

Abstract
Iron-sulfur clusters (Fe-S) are one of the most ancient, ubiquitous and versatile classes of metal cofactors found in nature. Proteins that contain Fe-S clusters constitute one of the largest families of proteins, with varied functions that include electron transport, regulation of gene expression, substrate binding and activation, radical generation, and, more recently discovered, DNA repair. Research during the past two decades has shown that mitochondria are central to the biogenesis of Fe-S clusters in eukaryotic cells via a conserved cluster assembly machinery (ISC assembly machinery) that also controls the synthesis of Fe-S clusters of cytosolic and nuclear proteins. Several key steps for synthesis and trafficking have been determined for mitochondrial Fe-S clusters, as well as the cytosol (CIA – cytosolic iron-sulfur protein assembly), but detailed mechanisms of cluster biosynthesis, transport, and exchange are not well established. Genetic mutations and the instability of certain steps in the biosynthesis and maturation of mitochondrial, cytosolic and nuclear Fe-S cluster proteins affects overall cellular iron homeostasis and can lead to severe metabolic, systemic, neurological and hematological diseases, often resulting in fatality. In this review we briefly summarize the current molecular understanding of both, mitochondrial ISC and CIA assembly machineries, and present a comprehensive overview of various associated inborn human disease states.
TOC image
Iron-sulfur cluster biogenesis and trafficking enables the function of a number of cellular proteins. As such, mutations or perturbations in these pathways lead to disease states.
Introduction
Iron-sulfur (Fe-S) clusters are small inorganic cofactors, composed of iron and sulfur. Protein-bound Fe-S clusters are among the most structurally and functionally versatile cofactors across all kingdoms of life.1 These cofactors are evolutionarily conserved and ubiquitous in nature,2 with the ability to be formed spontaneously in solution from Fe2+/Fe3+, RS-, and inorganic S2−, or from Fe2+, R-SH and sulfur under reducing conditions. However, Fe-S clusters are susceptible to oxidation in the presence of oxygen, presenting challenges for cluster synthesis and binding to proteins, due to atmospheric oxygen. In addition, the iron-mediated Fenton reaction poses a severe threat to the genome due to cytotoxicity via DNA damage.2a, 3 As such, highly conserved and controlled mechanisms are in place for cluster production and trafficking to target proteins to allow for their wide range of function as protein cofactors.
Over the last decade, several new roles associated with Fe-S clusters have emerged, both as a result of progress in identifying and purifying proteins with intact clusters, and also due to the higher level of sophistication of the biophysical techniques used in their characterization. Yet the presence of these clusters remains puzzling, given the challenges present with oxygen and the potential for cellular toxicity. This raises the fundamental question of why Fe-S clusters have been retained as a vital component of essential macromolecules ranging from proteins to nucleic acids, and highlights the need for mechanistic and biochemical studies to promote understanding of the role of these versatile cofactors. With recent advances, a variety of disease conditions have been associated with Fe-S cluster proteins that impact overall cluster trafficking. Herein, we briefly review the cluster biogenesis process and describe the associated inborn human disease phenotypes.
Mitochondrial Fe-S Cluster Biogenesis
Elucidation of the molecular and cellular basis of Fe-S cluster biogenesis and transfer to putative Fe-S cluster proteins has been an area of intense investigation since it was discovered that biogenesis is not spontaneous, but rather catalyzed.4 Three distinct pathways have been identified for bacterial Fe-S cluster biosynthesis.5 The ISC system is considered the major Fe-S cluster assembly system in humans,1a, 2b required not only for the maturation of mitochondrial but also cytosolic and nuclear Fe/S proteins. This machinery primarily mediates three steps of Fe-S cluster biosynthesis; namely, (1) cluster assembly on the scaffold protein, (2) chaperone-assisted release of the Fe-S cluster and a currently unclear mechanism of mitochondrial export, if necessary, and (3) subsequent transfer to putative target apo-proteins.1b, c Much of the research concerning mitochondrial Fe-S cluster biosynthesis has been carried out in yeast cells, human cells, and with human, yeast, or bacterial proteins in vitro. The overview presented here summarizes a compilation of this research, with the goal of primarily describing the current model for the human system.
1. Cluster assembly on the scaffold protein
The first step in Fe-S cluster biogenesis is the assembly of iron and sulfur on the scaffold protein IscU. Sulfur is provided by the eukaryotic cysteine desulfurase complex of NFS1-LYRM4 (human ortholog of the Nfs1-Isd11 complex), which converts cysteine to alanine through a persulfide intermediate (Fig. 1; Table 1)).6 The precise details of the assembly complex of Iscu-Isd11-Nfs are somewhat unclear, as larger complexes have been observed in yeast and bacteria, but the physiological significance of these oligomers is unknown.7 Redox chemistry ensues, although the details are poorly understood, possibly with the assistance of a redox-active ferredoxin.7d, 8 Mitochondria import ferrous iron in a membrane potential-dependent manner via the mammalian carrier proteins mitoferrin 1 and 2.9 This iron is then transferred to IscU through an iron donor protein called frataxin (Fxn),10 for complete assembly of a [2Fe-2S] cluster. Frataxin can also function as a regulatory partner in the Nfs1-Isd11-IscU complex to control not only sulfur production, but also iron entry during Fe-S cluster assembly, to accelerate the rate of Fe-S cluster biogenesis11,12
Figure 1.
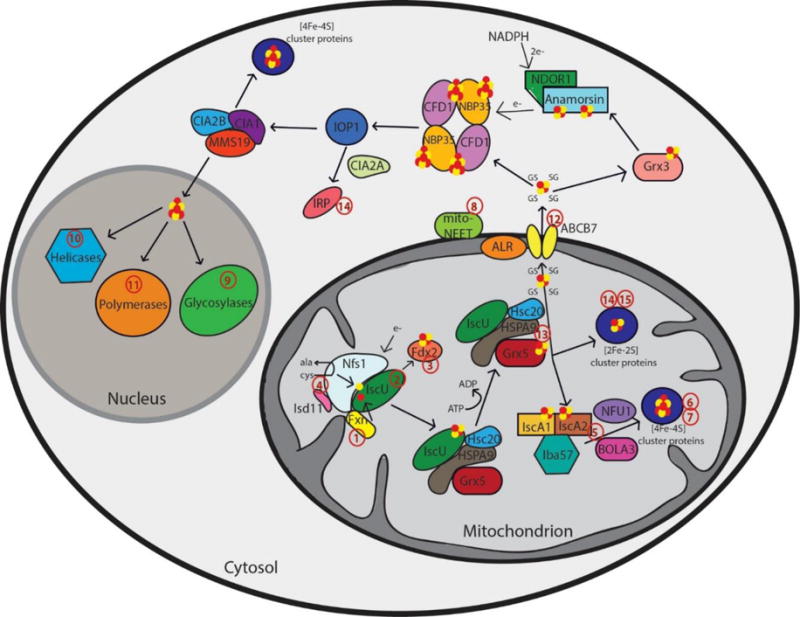
Schematic representation summarizing current models of mammalian Fe-S cluster biogenesis based on disease conditions. Nfs1, the cysteine desulfurase, with its cofactor protein Isd11 donates sulfur from L-cysteine for nascent Fe-S cluster assembly. Together, they bind to the primary scaffold protein IscU, the regulator protein frataxin (Fxn), which controls cysteine desulfurase activity and iron delivery, and a source of electrons. The newly formed IscU-bound [2Fe-2S] is subsequently transferred via a dedicated chaperone-co-chaperone system (Hsc20 – HSPA9) to Grx5. From Grx5, the Fe-S clusters are either directly inserted into mitochondrial [2Fe-2S] proteins or transferred via specific carrier systems such as the one composed of IscA1, IscA2 and Iba57 to mitochondrial [4Fe-4S] proteins. To date, the presence of another cluster type (such as glutathione complexed [2Fe-2S] cluster) under physiological conditions cannot not be excluded. For extra-mitochondrial Fe-S cluster, an uncharacterized compound (X) is exported for cytosolic and nuclear cluster trafficking via the Fe-S cluster export machinery consisting of ALR, glutathione and ABCB7. Species X has been proposed to be a glutathione-complexed [2Fe-2S] cluster and is depicted as the [2Fe-2S](GS)4 complex.17 Cytosolic cluster trafficking has been studied primarily in yeast. Fe-S clusters can be trafficked through Grx3 to anamorsin, which provides electrons to the cytosolic scaffold CFD1-NBP35. From there, [4Fe-4S] clusters are transferred via IOP1 to the CIA proteins and final target proteins, such as IRP. The CIA proteins can complex with MMS19 for cluster delivery to nuclear targets, like polymerases, helicases, and glycosylases. The red numbers in the figure correspond to the disease numbers in the review and in Table 1.
Table 1.
Iron-sulfur cluster proteins in relation to disease states. The number in the first column corresponds with the number in the review and figure 1. All of the proteins listed are shown in the general schematic of cluster biosynthesis, except Grx2 and FECH.
| Number | Protein | Function | Disease | Most Common Cause |
|---|---|---|---|---|
| 1 | Frataxin | Iron donor; allosteric effector of NFS1 | Friedreich’s ataxia (FRDA) | GAA-triplet expansion causing transcriptional silencing |
| 2 | IscU | Fe-S cluster scaffold | IscU myopathy | Splice mutation in intron 4 resulting in decreased protein levels |
| 3 | Fdx2 | Electron transport | Fdx2 myopathy | c. 1A>T mutation disrupting the initiation codon |
| 4 | Nfs1 | Sulfur donor for cluster assembly | Infantile mitochondrial complex II/III | p. R72Q decreasing transcripts and protein levels |
| 4 | ISD11 | Stability factor | Combined oxidative phosphorylation deficiency 19 (COXPD19) | p. R68L resulting in undetectable ISD11 protein levels |
| 5 | Nfu | Late acting [Fe-S] targeting factor | MMDS1 | p. G208C resulting in insufficient downstream holo targets |
| 5 | BOLA3 | Late acting [Fe-S] targeting factor | MMDS2 | Truncation mutation resulting in insufficient downstream holo targets |
| 5 | Iba57 | [4Fe-4S] cluster assembly factor | MMDS3 | p. Q314P causing protein misfolding and degradation |
| 5 | IscA1/2 | [4Fe-4S] cluster assembly | MMDS4 | p. G77S resulting in insufficient downstream holo targets |
| 6 | Ind1 | Assembly factor for complex I | Complex I deficiency | p. G56R and a c. 815-27T>C branch site mutation leading to aberrant splicing |
| 6 | SDHB | Respiratory complex II | Neurodegeneration and cancer | Mutations at Fe-S cluster ligating cysteines preventing cluster binding |
| 7 | SDHAF1 | Assembly factor for complex II | leukoencephalopathy | Mutations that disrupt binding interface for either Hsc20 or SDHB prevent cluster maturation on SDHB |
| 8 | MitoNEET | Maintenance of iron homeostasis | Association with cystic fibrosis, Parkinson’s disease, and cancer | Related to decrease in mRNA levels |
| 9 | MUTYH | Base excision repair of 8-oxoguanine:adenine mismatches | Colorectal polyposis | p. P281L resulting in low protein that cannot bind DNA |
| 10 | FANCJ | Genome maintenance and double-strand break repair | Fanconi anemia and breast cancer | p. M299I upregulates helicase activity or p.A349P that impairs DNA-protein interactions |
| 10 | XPD | Nucleotide excision repair and transcription | Xeroderma pigmentosum (XP), Cockayne’s syndrome (CS), Trichothiodystrophy (TTD), and Cerebro-oculo-facial-skeletal (COFS) syndrome | p. R112H that impairs transcriptional function and inactivates helicase activity |
| 10 | ChlR1 | Genome maintenance | Warsaw breakage syndrome | p.R263Q inhibits DNA binding and DNA-dependent hydrolysis |
| 10 | RTEL1 | Maintenance of telomeres | Cancer, dyskeratosis congenita (DC), and Hoyeraal-Hreidarsson syndrome (HHS) | Protein mutations that prevent telomere maintenance |
| 11 | POL δ and ε | DNA polymerases | Cancer | Unclear connection |
| 12 | ABCB7 | Fe-S cluster export from the mitochondria | X-linked sideroblastic anemia (XLSA) | Protein mutations that result in mitochondrial iron overload |
| 13 | Grx5 | [2Fe-2S] cluster carrier | Sideroblastic-like anemia | Aberrant splicing leading to decreased Fe-S cluster transfer and impaired iron homeostasis |
| 14 | IRP | Iron homeostasis | Parkinson’s disease | Erroneous regulation generating reactive oxygen species |
| 14 | Grx2 | Cluster carrier and redox regulation | Parkinson’s disease | Erroneous regulation of GSH levels |
| 15 | FECH | Heme biosynthesis | Erythropoietic protoporphyria | Over 130 different mutations that impair function |
2. Chaperone assisted release of the Fe-S cluster and mitochondrial export, if necessary
Cluster release from IscU to target proteins is mediated by a dedicated chaperone system comprising the Hsp70-type protein HSPA9 (homolog of yeast Ssq1 and bacterial HscA) and the co-chaperone Hsc20 (homolog of yeast Jac1 and bacterial HscB) (Fig. 1; Table 1).13 The chaperone complex binds and induces a conformational change in IscU, which enables release of the IscU-bound Fe-S cluster either to selected target proteins or to specific Fe-S cluster carrier proteins, with the proposed major target being the monothiol glutaredoxin, Grx5, a homodimeric protein, that binds a [2Fe-2S] cluster with two exogenous glutathione ligands for downstream cluster delivery and trafficking (Fig. 1).14
From Grx5, the [2Fe-2S] cluster can also be targeted to other downstream proteins within the mitochondria, or for export from the mitochondria to the cytosol via the ABC transporter ABCB7 (yeast Atm1) (Fig.1).15 The crystal structures of yeast Atm1 and a related bacterial ABC transporter that confers heavy metal tolerance16 indicate that the transporter exports a sulfur containing molecule from the mitochondria to the cytosol, which is critical for the CIA machinery.17 A role for glutathione in the ISC export process has been established in yeast cells, where a depletion of glutathione results in a phenotype similar to that of Atm1 deficiency that produces iron overload in mitochondria.18 Hence, glutathione is required for extra-mitochondrial Fe-S cluster biogenesis and therefore impacts cellular iron homeostasis.19 Prior reports have provided evidence for a tetrameric GSH-coordinated [2Fe-2S] cluster as a possible substrate for the exporter protein.17a In fact, mixing GSH-coordinated [2Fe-2S] complex with purified Atm1 stimulated its ATPase activity up to 1.8-fold.17b Structures were solved with potential glutathione and oxidized glutathione (GSSG) binding sites about 5 A° apart, which could readily accommodate a [2Fe-2S] cluster and provide support for a glutathione complexed Fe-S cluster.16 In vivo determination of these glutathione complexed [2Fe-2S] clusters can further strengthen the physiological existence of these small molecules in the cell. Another study has suggested the molecule GSSSG, i.e., the oxidized form of GSSH (glutathione persulfide) and GSH, as the Atm1 substrate and potential sulfur carrier.20 Nevertheless, an as yet ill-defined species X must be exported from the mitochondria in order to support proper Fe-S cluster trafficking in other cellular compartments.
