 14,500 rpm for 15 minutes at 4°C to extract protein. Standard methodology was used for western blot analysis. The following antibodies were used: c-MYB (ab109127, Abcam), HBB (16216-1-AP, Proteintech), hemoglobin γ (sc-21756, Santa Cruz Biotechnology) and GAPDH (sc-47724, Santa Cruz Biotechnology).
Plasmids
The following top and bottoms strands were synthetically made and annealed to make the shRNA template for HMI-LNCRNA: top strand 5′- GATCCGCTAG TATGTGAAGCACTTAGCTTCCTGTCAGACTAAGTGCTTCACATACTAGCTTTTTG-3′, bottom strand 5′-AATTCAAAAAGCTAGTATGTGAAGCACTTAGTCTGACAGGAAGCT AAGTGCTTCACATACTAGCG-3′) (135,096,650-135,096,670; hg38). Next, the shRNA template for HMI-LNCRNA was ligated to the lentivirus vector pGreenPuro (System Biosciences), upstream the EF1 promoter, and labeled HMI-lncRNA shRNA. pGreenPuro scrambled shRNA (System Biosciences) was used as the negative control. Both lentivectors co-express green fluorescent protein (GFP).
Lentivirus transduction
293T cells were transfected with scrambled shRNA and HMI-lncRNA shRNA using the LentiStarter 2.0 kit (System Biosciences), following manufacturer’s instructions, to generate lentiviral particles. HUDEP-2 cells were transduced at an MOI 50 with 5 ug/mL Polybrene, and centrifuged at room temperature for 30 min at 1250 × g. After 48 hours, transduction was repeated. 48 hours post-transduction, cells were cultured in expansion medium with 1 ug/mL Puromycin for up to 2 weeks.
Cell surface staining for FACS
Cells were harvested and washed in FACS buffer, and were resuspended in 100 μl of FACS buffer with antibody, and incubated on ice for 30 minutes. Next, cells were washed twice in FACS buffer by centrifugation at 300g for 5 minutes. Finally, cells were fixed in IC Fixation buffer (eBioscience) for 30 minutes before analyzing the cells using the BD FACScan. The following antibodies were used: CD71-PE (334105, BioLegend) and CD235-PerCP/Cy5.5 (306613, BioLegend).
Immunofluorescence (IF)
Fixed cells were washed in PBS, blocked in 5% BSA (in PBS) and incubated in primary antibody for 1 hour. Next, cells were washed, blocked in 5% normal goat serum and incubated in secondary fluorescent-labeled antibody for 30 minutes. Finally, cells were washed and covered with mounting medium containing DAPI. Images were taken with the Eclipse Ci (Nikon) microscope with a DS-Qi1Mc-03 mono-type (Nikon) camera, using NIS-Elements Br Imaging software (Nikon). The following antibodies were used: Hemoglobin γ (sc-21756, Santa Cruz), Monoclonal Mouse Anti-human β-globin (a gift from IsoLab), Anti-human alpha globin (Wallac Inc.), and Alexa Fluor-594-conjugated AffiniPure F(ab)2 Fragment Goat Anti-mouse IgG (H+L) (115-586-062, Jackson ImmunoResearch).
RESULTS
Determining the full length of HBS1L-MYB intergenic long noncoding RNA
To determine if the 50-bp transcript reported by ENCODE is part of a putative lncRNA, we used the sequence of this transcript as a guide to develop primers consecutively upstream and downstream of this region. cDNA from K562 cells, an immortalized human erythroid cell line, was used for PCR reactions until amplification failed. The longest PCR product amplified was 1180 bp (chr6:135,096,355–135,097,534; hg38) (Fig. 1A).
Figure 1. Determining the full length of the transcript found within the HBS1L-MYB intergenic region.
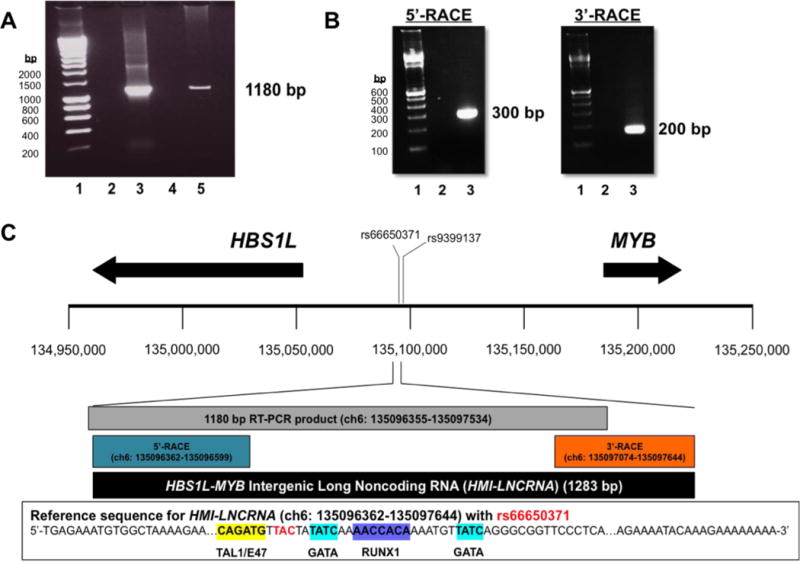
(A) PCR was done to amplify a 1180 bp region within the HBS1L-MYB intergenic region using cDNA from K652 cells. Samples were run on agarose gels. Lane 1: 100-bp DNA ladder; lane 2: negative control, PCR mix without DNA input; lane 3: positive control, PCR mix with genomic DNA; lane 4: PCR mix with cDNA generated without reverse transcriptase; and Lane 5: PCR mix with cDNA generated with reverse transcriptase. (B) Agarose gels show PCR products from the 5′-RACE reactions (using RNA from thymus) and 3′-RACE reactions (using RNA from K562 cells). For 5′-RACE, lane 1: 100-bp DNA ladder; lane 2: PCR mix with cDNA generated without Tobacco Acid Pyrophosphatase (TAP) treatment; and lane 3: PCR mix with cDNA generated with TAP treatment. For 3′-RACE, lane 1: 100-bp DNA ladder; lane 2: PCR mix with cDNA generated without reverse transcriptase; and lane 3: PCR mix with cDNA generated with reverse transcriptase. (C) Illustration of genomic region between chr6: 134,950,000–135,250,000 (hg38), showing approximate locations of the 3-bp deletion polymorphism rs66650371 and rs9399137 a SNP in LD with rs66650371, and HBS1L and MYB (arrows represent transcription direction and approximate length of genes). Based on DNA sequencing of RACE products, the 5′- and 3′-ends of the transcript were revealed to determine the full length of the transcript, which is 1283 bp in length and named the HBS1L-MYB Intergenic Long Noncoding RNA (HMI-LNCRNA). Located in the genomic sequence for HMI-LNCRNA are binding sites for erythroid-specific transcription factors TAL1/E47, GATA and RUNX1, and rs66650371. HMI-LNCRNA does not include rs9399137.
