

Abstract
Autism spectrum disorder (ASD) is a developmental disability characterized by impairments in social interaction and repetitive behavior, and is also associated with cognitive deficits. There is no current treatment that can ameliorate most of the ASD symptomatology; thus, identifying novel therapies is urgently needed. We used male BTBR T+ Itpr3tf/J (BTBR) mice, a model that reproduces most of the core behavioral phenotypes of ASD, to test the effects of systemic administration of insulin-like growth factor II (IGF-II), a polypeptide that crosses the blood–brain barrier and acts as a cognitive enhancer. We show that systemic IGF-II treatments reverse the typical defects in social interaction, cognitive/executive functions, and repetitive behaviors reflective of ASD-like phenotypes. In BTBR mice, IGF-II, via IGF-II receptor, but not via IGF-I receptor, reverses the abnormal levels of the AMPK-mTOR-S6K pathway and of active translation at synapses. Thus, IGF-II may represent a novel potential therapy for ASD.
SIGNIFICANCE STATEMENT Currently, there is no effective treatment for autism spectrum disorder (ASD), a developmental disability affecting a high number of children. Using a mouse model that expresses most of the key core as well as associated behavioral deficits of ASD, that are, social, cognitive, and repetitive behaviors, we report that a systemic administration of the polypeptide insulin-like growth factor II (IGF-II) reverses all these deficits. The effects of IGF-II occur via IGF-II receptors, and not IGF-I receptors, and target both basal and learning-dependent molecular abnormalities found in several ASD mice models, including those of identified genetic mutations. We suggest that IGF-II represents a potential novel therapeutic target for ASD.
Keywords: autism spectrum disorder, insulin-like growth factor II receptor, insulin-like growth factor II, memory, mouse model, mTOR
Introduction
Autism spectrum disorder (ASD), also called autism, is a neurodevelopmental disorder characterized by impairments in social interactions, deficits in communication, and restrictive and repetitive patterns of behaviors and interests (McFarlane et al., 2008; Volkmar et al., 2009; American Psychiatric Association, 2013).
Although only approximately half of the children diagnosed with ASD show cognitive impairments (Charman et al., 2011), most have severely impaired social cognitive abilities, as well as problems in dividing attention and processing the “whole”, and instead focus more on details. They also have low imitation skills, and an increased attention to inanimate objects rather than social ones (Davies et al., 2011; McPartland et al., 2011). Social cognitive abilities are based on learning through observation, and include an understanding of the intentions and emotions of others during interactions, suggesting that ASD subjects have deficits of learning and cognition (Frith and Frith, 2012). In agreement, studies have indicated that autism is characterized by deficits in learning and cognitive processing (Hill, 2004), and suggest a need for cognitive enhancement as a therapeutic intervention. Cognitive enhancers, in fact, may also provide rescuing of attention abilities, ability to understand social cues, and executive functions (Floresco and Jentsch, 2011; Lynch et al., 2011). Unfortunately, efficacious treatments for ASD are still lacking.
To identify novel treatments, preclinical investigations in animal models of ASD can be extremely helpful. An inbred strain, the BTBR T+ Itpr3tf/J (BTBR) mice, seems to recapitulate several core behavioral deficits of ASD and is thus considered a useful representative model for translational investigations (Moy et al., 2007; MacPherson et al., 2008; McFarlane et al., 2008; Roullet and Crawley, 2011). BTBR mice exhibit normal general health and physical abilities (Moy et al., 2007) but show cognitive impairments, which include deficits in fear conditioning, object recognition, spatial learning, and probabilistic reversal learning (Cabib et al., 2003; MacPherson et al., 2008; Amodeo et al., 2012, 2014). They also have social deficits including reduced social approach, social interaction, juvenile play, and social transmission of food preference (McFarlane et al., 2008; Ruskin et al., 2013). They also have reduced ultrasonic vocalizations in social settings and high levels of repetitive behaviors including self-grooming and marble burying (McFarlane et al., 2008; Scattoni et al., 2008; Silverman et al., 2010a; Pobbe et al., 2011). BTBR mice lack the corpus callosum, have anatomical alterations of the hippocampus, and spontaneous deletion of the disrupted in schizophrenia-1 (DISC-1) gene (Wahlsten et al., 2003; Stephenson et al., 2011). In common with other ASD mouse models (Zhou et al., 2009; Sato et al., 2012), the social deficits of BTBR mice ameliorate with rapamycin, which blocks the mammalian target of rapamycin (mTOR) pathway (Burket et al., 2014). These data suggest that dysregulation of active translation/protein concentration or autophagy are defective in BTBR, similar to what has been reported with other ASD mouse models (Kelleher and Bear, 2008). These dysregulations have been in fact implicated in several monogenic neurodevelopmental disorders linked to ASD (Ehninger and Silva, 2011; Sun et al., 2015, 2016), suggesting that deficits in the mTOR pathway are common to many types of ASD.
We and others have previously identified the polypeptide insulin growth factor II (IGF-II) as a potent cognitive enhancer when injected either into the hippocampus of rats or mice (Agis-Balboa et al., 2011; Chen et al., 2011), or subcutaneously in mice (Stern et al., 2014). The effect is rapid and persistent, and is mediated by IGF-II receptor (R) and not IGF-IR (Chen et al., 2011; Stern et al., 2014). Importantly, as IGF-II crosses the blood–brain barrier (Reinhardt and Bondy, 1994), it provides a unique opportunity for potential, rapid translational applications. The striking effect of IGF-II as an enhancer of cognitive and social learning as well as of executive functions in normal mice (Stern et al., 2014) prompted us to investigate the effect of systemically administered IGF-II on ASD-like behavioral phenotypes and on underlying mechanisms.
Materials and Methods
Animals.
A total of 230 adult male C57BL/6J (B6) (RID:IMSR_JAX:000664; https://www.jax.org/strain/000664) and 265 BTBR T+ Itpr3tf/J mice (6–10 weeks of age), purchased from The Jackson Laboratory (RID:IMSR_JAX:016926; https://www.jax.org/strain/016926), were used for this study. All mice were group housed, on a 12 h light/dark cycle, with ad libitum access to food and water. Experiments were performed during the light cycle. All protocols complied with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the NYU University Animal Welfare Committee.
Drugs.
Recombinant mouse IGF-II (R&D Systems) was dissolved in 0.1% BSA-PBS, pH 7.4, and injected at 30 μg/kg subcutaneously in 0.3 ml. Anti-IGF-IIR antibody (anti-IGF-IIR) or IgG control (both from R&D Systems) were dissolved in PBS at 20 ng/μl and 0.5 μl per side were injected into the hippocampus. This concentration blocks 90% of IGF-IIR binding in an in vitro binding assay (R&D Systems); it also blocks memory enhancement in rats and mice (Chen et al., 2011; Stern et al., 2014). The IGF-IR antagonist JB1 (Bachem Biosciences) was dissolved in PBS, and 0.5 μl of a solution at 20 ng/μl was injected in each side of the dorsal hippocampus. This concentration has been used successfully to block IGF-I activity in various tissues, including the brain (Quesada and Micevych, 2004).
Experimental design for IGF-II injection and behavioral tasks.
In all experiments, mice were handled for 2–3 min/d for 5 d before their first behavioral procedure. Each mouse was used for one to three behavioral tasks, and all behaviors were performed in the same order; the sequence went from the less adverse to the more adverse behavioral task. Animals were randomly assigned to receive either IGF-II or vehicle at the first session, after which the treatment was counterbalanced. Seven to 10 d between each behavioral procedure were allowed to obtain clearance from the drug. Untrained controls (naive) received the same handling and treatment, as detailed in the Results section, but not training or testing. Biochemistry and corticosterone (cort) studies were done on independent groups of mice, which were not used for multiple behaviors. For every behavior, multiple experiments using n = 3–4/group were repeated for data confirmation, with all conditions conducted in parallel. Different experimenters using different litters obtained similar results, which were finally pooled for statistical analyses.
Social interaction and social novelty memory in the open-field.
Social interaction and preference for social novelty were performed as previously described (Satoh et al., 2011; Stern et al., 2014). On two sides of a clean square cage (30.5 × 24.1 × 21.0 cm) a rectangular wire-holder containing either a stimulus mouse (C57BL/6J) or a novel object was placed. Stimulus mice were habituated to the wire-holders for 10 min the day before testing to prevent excessive movement. On the day of the experiment, the test mice were habituated to the cage with empty wire-holders for 10 min. During each subsequent phase the test mouse was placed into the cage and allowed to interact freely for 5 min with the following settings: in Phase 1, with a stranger mouse (novel Mouse 1) and a novel object placed under the wire holders. In Phase 2, the object was replaced with a novel mouse (Mouse 2). Finally, in Phase 3, 24 h later, Mouse 1 was placed under the wire-holder, and a third novel mouse (Mouse 3) was placed under the other wire-holder. Social interaction (also called sociability) was measured as the time spent interacting/sniffing with the mouse or object over 10 min. Immediate and long-term social novelty memories, which measured the ability to discriminate between the novel mouse and the familiar mouse, were measured as the percentage time spent interacting with the novel mouse. A percentage index preference was calculated as the time that the test mouse spent with the novel mouse divided by the total interaction time with both object and mouse (Test 1), or the two mice (Tests 2 and 3), and multiplied by 100. Two independent observers blind to mouse strain and treatment scored all the experiments.
Novel-object recognition.
Novel-object recognition (nOR) was adapted from Stern et al. (2014). One day before training, mice were placed in a clean square novel arena, free of bedding for 5 min. The following day, mice were trained in the same arena, containing two identical objects (Mega Bloks 120), by allowing them to interact freely for 3 min. Five minutes, 4 h, and/or 24 h later, the mice were returned to the arena in which one object had been replaced with a novel object, while the other remained constant. Each session was recorded and analyzed off-line. Time spent interacting/sniffing each object over 5 min was recorded in seconds, and memory was expressed as the percentage index preference for the novel object (novel object/novel object + old object × 100) over 5 min. Two independent observers blind to mouse strain and treatment scored all the experiments.
Contextual fear conditioning.
Mice were handled for 2–3 min per day for 5 d before training. The conditioning chamber consisted of a rectangular Perspex box (30.5 × 24.1 × 21.0 cm) with a metal grid floor (Model ENV-008 Med Associates) through which foot shocks were delivered via a constant current scrambler circuit. Contextual fear conditioning (CFC) training was performed as described previously (Guedea et al., 2011). After 2 min in the chamber, an unsignaled 2 s 0.7 mA footshock was delivered. After 1 additional minute in the chamber, the mouse was returned to its home cage. At testing, done at 24 h after training, the animal was placed back in to the conditioning chamber where it remained for 5 min in the absence of a footshock and was video-recorded for off-line analysis. Freezing, defined as lack of movement in addition to heartbeat and respiration, was recorded every 10th second by two independent trained observers unaware of the experimental conditions. Freezing was expressed in a score calculated as percentage of freezing behavior over the total number of observations.
Inhibitory avoidance.
Inhibitory avoidance (IA) was performed as described previously (Chen et al., 2011). The IA chamber (Med Associates) consisted of a rectangular Perspex box divided into a safe compartment and a shock compartment. The safe compartment was white and illuminated, whereas the shock compartment was black and dark. Foot shocks were delivered to the grid floor of the shock chamber via a constant current scrambler circuit. The apparatus was located in a sound-attenuated, non-illuminated room. During training sessions, each mouse was placed in the safe compartment with its head facing away from the door. After 10 s, the door separating the compartments was automatically opened, allowing the mouse access to the shock compartment; the mouse usually entered the shock (dark) compartment within 10–20 s of the door opening. The door closed 1 s after the mouse entered the shock compartment, and a brief foot shock (0.7 mA for 1 s) was administered. Latency to enter the shock compartment was taken in seconds as acquisition. The mouse was then returned to its home cage and tested for memory retention at the designated time point(s). Retention tests were done by placing the mouse back in the safe compartment and measuring its latency to enter the shock compartment. Foot shocks were not administered on the retention tests, and testing was terminated at 900 s. In reactivation (reconsolidation) experiments, mice were trained as described and memory was reactivated 24 h later. Reactivation consisted of placing the mouse in the light compartment for 30 s without the door opening. Mice were then tested 24 h and 7 d following reactivation.