3. Transfer to putative target apo-proteins
Recent studies have shown that a number of other proteins are further involved in Fe-S cluster biogenesis. Following release of the Fe-S cluster from the scaffold protein, it can either be directly delivered to [2Fe-2S] target proteins or converted to a [4Fe-4S] cluster.1b, c A current model suggests that [4Fe-4S] cluster maturation requires additional ISC proteins: IscA1 and IscA2, and the folate binding protein Iba57,21 in a process that requires sequential delivery of a [2Fe-2S], postulated to be from Grx5, with reductive coupling to form a [4Fe-4S] cluster (Fig. 1).22 However, knockdown of these IscA proteins does not eliminate production of [4Fe-4S] clusters, so other proteins and complexes may utilize a similar reductive coupling mechanism on target Fe-S cluster proteins.21b, 21d Other more specialized targeting factors have evolved to assist the maturation of dedicated subsets of [4Fe-4S] proteins and also for transfer of [2Fe-2S] clusters downstream of the scaffold assembly to final target proteins, such as Nfu, BolA3, Ind1, and SDHAF (Fig. 1; Table 1).1a However, the precise mapping of these pathways and all of the potential functions of these proteins remain unclear. For example, Nfu may act as an alternative scaffold protein to IscU, because Nfu can accommodate both a [2Fe-2S] cluster as well as a labile [4Fe–4S] cluster in vitro and pass these on to apo-proteins23
Cytosolic and Nuclear Fe-S Cluster Protein Biogenesis
The biogenesis of cytosolic and nuclear Fe-S proteins is heavily dependent on the conserved mitochondrial Fe-S cluster (ISC) assembly machinery in addition to the ABC transporter protein and cytosolic Fe-S cluster (CIA) assembly machinery. To date, nine components of the CIA machinery have been identified that play a role in mobilization of Fe-S cluster and its insertion into extra-mitochondrial apo-proteins. The assembly process has been primarily studied in yeast and comprises two mains steps:1a, 4c (1) [4Fe-4S] cluster assembly on the Cfd1-Nbp35 scaffold complex and (2) transfer of Fe-S clusters to target apo-proteins from the CIA scaffold complex.1b, c Yeast nomenclature has been used throughout this section and describes the proposed models based on the work in yeast Similar pathways are likely functional in humans too.
1. [4Fe-4S] cluster assembly on the CFD1-NBP35 scaffold complex
An initial step in cytosolic Fe–S protein biogenesis is the assembly and binding of a transient [4Fe–4S] cluster on the cytosolic scaffold protein complex, a heterotetramer comprised of the two P-loop NTPases Cfd1 and Nbp35, which bind two different kinds of [4Fe-4S] clusters (Fig. 1).4c, 24 A loosely bound [4Fe-4S] cluster at the C-terminus of both proteins bridges the two complex subunits together, whereas the other [4Fe-4S] cluster is tightly coordinated at the N-terminal domain of the Nbp35 protein. The lability of the C-terminus cluster is considered to be important for the second step, involving transfer of the cluster to target apo-proteins. Generation of the functionally essential N-terminal Fe-S cluster (N) of Nbp35 depends on the flavoprotein Tah18 (yeast homolog of NDOR1) and the Fe-S protein Dre2 (yeast homolog of CIAPIN1), which serve as an NADPH-dependent electron (e-) transfer chain (Fig. 1).25 Cytosolic-nuclear Fe-S protein biogenesis additionally requires the cytosolic multidomain glutaredoxin, Grx3, yet its precise function and site of action in the pathway remains to be determined,26 although studies from the yeast system suggest roles in iron regulation and maturation of other proteins such as ribonucleotide reductase.26b, 27 Accordingly, it is speculated that these cytosolic monothiol glutaredoxins play a general role in iron trafficking.
2. Transfer of Fe-S clusters to target apo-proteins from the CIA scaffold complex
The bridging Fe-S clusters are released from Cfd1–Nbp35 and transferred to apo-proteins in a reaction mediated by the Fe-S protein Nar1 and the CIA targeting complex Cia1–Cia2–Mms19 (Fig. 1).1a, 4c Nar1 coordinates two [4Fe–4S] cofactors that are similar to those in hydrogenases and bridges the early and late stage CIA machinery.61, 62 Cia1, Cia2 and Mms19 form dimeric and tetrameric complexes in both yeast and mammalian cells, and interact with target apo-proteins and assure specific Fe-S cluster insertion,28 including the crucial function of maturation of nuclear Fe-S proteins (Fig. 1). Several of the proteins involved in mitochondrial Fe-S cluster biogenesis have also been reported to be localized in the cytosol such as IscU, Nfu, Nfs1 and Isd11.29 However, their functional relevance is yet to be established.
Diseases Associated with Fe-S Cluster Biogenesis Proteins
A number of inborn human diseases have been identified with mutations associated either with the gene or the protein involved in Fe-S cluster biogenesis. In recent years, additional diseases have been identified with the increasing availability of information concerning the function and properties of Fe-S cluster proteins.1b, c Several of these disease states are discussed below (Table 1). Additional disease states related to Fe-S cluster maturation have been discussed in terms of infectious agents, but those will only be described briefly in this work.
Diseases Associated with Mitochondrial Fe-S Cluster Biogenesis
1. Friedreich’s ataxia (FRDA)
Friedreich’s ataxia (FRDA) is a neurodegenerative disease that is by far the most common disease linked to Fe-S cluster biosynthesis.30 FRDA patients carry a GAA-triplet expansion mutation in the first intron of the frataxin gene (Fxn). This expansion leads to transcriptional silencing of frataxin thereby resulting in severely decreased levels of the frataxin protein. Nonsense and missense mutations or deletions also result in the same symptoms for patients.30c, 31 No therapy for FRDA exists today, despite much effort to increase frataxin levels or mitochondrial function, or alleviating secondary symptoms by antioxidants or iron chelator strategies.32
2. IscU myopathy
IscU myopathy is characterized by severe exercise intolerance, tachycardia, fatigue and pain in active muscles.1a, 2b Patients have decreased mitochondrial Fe-S cluster enzyme activities, as well as iron deposits.33 Most affected patients are homozygous for a splice mutation in intron 4 of the IscU gene resulting in decreased IscU protein levels.33–34 A therapeutic approach consisting of antisense oligonucleotides, in order to induce skipping of the splice site responsible for the condition and restore normal mRNA splicing in fibroblasts, is currently being tested,35 as are treatment options that control miRNA levels to deal with the vascular symptoms and conditions.36 IscU has also recently been found as a target for regulation by the tumor suppressor protein p53 with implications for modulating iron homeostasis and iron levels in cancer.37
3. Fdx2 myopathy
In addition to IscU myopathy, a second novel mitochondrial myopathy has been found in a patient due to a disruption on the FDX1L gene, which encodes the mitochondrial protein ferredoxin 2 (Fdx2).38 The myopathy is due to a mutation (c. 1A>T) that disrupts the ATG initiation codon, resulting in low levels of Fdx2 in muscle and fibroblasts, and decreased activities of respiratory complexes I-III and mitochondrial aconitase.38 Symptoms resemble those of IscU myopathy but are not as severe, possibly indicating a functional rescue by the homolog Fdx1 or a bypass mechanism compared to the role of IscU in Fe-S biogenesis.38
4. ISD11-Nfs1
Recently, the importance of ISD11 (human LYRM4) and Nfs1 in cluster biogenesis as part of the de novo cluster assembly complex was highlighted as a result of their roles in disease states. For Nfs1, patients were found with a missense mutation (p. Arg72Glu) that results in infantile mitochondrial complex II/III deficiency.39 Levels of Nfs1 transcripts and protein levels were decreased in patient fibroblasts, as were the levels of respiratory complexes II and III, which could be indicative of an overall disruption in Fe-S cluster biogenesis and is also consistent with the absence of ISD11 association in co-immunoprecipitation assays.39 However, treatment with mitochondrial cofactor therapy was successful for one of the three patients in preventing lethality during infancy, suggesting a potential therapeutic option.39
The requirement for ISD11 in stabilizing Nfs1 has been confirmed in patients with mutations on ISD11, who demonstrated a similar phenotype in the form of respiratory complex deficiency, termed combined oxidative phosphorylation deficiency 19 (COXPD19).40 Interestingly, these patients also exhibited further downstream complications with deficiencies in complexes I, II and III, in addition to other mitochondrial and cytosolic Fe-S proteins.40a Both patients were homozygous for a missense mutation (p. Arg68Leu),40a which caused the levels of ISD11 to be undetectable and a decrease in respiratory chain complexes to below 40%. This mutation also impacted the interaction with Nfs1 by promoting aggregation and inhibiting desulfurase activity.40a Furthermore, IscU levels were also observed to decrease when the R68L mutation was present, suggesting that the complex is also essential for promoting the stability of IscU for overall Fe-S cluster biogenesis and trafficking.40b
5. Multiple Mitochondrial Dysfunction Syndrome (MMDS)
Multiple mitochondrial dysfunction syndrome (MMDS) describes a class of fatal mitochondrial diseases caused by mutations in proteins involved in Fe-S cluster biogenesis and trafficking. To date, four types of MMDS have been identified: namely, MMDS1 due to mutations on Nfu,41 MMDS2 for mutations with BOLA3,41b, 42 MMDS3 with mutations on Iba57,43 and MMDS4, with mutations on IscA2.44 Recently, an IscA1 variant has been described that results in MMDS-like symptoms, which may expand the class of disorders.45 All MMDS patients exhibit similar symptoms that include metabolic acidosis with hyperglycemia, and deficiency of respiratory complexes and lipoic acid bound enzymes, such as pyruvate dehydrogenase (PDH) and α-ketogluterate dehydrogenase (KGDH).41f, 46 Lipoate biogenesis relies on the mitochondrial [4Fe-4S] enzyme lipoate synthase (LIAS).
For MMDS1, five patients have been described with a homozygous mutation in Nfu (p.Arg182Gln), where the mutation is close to a splice site and results in skipping of exon 6 and no detectable protein levels.46 Most other patients were described with a homozygous mutation (p.Gly208Cys) that affects an amino acid close to a highly conserved Fe-S cluster binding domain of the Nfu protein. This mutation does not affect protein levels but does impair Fe-S cluster binding and transfer, most likely due to a perturbed monomer-dimer equilibrium that limits cellular reconstitution of Nfu.41f, 47 Most recently, a study of patients with the heterozygous gene for Nfu, one with the altered splice site or with the Gly208Cys missense mutation and the native allele, has revealed that low levels of the wild type Nfu mRNA were produced, which yielded sufficient functional LIAS to avoid lipoic acid deficiency, but the wild type transcripts were insufficient to support functional respiratory complex II.41d
In the case of MMDS2, 6 different patients have been identified with mutations on BOLA3.41b, 42 The first patient demonstrated a single base pair duplication that produced a frameshift with a premature stop codon and therefore loss of the full length protein.41b Two other patients have been characterized as having a missense mutation at a highly conserved residue (p.Ile67Asn)42b that could affect the overall protein fold based on the solution NMR structure of human BOLA3.48 The remaining three patients all demonstrated a truncating mutation of eight amino acids upstream of the first truncating mutation.42a To date, the molecular role of BOLA3 remains to be elucidated; however the availability of an NMR structure48–49 with additional biochemical characterization will provide increasing understanding of its role in MMDS.
Similar to BOLA3 in MMDS2, a wide variety of mutations on Iba57 have been identified for MMDS3. Initial reports described different patients with a range of phenotypes. The first case resulted in the most severe condition caused by a missense mutation (p. Gln314Pro) that appeared to result in protein misfolding with subsequent degradation to concentrations below critical levels.43a The mildest phenotype was observed in a patient with an Iba57 mutation that resulted in reduced mRNA levels because of aberrant splicing and a frameshift mutation that yielded a truncated form of Iba57.43c The intermediate phenotype of MMDS3 was due to a mutation on Iba57 that drastically reduced its ability to function in Fe-S biogenesis with diminished levels of [4Fe-4S] clusters.43b Most recently, four additional mutations on Iba57 have been observed with generally the same symptoms and phenotypes.43d The prevalence of such diverse mutations on Iba57 that all result in MMDS3 makes prediction of the disease difficult, based on genetic information, and strengthens the need for further biochemical characterization of Iba57 to understand why so many possible causes of the disease exist.
MMDS4 was the most recent class to be discovered, with six patients exhibiting a missense mutation (p.Gly77Ser) on IscA2, which is located in a highly conserved cluster binding domain. However, the exact effect of the mutation is unclear, since mRNA levels were not significantly impacted.44 Interestingly, patient fibroblasts also showed lower levels of IscA1 and Iba57 than control fibroblasts.44 How this mutation results in lower protein levels of these partner proteins remains unclear. Similarly, an IscA1 mutation (p.Glu87Lys) has been implicated in causing MMDS-like symptoms, which may result in a new class of the disorder, as one patient has been found with this mutation that is expected to lead to protein instability due to loss of a conserved salt-bridge.45
The class of MMDS disorders describes a newly discovered disease condition and further details on how mutations in key Fe-S biogenesis proteins influence cellular function, and their roles that result in disease, need to be determined in order to fully aid patients.
6. Respiratory complex 1
Fe-S clusters play essential roles in respiration and metabolism as cofactors in respiratory chain complexes. The manner in which Fe-S clusters are integrated into the respiratory complexes is currently unclear; however, respiratory chain complex maturation factor proteins have recently been identified that are required for correct formation of the complexes, either by promoting subunit association or cofactor integration.50
In particular, complex I of the respiratory complex, also known as NADH-CoQ oxidoreductase, is composed of 44 subunits, utilizes at least 10 assembly factors for correct complex formation, and accounts for one-third to one-quarter of mitochondrial disease, which occurs in about 1 out of every 5,000 births.51 One such assembly factor, Ind1 or NUBPL, has been implicated as an Fe-S cluster delivery protein that is specifically required for complex I assembly and activity in yeast,51 and mutations or perturbations of the Ind1 gene have resulted in a specific deficiency of complex I, while other respiratory complexes remain intact.52 Genetic sequencing has found two mutations in patients: namely, a p. Gly56Arg missense mutation and a c. 815-27T>C branch site mutation. Of the two, pathogenicity has been more attributed to the branch site mutation, since it results in aberrant splicing, very low levels of the NUBPL protein, and reduced levels of fully assembled complex I, as little as 20%.52–53 However, there remains the possibility that the missense mutation contributes to the pathogenicity of the branch site mutation. Furthermore, recent work in Arabidopsis thaliana suggests that Ind1 may also play a role in translational control,54 which may contribute to phenotypes observed in the pathogenic state. However, no work has been done in human cell lines to either evaluate this additional functional role or examine the possible role of Ind1 in Fe-S cluster delivery.