To obtain the full-length sequence of this RNA transcript, rapid amplification of cDNA ends (RACE) was employed to ascertain its 5′- and 3′-termini (Fig. 1B; Supplemental Figs. 1 and 2). RNA from K562 cells was used for the 3′-RACE, and RNA extracted from the thymus, which was more abundant for this IncRNA (Fig. 2), was used for the 5′-RACE. Moreover, both 3′- and 5′-RACE was repeated using RNA extracted from HUDEP-2 cells, and yielded identical results (Supplemental Fig. 3).
Figure 2. Expression pattern of HMI-LNCRNA among various human cells and tissue.
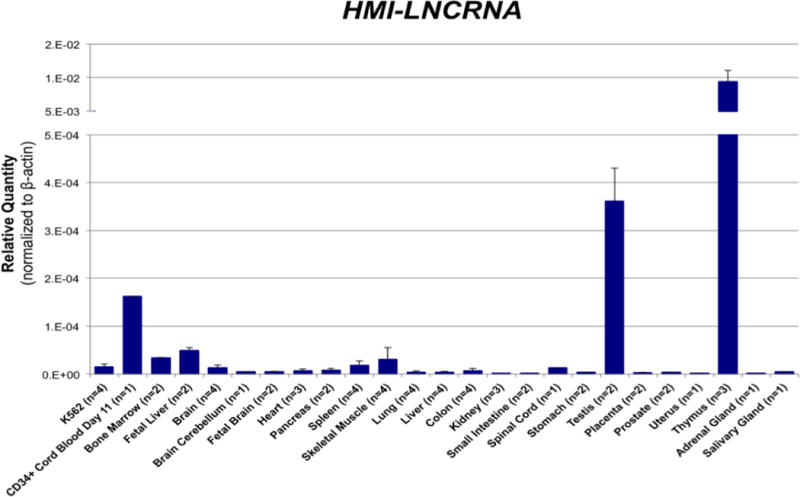
Relative quantity of HMI-LNCRNA was measured by qPCR in 25 different samples. CD34+ cord blood day 11 is erythroblasts harvested at Day 11 of two-phase expansion and differentiation culture of CD34+ mononuclear cells derived from cord blood (see Supplemental Fig. 6). Means are shown for samples with two or more independent samples. ACTB was used as the endogenous control.
Figure 1C illustrates the coordinates of the full-length lncRNA (chr6:135,096,362–135,097,644; hg38), and its nucleotide sequences are shown in Supplementary Table 1. It was 1283 bp in length, transcribed from a genomic site ~84.9 kb upstream from MYB and ~41.5 kb upstream from HBS1L, and herein called the HBS1L-MYB intergenic long noncoding RNA (HMI-LNCRNA). HMI-LNCRNA was transcribed from the enhancer for MYB and encompassed binding sites for erythroid transcription factors TAL1/E47, GATA, RUNX1, and within the transcript was the site of the HbF-associated 3-bp deletion polymorphism (rs66650371). HMI-LNCRNA was intron-less, had no 3′-end polyadenylation signal, and could be presumed to have a 5′-cap as 5′-RACE selected for only RNAs with a 5′-cap. It also contained no open reading frames longer than 300 nucleotides (data not shown); their presumed peptides were not catalogued in protein databases; and therefore they were unlikely to be translated [15].
Tissue-Specificity of HMI-LNCRNA
LncRNAs often are expressed in either one or a few cell types and are considered more tissue-specific than most mRNA [16]. The expression pattern of HMI-LNCRNA was analyzed by qPCR in 25 different human tissues and cells (Fig. 2). The thymus and testis had by far the highest expression. Erythroblasts derived from cord blood CD34+ mononuclear cells had high expression of HMI-LNCRNA, followed by other hematopoietic tissues and cells, such as the fetal liver, bone marrow, spleen and K562 cells. Non-hematopoietic tissues, except for testis where lncRNAs are especially common [17], had mostly minimal expression. HMI-LNCRNA exhibited a distinct association with cells of hematopoietic origin. Similar to HMI-LNCRNA, MYB was also expressed in the thymus and the other hematopoietic tissues and cells (Supplemental Fig. 4).