Marble burying.
Marble burying was used for testing repetitive and compulsive behavior (Angoa-Pérez et al., 2013). Empty home cages were filled with 10 cm of bedding, on top of which 12 marbles were placed in four rows of three marbles each. The fraction of time spent burying marbles was measured over 15 min under a red light. Digging was defined as coordinated movements of fore or hind limbs that displace the substrate. Sessions were video-recorded, and the time spent burying was quantified by two independent observers blind to strain and injection. Total number of marbles buried (>75% marble covered by bedding material) was determined at the end of the testing session.
Grooming behavior.
The conditioning chamber consisted of a rectangular box (30.5 × 24.1 × 21.0 cm) with 2 cm of bedding. Bedding was used based on previous reports suggesting that this would reduce anxiety and neophobia (Silverman et al., 2010a). Animal behavior was video-recorded for 10 min and analyzed off-line by two independent observers blind to strain and treatment.
Spontaneous alternation.
Spontaneous alternation was performed as previously described (Stern et al., 2014). The Y-maze consisted of three black polycarbonate arms (7.62 × 12.7 × 38.1 cm3). Mice were allowed to freely explore from the center of the maze for 10 min while being video-recorded for off-line analysis. Two independent observers, blind to strain and treatment, recorded entries into each arm and total arm entries. Spontaneous alternation was defined as successive entries into each of the three arms on overlapping triplet sets (e.g., ABC, BCA, CAB, etc.). Percentage alternation was defined as the ratio of actual alternations (total alternations) to possible alternations (total arm entries − 2) × 100.
Open-field.
Mice were allowed to freely explore an open-field arena (43.2 cm × 43.2 cm × 30.5 cm; Med Associates; ENV-515) designated into 16 quadrants for 5 min. Activity was analyzed with Ethovision-XT (Noldus Information Technology). Animals' activity was measured by scoring the number of times they moved from one square to another. The numbers of entries in the four center quadrants, as well as the time spent (in seconds) in the four center quadrants were measured. Illumination levels during testing were maintained at 195 lux.
Elevated plus maze.
Mice were placed in the center zone of a white 39-cm-arm-length elevated plus maze (EPM) arena (Columbus Instruments) and allowed to freely explore for 5 min. Subject movements were analyzed with Ethovision-XT (Noldus Information Technology). Illumination levels during testing were maintained at 195 lux.
Cort analysis.
Plasma cort levels collected from trunk blood of mice were analyzed using the Cayman EIA corticosterone kit (Cayman Chemical Company), according to the instructions of the manufacturer. Blood was collected either 20 min or 3 h post-IGF-II or vehicle injections.
Cannulae implants and drug injection.
Cannulae implants were adapted to mice and drug injection was performed as described previously (Chen et al., 2011; Stern et al., 2014). Mice were anesthetized with ketamine (100 mg/kg) and xylazine (75 mg/kg), and guide cannulae (C232GC; 22 gauge; Plastics One) were implanted to target the hippocampus (−1.7 mm posterior to bregma ±1.5 mm lateral to midline, −2.3 mm ventral to skull surface) using a stereotaxic apparatus (Kopf Instruments). Mice were allowed to recover for at least 5 d before undergoing behavioral experiments. Drugs were delivered in 0.50 μl over 3 min via injection through the cannula (26 gauge, extending 0.8 mm beyond the 1.5 mm guide) attached to polyethylene tubing (PE50) connected to a 10 μl Hamilton syringe and controlled by a microinfusion pump (Harvard Apparatus). Cannula placements were verified at the end of the behavioral experiments, following fixation of the brains in 10% formalin. Forty micrometer coronal sections were cut through the hippocampus and examined under a light microscope. All surgeries correctly targeted the hippocampus.
Total protein extracts, synaptoneurosomal extracts, and Western blot analyses.
Total protein extracts, synaptoneurosomal (SN) extracts, and Western blots analyses were performed as described previously (Chen et al., 2011). Dorsal hippocampi, medial prefrontal cortex (mPFC), striatum, or cerebellum were dissected rapidly in cold dissection buffer containing the following (in mm: 2.6 KCl, 1.23 sodium phosphate monobasic, 26 sodium bicarbonate, 5 kynurenic acid, 212 sucrose, 10 dextrose, 0.5 CaCl2, and 1 MgCl2), followed by homogenization in buffer containing 10 mm HEPES, 2 mm EDTA, 2 mm EGTA, 0.5 mm DTT, 2 mm NaF, 1 μm microcystin, 1 mm benzamidine, and phosphatase and protease inhibitor mixtures (Sigma-Aldrich). Tissues were homogenized and in part extracted with an addition of 0.2 m NaCl to generate total protein extracts and in part filtered sequentially through a 100 μm nylon mesh filter and a 5 μm nitrocellulose filter to generate SN preparations. SN fractions were obtained by resuspending pellets in homogenization buffer after centrifugation at 1000 × g for 10 min. Protein concentrations were determined using the Bio-Rad protein assay. For Western blot analyses, equal amounts of protein (10–20 μg, depending on the marker to be investigated) to which 10% β-mercaptoethanol was added were resolved using 10–15% denaturing SDS-PAGE and transferred to Immobilon-FL PVDF membranes (EMD Millipore) by electroblotting. The membrane was dried and then blocked with either 5% milk or 5% BSA in tris buffer saline (TBS). Primary antibodies were used at the following dilutions: anti-mTOR (1/1000; Cell Signaling Technology; RRID:AB_2105622), anti-phospho(Ser2448)-mTOR (1/1000; Cell Signaling Technology; RRID:AB_490932), anti-ULK (1/1000; Cell Signaling Technology; RRID:AB_2212518), anti-phospho(Ser757)-ULK (1/1000; Cell Signaling Technology; RRID:AB_10829226), anti-S6K (1/1000; Cell Signaling Technology; RRID:AB_10695156), anti-phospho(Thr389)-S6K (1/1000; Cell Signaling Technology; RRID:AB_1658214), anti-AMPK (1/1000; Cell Signaling Technology; RRID:AB_10695248), or anti-phospho(Thr172)-AMPK (1/1000; Cell Signaling Technology; RRID:AB_330330), LC3-B (1/1000; Novus; RRID:AB_1049142), p62 (1/1000; Cell Signaling Technology; RRID:AB_10624872), and anti-actin (1/10,000; Santa Cruz Biotechnology; RRID:AB_626632) All primary antibodies were diluted in TBS and used to incubate the blotted membranes with gently rocking overnight at 4°C. Membranes were then washed and incubated with secondary antibodies (anti-rabbit IRDye800CW and anti-mouse IRDye680; 1/10,000; Li-Cor) for 1 h at room temperature. Membranes were scanned on the Li-Cor Odyssey imager under nonsaturating conditions. Data were quantified using pixel intensities with the Odyssey software according to the protocols of the manufacturer (Li-Cor). Intensities were normalized to corresponding values of actin immunoreactivity and expressed as percentage fold-changes relative to the control group mean value.
In vivo SUnSET.
A protein synthesis assay was performed as previously described using the surface sensing of translation (SUnSET) method (Schmidt et al., 2009). In brief, puromycin (25 μg in 0.5 μl of PBS; Sigma-Aldrich) was injected bilaterally into dorsal hippocampus 60 min, immediately following an anti-IGF-IIR or IgG bilateral hippocampal injection. The animals were killed 60 min later. Additionally, mice received subcutaneous injections of IGF-II immediately following puromycin. Dorsal hippocampi were dissected; total and SN extracts were prepared as described above. Twenty-five micrograms of protein extract were resolved on a 4–20% gradient gel and transferred to Immunobilon-FL PVDF membranes (EMD Millipore) by electroblotting. The membranes were dried and then blocked with 5% BSA in TBS. Puromycin incorporation was revealed using an anti-puromycin monoclonal antibody (1/5000; Millipore; RRID:AB_2566826). Signal intensities were normalized to corresponding values for β-tubulin immunoreactivity (AB_10990122) in each sample. The mean value of each group was calculated and expressed as percentage fold-change relative to the control mean value.
Experimental design and statistical analyses.
For statistical analyses, unpaired t tests or one- or two-way ANOVA followed by Tukey or Bonferroni post hoc tests were used. Only male mice were used for all experiments. All tests were conducted using GraphPad Prism v7.0 software with significance set at p < 0.05. Neurobehavioral assessment was performed blindly with respect to experimental group and drug administration.
Results
IGF-II reverses social interaction deficits of BTBR mice and increases their social novelty memory
Detailed statistical analyses for all experiments shown in this paper can be found in the Extended Data tables (Figs. 1-4, 1-5, 1-6, 1-7, 2-2, 2-3, 3-1, 4-1, 5-2, 6-2, 6-3, and 7-1).
We tested the effects of a subcutaneous administration of IGF-II to adult male BTBR mice and compared them to B6 mice, which were used as controls. B6 is a standard inbred strain of mice that have normal social behavior, low repetitive behaviors, as well as good cognitive performance. For all experiments, the dose of 30 μg/kg and time of IGF-II administration were chosen on the basis of the previous study (Stern et al., 2014) in B6 mice, which established a dose–response curve and significant cognitive enhancing effects.
Because IGF-II increases social novelty memory in B6 mice (Stern et al., 2014), here we tested the effect of IGF-II on social interaction deficits as well as social novelty memory of BTBR mice. A subcutaneous injection of vehicle or IGF-II 20 min before testing social interaction showed that, while as expected (Moy et al., 2007; McFarlane et al., 2008) vehicle-injected B6 mice spent significantly more time with a stranger mouse compared with an object, vehicle-injected BTBR mice had no preference (group × test interaction: F(6,87) = 3.92, p = 0.0016; two-way ANOVA; Fig. 1A, Test 1). This sociability deficit of BTBR mice did not reflect an olfactory impairment or inability to detect novelty, because 5 min after Test 1 the same BTBR mice spent significantly more time investigating a novel mouse compared with the mouse just seen (Fig. 1A, Test 2). Notably, IGF-II did not change the social interaction of B6 mice, but completely rescued the social interaction deficit of BTBR mice (Fig. 1A, Test 1). Furthermore, 24 h after Test 1, whereas both B6 and BTBR mice injected with vehicle lost their social novelty memory, both IGF-II-injected groups still displayed significant social novelty memory (Fig. 1A, Test 3). Analyses of interaction time expressed in seconds showed similar effects (Fig. 1-1). No significant differences were detected in total exploration time between groups or treatment, leading to the conclusion that the effects of IGF-II were not due to changes in motivation or exploratory behavior (Fig. 1-3). Together, these data indicated that IGF-II rescues the social interaction deficits and increases social novelty memory in BTBR mice.
Figure 1.