7. Respiratory complex II
Respiratory complex II (or succinate dehydrogenase, SDH) is composed of four subunits (A-D). Subunits C and D anchor the complex to the membrane, while subunits A and B exhibit catalytic activity.55 All of the subunits have been implicated in disease states, because SDH plays vital cellular roles as a result of its involvement in both the Krebs cycle and oxidative phosphorylation. However, only subunit B (SDHB) contains iron-sulfur clusters.55a SDHB is the most commonly mutated subunit of the four,55a and mutations result primarily in either neurodegeneration, generally in the form of infantile leukodystrophy,56 or cancer, particularly renal cell carcinoma, paraganglioma, pheochromocytoma, or WT gastrointestinal stromal tumors.55, 57
In terms of the neurological impact, one patient has been identified with a missense mutation at an evolutionarily conserved residue (p.Asp48Val) near an iron-sulfur cluster binding site, which is hypothesized to be crucial for efficient electron transport through the respiratory chain and resulted in an almost complete loss of SDHB and decreased levels of SDHA.56 As with many mitochondrial disorders, there is no cure for SDH deficiency; however, the patient in this study was supplied with an oral treatment of coenzyme Q10 to yield a subjective improvement in strength.56
The remainder of patients with mutations on SDHB have been linked to some form of cancer. A recent analysis of the Leiden Open Variation Database showed that 37% of all the reported cancer cases linked to SDHB are due to mutations to the cluster cysteinyl ligands that prevented cluster binding, or in the L(I)YR motif, which blocks chaperone recognition by Hsc20 and the HSPA9 chaperone to prevent binding interactions with either of these proteins.58
As is the case for respiratory complex I, maturation factors for SDH have also been implicated in causing disease. In the case of SDHB, two assembly factors exist: SDHAF1 and SDHAF3.55b, 59 Both factors are believed to help protect SDHB from oxidative damage during the assembly of the complete SDH complex, by shielding the nascent SDHB Fe-S clusters from damage by reactive oxygen species.55b, 59 SDHAF1 also functions in concert with Fe-S cluster chaperones in recruiting the chaperone complex to SDHB via the L(I)YR motif,60 and was found to co-immunoprecipitate with Hsc20.58b As such, mutations on SDHAF1 that disrupt the binding interface for either SDHB or Hsc2060 have also been implicated in causing leukoencephalopathy,61 where accumulation of succinate in the white matter of the brain is caused by a loss of SDH activity. The accumulation of succinate was due to degradation of SDHB, when non-functional SDHAF1 was present; however, succinate levels could be reduced following treatment with riboflavin, which serves to increase the flavinylation of SDHA and allows it to convert succinate to fumarate to restore the activity of the Krebs cycle.60
8. MitoNEET
Outside of the traditional mitochondrial Fe-S cluster components and targets, the outer mitochondrial membrane protein, mitoNEET has been implicated in numerous diseases.62 MitoNEET was first discovered bound to thiazolidinedione, a drug used for the treatment of type 2 diabetes. Since then, its functional role has been under active investigation. Given its association with a diabetes drug, one of the physiological roles of mitoNEET is linked to diabetes, via control of lipid and glucose metabolism,63 most likely by modulation of lipid accumulation,64 which is related to insulin resistance. MitoNEET is also involved in controlling overall mitochondrial bioenergetics by regulating homeostasis and the ratio of energy stored to the energy spent,64 which connects with its role in causing obesity and potential regulation of mitophagy.63 In conjunction with mitochondrial homeostasis, overexpression of mitoNEET results in diminished levels of mitochondrial iron, while knockdown leads to iron overload and reduced activity of the electron transport chain, resulting in overall lower cellular energy.64 Accordingly, mitoNEET plays a key role in cellular and whole organism iron regulation and metabolism, and in maintenance of reactive oxygen species,62, 65 potentially by binding a [2Fe-2S] cluster for delivery and repair of cytosolic aconitase (IRP1),66 in a process that is under redox control.67
Given its broad role in metabolism, mitoNEET has also been associated with cystic fibrosis due to a decrease in mitoNEET mRNA,68 Parkinson’s disease due to an interaction with PARKIN,69 and cancer and tumorogenesis,62, 70 where mitoNEET was found to be upregulated in breast cancer cells.71 Despite the association of mitoNEET with broad cellular processes and diseases, many questions remain unanswered concerning its specific cellular mechanism and the connection between its role in Fe-S cluster biosynthesis and overall mitochondrial regulation. Furthermore, mitoNEET is still under consideration as a target for drug development for the treatment of cancer and diabetes based on overall mitochondrial dysfunction,62–63 with additional possibilities considering its likely roles in Parkinson’s and cystic fibrosis. As more is learned concerning mitoNEET and its physiological roles, better therapeutics may be developed to compensate for disruptions in its function.
Diseases Associated with Cytosolic Fe-S Cluster Proteins
Although a number of proteins have been identified as components of the cytosolic Fe-S cluster machinery,4b, 15, 72 many have only been discovered or characterized in depth over the last 5 years. For this reason, many of these proteins have not yet been linked to disease conditions. However, given their required roles in the biogenesis of essential Fe-S clusters and maintenance of iron homeostasis, there is the possibility of disease association as these proteins become better understood and disease states are examined in greater depth. For example, examination of proteins in the NAR family in Arabidopsis thaliana73 and yeast74 have indicated conserved roles in maintaining a response to oxidative stress, which may have downstream implications for genome maintenance,72 due to the connection to DNA damage repair pathways and corresponding involvement in aging disorders.
Likewise, the cytosolic Fe-S protein MMS19, has not specifically been associated with any diseases. However, this protein has been characterized as required for maturation of many nuclear Fe-S cluster containing proteins that are particularly involved in DNA replication and repair, such as FANCJ, XPD, RTEL, and POLD (discussed below).75 The essential nature of MMS19 in maturation of key nuclear proteins may link it to cancer and neurodegenerative phenotypes in the future.75b
Diseases Associated with Nuclear Fe-S Cluster Containing Proteins
With oxygen sensitivity and possible toxicity associated with Fe-S clusters, it was assumed that most nucleic acid enzymes did not contain Fe-S clusters. However, this view changed with the discovery of an Fe-S cluster in the DNA helicase XPD and associated protein members that are involved in DNA repair,76 and the possibility that Fe-S cluster containing enzymes find DNA damage by probing DNA integrity electronically.77 Today, all DNA polymerases involved in replication and also certain helicases that participate in Okazaki fragment processing contain some form of Fe-S clusters.2a, 78
9. Glycosylases
The glycosylase family provided the first protein example to be characterized with an Fe-S cluster by way of endonuclease III in E. coli;2a however, mammalian forms are now being characterized. Members of this family conduct Base Excision Repair (BER) to detect, remove, and replace damaged DNA bases. In particular, the human protein MUTYH serves to repair mismatches associated with 8-oxoguanine.79 A number of disease variants of MUTYH have been identified that result in cancer in the form of colorectal polyposis, termed MUTYH-associated polyposis (MAP).80 To date, one of the mutations (P281L) was found at a highly conserved proline in the Fe-S cluster binding domain, which impairs the ability to bind substrate DNA.80–81,82 A detailed understanding as to why variants, such as these, result in MAP will provide assistance to clinicians in patient-screening for cancer risk and subsequent treatment options.82
10. Helicases
Helicases constitute a second family of nuclear proteins that contain Fe-S clusters and function in DNA processing. Within the helicase family, the Fe-S cluster subset belongs to the SF2 grouping with shared homology of the helicase domain.2a, 83 A number of these proteins have disease implications and are therefore discussed below.
FANCJ, in conjunction with 15 other genes, is implicated in causing Fanconi anemia; a condition characterized by congenital defects, progressive bone marrow failure, susceptibility to cancer with chromosome breaks, and sensitivity to DNA damaging agents that result in intra-strand cross-links.84 Furthermore, FANCJ plays a role in genome stability by unwinding G-quadruplexes85 and by conducting double-strand break repair.86 Of the numerous mutations identified in the gene for FANCJ that are linked to various disease states, two of them (M299I and A349P) occur in the iron-sulfur cluster binding domain, where the M299I mutation has been found in breast cancer patients,87,83a, 88 and the A349P mutation causes Fanconi anemia.89 These mutational analyses have demonstrated the importance of the Fe-S cluster in the function of FANCJ and its role in downstream DNA processing activities in disease.83
A second Fe-S cluster-containing helicase is XPD, which serves key roles in nucleotide excision repair to remove photo-damaged DNA bases, and in transcription as a member of the TFIIH complex.83 Mutations in XPD have been linked to xeroderma pigmentosum (XP), Cockayne’s syndrome (CS), Trichothiodystrophy (TTD), and Cerebro-oculo-facial-skeletal (COFS) syndrome.83 To date, only one of these disorders has been linked to a mutation in the Fe-S cluster binding domain. In patients with TTD, a R112H mutation is present at a highly conserved residue that removes a hydrogen bond to a cluster-ligating cysteine,90 resulting in impaired interactions with proteins in the TFIIH complex and involvement in helicase inactivation.83a, 90a
The last two Fe-S cluster helicases that we will describe have been implicated in disease much more recently. The helicase ChlR1, or DDX11, is less understood than those already discussed. Its specific role in DNA processing and genome integrity is unclear; however, it may serve primarily to unwind intermediate DNA structures present during replication and recombination,91 with additional potential roles in sister chromatid cohesion, heterochromatin organization, viral genome maintenance, DNA repair, and G-quadruplex unwinding, via its helicase and ATPase activities92. Although its precise function is unknown, the first human patient with a defect in ChlR1 was identified in 2010 in a condition termed Warsaw breakage syndrome, due to either a splice site mutation93 or p. Arg263Gln mutation in ChlR1, which perturbs a conserved arginine residue in the Fe-S cluster binding domain.94
The last Fe-S cluster helicase that we will discuss is the Regulator of Telomere Length 1 (RTEL1), which functions to maintain telomere integrity and unwinds various DNA structures that are assembled in the process of replication, recombination, and repair.95 Thus far, a number of RTEL1 variants have been found that exhibit single nucleotide polymorphisms (SNPs) and increased susceptibility for brain tumors or hepatocellular carcinomas, suggesting a key role for the protein in tumorigenesis.95 Mutations of RTEL1 in coding regions have instead been implicated in premature aging disorders95, such as dyskeratosis congenita (DC)96 and Hoyeraal-Hreidarsson syndrome (HHS),97 which is considered a more severe form of DC. Both diseases are characterized by multi-system bone marrow failure, due to impaired telomere maintenance.95 One mutation (p. Glu275Lys) has been found in the Fe-S cluster domain,96b but the biochemical characterization and impact of this residue on the cluster has not yet been examined.
Further biochemical characterization is required to understand the Fe-S cluster helicase family, particularly to determine the role of the cluster in relation to disease phenotypes. Efforts in examining the molecular mechanisms will aid current potential therapies that use chemical rescue by small molecules to restore function to mutated helicases and treat disease conditions.2a
11. Polymerases
The last class of Fe-S cluster containing DNA processing enzymes that we will briefly discuss are polymerases. All replicative DNA polymerases, such as delta (δ) and epsilon (ε), contain essential [4Fe-4S] clusters2a, 98 that are matured via the CIA machinery.99 These two polymerases have implications in cancer;100 however, the precise functional role of the Fe-S cluster is unknown,2a and therefore its link to disease is unclear.
Conditions Resulting in Mitochondrial Iron Overload
Although the disorders below are linked primarily to the misregulation of iron, they occur specifically in Fe-S cluster proteins. Whether this link is due to a simple abundance of iron or to a more direct connection between Fe-S clusters and iron regulation, perhaps via the iron regulatory protein (IRP), remains uncertain and requires further research.
12. X-linked sideroblastic anemia (XLSA)
X-linked sideroblastic anemia is caused by mutations in the mitochondrial transporter protein ABCB7.101 The condition is characterized by early-onset ataxia, sideroblastic anemia and iron overload in affected tissues, primarily in the mitochondria, which suggests that the transporter is unable to function properly for trafficking of clusters in the cytosol and nucleus. Expanding on the previous disease symptoms, new phenotypes are being associated with ABCB7 mutations that give rise to cerebellar ataxia102 and ringed sideroblasts.103
13. Sideroblastic-like anemia and iron overload
A homozygous mutation in the exon 1 of the GRX5 gene causes problems with RNA splicing, resulting in sideroblastic-like anemia and iron overload condition.104 This condition causes moderate anemia, hepatosplenomegaly, and iron overload, and leads to a decreased level of mRNA for Grx5 but not a complete loss of the Grx5 protein function.14b, 104a The decreased protein levels result in impaired cluster transfer to downstream Fe-S proteins, such as IRP1 and ferrochelatase (FECH).104c An additional sideroblastic anemia patient has been found with a mutation in GRX5, which is a compound heterozygous mutation p. Lys101Gln and p. Leu148Ser.105 In vitro analysis of these mutations in Grx5 revealed that the L148S mutation results in similar effects as observed in cases of patients with diminished protein levels, where cluster transfer to downstream targets was impaired.106 The K101Q mutation yielded different effects, in that this variant was unable to bind a Fe-S cluster,106 and correlates with the fact that this residue participates in GSH binding,107 which is required as a cluster ligand. Aside from implications in sideroblastic anemia, patients have also been found to display variant non-ketotic hyperglycinemia, with symptoms similar to those observed for patients with MMDS2 due to mutations in BOLA3.42a In these three patients, Grx5 was found with a p. Lys51del mutation that impaired transfer to downstream targets and resulted in low levels of lipoate synthase activity,42a indicating that this mutation has functional consequences similar to the mRNA splicing defect and L148S variants106. Although Grx5 appears to function primarily in the early stage of Fe-S cluster trafficking,14a, b, 104a, 108 additional multifunctional cellular roles may exist given the variety of phenotypes observed in disease conditions related to Grx5.42a, 104c, 105–106
Most recently, a phenotype similar to that observed for sideroblastic anemia has been linked to mutations in the mitochondrial Fe-S cluster biosynthesis chaperone, HSPA9, which has also been implicated in key roles in erythroid differentiation.109 Mutations to HSPA9 result in lower mRNA expression levels with incomplete loss of function leading to the anemia,110 a phenotype which may be under a process of complex regulation.
Fe-S Cluster Proteins Linked to Neurological Disorders
Similar to the diseases related to iron overload, neurological disorders are often related to iron misregulation; however, it remains to be seen if there is a more direct connection between Fe-S clusters and these conditions. Recent work has indicated a link between mitochondrial dysfunction and neurodegenerative disorders, such as Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease.111 These neurodegenerative conditions are thought to exist as a feedback loop, where mitochondrial dysfunction causes increased production of reactive oxygen species, which decreases Fe-S cluster and heme biosynthesis and leads to IRP activation and iron overload in mitochondria.111–112 This accumulated iron in turn generates additional reactive oxygen species via Fenton chemistry. In the case of Alzheimer’s disease, these factors promote oligomerization of amyloid-β to form plaques113 and the formation of Fe3+-paired helical filament aggregates114 to further drive iron accumulation and decrease the levels of the soluble Tau protein.115 Similarly, in Huntington’s disease the Huntington protein (Htt) is believed to play a role in maintaining iron homeostasis via the transferrin receptor;111b, 116 when mutated in the disease state, Htt likely perturbs iron uptake and contributes to mitochondrial dysfunction.111b In the class of the neurodegenerative disorders, Parkinson’s disease has most been associated with iron-sulfur cluster proteins.