Expression pattern of HMI-LNCRNA and MYB in erythroid cells
Within erythroid cells, HMI-LNCRNA was expressed almost entirely in the nucleus (Supplemental Fig. 5), suggesting that it may function as a gene-regulating lncRNA. HMI-LNCRNA was much less abundant than MYB (Supplemental Fig. 6), which is a common characteristic of many lncRNAs compared to their protein-coding counterparts [18]. MYB is critical to erythropoiesis; high levels of expression favor proliferation of erythroid progenitor cells, while reduced levels promote erythroid maturation [19]. Additionally, downregulation of MYB in erythroid precursors was shown to increase HbF [8,12,13,20]. Therefore, it was important to determine if HMI-LNCRNA and MYB had similar expression patterns in erythroid cells and if this similarity was associated with differences in HBG and HBB expression.
Both HMI-LNCRNA and MYB transcript levels were compared in erythroblasts derived from culture of cord blood CD34+ cells, which expressed more HBG, to erythroblasts derived from culture of adult peripheral blood CD34+ cells, which expressed more HBB (Fig. 3A). Both HMI-LNCRNA and MYB were significantly higher in erythroblasts derived from adult peripheral blood CD34+ cells than in erythroblasts derived from cord blood CD34+ cells (Fig. 3A). With erythroid differentiation of erythroblasts derived from peripheral blood CD34+ cells, there was a decline in HMI-LNCRNA and in MYB expression (Fig. 3B; Supplemental Fig. 7). Next, the same comparison was made in HUDEP-1 cells, which mostly express HBG, and HUDEP-2 cells, which exclusively express HBB (Fig. 3C). HUDEP-2 cells had significantly higher expression of HMI-LNCRNA than HUDEP-1 cells; and there was no difference in MYB expression (Fig. 3C). During erythroid maturation of HUDEP-2 cells, both HMI-LNCRNA and MYB expression decreased similar to erythroblasts derived from peripheral blood CD34+ cells (Fig. 3D). These data show that HMI-LNCRNA expression is higher in erythroid cells which express more HBB, when compared to erythroid cells which express more HBG. With erythroid cell differentiation, the expression of both HMI-LNCRNA and MYB decreases.
Figure 3. HMI-LNCRNA and MYB expression pattern in erythroid cells derived from cord blood and peripheral blood CD34+ mononuclear cells, and in HUDEP-1 and HUDEP-2 cells.
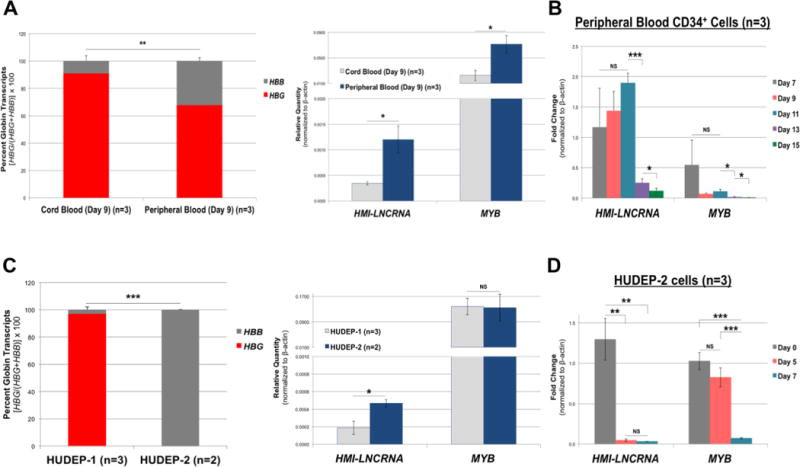
(A) Primary erythroid cells derived from cord blood (n=3) and adult peripheral blood (n=3) CD34+ mononuclear cells (both expanded for 7 days and differentiated for 2 days) were analyzed by qPCR analysis to determine percent HBG to HBB, and the relative quantity of MYB and HMI-LNCRNA. (B) Fold change of HMI-LNCRNA and MYB were determined during differentiation of CD34+ mononuclear cells derived from adult peripheral blood (n=3) at Days 7, 9, 11, 13 and 15. (C) Percent HBG to HBB, and the relative quantity of HMI-LNCRNA and MYB were determined by qPCR analysis in HUDEP-1 (n=3) and HUDEP-2 (n=2) cells (both maintained in expansion medium). (D) Fold change of HMI-LNCRNA and MYB were determined in HUDEP-2 cells at Days 0, 5 and 7 in cultures with differentiation medium. ACTB was used as the endogenous control. p-values: *< 0.05; **< 0.005; ***<0.0005; NS (not significant). p-values were obtained by Student T-test.
HMI-LNCRNA regulates hemoglobin expression in HUDEP-2 cells
To examine the possible role of HMI-LNCRNA in regulating MYB and hemoglobin expression, a lentiviral vector co-expressing GFP and a shRNA template targeting HMI-LNCRNA (HMI-lncRNA shRNA) was employed to downregulate the HMI-LNCRNA in HUDEP-2 cells. Figure 4A displays the timeline of the shRNA transduction experiments. After transduction, cells were cultured in expansion medium containing puromycin to select for lentivector-positive cells. Transduction of HUDEP-2 cells with HMI-lncRNA shRNA led to a 50% knockdown of HMI-LNCRNA when compared to naïve (non-transduced) cells and cells expressing scrambled shRNA (Fig. 4B). Reduction in HMI-LNCRNA led to a 200-fold increase in HBG mRNA, a 30% reduction in MYB mRNA, and no change in HBB or HBS1L mRNA when compared to naïve cells and cells transduced with scrambled shRNA (Fig. 4C). Percent HBG increased from less than 1% in naïve and scrambled shRNA cells to more than 20% of total HBG and HBB expression in cells with HMI-lncRNA shRNA (Fig. 4D). Furthermore, changes in HBG and MYB mRNA levels were associated with corresponding changes in HBG and c-MYB protein levels (Fig. 4E) shown by Western blot analyses. The up-regulation of HBG production was also confirmed by immunofluorescence staining using anti-HBG antibody (Fig. 4F). This immunocytological technique showed that high HBG expression were found in approximately 10% of more mature HUDEP-2 cells with knockdown of HMI-LNCRNA. There was no change in HBB or HBA protein expression (Supplemental Figs. 8 and 9). Down-regulation of MYB expression in HUDEP-2 cells transduced with HMI-lncRNA shRNA was inconsistent (Supplemental Fig. 10).