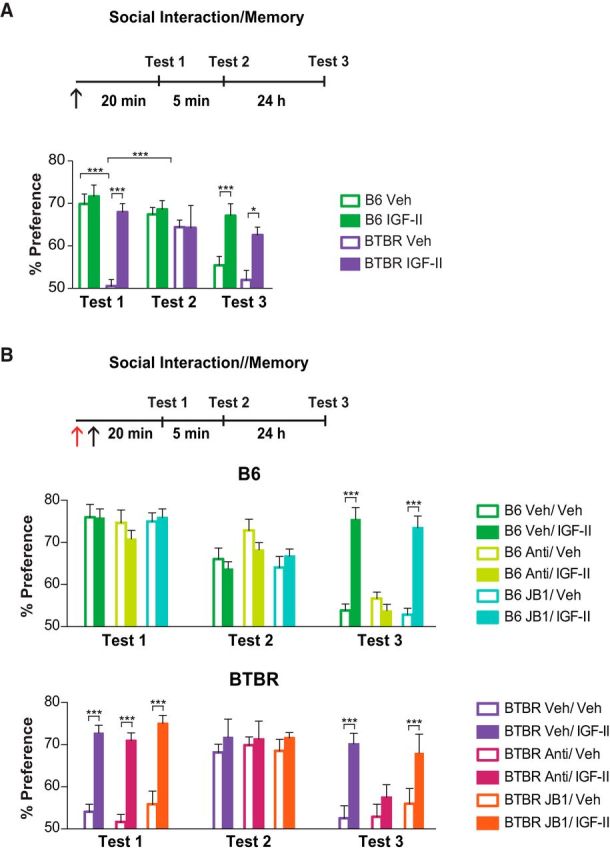
IGF-II reverses social interaction deficits and enhances social novelty memory in BTBR mice. Experimental timeline is shown above graphs. In all experiments mice received a subcutaneous injection (↑) of either vehicle (Veh) or IGF-II, 20 min before the first test. All data are expressed as the mean percentage (±SEM). n = 7–9 per group. Two-way ANOVA followed by Tukey or Bonferroni post hoc tests. *p < 0.05, ***p < 0.001. A, Percentage exploration preference for Mouse 1 versus novel object (Test 1), novel Mouse 2 versus Mouse 1 (Test 2), and novel Mouse 3 versus Mouse 1 (Test 3) of B6 and BTBR mice injected with Veh or IGF-II. B, Percentage exploration preference for Mouse 1 versus novel object (Test 1), novel Mouse 2 versus Mouse 1 (Test 2), and novel Mouse 3 versus Mouse 1 (Test 3) of B6 (top) and BTBR (bottom) mice that received a bilateral dorsal hippocampal injections (red ↑) of IGF-IR blocker (JB1), IGF-IIR functionally blocking antibody (Anti), IgG or Veh immediately before a systemic Veh or IGF-II injection. Time spent exploring Mouse 1 versus novel object (Test 1), novel Mouse 2 versus Mouse 1 (Test 2), and novel Mouse 3 versus Mouse 1 (Test 3) of B6 and BTBR mice injected with Veh or IGF-II. Exploration times are reported in the Extended Data (Figs. 1-1 to 1-3. Detailed statistical analyses are reported in the Extended Data tables (Figs. 1-4 to 1-7).
IGF-II injection reverses social interaction deficits and enhances social novelty memory in BTBR mice. Experimental timeline is shown above the graph. Mice received a s.c. injection (h) 20 min prior to social interaction test. All data are expressed as the mean (± s.e.m.) of total time spent interacting with mouse 1 or novel object (Test 1), novel mouse 2 or mouse 1 (Test 2), and novel mouse 3 or mouse 1 (Test 3) by B6 and BTBR mice injected with vehicle (Veh) or IGF-II. N=7-9 per group. Two-way analysis of variance (ANOVA) followed by Bonferroni post-hoc tests. *p<0.05, **p<0.01, *** p<0.001. See Extended Data table Figure 1-5 for detailed statistical analyses. Download Figure 1-1, DOCX file (16.7KB, docx)
IGF-II reverses social interaction deficits and enhances social novelty memory of BTBR mice via hippocampal IGF-IIR. Experimental timeline is shown above the graph. Mice received a s.c. injection (η) 20 min prior to social interaction test. All data are expressed as the mean (± s.e.m.). N=7-8 per group. Two-way analysis of variance (ANOVA) followed by Bonferroni post-hoc tests. *p<0.05, **p<0.01, *** p<0.001. Total time spent interacting with mouse 1 or novel object (Test 1), novel mouse 2 or mouse 1 (Test 2), and novel mouse 3 or mouse 1 (Test 3) of B6 and BTBR mice injected with vehicle (Veh) or IGF-II. See Extended Data table Figure 1-6 for detailed statistical analyses. Download Figure 1-2, DOCX file (17.2KB, docx)
IGF-II injection does not affect total exploration time in social interaction and social novelty memory. Experimental timeline is shown above the graph. Mice received a s.c. injection (η) 20 min prior to social interaction test. All data are expressed as the mean (± s.e.m.). N=7-9 per group. Two-way analysis of variance (ANOVA) followed by Tukey post-hoc tests. Total time spent interacting with novel mouse 1 and novel object (Test 1), novel mouse 2 and mouse 1 (Test 2), and novel mouse 3 and mouse 1 (Test 3) of B6 and BTBR mice injected with vehicle (Veh) or IGF-II. See Extended Data table Figure 1-7 for detailed statistical analyses. Download Figure 1-3, DOCX file (21.4KB, docx)
Statistical analyses related to Figure 1. Download Figure 1-4, DOCX file (16.4KB, docx)
Statistical analyses related to 1-1. Download Figure 1-5, DOCX file (24.9KB, docx)
Statistical analyses related to 1-2. Download Figure 1-6, DOCX file (15KB, docx)
Statistical analyses related to 1-3. Download Figure 1-7, DOCX file (17.3KB, docx)
IGF-II can bind to both IGF-IR and IGF-IIR, two distinct receptors, with different affinity for IGF-II (Nissley and Rechler, 1984). Because social novelty memory requires hippocampal functions, we then tested whether the rescuing effects on social interaction deficits and/or the enhancing effect on social novelty memory by systemic IGF-II administration occurs via IGF-IR or IGF-IIR in the dorsal hippocampus. An anti-IGF-IIR blocking antibody or the selective IGF-IR blocker, JB1, were injected bilaterally into the dorsal hippocampus 20 min before social interaction; immediately after, IGF-II was injected subcutaneously. Relative control injections consisted of IgG and vehicle solution, respectively. Neither blockers of IGF-IR or IGF-IIR affected social interaction in B6 mice. No effect was found also on the rescuing effect of IGF-II on social interaction deficit of BTBR mice (group × test interaction: F(22,252) = 57.19, p < 0.001; two-way ANOVA; Fig. 1B, Test 1). In contrast, blocking the IGF-IIR, but not the IGF-IR, completely blunted the effect of IGF-II in enhancing social novelty memory (Test 3) in both strains. When comparing time spent interacting with each object/mouse, compared with percentage, similar enhancement/rescuing effects of IGF-II were found. These effects were then blunted when an IGF-IIR, but not IGF-1R, blocker was administered into the hippocampus (Fig. 1-2). Thus, in line with a role of the hippocampus in social novelty memory but not in social interaction (McHugh et al., 2004; Felix-Ortiz and Tye, 2014; Hitti and Siegelbaum, 2014), the IGF-II effect only on social novelty memory required the selective role of hippocampal IGF-IIR but not IGF-IR.
IGF-II, via IGF-IIR, reverses memory deficits of BTBR mice
Approximately one-third of children diagnosed with ASD have a comorbid intellectual disability (Chakrabarti and Fombonne, 2005). To determine whether IGF-II rescues cognitive-like deficits, we next tested the effect of systemic IGF-II treatment on both non-aversive (nOR) and aversive (CFC and IA) memories. IGF-II injected 20 min before training had no effect on short-term nOR memory, tested 5 min after training (group × test interaction: F(3,46) = 0.708, p = 0.55; two-way ANOVA; Fig. 2A). However, IGF-II reversed the significant impairment of long-term nOR memory of BTBR mice tested 4 h following training (group × test interaction: F(6,84) = 54.52, p < 0.001; two-way ANOVA; Fig. 2B). As expected from this protocol of nOR, memory retention of both BTBR and B6 mice injected with vehicle significantly decayed 24 h following training; however, IGF-II injection significantly enhanced memory retention in both strains. No change in total exploration time among groups was found when comparing strains or injections, indicating that IGF-II did not alter motivation, exploration or motor performance (Fig. 2-1).
Figure 2.
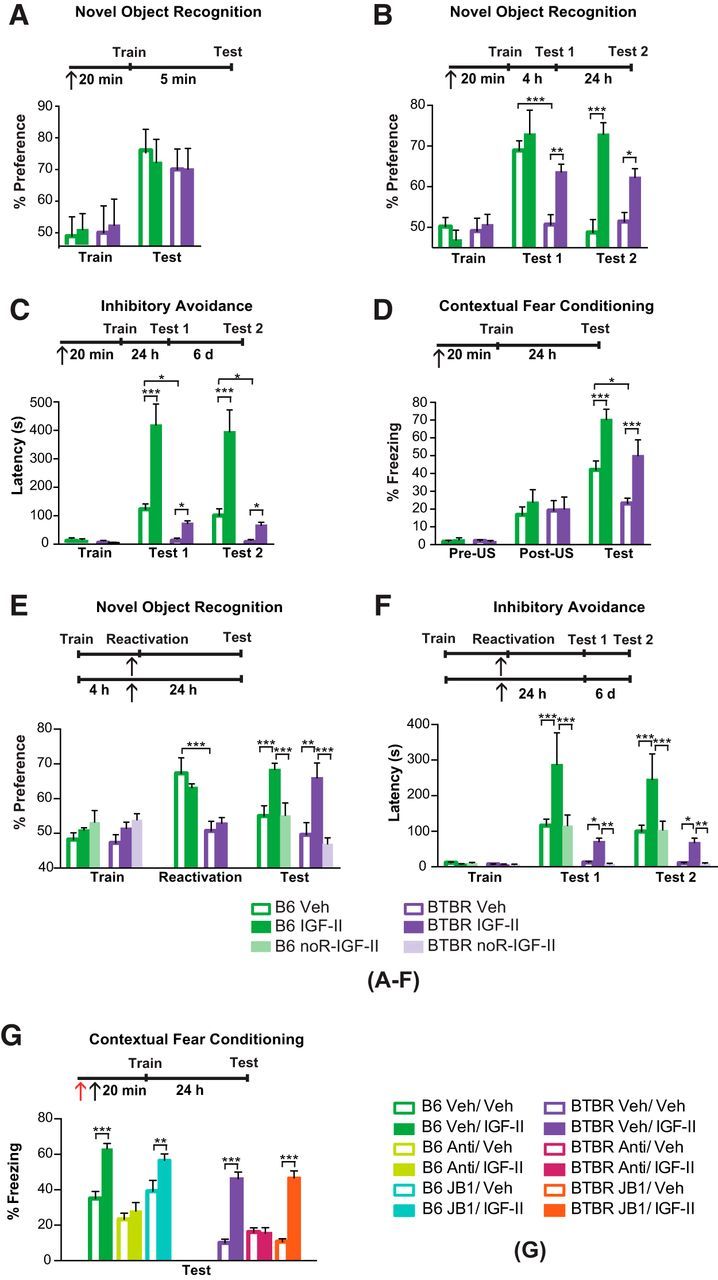
IGF-II reverses memory deficits of BTBR mice via hippocampal IGF-IIR. Experimental timelines are shown above graphs. In all experiments mice received a subcutaneous injection of either vehicle (Veh) or IGF-II (↑) 20 min before either training or memory reactivation as indicated. All data are expressed as the mean (±SEM). n = 7–8 per group. Two-way ANOVA followed by Tukey post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001. See Extended Data table (Fig. 2-2) for detailed statistical analyses. A, B, Percentage exploration preference for a novel object compared with a familiar object during novel object recognition training (Train) and testing conducted 5 min (A), 4 h (B, Test 1), or 24 h (B, Test 2) after training of B6 and BTBR mice. C, Latency of B6 and BTBR mice injected with Veh or IGF-II prior IA training (Train). Mice were tested at 24 h after training (Test 1) and again 6 d later (Test 2). D, Percentage of time spent freezing before (Pre-US) or after (Post-US) the shock delivery during contextual fear conditioning training and testing at 24 h after training (Test) of B6 and BTBR mice injected with Veh or IGF-II. E, Percentage exploration preference for a novel object compared with a familiar object during novel object recognition of B6 and BTBR mice injected with Veh or IGF-II 20 min before reactivation, which consisted in a full test session given 4 h after training (Train). Test was conducted 24 h after the reactivation. F, IA latency of B6 and BTBR mice during training (Train), Test 1 conducted 24 h after reactivation and Test 2 conducted 6 d after Test 1. Reactivation consisted in 30 s exposure to the context. G, Percentage of time spent freezing during CFC testing (Test) of B6 and BTBR mice, which received a bilateral dorsal hippocampal injection (↑) of IGF-IR blocker (JB1), IGF-IIR functionally blocking antibody (Anti), Veh, or IgG immediately before a subcutaneous injection of either Veh or IGF-II. Test was conducted 24 h after training. Exploration times are shown in the Extended Data (Fig. 2-1), and detailed statistical analyses are reported in the Extended Data tables (Figs. 2-2 and 2-3).