14. Parkinson’s Disease
Parkinson’s disease is a neurodegenerative disease affecting roughly 1:800 individuals with a survival rate of about 15 years after diagnosis. The degenerative process affects dopaminergic neurons leading to motor function deficits, such as resting tremor, rigidity, and postural instability.111–112 Age is considered to be the main risk factor for Parkinson’s-however, environmental toxins or genetic susceptibility are also implicated.117 Data suggests that patients with Parkinson’s also have a defect in ISC synthesis, partial inhibition of Complex I of the respiratory chain, and increased iron levels in dopaminergic neurons (in the substantia nigra),112, 118 again due to erroneous regulation of iron homeostasis by IRP.111–112 In addition to this, the pathway which delivers transferrin-bound iron to mitochondria and to complex I of the respiratory chain is found in the substantia nigra dopaminergic neurons, but also consists of transferrin receptor 2.119 In Parkinson’s, there is accumulation of oxidized transferrin inside mitochondria, which results in the release of ferrous iron from transferrin and the generation of hydroxyl radicals via Fenton chemistry.119a, 120 Furthermore, glutaredoxin 2 (Grx2) has been implicated in promoting the iron accumulation feedback loop in Parkinson’s disease based on its ability to regulate cellular redox status by acting on its substrate glutathione (GSH) to impact overall iron homeostasis.112, 121 As our understanding of these neurodegenerative disorders is expanded, therapeutic approaches are being explored as well. Thus far, some success has been observed in patients with Parkinson’s and other diseases in this area through the use of iron-chelation therapy.111b However, only one of the chelators has been successful in clinical trials,122 since many of them cause adverse side effects due to lack of targeting to specific tissues, resulting in systemic iron depletion.111b, 112 New compounds or approaches that include targeting with fewer broad effects could provide a successful treatment option.
15. Erythropoietic Protoporphyria
Proper Fe-S cluster biosynthesis is also crucial for the pathway of heme biosynthesis, since the last step in the process is catalyzed by ferrochelatase (FECH), which binds a [2Fe-2S] cluster.123 Mutations in FECH account for 98% of erythropoietic protoporphyria (EPP) with patients affected worldwide124 and prevalence as high as 45% in some countries.125 Symptoms of EPP include heightened and painful photosensitivity, and in some severe cases, liver damage occurs;124 all of which are due to the accumulation of the heme precursor, protoporphyrin IX from a decrease in FECH activity to less than 30%.126 The majority of patients present with the IVS3-48T>C low expression allele in conjunction with a missense mutation that causes a loss of FECH function, although other combinations have been observed, where patients can simply exhibit multiple loss of function mutations.124, 127 As of 2015, at least 130 different mutations on FECH have been implicated in EPP, with new mutations or sites being identified, as sequencing is extended and population studies are undertaken.127–128 Therefore, specific genotype-phenotype correlations and understanding of the specific molecular mechanism are challenging. Furthermore, patients with EPP were also found to display abnormal mRNA levels of mitoferrin-1,129 which has been found to complex with FECH and ABCB10 for heme biosynthesis130 and may also contribute to the phenotype of EPP. As familial screening becomes more common, doctors can be more aware of carriers to provide better treatment plans for patients,128 which typically involve avoiding sunlight and tools to reduce excess protophorphyrin for the less severe cases, and solutions, such as bone marrow transplants for long term solutions in the case of drastic phenotypes.124
Fe-S Cluster Protein Maturation in human infectious agents
Iron-sulfur clusters also play a vital role in prokaryotic metabolism, as catalysts, redox sensors or electron trafficking agents in a variety of important proteins. For example, across different strains of E. coli there are over 150 proteins that contain Fe-S clusters and over 50 different proteins in Mycobacterium tuberculosis.131 Fe-S clusters have consequently been exploited for bactericidal toxicity, where antibiotics promote cell respiration, forming endogenous superoxide and peroxide species, which degrade Fe-S clusters that serve important roles in bacterial cell cycles, and DNA and protein biogenesis.132 Fe-S clusters have also been targeted in terms of human pathogens in Pseudomonas aeruginosa. Deletion of the transcriptional regulator iscR leads to increased susceptibility to peroxides and severely decreased virulence in mouse and Drosophila models.133 Much work has been done in studying Fe-S clusters in Mycobacterium tuberculosis, a facultative pathogen that can live inside macrophages and lead to tuberculosis. These bacteria contain a family of WhiB-like 4Fe-4S cluster-containing transcriptional regulators that could be exploited as an attractive potential antibacterial target, since these regulators play a vital role in Mycobacterial cell division, lipid metabolism, oxidative stress, and antibiotic resistance.134
Summary and Perspectives
Since Fe-S clusters are required in a number of essential proteins or as necessary cofactors for a multitude of enzymatic reactions and physiological processes, conditions that perturb the process of cluster biogenesis and trafficking often lead to disease states. Additionally, a number of these perturbations lead to lethal consequences, making them challenging to investigate, which highlights the importance of understanding the molecular details of Fe-S cluster biosynthesis. Furthermore, recent work in bacteria, utilizing various double mutants and media supplementation, has demonstrated that these genetically engineered forms can grow, albeit slowly, without Fe-S clusters.135 The implications of this work in relation to human Fe-S cluster-based diseases are currently unclear, but may provide alternative treatment strategies in the future.
To date, the majority of inborn human diseases related to biogenesis or Fe-S cluster binding proteins are caused by mitochondrial proteins.1a, 2b No specific diseases have been associated with the cytosolic system, even though they have demonstrated roles in yeast viability (Table 1).1a, 4c, 15, 75, 98, 136 Furthermore, diseases have been linked to nuclear proteins that bind Fe-S clusters to function in DNA replication and repair;2a, 80, 82–84, 86, 90a, 93, 95, 100c, d however, the presence of the cluster and some of the specific cellular functions remain unclear, presenting challenges when trying to understand the role of the cluster in disease conditions. Aside from diseases associated with specific Fe-S cluster proteins, a subset of neurological disorders, such as Alzheimer’s and Parkinson’s, have been linked to the Fe-S cluster biogenesis pathway based on the presence of free iron and overall iron dysregulation in mitochondria.111 In addition, Fe-S cluster proteins from all of the cellular compartments have been implicated in a variety of cancers.3 The essential nature and necessary function of these metal cofactors dictates the need for a better understanding of their biogenesis and trafficking mechanisms to clarify the molecular basis of disease states and lead to potential therapeutic approaches.
Significance to metallomics.
Herein, we summarize current models for iron-sulfur cluster biogenesis, which reflects the complex and intricate series of cellular exchange pathways required for proper iron homeostasis and metal cofactor trafficking. Given the complexity of iron-sulfur cluster assembly and delivery, which begins in the mitochondria and includes a mechanism for nuclear delivery, malfuctions in these trafficking processes can lead to a number of disease conditions. In this review, we report on Fe-S cluster-based diseases that have been attributed to defects in cluster trafficking proteins and pathways and can lead to lethal consequences.
Acknowledgments
This work was supported by a grant from the National Institutes of Health [AI072443]. Christine Wachnowsky was supported by an NIH Chemistry/Biology Interface training grant (T32 GM095450) and an Ohio State University Presidential Fellowship.
Biographies
Christine Wachnowsky first studied biochemistry at Allegheny College in the lab of Dr. Margaret Nelson, focusing on cell differentiation in Dictyostelium discoideum. She earned her Ph.D. at the Ohio State University as part of the Ohio State Biochemistry Program. Christine conducted research in the laboratory of Dr. James A. Cowan on the topic of iron-sulfur cluster biosynthesis and trafficking in relation to disease states.

Insiya Fidai completed her Bachelor’s and Master’ in Biotechnology from the University of Mumbai, India. During her Masters, she worked on biocompatible synthesis of noble metal nanoparticles using various reducing sugars under the guidance of Dr. Pankaj Poddar at the National Chemical Laboratory, Pune, India. She then earned her Ph.D. at The Ohio State University as part of the Ohio State Biophysics Graduate Program. In Dr. Cowan’s laboratory, Insiya worked on ATCUN complexes as potential catalytic metallodrugs and role of iron-sulfur clusters in biogenesis, trafficking and storage, and implications for disease states.

James A. Cowan graduated with first class honours in Chemistry from the University of Glasgow, before moving to Cambridge University to pursue research in synthetic organic chemistry and chemical physics. Subsequently he took up a postdoctoral appointment at Caltech to pursue studies in bioinorganic chemistry before taking up a faculty position at The Ohio State University. Dr. Cowan’s research interests cover metal catalysis, metals in medicine, and the cellular chemistry of iron and metal cofactors

Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here].
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.(a) Maio N, Rouault TA. Iron–sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim Biophys Acta. 2015;1853:1493–1512. doi: 10.1016/j.bbamcr.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Braymer JJ, Lill R. Iron–sulfur cluster biogenesis and trafficking in mitochondria. J Biol Chem. 2017;292:12754–12763. doi: 10.1074/jbc.R117.787101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rouault TA, Maio N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J Biol Chem. 2017;292:12744–12753. doi: 10.1074/jbc.R117.789537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Fuss JO, Tsai CL, Ishida JP, Tainer JA. Emerging critical roles of Fe–S clusters in DNA replication and repair. Biochim Biophys Acta. 2015;1853:1253–1271. doi: 10.1016/j.bbamcr.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Stehling O, Wilbrecht C, Lill R. Mitochondrial iron-sulfur protein biogenesis and human disease. Biochimie. 2014;100:61–77. doi: 10.1016/j.biochi.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 3.Toyokuni S, Ito F, Yamashita K, Okazaki Y, Akatsuka S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic Biol Med. 2017;108:610–626. doi: 10.1016/j.freeradbiomed.2017.04.024. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460:831–8. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]; (b) Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, Uzarska MA, Webert H, Wilbrecht C, Muhlenhoff U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta. 2012;1823:1491–508. doi: 10.1016/j.bbamcr.2012.05.009. [DOI] [PubMed] [Google Scholar]; (c) Netz DJ, Mascarenhas J, Stehling O, Pierik AJ, Lill R. Maturation of cytosolic and nuclear iron-sulfur proteins. Trends Cell Biol. 2014;24:303–12. doi: 10.1016/j.tcb.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 5.(a) Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]; (b) Lill R, Mühlenhoff U. Iron-sulfur protein biogenesis in eukaryotes: components and mechanisms. Annu Rev Cell Dev Biol. 2006;22:457–486. doi: 10.1146/annurev.cellbio.22.010305.104538. [DOI] [PubMed] [Google Scholar]; (c) Ayala-Castro C, Saini A, Outten FW. Microbiol Mol Biol Rev. 2008;72:110. doi: 10.1128/MMBR.00034-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fontecave M, Ollagnier-de-Choudens S. Arch Biochem Biophys. 2008;474 doi: 10.1016/j.abb.2007.12.014. [DOI] [PubMed] [Google Scholar]; (e) Qi W, Cowan JA. Structural, mechanistic and coordination chemistry of relevance to the biosynthesis of iron–sulfur and related iron cofactors. Coord Chem Rev. 2011;255:688–699. doi: 10.1016/j.ccr.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Pandey A, Yoon H, Lyver ER, Dancis A, Pain D. Isd11p protein activates the mitochondrial cysteine desulfurase Nfs1p protein. J Biol Chem. 2011;286:38242–38252. doi: 10.1074/jbc.M111.288522. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; (b) Adam A, Bornhovd C, Prokisch H, Neupert W, Hell K. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 2006;25:174–183. doi: 10.1038/sj.emboj.7600905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Gakh O, Ranatunga W, Smith DY, IV, Ahlgren E-C, Al-Karadaghi S, Thompson JR, Isaya G. Architecture of the human mitochondrial iron-sulfur cluster assembly machinery. J Biol Chem. 2016;291:21296–29321. doi: 10.1074/jbc.M116.738542. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ranatunga W, Gakh O, Galeano BK, Smith DY, Söderberg CA, Al-Karadaghi S, Thompson JR, Isaya G. Architecture of the yeast mitochondrial iron-sulfur cluster assembly machinery: The sub-complex formed by the iron donor, Yfh1 protein, and the scaffold, Isu1 protein. J Biol Chem. 2016;291:10378–98. doi: 10.1074/jbc.M115.712414. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schmucker S, Martelli A, Colin F, Page A, Wattenhofer-Donzé M, Reutenauer L, Puccio H. Mammalian frataxin: An essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS One. 2011;6:e16199. doi: 10.1371/journal.pone.0016199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Webert H, Freibert SA, Gallo A, Heidenreich T, Linne U, Amlacher S, Hurt E, Mühlenhoff U, Banci L, Lill R. Functional reconstitution of mitochondrial Fe/S cluster synthesis on Isu1 reveals the involvement of ferredoxin. Nat Commun. 2014;5:5013. doi: 10.1038/ncomms6013. [DOI] [PubMed] [Google Scholar]
