

Abstract
PARK2 is the most common gene mutated in monogenic recessive familial cases of Parkinson's disease (PD). Pathogenic mutations cause a loss of function of the encoded protein Parkin. ParkinKO mice, however, poorly represent human PD symptoms as they only exhibit mild motor phenotypes, minor dopamine metabolism abnormalities, and no signs of dopaminergic neurodegeneration. Parkin has been shown to participate in mitochondrial turnover, by targeting damaged mitochondria with low membrane potential to mitophagy. We studied the role of Parkin on mitochondrial quality control in vivo by knocking out Parkin in the PD-mito-PstI mouse (males), where the mitochondrial DNA (mtDNA) undergoes double-strand breaks only in dopaminergic neurons. The lack of Parkin promoted earlier onset of dopaminergic neurodegeneration and motor defects in the PD-mito-PstI mice, but it did not worsen the pathology. The lack of Parkin affected mitochondrial morphology in dopaminergic axons and was associated with an increase in mtDNA levels (mutant and wild type). Unexpectedly, it did not cause a parallel increase in mitochondrial mass or mitophagy. Our results suggest that Parkin affects mtDNA levels in a mitophagy-independent manner.
SIGNIFICANCE STATEMENT Parkinson's disease is characterized by progressive motor symptoms due to the selective loss of dopaminergic neurons in the substantia nigra. Loss-of-function mutations of Parkin cause some monogenic forms of Parkinson's disease, possibly through its role in mitochondrial turnover and quality control. To study whether Parkin has a role in vivo in the context of mitochondrial damage, we knocked out Parkin in a mouse model in which the mitochondrial DNA is damaged in dopaminergic neurons. We found that the loss of Parkin did not exacerbate the parkinsonian pathology already present in the mice, but it was associated with an increase in mtDNA levels (mutant and wild-type) without altering mitochondrial mass. These results shed new light on the function of Parkin in vivo.
Keywords: deletion, double-strand breaks, mitochondria, mitophagy, mtDNA, Parkinson
Introduction
Parkinson's disease (PD) is the second most common progressive neurodegenerative disease after Alzheimer's disease. The classical motor symptoms are caused by striatal dopamine (DA) depletion, consequent to the progressive loss of dopaminergic neurons in the substantia nigra (SN; Dauer and Przedborski, 2003; Braak et al., 2004).
Over the last few decades, increasing relevance has been given to the role that mitochondrial defects play in the etiology of PD. Disruption of oxidative phosphorylation (OXPHOS), particularly Complex I, is believed to contribute to neuronal loss in PD (Schapira et al., 1990a,b). Nigrostriatal dopaminergic neurons in both PD and aged individuals also show Complex IV dysfunctions (Bender et al., 2006; Kraytsberg et al., 2006) and reduction of Complex I and Complex II subunits (Grünewald et al., 2016). Mitochondrial DNA (mtDNA) is also affected in PD patients: dopaminergic neurons in both individuals with PD and aged individuals have high levels of mtDNA deletions (Bender et al., 2006; Kraytsberg et al., 2006); single dopaminergic neurons from PD patients with Complex I and II defects, show low abundance of the mtDNA transcription factors TFAM (transcription factor A, mitochondrial) and TFB2M (mitochondrial transcription factor B2; Grünewald et al., 2016). Moreover, patients harboring mutations in polymerase γ, which cause mtDNA abnormalities, exhibit dopaminergic neurodegeneration and Lewy body accumulation (Reeve et al., 2013).