IGF-II injection does not affect total exploration time in novel object recognition test. Experimental timeline is shown above graph. Mice received a s.c. injection of either vehicle (Veh) or IGF-II (η) 20 min before training. All data is expressed as the mean (± s.e.m.). N=7-8 per group. Two-way analysis of variance (ANOVA) followed by Tukey post-hoc tests. Total interaction time of both objects during training (Train) and Testing conducted 4 h (Test 1) or 24 h (Test 2) after training of B6 and BTBR mice. See Extended Data table Figure 2-3 for detailed statistical analyses. Download Figure 2-1, DOCX file (28KB, docx)
Statistical analyses related to Figure 2. Download Figure 2-2, DOCX file (18.2KB, docx)
Statistical analyses related to 2-1. Download Figure 2-3, DOCX file (25.1KB, docx)
Compared with B6, BTBR mice also had impaired long-term IA memory retention at both 24 h and 7 d after training (main effect of test: F(1,46) = 120.7, p < 0.001; two-way ANOVA; Fig. 2C). Compared with vehicle, IGF-II reversed the impairment in BTBR mice and significantly enhanced memory in B6 mice at both time points. In agreement with previous studies (MacPherson et al., 2008), we found that vehicle-injected BTBR mice had also impaired long-term CFC memory retention compared with vehicle-injected B6 (main effect of group: F(5,84) = 47.41, p < 0.001; main effect of test: F(1,84) = 47.21, p < 0.001; two-way ANOVA; Fig. 2D). An IGF-II injection completely reversed the memory deficit in BTBR mice and, consistent with our previous findings (Stern et al., 2014), enhanced memory retention in B6 mice. No significant differences were detected in baseline freezing between the groups before, or immediately following, the footshock, indicating that IGF-II did not change baseline responses to the context or the footshock. Together, these results indicate that systemic administration of IGF-II significantly reverses deficits of both non-aversive and aversive types of long-term memory in an ASD mouse model.
IGF-II has been reported to also enhance rat and mouse memory reconsolidation, the process of destabilization and restabilization evoked by memory retrieval (Alberini, 2011; Stern et al., 2014). Targeting memory retrieval, reactivation and reconsolidation may offer several advantages for translational applications (Stern and Alberini, 2013). Here we tested whether IGF-II affects memory retrieval and/or reconsolidation in BTBR mice using the nOR and IA paradigms.
BTBR mice had no memory for a novel object 4 h after training, compared with B6 controls (group × test interaction: F(5,84) = 122.0, p < 0.001; two-way ANOVA; Fig. 2E). However, IGF-II injected 20 min before the 4 h testing (which served as a retrieval event, hence, reactivated the memory) significantly enhanced nOR memory at 24 h after training in both B6 and BTBR mice. The same treatment, in the absence of retrieval, had no effect on memory retention, proving that the enhancement produced by IGF-II required memory reactivation. Similarly, IGF-II injected 20 min before IA reactivation (consisting in 30 s exposure to the lit chamber), given 24 h after training, significantly enhanced IA memory retention tested 24 h later (Test 1) and 7 d later (Test 2) in both BTBR and B6 mice (group × test interaction: F(10,123) = 27.34, p < 0.001; two-way ANOVA; Fig. 2F). Similar to the effects observed in the non-aversive nOR task, the IGF-II enhancing effect was contingent upon reactivation. Furthermore, no effect was detected on retrieval per se in either paradigm. Thus, IGF-II enhances both non-aversive and aversive types of memory through retrieval-evoked mechanisms.
Finally, we chose one task, the CFC, to test whether IGF-I or IGF-II receptors were implicated in the enhancement and/or functional rescuing effects of IGF-II on long-term memory formation in BTBR and/or B6 mice. IGF-IR or IGF-IIR blockers were injected bilaterally into the dorsal hippocampus 20 min before training followed, immediately after, by a subcutaneous injection of IGF-II or vehicle. The IGF-II memory-enhancing effect in B6 mice and reversing effect in BTBR mice were completely blunted by the IGF-IIR blocking antibody but not by the IGF-I receptor blocker JB1 (group × strain interaction: F(5,84) = 2.698, p = 0.02; two-way ANOVA; Fig. 2G). In sum, the effect of IGF-II on contextual fear memory in both BTBR and B6 mice required the action of hippocampal IGF-IIR and not IGF-IR.
Together these results indicate that the deficits in aversive and non-aversive types of memory of the BTBR mice are reversed by an IGF-II injection given either before training or memory reactivation. The effect on CFC memory requires IGF-IIR but not IGF-IR in the hippocampus.
IGF-II reverses stereotyped repetitive behaviors of BTBR mice
The rapid effects on IGF-II on a variety of behavioral responses (Stern et al., 2014) led us to next investigate whether IGF-II affects stereotyped repetitive behaviors using three distinct tasks: marble burying, self-grooming, and spontaneous alternation.
Compared with vehicle-injected B6, vehicle-injected BTBR mice showed a significant deficit in marble burying, as they spent more time burying marbles (main effect of strain: F(1,28) = 4.01, p = 0.055; main effect of treatment: F(1,28) = 6.16, p = 0.019; two-way ANOVA) and buried a higher number of marbles (strain × treatment interaction: F(1,28) = 2.78, p = 0.037; two-way ANOVA; Fig. 3A,B). A subcutaneous IGF-II injection did not change the time spent burying or the number of marbles buried by B6 mice, but significantly and dramatically reduced both abnormally high behaviors in BTBR mice tested 20 min after injection.
Figure 3.
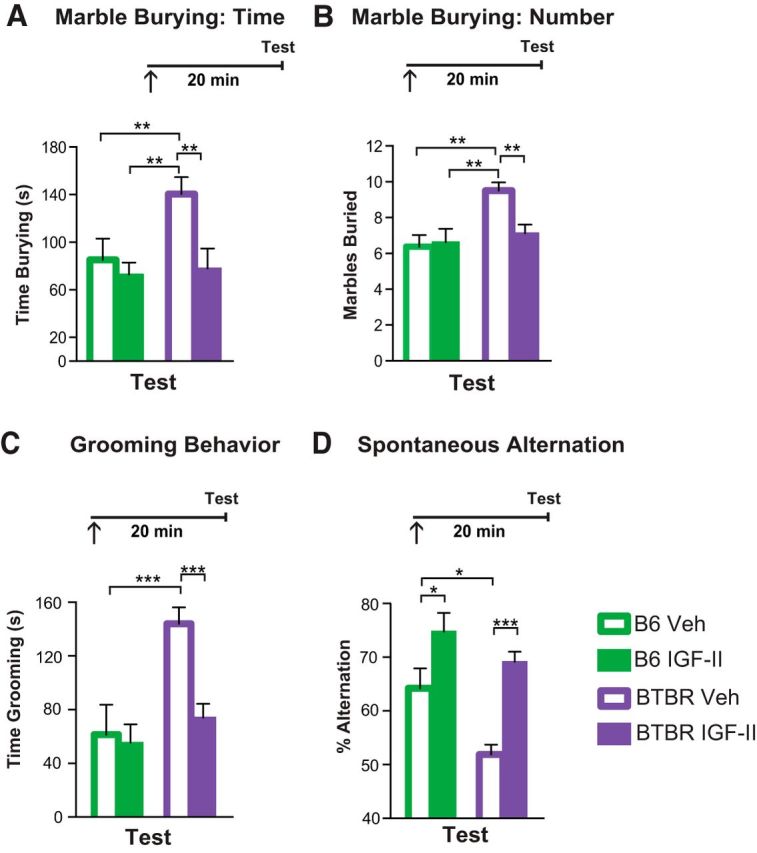
IGF-II reverses high repetitive behaviors of BTBR mice. Experimental timelines are shown above graphs. In all experiments mice received injections (↑) 20 min before testing. All data are expressed as the mean (±SEM). n = 7–8 per group. Two-way ANOVA followed by Tukey post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001. A, Time spent burying marbles by B6 and BTBR mice injected with vehicle (Veh) or IGF-II, 20 min before the test. B, Total number of marbles buried by B6 and BTBR mice injected with Veh or IGF-II before test. C, Time spent grooming (Test) 20 min following IGF-II or Veh injections in B6 and BTBR mice. D, Percentage of correct alternations in a Y-maze of B6 and BTBR mice injected with vehicle (Veh) or IGF-II before testing (Test). For detailed statistical analysis see the Extended Data table (Fig. 3-1).
Statistical analyses related to Figure 3. Download Figure 3-1, DOCX file (16.4KB, docx)
Vehicle-injected BTBR mice exhibited also significantly higher self-grooming behavior compared with vehicle-injected B6 mice. An IGF-II injection 20 min before the testing self-grooming behavior significantly reduced the self-grooming time in BTBR mice, bringing it to levels similar to those of B6 mice (strain × treatment interaction: F(1,28) = 34.74, p < 0.001; two-way ANOVA; Fig. 3C). IGF-II did not change self-grooming behavior in B6 mice.
Finally, using spontaneous alternation in a three-arm Y-maze, which is commonly used to test exploratory strategy in rodents, we found that vehicle-injected B6 mice, as expected, alternated their visits between the three arms (Stern et al., 2014), whereas vehicle-injected BTBR mice alternated more between recently visited arms before exploring the non-visited arm, in a repetitive fashion. Thus, vehicle-injected BTBR mice alternated significantly less than vehicle-injected B6 control mice. A subcutaneous IGF-II injection significantly increased correct alternations in B6 and reversed the repetitive behavior of BTBR mice (main effect of strain: F(1,18) = 9.64, p = 0.006; main effect of treatment: F(1,18) = 23.05, p = 0.001; two-way ANOVA; Fig. 3D).
In sum, a systemic IGF-II injection reversed abnormally high levels of several repetitive behaviors in BTBR mice, without changing the behaviors of control B6 mice.
IGF-II does not affect anxiety-related behaviors in BTBR mice, and decreases corticosterone levels only in B6 mice
It has been suggested that altered anxiety may contribute to the BTBR behavioral phenotypes (Pobbe et al., 2011), hence to the expression of ASD-like phenotypes (Silverman et al., 2010b). Here we tested the effects of a systemic injection of IGF-II on two paradigms commonly used to determine anxiety-like behavior: open-field and elevated plus maze (Bailey and Crawley, 2009). Compared with vehicle-injected B6 mice, vehicle-injected BTBR mice showed significantly less center entries (main effect of strain: F(1,28) = 105.7, p < 0.001; main effect of treatment: F(1,28) = 6.631, p = 0.01; two-way ANOVA; Fig. 4A), decreased time spent in the center of the open-field (main effect of strain: F(1,28) = 29.52, p < 0.001; main effect of treatment: F(1,28) = 11.76, p = 0.002; two-way ANOVA; Fig. 4B), and a significantly higher number of total entries (main effect of strain: F(1,28) = 9.42, p = 0.005; Fig. 4C). Together, these data suggest that BTBR mice have altered anxiety-like behavior compared with B6 mice. Compared with vehicle, IGF-II did not change the number of entries into the center, the time spent in the center, or the total amount of entries (Fig. 4A–C) of B6 or BTBR mice in the open-field arena, indicating that IGF-II has no effect on exploratory locomotion or general anxiety aspects.
Figure 4.