- 8.(a) Shi Y, Ghosh M, Kovtunovych G, Crooks DR, Rouault TA. Both human ferredoxins 1 and 2 and ferredoxin reductase are important for iron-sulfur cluster biogenesis. Biochim Biophys Acta. 2012;1823:484–92. doi: 10.1016/j.bbamcr.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu Y, Cowan JA. Iron sulfur cluster biosynthesis. Human NFU mediates sulfide delivery to ISU in the final step of [2Fe-2S] cluster assembly. Chem Commun. 2007:3192–4. doi: 10.1039/b704928e. [DOI] [PubMed] [Google Scholar]; (c) Wu S-p, Bellei M, Mansy SS, Battistuzzi G, Sola M, Cowan JA. Redox chemistry of the Schizosaccharomyces pombe ferredoxin electron-transfer domain and influence of Cys to Ser substitutions. J Inorg Biochem. 2011;105:806–811. doi: 10.1016/j.jinorgbio.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cai K, Tonelli M, Frederick RO, Markley JL. Human mitochondrial ferredoxin 1 (FDX1) and ferredoxin 2 (FDX2) both bind cysteine desulfurase and donate electrons for iron–sulfur cluster biosynthesis. Biochemistry. 2017 doi: 10.1021/acs.biochem.6b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Shaw G, Cope J, Li L, Corson K, Hersey C, Ackermann G, Gwynn B, Lambert A, Wingert R, Traver D, Trede N, Barut B, Zhou Y, Minet E, Donovan A, Brownlie A, Balzan R, Weiss M, Peters L, Kaplan J, Zon L, Paw B. Mitoferrin is essential for erythroid iron assimilation. Nature. 2006;440:96–100. doi: 10.1038/nature04512. [DOI] [PubMed] [Google Scholar]; (b) Paradkar P, Zumbrennen K, Paw B, Ward D, Kaplan J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol. 2009;29:1007–1016. doi: 10.1128/MCB.01685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Isaya G, O’Neill H, Gakh O, Park S, Mantcheva R, Mooney S. Functional studies of frataxin. Acta Paediatr. 2004;93:68–72. doi: 10.1111/j.1651-2227.2004.tb03061.x. [DOI] [PubMed] [Google Scholar]; (b) Nair M, Adinolfi S, Pastore C, Kelly G, Temussi P, Pastore A. Solution structure of the bacterial frataxin ortholog, CyaY: mapping the iron binding sites. Structure. 2004;12:2037–2048. doi: 10.1016/j.str.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 11.Colin F, Martelli A, Clemancey M, Latour J, Gambarelli S, Zeppieri L, Birck C, Page A, Puccio H, Choudens SOd. Mammalian frataxin controls sulfur production and iron entry during de novo Fe4S4 cluster assembly. J Am Chem Soc. 2013;135:733–740. doi: 10.1021/ja308736e. [DOI] [PubMed] [Google Scholar]
- 12.(a) Tsai C, Barondeau D. Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9139. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]; (b) Galeano BK, Ranatunga W, Gakh O, Smith DY, Thompson JR, Isaya G. Zinc and the iron donor frataxin regulate oligomerization of the scaffold protein to form new Fe–S cluster assembly centers. Metallomics. 2017 doi: 10.1039/c7mt00089h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Sheftel Alex D, Stehling Oliver, Pierik Antonio J, Elsässer Hans-Peter, Mühlenhoff Ulrich, Webert Holger, Hobler Anna, Hannemann Frank, Bernhardt Rita, Lill R. Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc Natl Acad Sci U S A. 2010;107:11775–11780. doi: 10.1073/pnas.1004250107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vickery L, Cupp-Vickery J. Molecular chaperones HscA/Ssq1 and HscB/ Jac1 and their roles in ironesulfur protein maturation. Crit Rev Biochem Mol Biol. 2007;42:95–111. doi: 10.1080/10409230701322298. [DOI] [PubMed] [Google Scholar]; (c) Uhrigshardt H, Singh A, Kovtunovych G, Ghosh M, Rouault T. Characterization of the human HSC20, an unusual DnaJ type III protein, involved in ironesulfur cluster biogenesis. Hum Mol Genet. 2010;19:3816–3834. doi: 10.1093/hmg/ddq301. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kim JH, Bothe JR, Alderson TR, Markley JL. Tangled web of interactions among proteins involved in iron–sulfur cluster assembly as unraveled by NMR, SAXS, chemical crosslinking, and functional studies. Biochim Biophys Acta. 2015;1853:1416–1428. doi: 10.1016/j.bbamcr.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Maio N, Rouault TA. Mammalian Fe–S proteins: definition of a consensus motif recognized by the co-chaperone HSC20. Metallomics. 2016;8:1032–1046. doi: 10.1039/c6mt00167j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Uzarska M, Dutkiewicz R, Freibert S, Lill R, Muhlenhoff U. The mitochondrial Hsp70 chaperone Ssq1 facilitates Fe/S cluster transfer from Isu1 to Grx5 by complex formation. Mol Biol Cell. 2013;24:1830–1841. doi: 10.1091/mbc.E12-09-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye H, Jeong S, Ghosh M, Kovtunovych G, Silvestri L, Ortillo D, Uchida N, Tisdale J, Camaschella C, Rouault T. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest. 2010;120:1749–1761. doi: 10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lillig C, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780:1340–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]; (d) Vranish JN, Das D, Barondeau DP. Real-time kinetic probes support monothiol glutaredoxins as intermediate carriers in Fe-S cluster biosynthetic pathways. ACS Chem Biol. 2016;11:3114–3121. doi: 10.1021/acschembio.6b00632. [DOI] [PubMed] [Google Scholar]
- 15.Lill R, Dutkiewicz R, Freibert SA, Heidenreich T, Mascarenhas J, Netz DJ, Paul VD, Pierik AJ, Richter N, Stümpfig M, Srinivasan V, Stehling O, Mühlenhoff U. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron–sulfur proteins. Eur J Cell Biol. 2015;94:280–291. doi: 10.1016/j.ejcb.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 16.(a) Lee JY, Yang JG, Zhitnitsky D, Lewinson O, Rees DC. Structural basis for heavy metal detoxification by an Atm1-type ABC exporter. Science. 2014;343:1133–1136. doi: 10.1126/science.1246489. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Srinivasan V, Pierik AJ, Lill R. Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science. 2014;343:1137–1140. doi: 10.1126/science.1246729. [DOI] [PubMed] [Google Scholar]
- 17.(a) Qi W, Li J, Chain CY, Pasquevich GA, Pasquevich AF, Cowan JA. Glutathione complexed Fe-S centers. J Am Chem Soc. 2012;134:10745–8. doi: 10.1021/ja302186j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li J, Cowan JA. Glutathione-coordinated [2Fe–2S] cluster: a viable physiological substrate for mitochondrial ABCB7 transport. Chem Commun. 2015;51:2253–2255. doi: 10.1039/c4cc09175b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Qi W, Li J, Cowan JA. A structural model for glutathione-complexed iron–sulfur cluster as a substrate for ABCB7-type transporters. Chem Commun. 2014;50:3795. doi: 10.1039/c3cc48239a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Cavadini P, Biasiotto G, Poli M, Levi S, Verardi R, Zanella I, Derosas M, Ingrassia R, Corrado M, Arosio P. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood. 2007;109:3552–3559. doi: 10.1182/blood-2006-08-041632. [DOI] [PubMed] [Google Scholar]; (b) Miao R, Kim H, Koppolu UMK, Ellis EA, Scott RA, Lindahl PA. Biophysical characterization of the iron in mitochondria from Atm1p-depleted Saccharomyces cerevisiae. Biochemistry. 2009;48:9556–9568. doi: 10.1021/bi901110n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar C, Igbaria A, D’Autreaux B, Planson AG, Junot C, Godat E, Bachhawat AK, Delaunay-Moisan A, Toledano MB. Glutathione revisited: a vitalfunction in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011;30:2044–2056. doi: 10.1038/emboj.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaedler TA, Thornton JD, Kruse I, Schwarzlander M, Meyer AJ, van Veen HW, Balk J. A conserved mitochondrial ATP-binding cassette transporter exports glutathione polysulfide for cytosolic metal cofactor assembly. J Biol Chem. 2014;289:23264–23274. doi: 10.1074/jbc.M114.553438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Gelling C, Dawes IW, Richhardt N, Lill R, Muhlenhoff U. Mitochondrial Iba57p is required for Fe/S cluster formation on aconitase and activation of radical SAM enzymes. Mol Cell Biol. 2008;28:1851–61. doi: 10.1128/MCB.01963-07. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Muhlenhoff U, Richter N, Pines O, Pierik AJ, Lill R. Specialized function of yeast Isa1 and Isa2 proteins in the maturation of mitochondrial [4Fe-4S] proteins. J Biol Chem. 2011;286:41205–41216. doi: 10.1074/jbc.M111.296152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sheftel AD, Wilbrecht C, Stehling O, Niggemeyer B, Elsasser HP, Muhlenhoff U, Lill R. The human mitochondrial ISCA1, ISCA2, and IBA57 proteins are required for [4Fee4S] protein maturation. Mol Biol Cell. 2012;23:1157–1166. doi: 10.1091/mbc.E11-09-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Beilschmidt LK, de Choudens SOllagnier, Fournier M, Sanakis I, Hograindleur M-A, Clémancey M, Blondin G, Schmucker S, Eisenmann A, Weiss A, Koebel P, Messaddeq N, Puccio H, Martelli A. ISCA1 is essential for mitochondrial Fe4S4 biogenesis in vivo. Nat Commun. 2017;8:15124. doi: 10.1038/ncomms15124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Kim KD, Chung WH, Kim HJ, Lee KC, Roe JH. Monothiol glutaredoxin Grx5 interacts with Fe-S scaffold proteins Isa1 and Isa2 and supports Fe-S assembly and DNA integrity in mitochondria of fission yeast. Biochem Biophys Res Commun. 2010;392:467–72. doi: 10.1016/j.bbrc.2010.01.051. [DOI] [PubMed] [Google Scholar]; (b) Banci L, Brancaccio D, Ciofi-Baffoni S, Del Conte R, Gadepalli R, Mikolajczyk M, Neri S, Piccioli M, Winkelmann J. [2Fe-2S] cluster transfer in iron-sulfur protein biogenesis. Proc Natl Acad Sci USA. 2014;111:6203–6208. doi: 10.1073/pnas.1400102111. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brancaccio D, Gallo A, Mikolajczyk M, Zovo K, Palumaa P, Novellino E, Piccioli M, Ciofi-Baffoni S, Banci L. Formation of [4Fe-4S] clusters in the mitochondrial iron–sulfur cluster assembly machinery. J Am Chem Soc. 2014;136:16240–16250. doi: 10.1021/ja507822j. [DOI] [PubMed] [Google Scholar]
- 23.(a) Tong WH, Jameson GNL, Huynh BH, Rouault TA. Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proceedings of the National Academy of Sciences. 2003;100:9762–9767. doi: 10.1073/pnas.1732541100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao H, Subramanian S, Couturier J, Naik SG, Kim SK, Leustek T, Knaff DB, Wu HC, Vignols F, Huynh BH, Rouhier N, Johnson MK. Arabidopsis thaliana Nfu2 accommodates [2Fe-2S] or [4Fe-4S] clusters and is competent for in vitro maturation of chloroplast [2Fe-2S] and [4Fe-4S] cluster-containing proteins. Biochemistry. 2013;52:6633–6645. doi: 10.1021/bi4007622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wachnowsky C, Fidai I, Cowan JA. Iron-sulfur cluster exchange reactions mediated by the human Nfu protein. J Biol Inorg Chem. 2016;21:825–836. doi: 10.1007/s00775-016-1381-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cai K, Liu G, Frederick Ronnie O, Xiao R, Montelione Gaetano T, Markley John L. Structural/functional properties of human NFU1, an intermediate [4Fe-4S] carrier in human mitochondrial iron-sulfur cluster biogenesis. Structure. 2016;24:2080–2091. doi: 10.1016/j.str.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Hausmann A, Netz DJA, Balk J, Pierik AJ, Muhlenhoff U, Lill R. The eukaryotic P loopNTPaseNbp35: an essential component of the cytosolic and nuclear iron–sulfur protein assembly machinery. Proc Natl Acad Sci U S A. 2005;102:3266–3271. doi: 10.1073/pnas.0406447102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Netz DJ, Pierik AJ, Stumpfig M, Muhlenhoff U, Lill R. The Cfd1-Nbp35 complex acts as a scaffold for iron-sulfur protein assembly in the yeast cytosol. Nat Chem Biol. 2007;3:278–86. doi: 10.1038/nchembio872. [DOI] [PubMed] [Google Scholar]; (c) Stehling O, Netz DJA, Niggemeyer B, Rosser R, Eisenstein RS, Puccio H, Pierik AJ, Lill R. Human Nbp35 is essential for both cytosolic iron-sulfur protein assembly and iron homeostasis. Mol Cell Biol. 2008;28:5517–5528. doi: 10.1128/MCB.00545-08. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Netz DJ, Pierik AJ, Stumpfig M, Bill E, Sharma AK, Pallesen LJ, Walden WE, Lill R. A bridging [4Fe–4S] cluster and nucleotide binding are essential for function of the Cfd1–Nbp35 complex as a scaffold in iron–sulfur protein maturation. J Biol Chem. 2012;287:12365–12378. doi: 10.1074/jbc.M111.328914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.(a) Banci L, Bertini I, Calderone V, Ciofi-Baffoni S, Giachetti A, Jaiswal D, Mikolajczyk M, Piccioli M, Winkelmann J. Molecular view of an electron transfer process essential for iron–sulfur protein biogenesis. Proc Natl Acad Sci U S A. 2013;110:7136–7141. doi: 10.1073/pnas.1302378110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Netz Daili JA, Stümpfig Martin, Doré Carole, Mühlenhoff Ulrich, Pierik AJ, Lill R. Tah18 transfers electrons to dre2 in cytosolic iron-sulfur protein biogenesis. Nature Chemical Biology. 2010;6:758–765. doi: 10.1038/nchembio.432. [DOI] [PubMed] [Google Scholar]
- 26.(a) Haunhorst P, Hanschmann EM, Brautigam L, Stehling O, Hoffmann B, Muhlenhoff U, Lill R, Berndt C, Lillig CH. Crucial function of vertebrate glutaredoxin 3 (PICOT) in iron homeostasis and hemoglobin maturation. Mol Biol Cell. 2013;24:1895–1903. doi: 10.1091/mbc.E12-09-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mühlenhoff U, Molik S, Godoy JR, Uzarska MA, Richter N, Seubert A, Zhang Y, Stubbe J, Pierrel F, Herrero E, Lillig CH, Lill R. Cytosolic Monothiol Glutaredoxins Function in Intracellular Iron Sensing and Trafficking via Their Bound Iron-Sulfur Cluster. Cell Metabolism. 2010;12:373–385. doi: 10.1016/j.cmet.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Outten CE, Albetel AN. Iron sensing and regulation in Saccharomyces cerevisiae: ironing out the mechanistic details. Curr Opin Microbiol. 2013;16:662–668. doi: 10.1016/j.mib.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Y, Liu L, Wu X, An X, Stubbe J, Huang M. Investigation of in vivo diferric tyrosyl radical formation in Saccharomyces cerevisiae Rnr2 protein: requirement of Rnr4 and contribution of Grx3/4 AND Dre2 proteins. J Biol Chem. 2011;286:41499–41509. doi: 10.1074/jbc.M111.294074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Seki M, Takeda Y, Iwai K, Tanaka K. IOP1 protein is an external component of the human cytosolic iron–sulfur cluster assembly (CIA) machinery and functions in the MMS19 protein-dependent CIA pathway. J Biol Chem. 2013;288:16680–16689. doi: 10.1074/jbc.M112.416602. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Srinivasan V, Netz DJ, Webert H, Mascarenhas J, Pierik AJ, Michel H, Lill R. Structure of the yeast WD40 domain protein Cia1, a component acting late in iron-sulfur protein biogenesis. Structure. 2007;15:1246–57. doi: 10.1016/j.str.2007.08.009. [DOI] [PubMed] [Google Scholar]; (c) Stehling O, Mascarenhas J, Vashisht AA, Sheftel AD, Niggemeyer B, Rosser R, Pierik AJ, Wohlschlegel JA, Lill R. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic–nuclear iron–sulfur proteins. Cell Metab. 2013;18:187–198. doi: 10.1016/j.cmet.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wietmarschen NV, Moradian A, Morin GB, Lansdorp PM, Uringa EJ. The mammalian proteins MMS19, MIP18, and ANT2 are involved in cytoplasmic iron–sulfur cluster protein assembly. J Biol Chem. 2012;287:43351–43358. doi: 10.1074/jbc.M112.431270. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Tong WH, Jameson GN, Huynh BH, Rouault TA. Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proc Natl Acad Sci USA. 2003;100:9762–7. doi: 10.1073/pnas.1732541100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tong WH, Rouault TA. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]; (c) Shi Y, Ghosh MC, Tong WH, Rouault TA. Human ISD11 is essential for both ironesulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum Mol Genet. 2009;18:3014–3025. doi: 10.1093/hmg/ddp239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Land T, Rouault TA. Targeting of a human ironesulfur cluster assembly enzyme, nifs, to different subcellular compartments is regulated through alternative AUG utilization. Mol Cell. 1998;2:807–815. doi: 10.1016/s1097-2765(00)80295-6. [DOI] [PubMed] [Google Scholar]