Figure 4. The effects of knocking down HMI-LNCRNA on hemoglobin, MYB and HBS1L in HUDEP-2 cells.
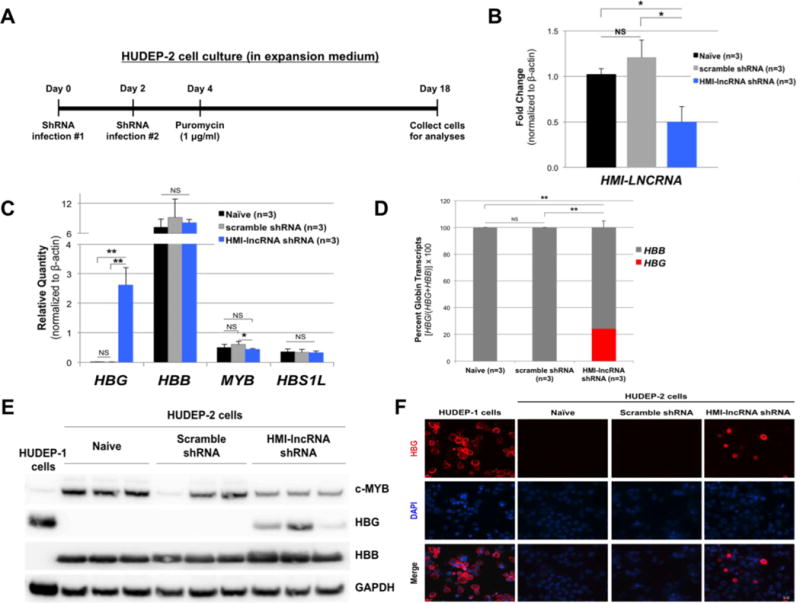
(A) Illustration of timeline for transduction and culture of HUDEP-2 cells. Cells were maintained in expansion medium. (B) Expression level of HMI-LNCRNA was determined by qPCR in HUDEP-2 cells that were not transduced (naïve), and transduced with either scrambled shRNA or HMI-lncRNA shRNA lentiviruses by qPCR. (C) HBG, HBB, MYB and HBS1L transcript levels, and (D) percent HBG and HBB out of the total of both transcripts were measured in these same samples. (E) Protein expression of c-MYB, HBG and HBB were analyzed by Western blot in naïve HUDEP-2 cells, and cells transduced with scrambled shRNA and HMI-lncRNA shRNA (HUDEP-1 cells were used as control). GAPDH was used as loading control. (F) Naïve HUDEP-2 cells, and cells transduced with scrambled and HMI-lncRNA shRNAs were stained with anti-HBG antibody, followed by secondary antibody labeled with Alexa Fluor-594 (in red) and DAPI to stain nuclei (in blue), and imaged with a fluorescent microscope at 40X magnification. For all qPCR analyses, ACTB was used as the endogenous control. p-values: *< 0.05; **< 0.005; ***<0.0005; NS (not significant). p-values were obtained by Student T-test.
The expression of key hemoglobin-regulating transcription factor genes (KLF1, BCL11A, ZBTB7A, CHD4, NR2C1, NR2C2 and KDM1α), which are all known to downregulate HBG, were either unchanged or slightly upregulated in HUDEP-2 cells transduced with HMI-lncRNA shRNA compared to controls (Fig. 5).
Figure 5. The effects of knocking down HMI-LNCRNA on erythroid-regulating transcription factors in HUDEP-2 cells.
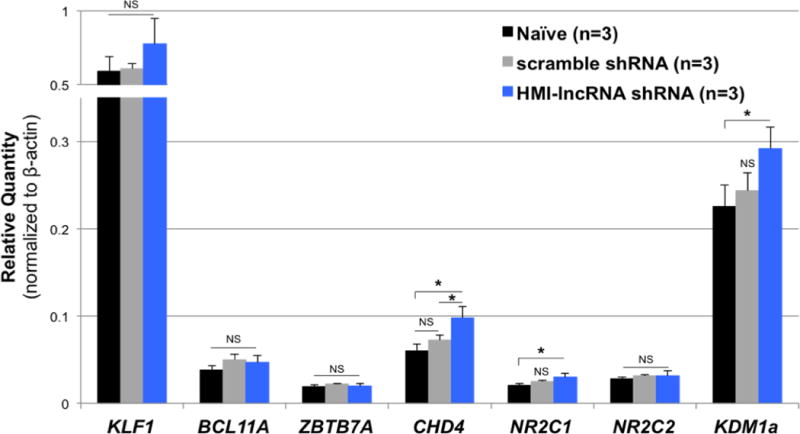
Relative expression of KLF1, BCL11A, ZBTB7A, CHD4, NR2C1, NR2C2 and KDM1a transcripts were analyzed by qPCR in naïve HUDEP-2 cells, and cells infected with scrambled shRNA and HMI-lncRNA shRNA. Beta actin was used as the endogenous control. p-values: *< 0.05; **< 0.005; ***<0.0005; NS (not significant). p-values were obtained by Student T-test.