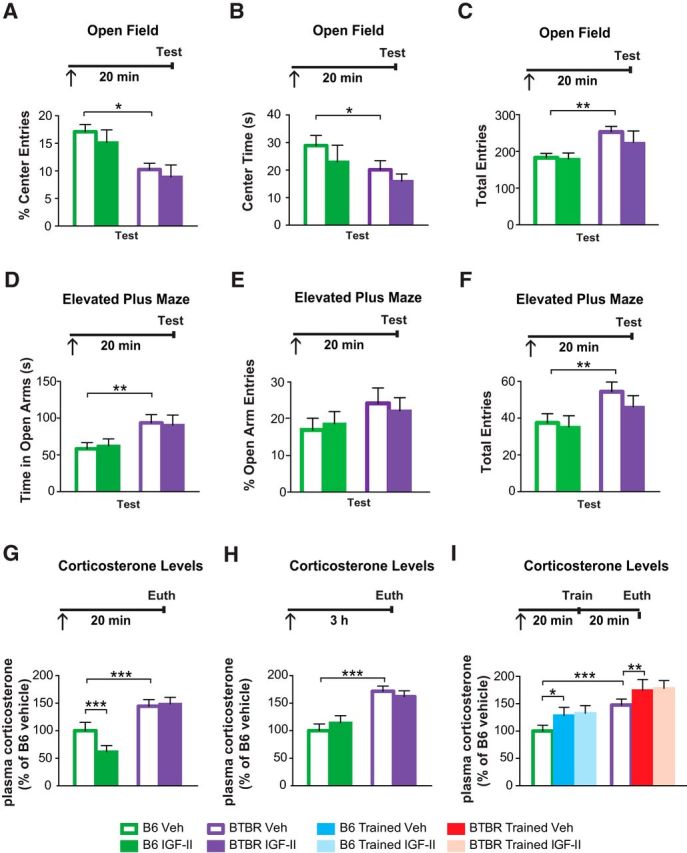
IGF-II does not change anxiety responses in BTBR mice. Experimental timelines are shown above graphs. In all experiments mice received a subcutaneous injection (↑) at the indicated time before behavioral assessment or euthanasia (Euth), as specified in each experiment. All data are expressed as the mean (±SEM). n = 7–8 per group. Two-way ANOVA followed by Tukey post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001. A, Percentage of center entries, (B) time spent in the center, and (C) total entries into the center of an open-field by B6 and BTBR mice following a subcutaneous vehicle (Veh) or IGF-II injection given 20 min before Test. D, Time spent in the open arms, (E) percentage of entries into the open arms, and (F) total entries into the open arms of an elevated plus maze by B6 and BTBR mice 20 min following Veh or IGF-II injection. G, H, Relative concentration of plasma corticosterone (normalized to the levels of B6 untrained mice) of untrained B6 and BTBR mice 20 min (G) or 3 h (H) following a subcutaneous injection of Veh or IGF-II. I, Relative concentration of plasma corticosterone (normalized to levels of B6 untrained mice) of untrained or CFC-trained B6 and BTBR mice, which received a subcutaneous injection of Veh or IGF-II and were killed (Euth) 20 min after training or at the matched time point for untrained mice. For detailed statistical analyses see the Extended Data table (Fig. 4-1).
Statistical analyses related to Figure 4. Download Figure 4-1, DOCX file (22.9KB, docx)
In the elevated plus maze, compared with vehicle-injected B6 mice, both vehicle- and IGF-II-injected BTBR mice spent more time in the open arms (strain × treatment interaction: F(1,28) = 34.74, p < 0.001; two-way ANOVA; Fig. 4D), but did not show a significant difference in the total number of entries into the open arm (Fig. 4E) indicating that BTBR mice have altered anxiety behavior compared with B6 mice. However, BTBR mice showed a significantly higher number of total entries in the elevated plus maze (main effect of strain: F(1,28) = 6.3, p = 0.019; Fig. 4F). No effect of IGF-II compared with vehicle injection was found in either B6 or BTBR mice. Hence, we concluded that IGF-II does not alter the regulation of anxiety behavior in BTBR mice measured by elevated plus maze (Fig. 4D–F).
We next tested whether the circulating level of the stress hormone corticosterone, which is generally used as an indicator of anxiety and stress responses (Kalinichev et al., 2002), is altered in BTBR mice. We also measured whether IGF-II treatment affects circulating cort by assessing the levels of cort at 20 min or 3 h following a systemic injection. We found that vehicle-injected BTBR mice, compared with B6 mice, had significantly higher circulating cort levels at both time points. IGF-II injection significantly reduced cort level 20 min (strain × treatment interaction: F(1,30) = 19.21, p = 0.001; two-way ANOVA) but not 3 h after injection in B6 mice (strain × treatment interaction: F(1,15) = 0.8987, p = 0.3582; two-way ANOVA), but had no effects on cort level of BTBR mice at either time points (Fig. 4G,H).
We then tested whether IGF-II alters the levels of circulating cort produced following an aversive experience. Toward this end, we measured circulating cort 20 min following CFC training. Training led to a significant increase in plasma cort level in both B6 and BTBR mice injected with vehicle (group × strain interaction: F(2,42) = 0.002, p = 0.9973; two-way ANOVA; Fig. 4I); however, IGF-II had no effect in changing cort levels in either strain.
We concluded that, although BTBR mice have significantly elevated circulating cort levels compared with control mice, IGF-II does not change them in either untrained conditions or following CFC training. Furthermore, compared with B6, BTBR mice have altered anxiety behavior as tested by locomotor activity in open-field and in the elevated plus maze. In either case IGF-II had no effect, suggesting that the regulation of major pathways of stress and anxiety is not targeted by IGF-II.
IGF-II reversed the deficits in the mTOR pathway and de novo translation in the hippocampus of BTBR mice
To identify which mechanisms are dysregulated in BTBR mice and are targeted by IGF-II, we first examined the endogenous expression levels of IGF-II and IGF-II receptor. We then focused on one brain region known to be significantly involved in cognitive-like functions, the dorsal hippocampus, to investigate the regulation of a molecular pathway significantly altered in ASD, which is the mTOR pathway.
The mTOR pathway, through the action of its complex 1 (mTORC1) is important for cap-dependent protein translation, autophagy, lipid synthesis in cell growth, proliferation, survival, and differentiation (Hay and Sonenberg, 2004), thus representing a crucial crossroad of cellular activation. The mTOR pathway has been found to be dysregulated in many neurological and psychiatric disorders, including Alzheimer's disease, schizophrenia, depression, epilepsy, and autism. Synaptic dysfunction caused by aberrant activation of the mTOR pathway, with an increased active translation, has been documented in several models of ASD (Kelleher and Bear, 2008; Richter and Klann, 2009; Crino, 2011; Ebert and Greenberg, 2013; Santini et al., 2013). Furthermore, inhibitors of the mTOR pathway, like rapamycin, have shown promising therapeutic results in several genetic models of ASD (Ehninger et al., 2008; Zhou et al., 2009). For example, decreasing the overactivation of the mTOR pathway rescued synaptic plasticity and learning and memory deficits in TSC2−/− mice and reduced anxiety, improved social behavior and controlled seizures in Pten mice (Ehninger and Silva, 2011; Sawicka and Zukin, 2012). Rapamycin has also been reported to rescue the impaired social behavior in BTBR mice (Burket et al., 2014), although no underlying mechanisms of this behavioral effect had been shown.
Here, we first quantified the relative IGF-II and/or IGF-IIR expression levels in BTBR mouse brain regions and compared them to those of B6 mice. Toward this end, we used total and SN protein extracts from dorsal hippocampi, mPFC, striatum, and cerebellum. SN preparations were validated (Fig. 5-1) by confirming their enrichment in PSD-95 and decrease in MAP-2 levels relative to total protein extracts.
As shown in Figure 5, levels of a mature (14 kDa) form of IGF-II did not differ between B6 and BTBR mice in either total or SN extracts of all brain regions investigated. However, BTBR brains showed a significant increase in the 21 kDa form of pro-IGF-II in the total extracts of mPFC (t(10) = 6.450, p < 0.001; unpaired t test) and striatum (t(10) = 2.451, p = 0.0497; unpaired t test) as well as in SN extracts of all brain regions investigated (hippocampus: t(14) = 4.119, p = 0.0010; mPFC: t(6) = 2.554, p = 0.0432; striatum: t(6) = 2.473, p = 0.0482; cerebellum: t(6) = 2.491, p = 0.0471; unpaired t tests). On the other hand, IGF-IIR levels were significantly decreased in the total extracts of hippocampus (t(12) = 2.146, p = 0.0499; unpaired t test) and striatum (t(6) = 3.099, p = 0.0269; unpaired t test) as well as in the SN fraction of all BTBR brain areas investigated compared with B6 controls (hippocampus: t(12) = 2.451, p = 0.0291; mPFC: t(6) = 3.327, p = 0.0159; striatum: t(6) = 2.612, p = 0.0400; cerebellum: t(10) = 2.616, p = 0.0398; unpaired t tests). IGF-IIR, also known as cation-independent-mannose-6-phosphate receptor regulates intracellular trafficking and lysosomal targeting (Ghosh et al., 2003). Collectively these data suggest that IGF-II-related mechanisms, such as endosomal trafficking and/or lysosome-targeted degradation, are affected in BTBR mice.
Figure 5.
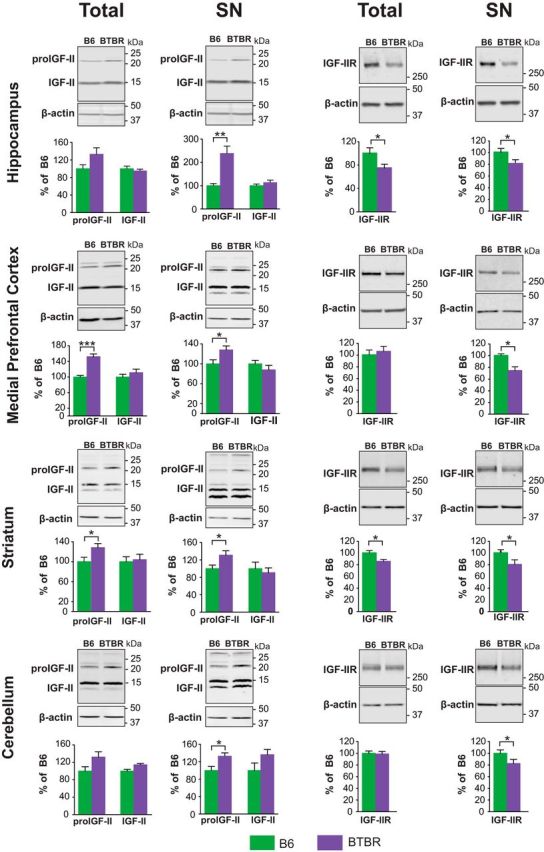
Increased proIGF-II and decreased IGF-IIR in BTBR compared with B6 mice. Western blot analyses of dorsal hippocampus, mPFC, striatum, and cerebellum, total and SN protein extracts. Each relative value was normalized against β-actin detected on the same blot. Representative blots are shown above their respective grouped data. Left: 20 kDa pro-IGF-II and 14 kDa IGF-II; Right: IGF-IIR in BTBR and B6 mice. All data are expressed as the mean (±SEM) and normalized to the level of B6 naive mice. n = 4–8 per group. Independent t test. *p < 0.05, **p < 0.01, ***p < 0.001. For SN comparisons to total extracts for MAP2 and PSD-95 see the Extended Data (Fig. 5-1), and for detailed statistical analyses see the Extended Data table (Fig. 5-2).
Biochemical characterization of the synaptoneurosomal fraction. Western blot analyses showed that synaptoneurosomal fraction (SN) is enriched for PSD95 compared to total lysate (T), whereas MAP2 in enriched in T compared to SN. Download Figure 5-1, TIF file (4.3MB, tif)
Statistical analyses related to Figure 5. Download Figure 5-2, TIF file (4.6MB, tif)
Given the alterations in IGF-II/IGF-IIR, which may target the autophagy/lysosomal systems, we investigated the expression levels of major components of the mTOR pathway in either untrained conditions or following CFC training. We then assessed the effect of systemic IGF-II injections on mechanisms of mTOR pathway. As the mTOR pathway regulates both de novo translation and macroautophagy (autophagy hereafter), a process that forms autophagosomes to capture and transport cytoplasmic components to lysosomes, we also investigated markers of both processes. As markers of translation we measured the levels of one of mTOR downstream effectors, the ribosomal protein p70S6 kinase1 (S6K), which has been previously found elevated in mouse models of autism as well as humans with monogenic syndromes with high incidence of ASD (Hoeffer and Klann, 2010; Bhattacharya et al., 2012). We also measured the phosphorylation of S6K at Thr389 (pS6K), a target of mTOR activation that reflects increased translation. As markers of autophagy, we measured the levels of the serine/threonine-protein kinase unc-51-like autophagy activating kinase 1 (ULK1), an enzyme required to initiate autophagy (Hara et al., 2008) and its phosphorylation in Ser757 (pULK1). We also determined the expression levels of AMP activated protein kinase (AMPK), a key kinase that directly binds ULK1 and activates autophagy (Lee et al., 2010). High mTOR activity prevents ULK1 activation by phosphorylating ULK1 in Ser757, hence disrupting the interaction between ULK1 and AMPK and preventing autophagy activation (Kim et al., 2011).