- 30.(a) Koeppen AH. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci. 2011;303:1–12. doi: 10.1016/j.jns.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Santos R, Lefevre S, Sliwa D, Seguin A, Camadro JM, Lesuisse E. Friedreich ataxia: Molecular mechanisms, redox considerations, and therapeutic opportunities. Antiox Redox Signal. 2010;13:651–690. doi: 10.1089/ars.2009.3015. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stamelou M. Late-onset cerebellar ataxia: Do not forget Friedreich’s. Mov Disord. 2016;31:7–8. doi: 10.1002/mds.26508. [DOI] [PubMed] [Google Scholar]
- 31.Schmucker S, Puccio H. Understanding the molecular mechanisms of Friedreich’s ataxia to develop therapeutic approaches. Hum Mol Genet. 2010;19:103–110. doi: 10.1093/hmg/ddq165. [DOI] [PubMed] [Google Scholar]
- 32.Cotticelli MG, Rasmussen L, Kushner NL, McKellip S, Sosa MI, Manouvakhova A, Feng S, White EL, Maddry JA, Heemskerk J, Oldt RJ, Surrey LF, Ochs R, Wilson RB. Primary and secondary drug screening assays for Friedreich ataxia. J Biomol Screen. 2012;17:303–313. doi: 10.1177/1087057111427949. [DOI] [PubMed] [Google Scholar]
- 33.Mochel F, Knight MA, Tong WH, Hernandez D, Ayyad K, Taivassalo T, Andersen PM, Singleton A, Rouault TA, Fischbeck KH, Haller RG. Splice mutation in the ironesulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet. 2008;82:652–660. doi: 10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsson A, Lind L, Thornell LE, Holmberg M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum Mol Genet. 2008;17:1666–1672. doi: 10.1093/hmg/ddn057. [DOI] [PubMed] [Google Scholar]
- 35.(a) Sanaker PS, Toompuu M, Hogan VE, He L, Tzoulis C, Lightowlers ZMC-, Taylor RW, Bindoff LA. Differences in RNA processing underlie the tissue specific phenotype of ISCU myopathy. Biochim Biophys Acta. 2010;1802:539–544. doi: 10.1016/j.bbadis.2010.02.010. [DOI] [PubMed] [Google Scholar]; (b) Kollberg G, Holme E. Antisense oligonucleotide therapeutics for iron-sulphur cluster deficiency myopathy. Neuromuscul Disord. 2009;19:833–836. doi: 10.1016/j.nmd.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 36.White K, Lu Y, Annis S, Hale AE, Chau BN, Dahlman JE, Hemann C, Opotowsky AR, Vargas SO, Rosas I, Perrella MA, Osorio JC, Haley KJ, Graham BB, Kumar R, Saggar R, Wallace WD, Ross DJ, Khan OF, Bader A, Gochuico BR, Matar M, Polach K, Johannessen NM, Prosser HM, Anderson DG, Langer R, Zweier JL, Bindoff LA, Systrom D, Waxman AB, Jin RC, Chan SY. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med. 2015;7:695–713. doi: 10.15252/emmm.201404511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Funauchi Y, Tanikawa C, Yi Lo PH, Mori J, Daigo Y, Takano A, Miyagi Y, Okawa A, Nakamura Y, Matsuda K. Regulation of iron homeostasis by the p53-ISCU pathway. Sci Rep. 2015;5:16497. doi: 10.1038/srep16497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spiegel R, Saada A, Halvardson J, Soiferman D, Shaag A, Edvardson S, Horovitz Y, Khayat M, Shalev SA, Feuk L, Elpeleg O. Deleterious mutation in FDX1L gene is associated with a novel mitochondrial muscle myopathy. Eur J Hum Genet. 2013;22:902–906. doi: 10.1038/ejhg.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farhan SMK, Wang J, Robinson JF, Lahiry P, Siu VM, Prasad C, Kronick JB, Ramsay DA, Rupar CA, Hegele RA. Exome sequencing identifies NFS1 deficiency in a novel Fe-S cluster disease, infantile mitochondrial complex II/III deficiency. Mol Genet Genomic Med. 2014;2:73–80. doi: 10.1002/mgg3.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.(a) Lim SC, Friemel M, Marum JE, Tucker EJ, Bruno DL, Riley LG, Christodoulou J, Kirk EP, Boneh A, DeGennaro CM, Springer M, Mootha VK, Rouault TA, Leimkuhler S, Thorburn DR, Compton AG. Mutations in LYRM4, encoding iron-sulfur cluster biogenesis factor ISD11, cause deficiency of multiple respiratory chain complexes. Hum Mol Genet. 2013;22:4460–4473. doi: 10.1093/hmg/ddt295. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saha PP, Srivastava S, Kumar P, K S, Sinha D, D’Silva P. Mapping key residues of ISD11 critical for NFS1-ISD11 subcomplex stability. J Biol Chem. 2015;290:25876–25890. doi: 10.1074/jbc.M115.678508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Ahting U, Mayr JA, Vanlander AV, Hardy SA, Santra S, Makowski C, Alston CL, Zimmermann FA, Abela L, Plecko B, Rohrbach M, Spranger S, Seneca S, Rolinski B, Hagendorff A, Hempel M, Sperl W, Meitinger T, Smet J, Taylor RW, Van Coster R, Freisinger P, Prokisch H, Haack TB. Clinical, biochemical, and genetic spectrum of seven patients with NFU1 deficiency. Front Genet. 2015;06 doi: 10.3389/fgene.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cameron JM, Janer A, Levandovskiy V, Mackay N, Rouault TA, Tong WH, Ogilvie I, Shoubridge EA, Robinson BH. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89:486–95. doi: 10.1016/j.ajhg.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ferrer-Cortès X, Font A, Bujan N, Navarro-Sastre A, Matalonga L, Arranz JA, Riudor E, del Toro M, Garcia-Cazorla A, Campistol J, Briones P, Ribes A, Tort F. Protein expression profiles in patients carrying NFU1 mutations. Contribution to the pathophysiology of the disease. J Inherit Metab Dis. 2012;36:841–847. doi: 10.1007/s10545-012-9565-z. [DOI] [PubMed] [Google Scholar]; (d) Ferrer-Cortès X, Narbona J, Bujan N, Matalonga L, Del Toro M, Arranz JA, Riudor E, Garcia-Cazorla A, Jou C, O’Callaghan M, Pineda M, Montero R, Arias A, García-Villoria J, Alston CL, Taylor RW, Briones P, Ribes A, Tort F. A leaky splicing mutation in NFU1 is associated with a particular biochemical phenotype. Consequences for the diagnosis. Mitochondrion. 2016;26:72–80. doi: 10.1016/j.mito.2015.12.004. [DOI] [PubMed] [Google Scholar]; (e) Invernizzi F, Ardissone A, Lamantea E, Garavaglia B, Zeviani M, Farina L, Ghezzi D, Moroni I. Cavitating leukoencephalopathy with multiple mitochondrial dysfunction syndrome and NFU1 mutations. Front Genet. 2014;5 doi: 10.3389/fgene.2014.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Navarro-Sastre A, Tort F, Stehling O, Uzarska Marta A, Arranz José A, del Toro M, Labayru MT, Landa J, Font A, Garcia-Villoria J, Merinero B, Ugarte M, Gutierrez-Solana Luis G, Campistol J, Garcia-Cazorla A, Vaquerizo J, Riudor E, Briones P, Elpeleg O, Ribes A, Lill R. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–667. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Tonduti D, Dorboz I, Imbard A, Slama A, Boutron A, Pichard S, Elmaleh M, Vallée L, Benoist J, Ogier H, Boespflug-Tanguy O. New spastic paraplegia phenotype associated to mutation of NFU1. Orphanet J Rare Dis. 2015;10:13. doi: 10.1186/s13023-015-0237-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.(a) Baker PR, Friederich MW, Swanson MA, Shaikh T, Bhattacharya K, Scharer GH, Aicher J, Creadon-Swindell G, Geiger E, MacLean KN, Lee WT, Deshpande C, Freckmann ML, Shih LY, Wasserstein M, Rasmussen MB, Lund AM, Procopis P, Cameron JM, Robinson BH, Brown GK, Brown RM, Compton AG, Dieckmann CL, Collard R, Coughlin CR, Spector E, Wempe MF, Van Hove JLK. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. Brain. 2013;137:366–379. doi: 10.1093/brain/awt328. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Haack TB, Rolinski B, Haberberger B, Zimmermann F, Schum J, Strecker V, Graf E, Athing U, Hoppen T, Wittig I, Sperl W, Freisinger P, Mayr JA, Strom TM, Meitinger T, Prokisch H. Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J Inherit Metab Dis. 2012;36:55–62. doi: 10.1007/s10545-012-9489-7. [DOI] [PubMed] [Google Scholar]
- 43.(a) Bolar NAjit, Vanlander AV, Wilbrecht C, Van der Aa N, Smet J, Paepe BDE, Vandeweyer G, Kooy F, Eyskens F, Latter EDE, Delanghe G, Govaert P, Leroy JG, Loeys B, Lill R, Van Laer L, Van Coster R. Mutation of the iron-sulfur cluster assembly gene IBA57 causes severe myopathy and encephalopathy. Hum Mol Genet. 2013;22:2590–2602. doi: 10.1093/hmg/ddt107. [DOI] [PubMed] [Google Scholar]; (b) Debray FG, Stümpfig C, Vanlander AV, Dideberg V, Josse C, Caberg JH, Boemer F, Bours V, Stevens R, Seneca S, Smet J, Lill R, van Coster R. Mutation of the iron-sulfur cluster assembly gene IBA57 causes fatal infantile leukodystrophy. J Inherit Metab Dis. 2015;38:1147–1153. doi: 10.1007/s10545-015-9857-1. [DOI] [PubMed] [Google Scholar]; (c) Lossos A, Stümpfig C, Stevanin G, Gaussen M, Zimmerman BE, Mundwiller E, Asulin M, Chamma L, Sheffer R, Misk A, Dotan S, Gomori JM, Ponger P, Brice A, Lerer I, Meiner V, Lill R. Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology. 2015;84 doi: 10.1212/WNL.0000000000001270. [DOI] [PubMed] [Google Scholar]; (d) Torraco A, Ardissone A, Invernizzi F, Rizza T, Fiermonte G, Niceta M, Zanetti N, Martinelli D, Vozza A, Verrigni D, Di Nottia M, Lamantea E, Diodato D, Tartaglia M, Dionisi-Vici C, Moroni I, Farina L, Bertini E, Ghezzi D, Carrozzo R. Novel mutations in IBA57 are associated with leukodystrophy and variable clinical phenotypes. J Neurol. 2016 doi: 10.1007/s00415-016-8312-z. [DOI] [PubMed] [Google Scholar]
- 44.Al-Hassnan ZN, Al-Dosary M, Alfadhel M, Faqeih EA, Alsagob M, Kenana R, Almass R, Al-Harazi OS, Al-Hindi H, Malibari OI, Almutari FB, Tulbah S, Alhadeq F, Al-Sheddi T, Alamro R, AlAsmari A, Almuntashri M, Alshaalan H, Al-Mohanna FA, Colak D, Kaya N. ISCA2 mutation causes infantile neurodegenerative mitochondrial disorder. J Med Genet. 2015;52:186–194. doi: 10.1136/jmedgenet-2014-102592. [DOI] [PubMed] [Google Scholar]
- 45.Shukla A, Hebbar M, Srivastava A, Kadavigere R, Upadhyai P, Kanthi A, Brandau O, Bielas S, Girisha KM. Homozygous p.(Glu87Lys) variant in ISCA1 is associated with a multiple mitochondrial dysfunctions syndrome. J Hum Genet. 2017 doi: 10.1038/jhg.2017.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cameron Jessie M, Janer A, Levandovskiy V, MacKay N, Rouault Tracey A, Tong W-H, Ogilvie I, Shoubridge Eric A, Robinson Brian H. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89:486–495. doi: 10.1016/j.ajhg.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wachnowsky C, Wesley NA, Fidai I, Cowan JA. Understanding the molecular basis for Multiple Mitochondrial Dysfunctions Syndrome 1 (MMDS1) - Impact of a disease-causing Gly208Cys substitution on structure and activity of NFU1 in the Fe/S cluster biosynthetic pathway. J Mol Biol. 2017;429:790–807. doi: 10.1016/j.jmb.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uzarska MA, Nasta V, Weiler BD, Spantgar F, Ciofi-Baffoni S, Saviello MR, Gonnelli L, Muehlenhoff U, Banci L, Lill R. Mitochondrial Bol1 and Bol3 function as assembly factors for specific iron-sulfur proteins. eLife. 2016;5:e15991. doi: 10.7554/eLife.16673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melber A, Na U, Vashisht A, Weiler BD, Lill R, Wohlschlegel JA, Winge DR. Role of Nfu1 and Bol3 in Iiron-sulfur cluster transfer to mitochondrial clients. eLife. 2016;5:e15991. doi: 10.7554/eLife.15991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maio N, Kim KS, Singh A, Rouault TA. A single adaptable cochaperone-scaffold complex delivers nascent iron-sulfur clusters to mammalian respiratory chain complexes I–III. Cell Metab. 2017;25:945–953.e6. doi: 10.1016/j.cmet.2017.03.010. [DOI] [PubMed] [Google Scholar]
- 51.Scheffler IE. Mitochondrial disease associated with complex I (NADH-CoQ oxidoreductase) deficiency. J Inherit Metab Dis. 2014;38:405–415. doi: 10.1007/s10545-014-9768-6. [DOI] [PubMed] [Google Scholar]