PARK2 is one of the genes mutated in the rare cases of familial PD (Kitada et al., 1998), and its mutations are the most frequent cause of autosomal recessive, early-onset, and juvenile PD. Parkin, its encoded protein, is also downregulated in sporadic PD, supporting its importance in the pathophysiology of this disease (LaVoie et al., 2005; Shin et al., 2011). Parkin is a ubiquitin ligase involved in many mitochondrial pathways, as follows: it induces the clearance of dysfunctional mitochondria through mitophagy (Koh and Chung, 2010; Narendra and Youle, 2011), it influences mitochondrial biogenesis by ubiquitination of PARIS (PGC-1α [Peroxisome proliferator-activated receptor gamma coactivator 1-alpha] transcriptional repressor; Lee et al., 2017); and it can regulate mitochondrial movement by regulating Miro (Wang et al., 2011; Gaweda-Walerych and Zekanowski, 2013). The crucial role of Parkin in mitochondrial functions has been extensively demonstrated in vitro, but in vivo studies have been less conclusive, with controversial results depending on the animal model involved. Parkin mutant Drosophila displays flight muscle defects, locomotive behavioral problems, male sterility, and reduced life span (Greene et al., 2003). Parkin mutant flies are also more susceptible to oxidative stress, some dopaminergic neurons display shrinkage and abnormal morphology, and mitochondria are defective and swollen in muscle cells (Pickrell and Youle, 2015). On the other side, ParkinKO mice do not show typical signs of PD, including motor phenotypes, DA metabolism abnormalities, or nigrostriatal degeneration (Goldberg et al., 2003; Perez and Palmiter, 2005; Kitada et al., 2009). Accordingly, ParkinKO mice do not have significant mitochondrial defects (Damiano et al., 2014). One possible explanation for this phenomenon is that a compensatory or an alternative mitochondrial quality control mechanism takes over during development. The fact that nigrostriatal degeneration does occur whether Parkin is conditionally silenced after birth validates this hypothesis (Shin et al., 2011). Another possibility is that age-related factors make it difficult to reproduce the human phenotype in mice. Both genetic and environmental factors influence the development of PD, and aging is the most important risk factor in PD. mtDNA damage also accumulates with aging, so the short life span of a mouse would not allow for the accumulation of mitochondrial dysfunction, which normally occurs in patients (Bratic and Larsson, 2013). ParkinKO mice would not recapitulate PD phenotypes because they lack the pathogenic process of human aging and, in particular, the accumulation of damaged mtDNA in dopaminergic neurons. To investigate this possibility, we knocked out Parkin in the PD-mito-PstI mouse, where mtDNA damage accumulates specifically in dopaminergic neurons. Using this genetic mouse model, we combined a heritable cause of PD (Parkin loss of function) with an acquired insult (mtDNA damage) that has been observed in normal aging.
Materials and Methods
Animals.
All mice procedures were performed according to a protocol approved by the University of Miami. Mice were housed in a virus antigen-free facility at the University of Miami in a 12 h light/dark cycle at room temperature and fed ad libitum with a standard rodent diet.
The generation of PD-mito-PstI transgenic was previously described (Pickrell et al., 2011a). Briefly, a mammalian version of the bacterial PstI gene was positioned downstream of a 5′ mitochondrial targeting sequence from human COX VIII gene (cytochrome c oxidase subunit VIII). The intervening sequence 8 (IVS8) was introduced between the tetracycline response element (TRE) promoter sequence and the mito-PstI coding sequence. Mice were crossed with transgenic mice harboring one allele for a dopamine transporter (DAT)-driven tetracycline transactivator (tTA) protein (Cagniard et al., 2006). PD-mito-PstI animals with or without Parkin were always compared with control animals harboring the DAT-tTA allele with or without Parkin (DAT+ PstI−).
PD-mito-PstI mice were crossed with ParkinKO mice [B6.129S4-Park2tm1Shn/J (https://www.jax.org/strain/006582), The Jackson Laboratory] in which most of exon 3 was replaced in frame by the coding sequence of EGFP. Exon 3 skipping causes a missense mutation and premature termination at a stop codon in exon 5 following 49 additional out-of-frame amino acid residues (Goldberg et al., 2003). The nuclear background of all the mouse models described here was C57BL/6J (backcrossed at least 10 generations). Male mice were used in this study.
For the mitochondrial morphology studies, mice were crossed with mito-eYFP mice that express a TRE promoter-regulated mitochondrial-targeted enhanced yellow fluorescent protein [eYFP; Chandrasekaran et al., 2006; C57BL/6-Tg(tetO-COX8A/EYFP)1Ksn/J (https://www.jax.org/strain/006618), The Jackson Laboratory].
Pole test.