Hemoglobin expression during erythroid differentiation of HUDEP-2 cells with knockdown of HMI-LNCRNA
In addition to analyzing the effect of downregulating HMI-LNCRNA in non-differentiated HUDEP-2 cells, after the two-week selection for transduced cells with puromycin treatment, the cells underwent erythroid maturation for up to seven days as shown in Figure 6A. HBG was significantly higher at days 0, 5 and 7 in HUDEP-2 cells with HMI-lncRNA shRNA (Fig. 6B), and HBG expression remained at about 20% of total HBG and HBB transcripts at each time point (Fig. 6C). The increased HBG expression was also corroborated at the protein level (Supplementary Figs. 10 and 11).
Figure 6. Hemoglobin expression during erythroid differentiation of HUDEP-2 cells with knockdown of HMI-LNCRNA.
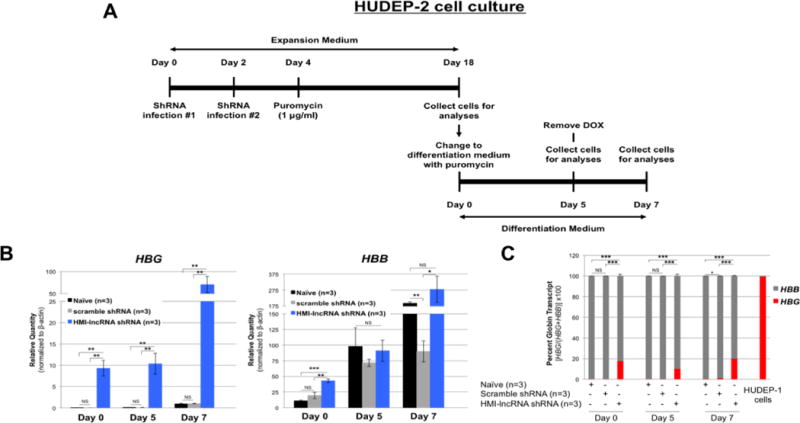
(A) Illustration of timeline for infection and culture of HUDEP-2 cells. Cells were maintained in expansion medium for 2 weeks after transduction, and then placed in differentiation medium for up to 7 days. Doxycycline (DOX) was removed at Day 5 to promote erythroid maturation. (B) Relative quantity for HBG and HBB transcripts were analyzed by qPCR in naïve HUDEP-2 cells (n=3), and cells transduced with scrambled shRNA (n=3) and HMI-lncRNA shRNA (n=3) at Day 0, 5 and 7 of differentiation. (C) Percent hemoglobin of HBG to HBB was determined by qPCR. For all qPCR analyses, ACTB was used as the endogenous control. p-values: *< 0.05; **< 0.005; ***<0.0005; NS (not significant). p-values were obtained by Student T-test.
Role of HMI-LNCRNA in modulating erythroid maturation in HUDEP-2 cells
HUDEP-2 cells resemble basophilic erythroblasts expressing both transferrin receptor (CD71), a cell surface marker for immature erythroblasts, and glycophorin A (CD235), a cell surface marker for more mature erythroblasts [6]. The percentage of cells positive for these markers were examined by flow cytometry in naïve cells, cells expressing scrambled shRNA and cells expressing HMI-lncRNA shRNA. Within both control groups, approximately 55–65% of the cells were positive for both CD71 and CD235 (Fig. 7). In contrast, about 85% of cells transduced with HMI-lncRNA shRNA were positive for both markers. These observations suggest that downregulation of HMI-LNCRNA promotes maturation of HUDEP-2 cells.
Figure 7. The effects of knocking down HMI-LNCRNA on erythroid maturation of HUDEP-2 cells.
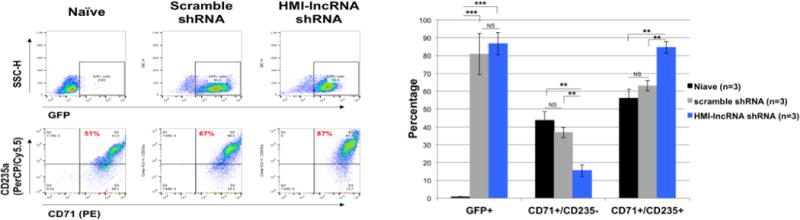
Naïve HUDEP-2 cells, and cells transduced with scrambled shRNA and HMI-lncRNA shRNA (cultured in expansion medium) were stained with PE-labeled transferrin receptor (CD71) and PerCP/Cy5.5-labeled glycophorin-A (CD235) antibodies, and analyzed by flow cytometry to discriminate between cells that are positive and negative for GFP, CD71 and CD235. Data was analyzed with FlowJo. Average percent GFP+, CD71+/CD235- and CD71+/CD235+ for each group was plotted on a bar graph. p-values: *< 0.05; **< 0.005; ***<0.0005; NS (not significant). p-values were obtained by Student T-test.
DISCUSSION
A novel 1283 bp lncRNA in erythroid cells is transcribed from the distal enhancer of MYB on chr6q23 highlighted by the 3-bp deletion polymorphism (rs66650371). Knockdown of HMI-LNCRNA to half of its normal level increased HBG expression by 200-fold and this is reflected by a marked increase in HBG production.
Long non-coding RNAs are important regulators of multiple cellular processes, biomarkers for disease prognosis and possible therapeutic targets [21–24]. Many tissue specific lncRNAs are expressed during hematopoiesis and erythropoiesis [25–29]. To date, long non-coding RNAs that can modulate globin gene expression have not been described.