- 52.Kevelam SH, Rodenburg RJ, Wolf NI, Ferreira P, Lunsing RJ, Nijtmans LG, Mitchell A, Arroyo HA, Rating D, Vanderver A, van Berkel CGM, Abbink TEM, Heutink P, van der Knapp MS. NUBPL mutations in patients with complex I deficiency and a distinct MRI pattern. Neurology. 2013;80:1577–83. doi: 10.1212/WNL.0b013e31828f1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.(a) Tucker EJ, Mimaki M, Compton AG, McKenzie M, Ryan MT, Thorburn DR. Next-generation sequencing in molecular diagnosis:NUBPLmutations highlight the challenges of variant detection and interpretation. Hum Mutat. 2012;33:411–418. doi: 10.1002/humu.21654. [DOI] [PubMed] [Google Scholar]; (b) Wydro MM, Balk J. Insights into the pathogenic character of a common NUBPL branch-site mutation associated with mitochondrial disease and complex I deficiency using a yeast model. Dis Model Mech. 2013;6:1279–1284. doi: 10.1242/dmm.012682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wydro MM, Sharma P, Foster JM, Bych K, Meyer EH, Balk J. The evolutionarily conserved iron-sulfur protein INDH is required for complex I assembly and mitochondrial translation in. Arabidopsis Plant Cell. 2013;25:4014–4027. doi: 10.1105/tpc.113.117283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.(a) Hoekstra AS, Bayley JP. The role of complex II in disease. Biochim Biophys Acta. 2013;1827:543–551. doi: 10.1016/j.bbabio.2012.11.005. [DOI] [PubMed] [Google Scholar]; (b) Van Vranken JG, Na U, Winge DR, Rutter J. Protein-mediated assembly of succinate dehydrogenase and its cofactors. Crit Rev Biochem Mol Biol. 2014;50:168–180. doi: 10.3109/10409238.2014.990556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alston CL, Davison JE, Meloni F, van der Westhuizen FH, He L, Hornig-Do HT, Peet AC, Gissen P, Goffrini P, Ferrero I, Wassmer E, McFarland R, Taylor RW. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J Med Genet. 2012;49:569–577. doi: 10.1136/jmedgenet-2012-101146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pantaleo MA, Astolfi A, Urbini M, Nannini M, Paterini P, Indio V, Saponara M, Formica S, Ceccarelli C, Casadio R, Rossi G, Bertolini F, Santini D, Pirini MG, Fiorentino M, Basso U, Biasco G. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet. 2013;22:32–39. doi: 10.1038/ejhg.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.(a) Saxena N, Maio N, Crooks DR, Ricketts CJ, Yang Y, Wei MH, Fan TWM, Lane AN, Sourbier C, Singh A, Killian JK, Meltzer PS, Vocke CD, Rouault TA, Linehan WM. SDHB-deficient cancers: The role of mutations that impair iron sulfur cluster delivery. J Natl Cancer Inst. 2016;108:djv287. doi: 10.1093/jnci/djv287. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maio N, Singh A, Uhrigshardt H, Saxena N, Tong W-H, Rouault Tracey A. Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell Metab. 2014;19:445–457. doi: 10.1016/j.cmet.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Na U, Yu W, Cox J, Bricker Daniel K, Brockmann K, Rutter J, Thummel Carl S, Winge Dennis R. The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron-sulfur subunit of succinate dehydrogenase. Cell Metab. 2014;20:253–266. doi: 10.1016/j.cmet.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maio N, Ghezzi D, Verrigni D, Rizza T, Bertini E, Martinelli D, Zeviani M, Singh A, Carrozzo R, Rouault Tracey A. Disease-causing SDHAF1 mutations impair transfer of Fe-S clusters to SDHB. Cell Metab. 2016;23:292–302. doi: 10.1016/j.cmet.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ohlenbusch A, Edvardson S, Skorpen J, Bjornstad A, Saada A, Elpeleg O, Gärtner J, Brockmann K. Leukoencephalopathy with accumulated succinate is indicative of SDHAF1 related complex II deficiency. Orphanet J Rare Dis. 2012;7 doi: 10.1186/1750-1172-7-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamir S, Paddock ML, Darash-Yahana-Baram M, Holt SH, Sohn YS, Agranat L, Michaeli D, Stofleth JT, Lipper CH, Morcos F, Cabantchik IZ, Onuchic JN, Jennings PA, Mittler R, Nechushtai R. Structure–function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim Biophys Acta. 2015;1853:1294–1315. doi: 10.1016/j.bbamcr.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 63.Geldenhuys WJ, Leeper TC, Carroll RT. mitoNEET as a novel drug target for mitochondrial dysfunction. Drug Discov Today. 2014;19:1601–1606. doi: 10.1016/j.drudis.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 64.Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, Scherer PE. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nature Med. 2012;18:1539–1549. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nechushtai R, Conlan AR, Harir Y, Song L, Yogev O, Eisenberg-Domovich Y, Livnah O, Michaeli D, Rosen R, Ma V, Luo Y, Zuris JA, Paddock ML, Cabantchik ZI, Jennings PA, Mittler R. Characterization of Arabidopsis NEET reveals an ancient role for NEET proteins in iron metabolism. Plant Cell. 2012;24:2139–2154. doi: 10.1105/tpc.112.097634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ferecatu I, Goncalves S, Golinelli-Cohen MP, Clemancey M, Martelli A, Riquier S, Guittet E, Latour JM, Puccio H, Drapier JC, Lescop E, Bouton C. The diabetes drug target mitoNEET governs a novel trafficking pathway to rebuild an Fe-S cluster into cytosolic aconitase/iron regulatory protein 1. J Biol Chem. 2014;289:28070–28086. doi: 10.1074/jbc.M114.548438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Golinelli-Cohen MP, Lescop E, Mons C, Gonçalves S, Clémancey M, Santolini J, Guittet E, Blondin G, Latour JM, Bouton C. Redox control of the human iron-sulfur repair protein mitoNEET activity via its iron-sulfur cluster. J Biol Chem. 2016;291:7583–7593. doi: 10.1074/jbc.M115.711218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taminelli GL, Sotomayor V, Valdivieso AG, Teiber ML, Marın MAC, Santa-Coloma TA. CISD1 codifies a mitochondrial protein upregulated by the CFTR channel. Biophys Biochem Res Commun. 2008;365:856–862. doi: 10.1016/j.bbrc.2007.11.076. [DOI] [PubMed] [Google Scholar]
- 69.Okatsu K, Iemura S-I, Koyano F, Go E, Kimura M, Natsume T, Tanaka K, Matsuda N. Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem Biophys Res Commun. 2012;428:197–202. doi: 10.1016/j.bbrc.2012.10.041. [DOI] [PubMed] [Google Scholar]
- 70.(a) Salem AF, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle. 2012;11:4174–80. doi: 10.4161/cc.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bai F, Morcos F, Sohn YS, Darash-Yahana M, Rezende CO, Lipper CH, Paddock ML, Song L, Luo Y, Holt SH, Tamir S, Theodorakis EA, Jennings PA, Onuchica JN, Mittler R, Nechushtai R. The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer. Proc Natl Acad Sci. 2015;112:3698–3703. doi: 10.1073/pnas.1502960112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sohn YS, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, Harir Y, Holt SH, Shulaev V, Paddock ML, Hochberg A, Cabanchick IZ, Onuchic JN, Jennings PA, Nechushtaia R, Mittler R. NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc Natl Acad Sci. 2013;110:14676–14681. doi: 10.1073/pnas.1313198110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paul VD, Lill R. Biogenesis of cytosolic and nuclear iron–sulfur proteins and their role in genome stability. Biochim Biophys Acta. 2015;1853:1528–1539. doi: 10.1016/j.bbamcr.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 73.Nakamura M, Buzas DM, Kato A, Fujita M, Kurata N, Kinoshita T. The role of Arabidopsis thaliana NAR1, a cytosolic iron-sulfur cluster assembly component, in gametophytic gene expression and oxidative stress responses in vegetative tissue. New Phytol. 2013;199:925–935. doi: 10.1111/nph.12350. [DOI] [PubMed] [Google Scholar]
- 74.Corbin MV, Rockx DAP, Oostra AB, Joenje H, Dorsman JC. The iron-sulfur cluster assembly network component NARFL is a key element in the cellular defense against oxidative stress. Free Radic Biol Med. 2015;89:863–872. doi: 10.1016/j.freeradbiomed.2015.08.026. [DOI] [PubMed] [Google Scholar]
- 75.(a) Gari K, Ortiz AML, Borel V, Flynn H, Skehel JM, Boulton SJ. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science. 2012;337:243–5. doi: 10.1126/science.1219664. [DOI] [PubMed] [Google Scholar]; (b) Stehling O, Vashisht AA, Mascarenhas J, Jonsson ZO, Sharma T, Netz DJA, Pierik AJ, Wohlschlegel JA, Lill R. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science. 2012;337:195–199. doi: 10.1126/science.1219723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rudolf J, Makrantoni V, Ingledew W, Stark M, White M. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell. 2006 doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 77.Service R. Science. 2014;346:1284–1287. doi: 10.1126/science.346.6215.1284. [DOI] [PubMed] [Google Scholar]
- 78.(a) Pokharel S, Campbell J. Cross talk between the nuclease and helicase activities of Dna2: role of an essential iron-sulfur cluster domain. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks534. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Netz D, Stith C, Stümpfig M, Köpf G, Vogel D, Genau HEA. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2011;8:125–132. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Oliveira AHS, da Silva AE, de Oliveira IM, Henriques JAP, Agnez-Lima LF. MutY-glycosylase: An overview on mutagenesis and activities beyond the GO system. Mutat Res. 2014;769:119–131. doi: 10.1016/j.mrfmmm.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 80.Mazzei F, Viel A, Bignami M. Role of MUTYH in human cancer. Mutat Res. 2013;743–744:33–43. doi: 10.1016/j.mrfmmm.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 81.Out AA, Tops CMJ, Nielsen M, Weiss MM, van Minderhout IJHM, Fokkema IFAC, Buisine MP, Claes K, Colas C, Fodde R, Fostira F, Franken PF, Gaustadnes M, Heinimann K, Hodgson SV, Hogervorst FBL, Holinski-Feder E, Lagerstedt-Robinson K, Olschwang S, van den Ouweland AMW, Redeker EJW, Scott RJ, Vankeirsbilck B, Grønlund RV, Wijnen JT, Wikman FP, Aretz S, Sampson JR, Devilee P, den Dunnen JT, Hes FJ. Leiden open variation database of the MUTYH gene. Hum Mutat. 2010;31:1205–15. doi: 10.1002/humu.21343. [DOI] [PubMed] [Google Scholar]
- 82.Brinkmeyer MK, David SS. Distinct functional consequences of MUTYH variants associated with colorectal cancer: Damaged DNA affinity, glycosylase activity and interaction with PCNA and Hus1. DNA Repair. 2015;34:39–51. doi: 10.1016/j.dnarep.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.(a) Suhasini AN, Brosh RM. Disease-causing missense mutations in human DNA helicase disorders. Mutat Res. 2013;752:138–152. doi: 10.1016/j.mrrev.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Suhasini AN, Brosh RM. DNA helicases asociated with genetic instability, cancer, and aging. Adv Exp Med Biol. 2013;973:123–144. doi: 10.1007/978-1-4614-5037-5_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: Structural and functional insights into a complex disorder. Annu Rev Biophys. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- 85.Bharti SK, Sommers JA, George F, Kuper J, Hamon F, Shin-ya K, Teulade-Fichou MP, Kisker C, Brosh RM. Specialization among iron-sulfur cluster helicases to resolve G-quadruplex DNA structures that threaten genomic stability. J Biol Chem. 2013;288:28217–28229. doi: 10.1074/jbc.M113.496463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.(a) Cantor SB, Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future Oncol. 2011;7:253–261. doi: 10.2217/fon.10.191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rafnar T, Gudbjartsson DF, Sulem P, Jonasdottir A, Sigurdsson A, Jonasdottir A, Besenbacher S, Lundin P, Stacey SN, Gudmundsson J, Magnusson OT, le Roux L, Orlygsdottir G, Helgadottir HT, Johannsdottir H, Gylfason A, Tryggvadottir L, Jonasson JG, de Juan A, Ortega E, Ramon-Cajal JM, García-Prats MD, Mayordomo C, Panadero A, Rivera F, Aben KKH, van Altena AM, Massuger LFAG, Aavikko M, Kujala PM, Staff S, Aaltonen LA, Olafsdottir K, Bjornsson J, Kong A, Salvarsdottir A, Saemundsson H, Olafsson K, Benediktsdottir KR, Gulcher J, Masson G, Kiemeney LA, Mayordomo JI, Thorsteinsdottir U, Stefansson K. Mutations in BRIP1 confer high risk of ovarian cancer. Nature Genet. 2011;43:1104–1107. doi: 10.1038/ng.955. [DOI] [PubMed] [Google Scholar]
- 87.Gupta R, Sharma S, Doherty KM, Sommers JA, Cantor SB, Brosh RM. Inhibition of BACH1 (FANCJ) helicase by backbone discontinuity is overcome by increased motor ATPase or length of loading strand. Nucleic Acids Res. 2006;34:6673–6683. doi: 10.1093/nar/gkl964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sommers JA, Rawtani N, Gupta R, Bugreev DV, Mazin AV, Cantor SB, Brosh RM. FANCJ uses its motor ATPase to destabilize protein-DNA complexes, unwind triplexes, and inhibit RAD51 strand exchange. J Biol Chem. 2009;284:7505–7517. doi: 10.1074/jbc.M809019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.(a) Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nature Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]; (b) Wu Y, Sommers JA, Suhasini AN, Leonard T, Deakyne JS, Mazin AV, Shin-ya K, Kitao H, Brosh RM. Fanconi anemia group J mutation abolishes its DNA repair function by uncoupling DNA translocation from helicase activity or disruption of protein-DNA complexes. Blood. 2010;116:3780–3791. doi: 10.1182/blood-2009-11-256016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.(a) Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: Insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. 2012;132:785–796. doi: 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu Y, Sommers JA, Khan I, de Winter JP, Brosh J, Robert M. Biochemical characterization of Warsaw breakage syndrome helicase. J Biol Chem. 2011;287:1007–1021. doi: 10.1074/jbc.M111.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bharti SK, Khan I, Banerjee T, Sommers JA, Wu Y, Brosh RM. Molecular functions and cellular roles of the ChlR1 (DDX11) helicase defective in the rare cohesinopathy Warsaw breakage syndrome. Cell Mol Life Sci. 2014;71:2625–2639. doi: 10.1007/s00018-014-1569-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.van der Lelij P, Chrzanowska KH, Godthelp BC, Rooimans MA, Oostra ABS, Markus, Zdzienicka MZ, Joenje H, de Winter JP. Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet. 2010;86:262–266. doi: 10.1016/j.ajhg.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Capo-Chichi J-M, Bharti SK, Sommers JA, Yammine T, Chouery E, Patry L, Rouleau GA, Samuels ME, Hamdan FF, Michaud JL, Brosh RM, Jr, Mégarbane A, Kibar Z. Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Human Mutat. 2013;34:103–107. doi: 10.1002/humu.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vannier JB, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends Cell Biol. 2014;24:416–425. doi: 10.1016/j.tcb.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 96.(a) Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, Alter BP, Savage SA. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum Genet. 2013;132:473–480. doi: 10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Walne Amanda J, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92:448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.(a) Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, Weizman OE, Schertzer M, Wang Z, Vladimirova O, Schug J, Aker M, Londoño-Vallejo A, Kaestner KH, Lieberman PM, Tzfati Y. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal–Hreidarsson syndrome. Proc Natl Acad Sci. 2013;110:E3408–16. doi: 10.1073/pnas.1300600110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, Carpentier W, Nitschke P, Picard C, Couillault G, Soulier J, Fischer A, Callebaut I, Jabado N, Londono-Vallejo A, de Villartay JP, Revy P. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22:3239–3249. doi: 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- 98.Netz DJA, Stith CM, Stümpfig M, Köpf G, Vogel D, Genau HM, Stodola JL, Lill R, Burgers PMJ, Pierik AJ. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat Chem Biol. 2011;8:125–132. doi: 10.1038/nchembio.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moiseeva TN, Gamper AM, Hood BL, Conrads TP, Bakkenist CJ. Human DNA polymerase ε is phosphorylated at serine-1940 after DNA damage and interacts with the iron-sulfur complex chaperones CIAO1 and MMS19. DNA Repair. 2016;43:9–17. doi: 10.1016/j.dnarep.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.(a) Heitzer E, Tomlinson I. Replicative DNA polymerase mutations in cancer. Curr Opin Genet Dev. 2014;24:107–113. doi: 10.1016/j.gde.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kane DP, Shcherbakova PV. A common cancer-associated DNA polymerase ε mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading. Cancer Res. 2014;74:1895–1901. doi: 10.1158/0008-5472.CAN-13-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nicolas E, Golemis EA, Arora S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene. 2016;590:128–141. doi: 10.1016/j.gene.2016.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Church DN, Briggs SEW, Palles C, Domingo E, Kearsey SJ, Grimes JM, Gorman M, Martin L, Howarth KM, Hodgson SV, Kaur K, Taylor J, Tomlinson IPM. DNA polymerase ε and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. 2013;22:2820–2828. doi: 10.1093/hmg/ddt131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fujiwara T, Harigae H. Pathophysiology and genetic mutations in congenital sideroblastic anemia. Pediatr Int. 2013;55:675–679. doi: 10.1111/ped.12217. [DOI] [PubMed] [Google Scholar]