Pole test for motor coordination/nigrostriatal dysfunction of mice was previously described (Matsuura et al., 1997). Animals were hung upright on a vertical (diameter, 8 mm; length, 55 mm) pole and were given 3 min to change orientation and descend. Animals were given three trials, and the average latency to descend to the base was recorded. Failure to descend or fall from the pole was given a maximum time of 3 min.
Rotarod.
Motor coordination was evaluated with a Rotarod (IITC Life Science) designed for mice. Animals were tested with three runs on a given day with one run for practice. A resting period of 120 s between each run was given. Animals were required to position limbs to stay on a rotating rod accelerating from 6 to 20 rpm over a 180 s time period. Mice that completed the task received a final latency time of 180 s.
Activity monitoring.
Spontaneous self-initiated movement was recorded using an activity cage setup (Columbus Instruments) designed for mice. Animals were housed individually in a clean cage environment 30 min before their dark cycle and then monitored for a 12 h period undisturbed. Ambulatory movement was defined by the number of infrared beam breaks that occurred inside of the cage for each 30 min period recorded.
Open field test.
Open field (Med Associates) consists of a chamber and a system of 16 infrared transmitters that record the position of the animal in the three-dimensional space. With this system, we recorded the horizontal movement and the rearing activity. Animals were placed in the chamber 30 min before the test, and the locomotor activities were recorded for 15 min.
Stereological neuron counting.
Anesthetized mice were killed using cervical dislocation. Brains were isolated, and the regions of interest were dissected using a Mouse Brain Slicer Matrix (for midbrain, the region between −1 and −4 mm from bregma; for striatum, the region between −1 and +3 mm from bregma). Brain segments were submerged in 4% paraformaldehyde (PFA) at 4°C overnight and then cryoprotected in a 30% sucrose solution. Brains were embedded in Tissue-Tek O.C.T. compound (Sakura) and frozen by submersion into 2-methylbutane cooled in liquid nitrogen. Sections were cut on a cryostat (30 μm sections were used), and, starting from bregma −1 mm, the first 30 slides were discarded. Dopaminergic neurons were counted in every fifth section, with 10–12 total sections counted per individual animal. Slides were permeabilized with 0.4% Triton X-100, blocked for 1 h at room temperature with normal goat serum (KPL), and incubated with primary antibody (Ab) anti-tyrosine hydroxylase (TH; catalog #T1299, Sigma-Aldrich; RRID:AB_477560) at 1:500. Secondary mouse-conjugated HRP antibody (KPL) was used for 1 h at room temperature. Slides were subsequently incubated with streptavidin-peroxidase (KPL) for 30 min, visualized with 0.05% diaminobenzidine (DAB) for 7 min, and mounted with glycerol.
Stereology workstation program (StereoInvestigator, MicroBrightField) was used to quantify TH+ neurons. According to the program, we set a guard zone of 3 μm from the top and from the bottom of each section that was not considered in the counting. The A9 area was outlined using the 10× objective, and neurons were counted at 60× magnification on an Olympus BX51 Microscope. A scan grid size was determined to have at least 10 grid sites per section. The estimated numbers of dopaminergic neurons were calculated by counting TH+ cells present in the unbiased virtual inclusion counting frames. We accepted a coefficient of error (Gundersen) of <0.15.
Western blotting.
Protein extracts were prepared from isolated striatum samples homogenized with a hand-held rotor (VWR) in PBS containing a protease inhibitor cocktail (Roche). Samples were then snap frozen twice in liquid nitrogen and stored at −80°C until use. Upon use, SDS was added to the homogenate at a final concentration of 4%. Homogenates were then sonicated and centrifuged at 10,000 × g for 5 min, and the supernatant was collected for analysis. Proteins were run on 4–20% SDS-polyacrylamide gradient gel (Bio-Rad). The gel was blotted on polyvinylidene fluoride (Bio-Rad) membrane. Membranes were blocked in 1:1 Odyssey blocking solution (LI-COR) for 1 h at room temperature.