HUDEP-2 cells, which do not have the 3-bp deletion at rs66650371 (data not shown) and synthesize almost exclusively HBB have been used to study the regulation of HBG expression [6,13,14]. It will be necessary to replicate the present experiments in cultured erythroblasts derived from primary CD34+ progenitor cells from healthy individuals and SCD patients. To determine if upregulation of HMI-LNCRNA could reduce HBG expression, we overexpressed this lncRNA in HUDEP-1 cells that express primarily HBG but saw no change (supplemental Fig. 12).
The expression of HBG in HUDEP-2 cells was heterocellular. With erythroid maturation, the proportion of these HUDEP-2 cells that expressed HBG increased from approximately 10% to 25% (Fig. 4F; Supplementary Fig. 11). It remains to be determined if additional down-regulation of HMI-LNCRNA or allowing these HUDEP-2 cells to become fully mature erythroblasts might lead to more cells expressing HBG. It is possible that transduction of HUDEP-2 cells with lentiviral particles was not homogeneous, thus accounting for the heterocellular expression of HBG. The genomic sites for the lentiviral particle integration might also be another factor that could modulate HBG expression [30]. Furthermore, HBG expression in cultured erythroid cells derived from single adult erythroid progenitors is stochastic [31].
The mechanism whereby HMI-LNCRNA augments HBG expression requires further study. LncRNAs may regulate gene expression by providing the necessary scaffold which transcription factor and other protein complexes can be associated with and be guided to cogent promoters[21,22]. They may recruit chromatin modification enzymes for epigenetic remodeling processes [22,32]. They may also act by promoting intra-chromosomal looping to foster interaction between enhancers and promoters [33,34]. Since HMI-LNCRNA is transcribed from the distal enhancer for MYB, it is highly probable that HMI-LNCRNA is an enhancer RNA (eRNA), and functions by facilitating the interaction between the distal enhancer and the MYB promoter. We hypothesized that downregulation of HMI-LNCRNA would reduce interaction of the distal enhancer with the promoter of MYB and reduce its transcription, which would ultimately lead to increase in HBG expression. However, decreased expression of MYB was not consistently observed in HUDEP-2 cells transduced with HMI-LncRNA shRNA to downregulate HMI-LNCRNA to 50% of normal level (Fig. 4E; Supplementary Fig. 10). Conceivably, additional down-regulation of HMI-LNCRNA may result in consistently decreased MYB expression. Whether or not interaction between distal enhancer and promoter of MYB is attenuated with HMI-LNCRNA down-regulation should be informative. Alternatively, HMI-LNCRNA might function through a yet undescribed molecular mechanism to modulate HBG expression. An important future study is to determine genomic or protein binding sites for HMI-LNCRNA [35].
Increasing HbF levels in SCD and β-thalassemia major is a proven approach to treatment. Hydroxyurea, the first FDA-approved drug for SCD, can induce high HbF level. Unfortunately, not all treated patients responded to this agent with sufficient increase in HbF to effect substantial clinical improvement. There has been remarkable progress on gene therapy by different approaches [36–38], and many different agents are also being investigated for their efficacy to induce HbF expression [39–44]. Down-regulating HMI-LNCRNA can possibly be an additional therapeutic target for HbF induction. If the increment in HbF increase found in vitro can be recapitulated in vivo in SCD and β-thalassemia major, it should be clinically beneficial [45].
Supplementary Material
Acknowledgments
We thank Dr. Richard Myers and Dr. Wenqian Hu for the helpful discussions, comments and guidance, especially at the earlier stages of this project. This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (U01HL107443-01 and 1F31HL126462, and 5T32HL007501-31).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
T.A.M. performed all experiments; I.W. carried out initial experiments; H-Y.L. did the immunofluorescence experiments and provided expertise in tissue cultures; R.K. and Y.N. provided HUDEP-2 and HUDEP-1 cells; J.J.F., M.H.S. and D.H.K.C. conceived of the idea, and together with T.A.M., H-Y.L., G.J.M., and S.C. designed experiments. T.A.M., M.H.S. and D.H.K.C. wrote the manuscript, and all authors have read and approved of the manuscript.
All authors have no conflict of interest disclosure, nor financial interests.
References
- 1.Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017;376:1561–1573. doi: 10.1056/NEJMra1510865. [DOI] [PubMed] [Google Scholar]
- 2.Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, Chui DHK, Steinberg MH. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19–27. doi: 10.1182/blood-2011-03-325258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009;18:R216–R223. doi: 10.1093/hmg/ddp401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sankaran VG, Orkin SH. The Switch from Fetal to Adult Hemoglobin. Cold Spring Harb Perspect Med. 2013;3:a011643. doi: 10.1101/cshperspect.a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ, Sabath DE, Groudine M, Orkin SH, Premawardhena A, Bender MA. A Functional Element Necessary for Fetal Hemoglobin Silencing. N Engl J Med. 2011;365:807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, Fisher C, Suciu M, Martyn GE, Norton LJ, Zhu C, Kurita R, Nakamura Y, Xu J, Higgs DR, Crossley M, Bauer DE, Orkin SH, Kharchenko PV, Maeda T. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang P, Keller CA, Giardine B, Grevet JD, Davies JOJ, Hughes JR, Kurita R, Nakamura Y, Hardison RC, Blobel GA. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 2017;31:1704–1713. doi: 10.1101/gad.303461.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang J, Best S, Menzel S, Silver N, Lai MI, Surdulescu GL, Tim D, Thein SL, Dc W, Spector TD. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood. 2006;108:1077–1083. doi: 10.1182/blood-2006-01-008912. [DOI] [PubMed] [Google Scholar]