- 102.Protasova MS, Grigorenko AP, Tyazhelova TV, Andreeva TV, Reshetov DA, Gusev FE, Laptenko AE, Kuznetsova IL, Goltsov AY, Klyushnikov SA, Illarioshkin SN, Rogaev EI. Whole-genome sequencing identifies a novel ABCB7 gene mutation for X-linked congenital cerebellar ataxia in a large family of Mongolian ancestry. Eur J Hum Genet. 2015;24:550–555. doi: 10.1038/ejhg.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sato K, Torimoto Y, Hosoki T, Ikuta K, Takahashi H, Yamamoto M, Ito S, Okamura N, Ichiki K, Tanaka H, Shindo M, Hirai K, Mizukami Y, Otake T, Fujiya M, Sasaki K, Kohgo Y. Loss of ABCB7 gene: pathogenesis of mitochondrial iron accumulation in erythroblasts in refractory anemia with ringed sideroblast with isodicentric (X)(q13) Int J Hematol. 2011;93:311–318. doi: 10.1007/s12185-011-0786-y. [DOI] [PubMed] [Google Scholar]
- 104.(a) Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–8. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]; (b) Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, Weber GJ, Dooley K, Davidson AJ, Schmid B, Paw BH, Shaw GC, Kingsley P, Palis J, Schubert H, Chen O, Kaplan J, Zon LI. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–39. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]; (c) Ye H, Jeong SY, Ghosh MC, Kovtunovych G, Silvestri L, Ortillo D, Uchida N, Tisdale J, Camaschella C, Rouault TA. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J Clin Invest. 2010;120:1749–1761. doi: 10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu G, Guo S, Anderson GJ, Camaschella C, Han B, Nie G. Heterozygous missense mutations in the GLRX5 gene cause sideroblastic anemia in a Chinese patient. Blood. 2014;124:2750–1. doi: 10.1182/blood-2014-08-598508. [DOI] [PubMed] [Google Scholar]
- 106.Liu G, Wang Y, Anderson GJ, Camaschella C, Chang Y, Nie G. Functional analysis of GLRX5 mutants reveals distinct functionalities of GLRX5 protein. J Cell Biochem. 2016;117:207–217. doi: 10.1002/jcb.25267. [DOI] [PubMed] [Google Scholar]
- 107.Johansson C, Roos Annette K, Montano Sergio J, Sengupta R, Filippakopoulos P, Guo K, Delft Fvon, Holmgren A, Oppermann U, Kavanagh Kathryn L. The crystal structure of human GLRX5: iron–sulfur cluster co-ordination, tetrameric assembly and monomer activity. Biochemical Journal. 2011;433:303–311. doi: 10.1042/BJ20101286. [DOI] [PubMed] [Google Scholar]
- 108.Wingert RA, Galloway JL, Barut B, Foott H, Fraenkel P, Axe JL, Weber GJ, Dooley K, Davidson AJ, Schmidt B, Paw BH, Shaw GC, Kingsley P, Palis J, Schubert H, Chen O, Kaplan J, C. The Tübingen Screen. Zon LI. Deficiency of glutaredoxin 5 reveals Fe–S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 109.Shan Y, Cortopassi G. Mitochondrial Hspa9/Mortalin regulates erythroid differentiation via iron-sulfur cluster assembly. Mitochondrion. 2016;26:94–103. doi: 10.1016/j.mito.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 110.Schmitz-Abe K, Ciesielski SJ, Schmidt PJ, Campagna DR, Rahimov F, Schilke BA, Cuijpers M, Rieneck K, Lausen B, Linenberger ML, Sendamarai AK, Guo C, Hofmann I, Newburger PE, Matthews D, Shimamura A, Snijders PJLM, Towne MC, Niemeyer CM, Watson HG, Dziegiel MH, Heeney MM, May A, Bottomley SS, Swinkels DW, Markianos K, Craig EA, Fleming MD. Congenital sideroblastic anemia due to mutations in the mitochondrial HSP70 homologue HSPA9. Blood. 2015;126:2734–8. doi: 10.1182/blood-2015-09-659854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.(a) Urrutia PJ, Mena NP, Nunez MT. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol. 2014;5:38. doi: 10.3389/fphar.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mena NP, Urrutia PJ, Lourido F, Carrasco CM, Núñez MT. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion. 2015;21:92–105. doi: 10.1016/j.mito.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 112.Muñoz Y, Carrasco CM, Campos JD, Aguirre P, Núñez MT. Parkinson’s disease: The mitochondria-iron link. Parkinsons Dis. 2016;2016:1–21. doi: 10.1155/2016/7049108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.(a) Hefti F, Goure WF, Jerecic J, Iverson KS, Walicke PA, Krafft GA. The case for soluble Aβ oligomers as a drug target in Alzheimer’s disease. Trends Pharmacol Sci. 2013;34:261–266. doi: 10.1016/j.tips.2013.03.002. [DOI] [PubMed] [Google Scholar]; (b) Gu XM, Huang HC, Jiang ZF. Mitochondrial dysfunction and cellular metabolic deficiency in Alzheimer’s disease. Neurosci Bull. 2012;28:631–640. doi: 10.1007/s12264-012-1270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guo C, Wang P, Zhong ML, Wang T, Huang XS, Li JY, Wang ZY. Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem Int. 2013;62:165–172. doi: 10.1016/j.neuint.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 115.Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BXW, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18:291–295. doi: 10.1038/nm.2613. [DOI] [PubMed] [Google Scholar]
- 116.(a) Lumsden AL, Henshall TL, Dayan S, Lardelli MT, Richards RI. Huntingtin-deficient zebrafish exhibit defects in iron utilization and development. Hum Mol Genet. 2007;16:1905–1920. doi: 10.1093/hmg/ddm138. [DOI] [PubMed] [Google Scholar]; (b) Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, MacDonald ME. Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. 2000;9:2789–2797. doi: 10.1093/hmg/9.19.2789. [DOI] [PubMed] [Google Scholar]
- 117.Lees AJ, Hardy J, Revesz T. Parkinson’sdisease. Lancet. 2009;373:2055–2066. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 118.(a) Berg D, Hochstrasser H. Iron metabolism in Parkinsonian syndromes. Mov Disord. 2006;21:1299–1310. doi: 10.1002/mds.21020. [DOI] [PubMed] [Google Scholar]; (b) Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, et al. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 2007;68:1820–1825. doi: 10.1212/01.wnl.0000262033.01945.9a. [DOI] [PubMed] [Google Scholar]
- 119.(a) Mastroberardino PG, Hoffman EK, Horowitz MP, Betarbet R, Taylor G, Cheng D, et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol Dis. 2009;34:417–431. doi: 10.1016/j.nbd.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rhodes SL, Buchanan DD, Ahmed I, Taylor KD, Loriot MA, Sinsheimer JS, Bronstein JM, Elbaz A, Mellick GD, Rotter JI, Ritz B. Pooled analysis of iron-related genes in Parkinson’s disease: Association with transferrin. Neurobiol Dis. 2014;62:172–178. doi: 10.1016/j.nbd.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Horowitz MP, Greenamyre JT. Mitochondrial iron metabolism and its role in neurodegeneration. J Alzheimers Dis. 2010;20:551–568. doi: 10.3233/JAD-2010-100354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee DW, Kaur D, Chinta SJ, Rajagopalan S, Andersen JK. A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: Implications for Parkinson’s disease. Antiox Redox Signal. 2009;11:2083–2094. doi: 10.1089/ars.2009.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.(a) Katsuyama M, Iwata K, Ibi M, Matsuno K, Matsumoto M, Yabe-Nishimura C. Clioquinol induces DNA double-strand breaks, activation of ATM, and subsequent activation of p53 signaling. Toxicology. 2012;299:55–59. doi: 10.1016/j.tox.2012.05.013. [DOI] [PubMed] [Google Scholar]; (b) Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, Jonneaux A, Ryckewaert G, Garçon G, Rouaix N, Duhamel A, Jissendi P, Dujardin K, Auger F, Ravasi L, Hopes L, Grolez G, Firdaus W, Sablonnière B, Strubi-Vuillaume I, Zahr N, Destée A, Corvol JC, Pöltl D, Leist M, Rose C, Defebvre L, Marchetti P, Cabantchik ZI, Bordet R. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antiox Redox Signal. 2014;21:195–210. doi: 10.1089/ars.2013.5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Barupala DP, Dzul SP, Riggs-Gelasco PJ, Stemmler TL. Synthesis, delivery and regulation of eukaryotic heme and Fe–S cluster cofactors. Arch Biochem Biophys. 2016;592:60–75. doi: 10.1016/j.abb.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lecha M, Puy H, Deybach JC. Erythropoietic protoporphyria. Orphanet J Rare Dis. 2009;4:19. doi: 10.1186/1750-1172-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nakano H, Nakano A, Toyomaki Y, Ohashi S, Harada K, Moritsugu R, Takeda H, Kawada A, Mitsuhashi Y, Hanada K. Novel ferrochelatase mutations in Japanese patients with erythropoietic protoporphyria: High frequency of the splice site modulator IVS3–48C polymorphism in the Japanese population. J Invest Dermatol. 2006;126:2717–2719. doi: 10.1038/sj.jid.5700456. [DOI] [PubMed] [Google Scholar]
- 126.Kong XF, Ye J, Gao DY, Gong QM, Zhang DH, Lu ZM, Lu YM, Zhang XX. Identification of a ferrochelatase mutation in a Chinese family with erythropoietic protoporphyria. J Hepatol. 2008;48:375–379. doi: 10.1016/j.jhep.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 127.Balwani M, Doheny D. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med. 2013;19:1. doi: 10.2119/molmed.2012.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Farrag MS, Kucerova J, Šlachtova L, Šeda O, Šperl J, Martasek P. A novel mutation in the FECH Gene in a Czech family with erythropoietic protoporphyria and a population study of IVS3-48C variant contributing to the disease. Folia Biol (Praha) 2015;61:227–32. doi: 10.14712/fb2015061060227. [DOI] [PubMed] [Google Scholar]
- 129.Wang Y, Langer NB, Shaw GC, Yang G, Li L, Kaplan J, Paw BH, Bloomer JR. Abnormal mitoferrin-1 expression in patients with erythropoietic protoporphyria. Exp Hematol. 2011;39:784–794. doi: 10.1016/j.exphem.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.(a) Chen W, Dailey HA, Paw BH. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood. 2010;116:628–630. doi: 10.1182/blood-2009-12-259614. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamamoto M, Arimura H, Fukushige T, Minami K, Nishizawa Y, Tanimoto A, Kanekura T, Nakagawa M, Akiyama SI, Furukawa T. Abcb10 Role in Heme Biosynthesis In Vivo: Abcb10 Knockout in Mice Causes Anemia with Protoporphyrin IX and Iron Accumulation. Mol Cell Biol. 2014;34:1077–1084. doi: 10.1128/MCB.00865-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Troadec MB, Warner D, Wallace J, Thomas K, Spangrude GJ, Phillips J, Khalimonchuk O, Paw BH, Ward DM, Kaplan J. Targeted deletion of the mouse Mitoferrin1 gene: from anemia to protoporphyria. Blood. 2011;117:5494–5502. doi: 10.1182/blood-2010-11-319483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Beinert H. Iron–sulfur proteins: ancient structures, still full of surprises. J Biol Inorg Chem. 2000;5:2–15. doi: 10.1007/s007750050002. [DOI] [PubMed] [Google Scholar]
- 132.(a) Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol. 2010:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]; (c) Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.(a) Choi YS, Shin DH, Chung IY, Kim SH, Heo YJ, Cho YH. Identification of Pseudomonas aeruginosa genes crucial for hydrogen peroxide resistance. J Microbiol Biotechnol. 2007;17:1344–1352. [PubMed] [Google Scholar]; (b) Kim SH, Lee BY, Lau GW, Cho YH. IscR modulates catalase A (KatA) activity, peroxide resistance, and full virulence of Pseudomonas aeruginosa PA14. J Microbiol Biotechnol. 2009;19:1520–1526. doi: 10.4014/jmb.0906.06028. [DOI] [PubMed] [Google Scholar]
- 134.(a) Morris RP, Nguyen L, Gatfield J, Visconti K, Nguyen K, Schnappinger D, Ehrt S, Liu Y, Heifets L, Pieters J, Schoolnik G, Thompson CJ. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2005;102:12200–12205. doi: 10.1073/pnas.0505446102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rybniker J, Nowag A, Van Gumpel E, Nissen N, Robinson N, Plum G, Hartmann P. Insights into the function of the WhiB-like protein of mycobacteriophage TM4, a transcriptional inhibitor of WhiB2. Mol Microbiol. 2010;77:642–657. doi: 10.1111/j.1365-2958.2010.07235.x. [DOI] [PubMed] [Google Scholar]; (c) Saini V, Farhana A, Steyn AJ. Mycobacterium tuberculosis WhiB3: a novel iron-sulfur cluster protein that regulates redox homeostasis and virulence. Antioxid Redox Signal. 2012;16:687–697. doi: 10.1089/ars.2011.4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rocha AG, Dancis A. Life without Fe-S clusters. Mol Microbiol. 2016;99:821–826. doi: 10.1111/mmi.13273. [DOI] [PubMed] [Google Scholar]
- 136.(a) Netz DJA, Pierik AJ, Stumpfig M, Bill E, Sharma AK, Pallesen LJ, Walden WE, Lill R. A bridging [4Fe-4S] cluster and nucleotide binding are essential for function of the Cfd1-Nbp35 complex as a scaffold in iron-sulfur protein maturation. J Biol Chem. 2012;287:12365–12378. doi: 10.1074/jbc.M111.328914. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Soler N, Delagoutte E, Miron S, Facca C, Baïlle D, d’Autreaux B, Craescu G, Frapart Y-M, Mansuy D, Baldacci G, Huang M-E, Vernis L. Interaction between the reductase Tah18 and highly conserved Fe-S containing Dre2 C-terminus is essential for yeast viability. Mol Microbiol. 2011;82:54–67. doi: 10.1111/j.1365-2958.2011.07788.x. [DOI] [PubMed] [Google Scholar]; (c) Vernis L, Facca C, Delagoutte E, Soler N, Chanet R, Guiard B, Faye G, Baldacci G. A newly identified essential complex, Dre2-Tah18, controls mitochondria integrity and cell death after oxidative stress in yeast. PLoS One. 2009;4:e4376. doi: 10.1371/journal.pone.0004376. [DOI] [PMC free article] [PubMed] [Google Scholar]