Relative quantifications of Western blot analyses of SN fractions revealed that vehicle-injected BTBR mice, compared with vehicle-injected B6 mice, had significantly higher levels of mTOR, pmTOR, S6K, pS6K, AMPK, pAMPK, ULK1, and pULK1 (Fig. 6A). Compared with vehicle, a subcutaneous IGF-II injection 80 min before tissue collection, significantly reduced the excess of synaptic mTOR (strain × treatment interaction: F(1,20) = 15.89, p = 0.0007; two-way ANOVA), pmTOR (strain × treatment interaction: F(1,20) = 7.109, p = 0.0148; two-way ANOVA), pS6K (strain × treatment interaction: F(1,20) = 40.19, p < 0.0001; two-way ANOVA), pAMPK (strain × treatment interaction: F(1,20) = 18.42, p = 0.0004; two-way ANOVA), ULK1 (main effect of strain: F(1,20) = 4.422, p = 0.0483; main effect of treatment: F(1,20) = 16.7, p = 0.0006; two-way ANOVA), and pULK1 (strain × treatment interaction: F(1,20) = 8.312, p = 0.0092; two-way ANOVA) in the hippocampus of BTBR mice, without changing their levels in the hippocampus of B6 mice (Fig. 6A). These data suggest that BTBR mice have a dysregulation of the mTOR pathway, and that this dysregulation is significantly reversed by systemic IGF-II treatment.
Figure 6.

IGF-II reduces the abnormally high levels of the mTOR-S6K pathway in the brain of BTBR mice. Experimental timelines are shown above graphs. In all experiments mice received a subcutaneous injection (↑) at the time specified in each panel. All data are expressed as the mean (±SEM) and normalized to the level of B6 vehicle-injected mice. n = 6–8 per group. Two-way ANOVA followed by Tukey post hoc tests. *p < 0.05, **p < 0.01, ***p < 0.001. Representative blots are shown beside their respective grouped data. A, Western blot analyses of mTOR, pmTOR, S6K, pS6K, AMPK, pAMPK, ULK1, and pULK1 of SN protein extracts obtained from dorsal hippocampi of untrained B6 and BTBR mice 80 min following a subcutaneous vehicle (Veh) or IGF-II injection. Each relative value was normalized to β-actin detected on the same blot. B, Western blot analyses of mTOR, pmTOR, S6K, pS6K, AMPK, pAMPK, ULK1, and pULK1 of SN protein extracts obtained from dorsal hippocampi of untrained and trained B6 and BTBR mice that received a subcutaneous vehicle (Veh) or IGF-II injection 20 min before training and were killed 1 h after training or at the matched time point for untrained controls. Each relative value was normalized to β-actin detected on the same blot. For Western blot analyses of LC3-BI, LC3-BII, and p62 from dorsal hippocampi of naive B6 and BTBR mice; see the Extended Data (Fig. 6-1). For detailed statistical analyses see the Extended Data tables (Figs. 6-2 and 6-3).
Similar levels of LC3-BI, LC3-BII, and p62 in B6 and BTBR mice. Experimental timeline is shown above the graph. Mice received a s.c. injection (η) 80 min prior to euthanasia (Euth). Western blot analyses of LC3-BI, LC3-BII, and p62 of synaptoneursomal protein extracts obtained from dorsal hippocampi of naive B6 and BTBR mice. All data are expressed as the mean (± s.e.m.) and normalized to B6 naive mice. N=6-8 per group. Independent t-test. See Extended Data table Figure 6-3 for detailed statistical analyses. Download Figure 6-1, TIF file (5MB, tif)
Statistical analyses related to Figure 6. Download Figure 6-2, EPS file (315.7KB, eps)
Statistical analyses related to 6-1. Download Figure 6-3, EPS file (1.1MB, eps)
We then conducted similar analyses in mice that received a vehicle or IGF-II injection 20 min before CFC training and were killed 1 h post-training.
Like the untrained groups, trained vehicle-injected BTBR mice had significantly higher levels of mTOR, pmTOR, S6K, pS6K, AMPK, pAMPK, ULK1, and pULK1, compared with trained vehicle-injected B6 (Fig. 6B). IGF-II systemic injection significantly reduced the levels of mTOR (group × strain interaction: F(2,30) = 9.285, p = 0.0007; two-way ANOVA), S6K (group × strain interaction: F(2,30) = 23.96, p < 0.0001; two-way ANOVA), pS6K (group × strain interaction: F(2,30) = 21.6, p < 0.0001; two-way ANOVA), AMPK (group × strain interaction: F(2,30) = 9.915, p = 0.0003; two-way ANOVA), and pULK1 (main effect of group: F(2,30) = 3.492, p = 0.0433; two-way ANOVA) in BTBR mice. In B6 mice, IGF-II injection significantly increased pmTOR in trained mice, but did not affect any of the other markers.
The increase in ULK1 levels suggested a decrease in autophagy. As previous studies (Tang et al., 2014) reported that ASD patient brains have significant decreased levels of microtubule-associated protein 1A/1B-light chain 3 II (LC3-II), a marker of autophagosome formation, and significant increased levels of p62, an autophagy protein substrate, we measured the relative levels of LC3-B and p62 in BTBR and B6 hippocampi. We found no differences (Fig. 6-1).
Given the altered levels of mTOR regulation markers in BTBR mice, suggesting an increased translation, we explored the rate of in vivo hippocampal active translation using the SUnSET technique (Schmidt et al., 2009). This method is based on measuring puromycin incorporation into newly synthesized proteins. Relative quantifications of Western blot analyses of total dorsal hippocampal and SN extracts obtained from mice injected bilaterally into the dorsal hippocampus with puromycin revealed that BTBR mice have a significantly increased puromycin labeling compared with B6 mice (Fig. 7A) in both total and SN extracts (SN: t(10) = 4.067, p = 0.0023; Total: t(10) = 4.305, p = 0.0012; unpaired t tests). A systemic injection of IGF-II immediately after the hippocampal puromycin injection significantly reduced the active translation in both total and SN fraction in BTBR mice, but did not change those of B6 controls (SN: main effect of strain: F(1,20) = 12.36, p = 0.0022; Total: strain × drug interaction: F(1,20) = 8.297, p = 0.0092; two-way ANOVA; Fig. 7B). Focusing then on synaptic mechanisms, we found that a bilateral hippocampal injection of anti-IGF-IIR antibody, immediately before puromycin injection, blocked the IGF-II effect on synaptic translation in the BTBR mice (F(3,27) = 3.436, p = 0.0329; ANOVA; Fig. 7C). We concluded that BTBR mice exhibit exaggerated active translation, which is reduced with IGF-II via IGF-IIR.
Figure 7.

IGF-II reduces overactive translation in the hippocampus of BTBR mice. Experimental timelines are shown above graphs. Active translation was measured by puromycin incorporation, which was injected bilaterally into the dorsal hippocampus (yellow ↑). Immediately after the puromycin injection mice received a subcutaneous injection (↑) of IGF-II or vehicle (Veh; B) in the presence or absence of anti-IGF-II receptor blocking antibody (C). Representative blots are shown beside their respective grouped data. Each relative value was normalized against β-tubulin (Tu) detected on the same blot. All data are expressed as the mean (±SEM) of B6 naive (A), B6 vehicle-injected (B), or BTBR vehicle-injected (C) mice. n = 6–8 per group. −Control represents the labeling of control samples without puromycin. A, B, Two-way ANOVA followed by Tukey post hoc tests. C, One-way ANOVA followed by Tukey post hoc tests. *p < 0.05, **p < 0.01. A, Western blot analyses of puromycin (P) incorporation in SN (top) and total protein (bottom) extracts obtained from dorsal hippocampi of untrained B6 and BTBR mice killed 60 min after P injection. B, Western blot analyses of P incorporation in SN (top) and total protein (bottom) extracts obtained from dorsal hippocampus of B6 and BTBR mice injected subcutaneously with either Veh or IGF-II and killed 60 min after the injection. C, Western blot analyses of P incorporation in SN protein extracts obtained from dorsal hippocampi of BTBR mice injected bilaterally into the dorsal hippocampus with an IGF-IIR functionally blocking antibody (Anti; red ↑) or IgG immediately before a subcutaneous injection of Veh or IGF-II and killed 60 min after the subcutaneous injection. For detailed statistical analyses see the Extended Data table (Fig. 7-1).
Statistical analyses related to Figure 7. Download Figure 7-1, EPS file (462.8KB, eps)
Together these results indicate that the hippocampi of BTBR mice, compared with B6 controls, have altered endogenous levels of IGF-IIR and IGF-II processing or release. They also have a dramatic upregulation of the AMPK and mTOR-S6K pathways, as well as abnormally high levels of active translation in the hippocampus. BTBR mice also show a synaptic inhibition of the mTOR-ULK1 pathway, thus suggesting decreased autophagy. Systemic IGF-II administration reverses all these abnormal regulations and restores them to levels comparable to those of control B6 mice in both naive conditions as well as after learning.
Discussion
Identifying novel compounds that effectively reverse ASD deficits is a critical and urgent need
Here we showed that an acute systemic injection of IGF-II reverses many major ASD-like behaviors, and specifically defects in cognitive, social, and repetitive behaviors in the BTBR mice. We also identified the mTOR pathway and resulting increased active translation as critical target mechanisms dysregulated in the brain of BTBR mice and the reversing effects of IGF-II treatment. As the mTOR pathway is altered in a number of ASD animal models, our data suggest that the beneficial effects of IGF-II may extend to several autism spectrum disorders.
Because the effect of IGF-II was tested in young adult mice, the results imply that the treatment is effective after the deficits have been established.
As reported by Stern et al. (2014), a systemic treatment with IGF-II in control B6 mice has no aversive effects detected by a battery of physical, behavioral, sensorimotor, and motor tests. In agreement, here we found that IGF-II had no effects on BTBR locomotor activity, open-field, and elevated plus maze, indicating that IGF-II does not change the baseline levels of motor, exploratory, or some anxiety behaviors.
Other cognitive enhancers previously tested in rodent models of ASD and found to affect some of the ASD phenotypes are as follows: (1) the class of AMPA receptor modulators Ampakines (Lynch, 2006), which partially rescued social deficits of BTBRs (Silverman et al., 2013); (2) modulators of mGluR5, like CDPPB, which alleviated cognitive impairments in a mouse model of tuberous sclerosis (Auerbach et al., 2011) and rescued social deficits in Shank2−/− mice (Won et al., 2012); (3) the GluR5 modulators MPEP and GRN-529, which ameliorated repetitive behavior and social deficits (Silverman et al., 2010a, 2012); and (4) IGF-I, which was found to partially rescue lethality, hypoactivity, and respiratory problems and to normalize impaired spine density, synaptic transmission, and cortical plasticity in a Rett syndrome mouse model (Tropea et al., 2009). Also IGF-I ameliorated deficits of hippocampal LTP and motor performance in mice with targeted disruption of Shank3 (Bozdagi et al., 2013). IGF-I together with Shank3 has also been found to restore synaptic deficits in neurons generated from pluripotent stem cells obtained from 22q13 deletion syndrome patients (Shcheglovitov et al., 2013). These data may imply that IGF-I and IGF-II systems cross-react or overlap in targeting mechanistic deficits of ASD. However, as for the previously established effect of IGF-II as a memory enhancer (Chen et al., 2011), here we showed that the behavioral effects of IGF-II in BTBR mice, including an effect on social and contextual memories, occur selectively via IGF-IIR and not IGF-IR. Thus, we suggest that IGF-II via IGF-IIR has a unique effect on plasticity and neural functions, which is distinct from that of the IGF-I system.
Other drugs that have been reported to ameliorate some of the behavioral deficits in models of ASD include rapamycin in the Pten conditional mouse model of autism (Zhou et al., 2009), d-cycloserine in the neuroligin-1 knock-out mice (Blundell et al., 2010), and risperidone and 5HT receptor antagonists in BTBR mice (Amodeo et al., 2014). Finally, nasal administrations of the hypothalamic hormone oxytocin have been shown to improve social interactions and communications as well as repetitive behavior in ASD patients (Andari et al., 2010).