Primary antibodies used were as follows: mouse anti-TH 1:1000 dilution (catalog #T1299, Sigma-Aldrich; RRID:AB_477560), rabbit anti-DAT 1:1000 (catalog #D6944, Sigma-Aldrich; RRID:AB_1840807), mouse α-tubulin 1:2000 (catalog #T9026, Sigma-Aldrich; RRID:AB_477593), rabbit anti-MAO 1:1000 (catalog #M1821, Sigma-Aldrich; RRID:AB_1841010), mouse anti COMT 1:1000 (catalog #611970, BD Biosciences; RRID:AB_399391), rabbit anti-Porin 1:2000 (catalog #4866, Cell Signaling Technology; RRID:AB_2272627), mouse anti OXPHOS cocktail 1:1000 (catalog #ab110413, Abcam; RRID:AB_2629281), rabbit β-actin 1:5000 (catalog #A2066, Sigma-Aldrich; RRID:AB_476693), mouse anti p62 1:1000 (catalog #ab56416, Abcam; RRID:AB_945626), rabbit anti-LC3b 1:1000 (catalog #2775 and #2775S, Cell Signaling Technology; RRID:AB_915950), optic atrophy protein 1 (OPA1; catalog #612606, BD Biosciences; RRID:AB_399888), Mitofusin 2 (Mfn2; catalog #Ab56889, Abcam; RRID:AB_2142629), Tim23 (catalog #611222, BD Biosciences; RRID:AB_398754), PARIS (catalog #MABN476, Sigma-Aldrich; RRID: AB_2688024). Secondary antibodies used were infrared-conjugated antibodies anti-rabbit-700/anti-mouse-800 (Rockland) at 1:3000 to 1:5000 concentrations, respectively, and incubated at room temperature for 1 h.
Blots were visualized with an Odyssey Infrared Imaging System (LI-COR). Optical density measurements were taken by default software supplied by LI-COR on blots.
Dopamine and metabolite quantification.
Dopamine and metabolite quantification measurements were performed at the Vanderbilt University Neurochemistry Core Laboratory. Neurotransmitter and metabolite levels were normalized to total protein. Determinations were achieved using two dedicated Waters high-performance liquid chromatography systems equipped with autosamplers and a Decade II electrochemical detector.
Laser capture microdissection.
Twenty micrometer sections were immunostained for TH (catalog #T1299, Sigma-Aldrich; RRID: AB_477560) using HRP-conjugated secondary Ab and chromogenic substrate (DAB). After dehydration, they were cleared with xylenes and air dried. Sections were dried overnight in a chamber with desiccant chips (1 g each). TH+ neurons in the SN region were pooled from 10 slides per animal using an LMD Laser Microdissection Microscope (Leica). DNA was extracted using the QIAmp DNA Micro Kit (Qiagen).
Real-time PCR.
Total DNA was extracted with standard phenol/chloroform/ethanol precipitation from striatum and from midbrain. To determine the relative quantity of mtDNA in each sample, we used the comparative Ct method (Schmittgen and Livak, 2008) normalizing to a genomic DNA region. To estimate the levels of total mtDNA (full-length and recombined), we designed primers to amplify mtDNA regions distal to both PstI sites (COXI F, AGGCTTCACCCTAGATGACACA; COXI R, GTAGCGTCGTGGTATTCCTGAA; and ND1 F, CAGCCTGACCCATAGCCATA; ND1 R, ATTCTCCTTCTGTCAGGTCGAA). To determine the relative quantity of only the full-length mtDNA in each sample, we designed primers to amplify an mtDNA region inside PstI sites (ND4 F, GGAACCAAACTGAACGCCTA; ND4 R, ATGAGGGCAATTAGCAGTGG). Primers for genomic DNA were as follows: β-Actin F, GCGCAAGTACTCTGTGTGGA; and β-Actin R, CATCGTACTCCTGCTTGCTG.
For quantification of mito-PstI deletions (recombinant mtDNA), we used primers flanking the PstI breakpoint, normalized for primers amplifying an mtDNA region not affected by the PstI cleavage as follows: PstI F, TTGCCCACTTCCTTCCACAAG; PstI R, GATGTCTCCGATGCGGTTAT.
Targets were amplified with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) using the CFX96 Realtime PCR system (Bio-Rad) under specified manufacturer conditions. Relative quantity was corrected for relative PCR amplification efficiency using Bio-Rad CFX Manager Software.
Southern blotting.