- 9.Menzel S, Jiang J, Silver N, Gallagher J, Cunningham J, Surdulescu G, Lathrop M, Farrall M, Spector TD, Thein SL. The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood. 2007;110:3624–3626. doi: 10.1182/blood-2007-05-093419. [DOI] [PubMed] [Google Scholar]
- 10.Farrell JJ, Sherva RM, Chen ZY, Luo HY, Chu BF, Ha SY, Li CK, Lee ACW, Li RCH, Li CK, Yuen HL, So JCC, Ma ESK, Chan LC, Chan V, Sebastiani P, Farrer LA, Baldwin CT, Steinberg MH, Chui DHK. A 3-bp deletion in the HBS1L-MYB intergenic region on chromosome 6q23 is associated with HbF expression. Blood. 2011;117:4935–4945. doi: 10.1182/blood-2010-11-317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suzuki M, Yamazaki H, Mukai HY, Motohashi H, Shi L, Tanabe O, Engel JD, Yamamoto M. Disruption of the Hbs1l-Myb Locus Causes Hereditary Persistence of Fetal Hemoglobin in a Mouse Model. Mol Cell Biol. 2013;33:1687–1695. doi: 10.1128/MCB.01617-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, Van IJcken W, Lenhard B, Rooks H, Best S, Menzel S, Grosveld F, Thein SL, Soler E. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest. 2014;124:1699–1710. doi: 10.1172/JCI71520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Canver M, Lessard S, Pinello L, Ilboudo Y, Wu Y, Stern E, Needleman F, Galactéros A, Brugnara C, Kutlar A, Chen D, Das P, Kurita R, Nakamura Y, Yuan G, Lettre G, Bauer D, Orkin S. Variant-aware saturating mutagenesis using multiple Cas9 nucleases identifies regulatory elements at trait-associated loci. Nat Genet. 2017;49:625–634. doi: 10.1038/ng.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurita R, Suda N, Sudo K, Miharada K, Hiroyama T, Miyoshi H, Tani K, Nakamura Y. Establishment of Immortalized Human Erythroid Progenitor Cell Lines Able to Produce Enucleated Red Blood Cells. PLoS One. 2013;8:e59890. doi: 10.1371/journal.pone.0059890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paralkar VR, Taborda CC, Huang P, Yao Y, Kossenkov AV, Prasad R, Luan J, Davies JOJ, Hughes JR, Hardison RC, Blobel GA, Weiss MJ. Unlinking an lncRNA from Its Associated cis Element. Mol Cell. 2016;62:104–110. doi: 10.1016/j.molcel.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grützner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014;505:635–640. doi: 10.1038/nature12943. [DOI] [PubMed] [Google Scholar]
- 18.Kornienko AE, Dotter CP, Guenzl PM, Gisslinger H, Gisslinger B, Cleary C, Kralovics R, Pauler FM, Barlow DP. Long non-coding RNAs display higher natural expression variation than protein-coding genes in healthy humans. Genome Biol. 2016;17:1–23. doi: 10.1186/s13059-016-0873-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greig KT, Carotta S, Nutt SL. Critical roles for c-Myb in hematopoietic progenitor cells. Semin Immunol. 2008;20:247–256. doi: 10.1016/j.smim.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Sankaran VG, Menne TF, Šćepanović D, Vergilio JA, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci U S A. 2011;108:1519–1524. doi: 10.1073/pnas.1018384108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ørom UA, Shiekhattar R. Noncoding RNAs and enhancers: complications of a long-distance relationship. Trends Genet. 2011;27:433–439. doi: 10.1016/j.tig.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rinn JL, Chang HY. Genome Regulation by Long Noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothschild G, Basu U, Feng Y, Al E, Mullokandov G, Al E, Hennessy E, O’Driscoll L, Siomi MC, Carmichael GG, Quinn JJ, Chang HY, Hung T, Chang HY, Qureshi IA, Mehler MF, Antoniou D, Al E, Kung JT, Al E, Pefanis E, Al E, Pefanis E, Al E, Andersson R, Al E, Li W, Al E, De Santa F, Al E, Whyte WA, Al E, Pott S, Lieb JD, Sigova AA, Al E, Lemay JF, Al E, Kim TK, Al E, Qian J, Al E, Chapuy B, Al E, Rinn JL, Al E, Bhan A, Mandal SS, Wang X, Al E, Orom UA, Al E, Wang KC, Al E, Lieberman-Aiden E, Al E, Dixon JR, Al E, Gorkin DU, Al E, Sun JB, Al E, Pavri R, Al E, Kodgire P, Al E, Maul RW, Al E, Schwer B, Al E, Zheng S, Al E, Madabhushi R, Al E, Lu Z, Al E, Lu Z, Al E, Pannunzio NR, Lieber MR, Lieber MR, Zhang H, Al E, Dixon JR, Al E, Cavalli G, Misteli T, Matharu N, Ahituv N, Allen BL, Taatjes DJ, Lai F, Al E, Fudenberg G, Al E, Hnisz D, Al E, Rowley MJ, Corces VG, Lupianez DG, Al E, Ong CT, Corces VG, Dekker J, Misteli T, Bouwman BAM, de Laat W, Merkenschlager M, Nora EP, Bochman ML, Al E, Kouzine F, Al E, Hamperl S, Cimprich KA, Kim N, Jinks-Robertson S, Grzechnik P, Al E, Chan YA, Al E, Garcia-Muse T, Aguilera A, Santos-Pereira JM, Aguilera A, Wiedemann E-M, Al E, Flynn RA, Al E, Meng FL, Al E, Lenoir GM, Al E, Boxer LM, Dang CV, Hobson DJ, Al E, Kagey MH, Al E, Guo Y, Al E, Rao SS, Al E, Lupianez DG, Al E, Rudan MV, Hadjur S, Rudan MV, Al E, Achour M, Al E, Loven J, Al E, Mansour MR, Al E, Peeters JG, Al E, Vahedi G, Al E, Krantz ID, Al E. Lingering Questions about Enhancer RNA and Enhancer Transcription-Coupled Genomic Instability. Trends Genet. 