Although the treatments thus far experimented in both animal models and humans seem to ameliorate only some of the ASD symptoms and impairments, our data suggest that IGF-II may have a global effect on at least three major phenotypic deficits and its effect is generalized to etiologically distinct ASD models.
The effect of IGF-II in both models manifests very rapidly, showing recovery of functions like from impairments in social interaction and repetitive behaviors as early as 20 min after administration. The effects are persistent, as shown by the recovery of memory, social, and repetitive impairments in BTBR mice found to continue at 24 h post-treatment. These results suggest that the mechanisms targeted by IGF-II are rapid but influence long-term molecular and cellular regulations.
We found that BTBR, similar to other ASD mouse models (Tang et al., 2014, Huber et al., 2015), have impaired regulation of the mTOR-S6K pathway, and consequently, abnormally high active protein translation. IGF-II reversed this over activation, suggesting that the treatment may restore synaptic homeostasis by decreasing protein translation and/or increasing protein degradation. As the IGF-IIR but not IGF-IR is responsible for the IGF-II effects, we speculate that the balance between autophagy, lysosomal degradation, and translation may be an important target mechanism of IGF-II treatment.
How can the multiple effects of IGF-II on memory, social interaction, and repetitive behaviors be explained? As suggested by the effect of IGF-II on the mTOR pathway and protein translation, which in fact targets several cellular functions, we speculate that the action of IGF-II encompasses many cellular processes. These include synaptic plasticity, neuromodulation, and/or receptor trafficking or composition. It has been recently reported that exogenous IGF-II restores synapse density and promotes spine maturation in IKK/NF-κB signaling-deficient neurons within 24 h of treatment and that this process requires an IGF-IIR-mediated MEK/ERK activation (Schmeisser et al., 2012). In agreement with this possibility many of the genes mutated in ASD affect critical mechanisms of activity-dependent pathways that regulate synaptic development and plasticity (Ebert and Greenberg, 2013). Furthermore, modulation of glycine transport, GABA, or acetylcholine, all of which are under consideration as cognitive enhancers (Sarter et al., 2009; Lynch et al., 2011), have been previously found to be regulated upon IGF-II or IGF-IIR treatments in vitro (Hawkes et al., 2006; Amritraj et al., 2010). The imbalance of either of these neuromodulators is considered to be a potential mechanism for ASD (Deutsch et al., 2010; Won et al., 2013).
As our treatment consisted of a single dose, it remains to be established whether repeated treatments or chronic administration of IGF-II are effective and perhaps even extend in time the positive effects of an acute IGF-II dose.
We conclude that IGF-II and IGF-IIR may represent novel potential targets for treating ASD.
Footnotes
This work is supported by Grants R37-MH065635 and R01-MH074736 awarded to C.M.A., T32-MH019524 to A.B.S., and F31-MH090636 awarded to S.A.S. We thank Gabriella Pollonini and Susan Sheng for technical help.
The authors declare no competing financial interests.
References
- Agis-Balboa RC, Arcos-Diaz D, Wittnam J, Govindarajan N, Blom K, Burkhardt S, Haladyniak U, Agbemenyah HY, Zovoilis A, Salinas-Riester G, Opitz L, Sananbenesi F, Fischer A (2011) A hippocampal insulin-growth factor 2 pathway regulates the extinction of fear memories. EMBO J 30:4071–4083. 10.1038/emboj.2011.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM. (2011) The role of reconsolidation and the dynamic process of long-term memory formation and storage. Front Behav Neurosci 5:12. 10.3389/fnbeh.2011.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association 2013. Diagnostic and statistical manual of mental disorders, Ed 5 Arlington, VA: American Psychiatric Publishing. [Google Scholar]
- Amodeo DA, Jones JH, Sweeney JA, Ragozzino ME (2012) Differences in BTBR T+ tf/J and C57BL/6J mice on probabilistic reversal learning and stereotyped behaviors. Behav Brain Res 227:64–72. 10.1016/j.bbr.2011.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodeo DA, Jones JH, Sweeney JA, Ragozzino ME (2014) Risperidone and the 5-HT2A receptor antagonist M100907 improve probabilistic reversal learning in BTBR T + tf/J mice. Autism Res 7:555–567. 10.1002/aur.1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amritraj A, Rauw G, Baker GB, Kar S (2010) Leu27 insulin-like growth factor-II, an insulin-like growth factor-II analog, attenuates depolarization-evoked GABA release from adult rat hippocampal and cortical slices. Neuroscience 170:722–730. 10.1016/j.neuroscience.2010.07.026 [DOI] [PubMed] [Google Scholar]
- Andari E, Duhamel JR, Zalla T, Herbrecht E, Leboyer M, Sirigu A (2010) Promoting social behavior with oxytocin in high-functioning autism spectrum disorders. Proc Natl Acad Sci U S A 107:4389–4394. 10.1073/pnas.0910249107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angoa-Pérez M, Kane MJ, Briggs DI, Francescutti DM, Kuhn DM (2013) Marble burying and Nestlet shredding as tests of repetitive, compulsive-like behaviors in mice. J Vis Exp 82:50978. 10.3791/50978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach BD, Osterweil EK, Bear MF (2011) Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480:63–68. 10.1038/nature10658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey KR, Crawley JN (2009) Anxiety-related behaviors in mice. In: Methods of behavior analysis in neuroscience, Ed 2 (Buccafusco JJ, ed). Boca Raton, FL: CRC/Taylor and Francis. [Google Scholar]
- Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E (2012) Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron 76:325–337. 10.1016/j.neuron.2012.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J, Blaiss CA, Etherton MR, Espinosa F, Tabuchi K, Walz C, Bolliger MF, Südhof TC, Powell CM (2010) Neuroligin-1 deletion results in impaired spatial memory and increased repetitive behavior. J Neurosci 30:2115–2129. 10.1523/JNEUROSCI.4517-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Tavassoli T, Buxbaum JD (2013) Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol Autism 4:9. 10.1186/2040-2392-4-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burket JA, Benson AD, Tang AH, Deutsch SI (2014) Rapamycin improves sociability in the BTBR T+Itpr3tf/J mouse model of autism spectrum disorders. Brain Res Bull 100:70–75. 10.1016/j.brainresbull.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabib S, Pascucci T, Ventura R, Romano V, Puglisi-Allegra S (2003) The behavioral profile of severe mental retardation in a genetic mouse model of phenylketonuria. Behav Genet 33:301–310. 10.1023/A:1023498508987 [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Fombonne E (2005) Pervasive developmental disorders in preschool children: confirmation of high prevalence. Am J Psychiatry 162:1133–1141. 10.1176/appi.ajp.162.6.1133 [DOI] [PubMed] [Google Scholar]
- Charman T, Pickles A, Simonoff E, Chandler S, Loucas T, Baird G (2011) IQ in children with autism spectrum disorders: data from the special needs and autism project (SNAP). Psychol Med 41:619–627. 10.1017/S0033291710000991 [DOI] [PubMed] [Google Scholar]
- Chen DY, Stern SA, Garcia-Osta A, Saunier-Rebori B, Pollonini G, Bambah-Mukku D, Blitzer RD, Alberini CM (2011) A critical role for IGF-II in memory consolidation and enhancement. Nature 469:491–497. 10.1038/nature09667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB. (2011) mTOR: a pathogenic signaling pathway in developmental brain malformations. Trends Mol Med 17:734–742. 10.1016/j.molmed.2011.07.008 [DOI] [PubMed] [Google Scholar]
- Davies MS, Dapretto M, Sigman M, Sepeta L, Bookheimer SY (2011) Neural bases of gaze and emotion processing in children with autism spectrum disorders. Brain Behav 1:1–11. 10.1002/brb3.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch SI, Urbano MR, Neumann SA, Burket JA, Katz E (2010) Cholinergic abnormalities in autism: is there a rationale for selective nicotinic agonist interventions? Clin Neuropharmacol 33:114–120. 10.1097/WNF.0b013e3181d6f7ad [DOI] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME (2013) Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337. 10.1038/nature11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Silva AJ (2011) Rapamycin for treating tuberous sclerosis and autism spectrum disorders. Trends Mol Med 17:78–87. 10.1016/j.molmed.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ (2008) Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med 14: 843–848. 10.1038/nm1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix-Ortiz AC, Tye KM (2014) Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci 34:586–595. 10.1523/JNEUROSCI.4257-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, Jentsch JD (2011) Pharmacological enhancement of memory and executive functioning in laboratory animals. Neuropsychopharmacology 36:227–250. 10.1038/npp.2010.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith CD, Frith U (2012) Mechanisms of social cognition. Annu Rev Psychol 63:287–313. 10.1146/annurev-psych-120710-100449 [DOI] [PubMed] [Google Scholar]
- Ghosh P, Dahms NM, Kornfeld S (2003) Mannose 6-phosphate receptors: new twists in the tale. Nat Rev Mol Cell Biol 4:202–212. 10.1038/nrm1050 [DOI] [PubMed] [Google Scholar]
- Guedea AL, Schrick C, Guzman YF, Leaderbrand K, Jovasevic V, Corcoran KA, Tronson NC, Radulovic J (2011) ERK-associated changes of AP-1 proteins during fear extinction. Mol Cell Neurosci 47:137–144. 10.1016/j.mcn.2011.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N (2008) FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181:497–510. 10.1083/jcb.200712064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes C, Jhamandas JH, Harris KH, Fu W, MacDonald RG, Kar S (2006) Single transmembrane domain insulin-like growth factor-II/mannose-6-phosphate receptor regulates central cholinergic function by activating a G-protein-sensitive, protein kinase C-dependent pathway. J Neurosci 26:585–596. 10.1523/JNEUROSCI.2730-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18:1926–1945. 10.1101/gad.1212704 [DOI] [PubMed] [Google Scholar]
- Hill EL. (2004) Executive dysfunction in autism. Trends Cogn Sci 8:26–32. 10.1016/j.tics.2003.11.003 [DOI] [PubMed] [Google Scholar]
- Hitti FL, Siegelbaum SA (2014) The hippocampal CA2 region is essential for social memory. Nature 508:88–92. 10.1038/nature13028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Klann E (2010) mTOR signaling: at the crossroads of plasticity, memory, and disease. Trends Neurosci 33:67–75. 10.1016/j.tins.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Klann E, Costa-Mattioli M, Zukin RS (2015) Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci 35:13836–13842. 10.1523/JNEUROSCI.2656-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinichev M, Easterling KW, Plotsky PM, Holtzman SG (2002) Long-lasting changes in stress-induced corticosterone response and anxiety-like behaviors as a consequence of neonatal maternal separation in Long–Evans rats. Pharmacol Biochem Behav 73:131–140. 10.1016/S0091-3057(02)00781-5 [DOI] [PubMed] [Google Scholar]
- Kelleher RJ 3rd, Bear MF (2008) The autistic neuron: troubled translation? Cell 135:401–406. 10.1016/j.cell.2008.10.017 [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Park S, Takahashi Y, Wang HG (2010) The association of AMPK with ULK1 regulates autophagy. PLoS One 5:e15394. 10.1371/journal.pone.0015394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G. (2006) Glutamate-based therapeutic approaches: ampakines. Curr Opin Pharmacol 6: 82–88. 10.1016/j.coph.2005.09.005 [DOI] [PubMed] [Google Scholar]
- Lynch G, Palmer LC, Gall CM (2011) The likelihood of cognitive enhancement. Pharmacol Biochem Behav 99:116–129. 10.1016/j.pbb.2010.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson P, McGaffigan R, Wahlsten D, Nguyen PV (2008) Impaired fear memory, altered object memory and modified hippocampal synaptic plasticity in split-brain mice. Brain Res 1210:179–188. 10.1016/j.brainres.2008.03.008 [DOI] [PubMed] [Google Scholar]
- McFarlane HG, Kusek GK, Yang M, Phoenix JL, Bolivar VJ, Crawley JN (2008) Autism-like behavioral phenotypes in BTBR T+tf/J mice. Genes Brain Behav 7:152–163. 10.1111/j.1601-183X.2007.00330.x [DOI] [PubMed] [Google Scholar]