2017;33:143–154. doi: 10.1016/j.tig.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16:167–179. doi: 10.1038/nrd.2016.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi L, Lin YH, Sierant MC, Zhu F, Cui S, Guan Y, Sartor MA, Tanabe O, Lim KC, Engel JD. Developmental transcriptome analysis of human erythropoiesis. Hum Mol Genet. 2014;23:4528–4542. doi: 10.1093/hmg/ddu167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alvarez-Dominguez JR, Hu W, Yuan B, Shi J, Park SS, Gromatzky AA, Van Oudenaarden A, Lodish HF, Van Oudenaarden A, Lodish HF. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood. 2014;123:570–581. doi: 10.1182/blood-2013-10-530683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paralkar VR, Mishra T, Luan J, Yao Y, Kossenkov AV, Stacie M, Dunagin M, Pimkin M, Gore M, Sun D, Konuthula N, An X, Mohandas N, Bodine DM, Hardison RC, Weiss MJ, Dc W, Anderson SM. Lineage and species-specific long noncoding RNAs during erythro-megakaryocytic development. Blood. 2014;123:1927–1937. doi: 10.1182/blood-2013-12-544494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu W, Yuan B, Flygare J, Lodish HF. Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes Dev. 2011;25:2573–2578. doi: 10.1101/gad.178780.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez-Dominguez JR, Knoll M, Gromatzky AA, Lodish HF. The Super-Enhancer-Derived alncRNA-EC7/Bloodlinc Potentiates Red Blood Cell Development in trans. Cell Rep. 2017;19:2503–2514. doi: 10.1016/j.celrep.2017.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorolla A, Tallack MR, Oey H, Harten SK, Clemens-Daxinger L, Magor GW, Combes AN, Ilsley M, Whitelaw E, Perkins AC. Identification of novel hypomorphic and null mutations in Klf1 derived from a genetic screen for modifiers of α-globin transgene variegation. Genomics. 2015;105:116–122. doi: 10.1016/j.ygeno.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 31.Stamatoyannopoulos G, Kurnitt DM, Papayannopoulou T. Stochastic expression of fetal hemoglobin in adult erythroid cells. Proc Natl Acad Sci U S A. 1981;78:7005–7009. doi: 10.1073/pnas.78.11.7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Morales DR, Thomas K, Presser A, Bernstein BE, Van Oudenaarden A, Regev A, Lander ES, Rinn JL. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–11672. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batista PJ, Chang HY. Long noncoding RNAs: Cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergmann JH, Spector DL. Long non-coding RNAs: modulators of nuclear structure and function. Curr Opin Cell Biol. 2014;26:10–18. doi: 10.1016/j.ceb.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE, Yamamoto KR. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A. 2011;108:20497–20502. doi: 10.1073/pnas.1113536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canver MC, Orkin SH. Customizing the genome as therapy for the beta hemoglobinopathies. Blood. 2016;127:2536–2545. doi: 10.1182/blood-2016-01-678128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, Hughes JR, Hardison RC, Blobel GA, Li C, Weiss MJ. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ribeil J-A, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, El Nemer W, Bartolucci P, Weber L, Puy H, Meritet J-F, Grevent D, Beuzard Y, Chretien S, Lefebvre T, Ross RW, Negre O, Veres G, Sandler L, Soni S, de Montalembert M, Blanche S, Leboulch P, Cavazzana M. Gene Therapy in a Patient with Sickle Cell Disease. N Engl J Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 39.Saunthararajah Y, Molokie R, Saraf S, Sidhwani S, Gowhari M, Vara S, Lavelle D, DeSimone J. Clinical effectiveness of decitabine in severe sickle cell disease. Br J Haematol. 2008;141:126–129. doi: 10.1111/j.1365-2141.2008.07027.x. [DOI] [PubMed] [Google Scholar]
- 40.Esrick EB, McConkey M, Lin K, Frisbee A, Ebert BL. Inactivation of HDAC1 or HDAC2 induces gamma globin expression without altering cell cycle or proliferation. Am J Hematol. 2015;90:624–628. doi: 10.1002/ajh.24019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui S, Lim KC, Shi L, Lee M, Jearawiriyapaisarn N, Myers G, Campbell A, Harro D, Iwase S, Trievel RC, Rivers A, Desimone J, Lavelle D, Saunthararajah Y, Engel JD. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood. 2015;126:386–396. doi: 10.1182/blood-2015-02-626259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shearstone JR, Golonzhka O, Chonkar A, Tamang D, Van Duzer JH, Jones SS, Jarpe MB. Chemical Inhibition of Histone Deacetylases 1 and 2 Induces Fetal Hemoglobin through Activation of GATA2. PLoS One. 2016;11:e0153767. doi: 10.1371/journal.pone.0153767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dulmovits BM, Appiah-Kubi AO, Papoin J, Hale J, He M, Al-Abed Y, Didier S, Gould M, Husain-Krautter S, Singh SA, Chan KWH, Vlachos A, Allen SL, Taylor N, Marambaud P, An X, Gallagher PG, Mohandas N, Lipton JM, Liu JM, Blanc L. Pomalidomide reverses γ-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood. 2016;127:1481–1492. doi: 10.1182/blood-2015-09-667923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dai Y, Chen T, Ijaz H, Cho EH, Steinberg MH. SIRT1 Activates the Expression of Fetal Hemoglobin Genes. Am J Hematol. doi: 10.1002/ajh.24879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinberg MH, Chui DHK, Dover GJ, Sebastiani P, Alsultan A. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123:481–486. doi: 10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.