- McHugh SB, Deacon RM, Rawlins JN, Bannerman DM (2004) Amygdala and ventral hippocampus contribute differentially to mechanisms of fear and anxiety. Behav Neurosci 118:63–78. 10.1037/0735-7044.118.1.63 [DOI] [PubMed] [Google Scholar]
- McPartland JC, Webb SJ, Keehn B, Dawson G (2011) Patterns of visual attention to faces and objects in autism spectrum disorder. J Autism Dev Disord 41:148–157. 10.1007/s10803-010-1033-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, Wilson LM, Threadgill DW, Lauder JM, Magnuson TR, Crawley JN (2007) Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res 176:4–20. 10.1016/j.bbr.2006.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissley SP, Rechler MM (1984) Insulin-like growth factors: biosynthesis, receptors, and carrier proteins. In: Hormonal proteins and peptides, pp 127–201. New York, NY: Academic. [Google Scholar]
- Pobbe RL, Defensor EB, Pearson BL, Bolivar VJ, Blanchard DC, Blanchard RJ (2011) General and social anxiety in the BTBR T+ tf/J mouse strain. Behav Brain Res 216:446–451. 10.1016/j.bbr.2010.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada A, Micevych PE (2004) Estrogen interacts with the IGF-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxdopamine lesions. J Neurosci Res 75:107–116. 10.1002/jnr.10833 [DOI] [PubMed] [Google Scholar]
- Reinhardt RR, Bondy CA (1994) Insulin-like growth factors cross the blood–brain barrier. Endocrinology 135:1753–1761. 10.1210/endo.135.5.7525251 [DOI] [PubMed] [Google Scholar]
- Richter JD, Klann E (2009) Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev 23:1–11. 10.1101/gad.1735809 [DOI] [PubMed] [Google Scholar]
- Roullet FI, Crawley JN (2011) Mouse models of autism: testing hypotheses about molecular mechanisms. Curr Top Behav Neurosci 7:187–212. 10.1007/7854_2010_113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin DN, Svedova J, Cote JL, Sandau U, Rho JM, Kawamura M Jr, Boison D, Masino SA (2013) Ketogenic diet improves core symptoms of autism in BTBR mice. PLoS One 8:e65021. 10.1371/journal.pone.0065021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E (2013) Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493:411–415. 10.1038/nature11782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe WM (2009) nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms. Biochem Pharmacol 78:658–667. 10.1016/j.bcp.2009.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M (2012) Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun 3:1292. 10.1038/ncomms2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Nakata T, Kobayashi Y, Yamada K, Ikeda T, Takeuchi A, Hiramoto T, Watanabe Y, Kazama T (2011) ERK2 contributes to the control of social behaviors in mice. J Neurosci 31:11953–11967. 10.1523/JNEUROSCI.2349-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicka K, Zukin RS (2012) Dysregulation of mTOR signaling in neuropsychiatric disorders: therapeutic implications. Neuropsychopharmacology 37:305–306. 10.1038/npp.2011.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scattoni ML, Gandhy SU, Ricceri L, Crawley JN (2008) Unusual repertoire of vocalizations in the BTBR T+tf/J mouse model of autism. PloS One 3:e3067. 10.1371/journal.pone.0003067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ, Baumann B, Johannsen S, Vindedal GF, Jensen V, Hvalby ØC, Sprengel R, Seither J, Maqbool A, Magnutzki A, Lattke M, Oswald F, Boeckers TM, Wirth T (2012) IκB kinase/nuclear factor κB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J Neurosci 32: 5688–5703. 10.1523/JNEUROSCI.0111-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET: a nonradioactive method to monitor protein synthesis. Nat Methods 6:275–277. 10.1038/nmeth.1314 [DOI] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer, Hallmayer JF, Dolmetsch RE (2013) SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503:267–271. 10.1038/nature12618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Tolu SS, Barkan CL, Crawley JN (2010a) Repetitive self-grooming behavior in the BTBR mouse model of autism is blocked by the mGluR5 antagonist MPEP. Neuropsychopharmacology 35:976–989. 10.1038/npp.2009.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Turner SM, Katz AM, Bell DB, Koenig JI, Crawley JN (2010b) Low stress reactivity and neuroendocrine factors in the BTBR T+tf/J mouse model of autism. Neuroscience 171:1197–1208. 10.1016/j.neuroscience.2010.09.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Smith DG, Rizzo SJ, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, Crawley JN (2012) Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci Transl Med 4:131ra51. 10.1126/scitranslmed.3003501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Oliver CF, Karras MN, Gastrell PT, Crawley JN (2013) AMPAKINE enhancement of social interaction in the BTBR mouse model of autism. Neuropharmacology 64:268–282. 10.1016/j.neuropharm.2012.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson DT, O'Neill SM, Narayan S, Tiwari A, Arnold E, Samaroo HD, Du F, Ring RH, Campbell B, Pletcher M, Vaidya VA, Morton D (2011) Histopathologic characterization of the BTBR mouse model of autistic-like behavior reveals selective changes in neurodevelopmental proteins and adult hippocampal neurogenesis. Mol Autism 2:7. 10.1186/2040-2392-2-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern SA, Alberini CM (2013) Mechanisms of memory enhancement. Wiley Interdiscip Rev Syst Biol Med 5:37–53. 10.1002/wsbm.1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern SA, Chen DY, Alberini CM (2014) The effect of insulin and insulin-like growth factors on hippocampus- and amygdala-dependent long-term memory formation. Learn Mem 21:556–563. 10.1101/lm.029348.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Liu Y, Moreno S, Baudry M, Bi X (2015) Imbalanced mechanistic target of rapamycin C1 and C2 activity in the cerebellum of Angelman syndrome mice impairs motor function. J Neurosci 35:4706–4718. 10.1523/JNEUROSCI.4276-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Liu Y, Tran J, O'Neal P, Baudry M, Bi X (2016) mTORC1–S6K1 inhibition or mTORC2 activation improves hippocampal synaptic plasticity and learning in Angelman syndrome mice. Cell Mol Life Sci 73:4303–4314. 10.1007/s00018-016-2269-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Gudsnuk K, Kuo S, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS, Kanter E, Castagna C, Yamamoto A, Yue Z, Arancio O, Peterson BS, Champagne F, Dwork AJ, Goldman J, Sulzer D (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83:1131–1143. 10.1016/j.neuron.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M (2009) Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A 106:2029–2034. 10.1073/pnas.0812394106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmar FR, State M, Klin A (2009) Autism and autism spectrum disorders: diagnostic issues for the coming decade. J Child Psychol Psychiatry 50:108–115. 10.1111/j.1469-7610.2008.02010.x [DOI] [PubMed] [Google Scholar]
- Wahlsten D, Metten P, Crabbe JC (2003) Survey of 21 inbred mouse strains in two laboratories reveals that BTBR T/+ tf/tf has severely reduced hippocampal commissure and absent corpus callosum. Brain Res 971:47–54. 10.1016/S0006-8993(03)02354-0 [DOI] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E (2012) Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486: 261–265. 10.1038/nature11208 [DOI] [PubMed] [Google Scholar]
- Won H, Mah W, Kim E (2013) Autism spectrum disorder causes, mechanisms, and treatments: focus on neuronal synapses. Front Mol Neurosci 6:19. 10.3389/fnmol.2013.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF (2009) Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 29:1773–1783. 10.1523/JNEUROSCI.5685-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
IGF-II injection reverses social interaction deficits and enhances social novelty memory in BTBR mice. Experimental timeline is shown above the graph. Mice received a s.c. injection (h) 20 min prior to social interaction test. All data are expressed as the mean (± s.e.m.) of total time spent interacting with mouse 1 or novel object (Test 1), novel mouse 2 or mouse 1 (Test 2), and novel mouse 3 or mouse 1 (Test 3) by B6 and BTBR mice injected with vehicle (Veh) or IGF-II. N=7-9 per group. Two-way analysis of variance (ANOVA) followed by Bonferroni post-hoc tests. *p<0.05, **p<0.01, *** p<0.001. See Extended Data table Figure 1-5 for detailed statistical analyses. Download Figure 1-1, DOCX file (16.7KB, docx)
IGF-II reverses social interaction deficits and enhances social novelty memory of BTBR mice via hippocampal IGF-IIR. Experimental timeline is shown above the graph. Mice received a s.c. injection (η) 20 min prior to social interaction test. All data are expressed as the mean (± s.e.m.). N=7-8 per group. Two-way analysis of variance (ANOVA) followed by Bonferroni post-hoc tests. *p<0.05, **p<0.01, *** p<0.001. Total time spent interacting with mouse 1 or novel object (Test 1), novel mouse 2 or mouse 1 (Test 2), and novel mouse 3 or mouse 1 (Test 3) of B6 and BTBR mice injected with vehicle (Veh) or IGF-II. See Extended Data table Figure 1-6 for detailed statistical analyses. Download Figure 1-2, DOCX file (17.2KB, docx)
IGF-II injection does not affect total exploration time in social interaction and social novelty memory. Experimental timeline is shown above the graph. Mice received a s.c. injection (η) 20 min prior to social interaction test. All data are expressed as the mean (± s.e.m.). N=7-9 per group. Two-way analysis of variance (ANOVA) followed by Tukey post-hoc tests. Total time spent interacting with novel mouse 1 and novel object (Test 1), novel mouse 2 and mouse 1 (Test 2), and novel mouse 3 and mouse 1 (Test 3) of B6 and BTBR mice injected with vehicle (Veh) or IGF-II. See Extended Data table Figure 1-7 for detailed statistical analyses. Download Figure 1-3, DOCX file (21.4KB, docx)
Statistical analyses related to Figure 1. Download Figure 1-4, DOCX file (16.4KB, docx)
Statistical analyses related to 1-1. Download Figure 1-5, DOCX file (24.9KB, docx)
Statistical analyses related to 1-2. Download Figure 1-6, DOCX file (15KB, docx)
Statistical analyses related to 1-3. Download Figure 1-7, DOCX file (17.3KB, docx)
IGF-II injection does not affect total exploration time in novel object recognition test. Experimental timeline is shown above graph. Mice received a s.c. injection of either vehicle (Veh) or IGF-II (η) 20 min before training. All data is expressed as the mean (± s.e.m.). N=7-8 per group. Two-way analysis of variance (ANOVA) followed by Tukey post-hoc tests. Total interaction time of both objects during training (Train) and Testing conducted 4 h (Test 1) or 24 h (Test 2) after training of B6 and BTBR mice. See Extended Data table Figure 2-3 for detailed statistical analyses. Download Figure 2-1, DOCX file (28KB, docx)
Statistical analyses related to Figure 2. Download Figure 2-2, DOCX file (18.2KB, docx)
Statistical analyses related to 2-1. Download Figure 2-3, DOCX file (25.1KB, docx)
Statistical analyses related to Figure 3. Download Figure 3-1, DOCX file (16.4KB, docx)
Statistical analyses related to Figure 4. Download Figure 4-1, DOCX file (22.9KB, docx)
Biochemical characterization of the synaptoneurosomal fraction. Western blot analyses showed that synaptoneurosomal fraction (SN) is enriched for PSD95 compared to total lysate (T), whereas MAP2 in enriched in T compared to SN. Download Figure 5-1, TIF file (4.3MB, tif)
Statistical analyses related to Figure 5. Download Figure 5-2, TIF file (4.6MB, tif)
Similar levels of LC3-BI, LC3-BII, and p62 in B6 and BTBR mice. Experimental timeline is shown above the graph. Mice received a s.c. injection (η) 80 min prior to euthanasia (Euth). Western blot analyses of LC3-BI, LC3-BII, and p62 of synaptoneursomal protein extracts obtained from dorsal hippocampi of naive B6 and BTBR mice. All data are expressed as the mean (± s.e.m.) and normalized to B6 naive mice. N=6-8 per group. Independent t-test. See Extended Data table Figure 6-3 for detailed statistical analyses. Download Figure 6-1, TIF file (5MB, tif)
Statistical analyses related to Figure 6. Download Figure 6-2, EPS file (315.7KB, eps)
Statistical analyses related to 6-1. Download Figure 6-3, EPS file (1.1MB, eps)
Statistical analyses related to Figure 7. Download Figure 7-1, EPS file (462.8KB, eps)