Two micrograms of total DNA was digested with EagI HF (NEB), separated on 0.7% agarose gel, and transferred to a Zeta-probe GT Membrane (Bio-Rad). Probes to detect mtDNA were amplified from genomic DNA isolated from striata of a C57BL/6J wild-type mouse with Q5 Hot Start High-Fidelity DNA Polymerase (NEB). Primers used to amplify the mtDNA probe, as follows: 15660 F, CTATCCCCTTCCCCATTTGGTCTATTAAT; and 1078 R, GTCATGAAATCTTCTGGGTGTAGG.
The following primers were used to amplify the nuclear DNA probe: 18S F, CCCGGGGAGGTAGTGACGAAAAAT; and 18S R, CCGGACATCTAAGGGCATCACAG.
Amplified DNA was purified, labeled with [α-32P]dCTP using a Random Primed DNA Labeling Kit (Roche Applied Science) and cleaned with G-50 Sephadex Quick Spin Columns (GE Healthcare). Detection and densitometric quantification of mtDNA and 18S recombinant DNA signals was performed with the Cyclone Plus Phosphor Imager equipped with the Optiquant software (PerkinElmer).
Long-range PCR.
Long-range PCR to detect DNA lesions followed the protocol of Furda et al. (2012). In brief, striatal DNA was isolated using a DNeasy Blood and Tissue Kit (Qiagen). Five hundred nanograms of DNA was digested for 2 h with HaeII enzyme at 37°C to linearize mtDNA. Fifteen nanograms of DNA was amplified to generate a 10 kb fragment using a Long-Range PCR Polymerase (Takara). Values from 10 kb amplification were normalized to values from a 117 bp mtDNA product.
The primers for 10 kB fragment amplification were as follows: 3278 F, GCCAGCCTGACCCATAGCCATAATAT; and 13337 R, GAGAGATTTTATGGGTGTAATGCGG.
The primers for 117 bp fragment amplification were as follows: 13597 F, CCCAGCTACTACCATCATTCAAGT; and 13688 R, GATGGTTTGGGAGATTGGTTGATGT.
Mitochondrial counting analysis.
FIJI software was used to evaluate mitochondrial size and number in dopaminergic axons. Mice expressing mito-eYFP were perfused with 4% PFA, brains were removed, and 50 μm sections were cut with a vibratome. Z-stack images were taken at 40× with a minimum of 10 layers per figure. The Z-stack file was then opened in FIJI. An overlay of the images was created and converted to 8 bit, and the colors were inverted. Threshold was set at 230. Single axons were selected freehand, and particles were analyzed (size 1 to infinity). Neurite length was then measured with the Simple Neurite Tracer plugin. Mitochondria numbers were normalized per axon length. Mitochondria sizes were divided into four groups (1–5, 5–10, 10–20, and >20 μm2). Three to five neurites were analyzed per slice, and three to five slices were analyzed per mouse with a range of particles/mouse between 243 and 750.
Experimental design and statistical analysis.
The experimental design for different experiments is described in the previous subsections. Post hoc power for behavioral studies was 0.66. A one-way ANOVA was performed for multiple comparisons with Tukey post hoc analysis; p ≤ 0.05 determined significance. When two groups of animals were compared, a t test was performed (indicated as a “T” in the figures). The number of observations per animal used in each experimental series was included in the figure legends (*p < 0.05, **p < 0.01, ***p < 0.005). The exact p values were indicated in the manuscript. GraphPad (GraphPad Software) was used to perform statistical analysis. Detailed statistical analyses are described in Figures 1-2, 1-3, 1-4, 1-5, 3-2, 4-2, 4-3, and 5-1.
Results
Creating a mouse model with Park2 knocked out in neurons with damaged mtDNA
PD-mito-PstI mice express a mitochondrial-targeted endonuclease (mito-PstI) under the indirect control of the DAT promoter. Mito-PstI causes two sequence-specific double-strand breaks (DSBs) in the mtDNA of dopaminergic neurons (Pickrell et al., 2011a), leading to mtDNA depletion and, to a lesser extent, to the formation of recombinant mtDNA molecules (Srivastava and Moraes, 2005; Fukui and Moraes, 2009; Pickrell et al., 2011b). PD-mito-PstI mice show a clear and progressive parkinsonian phenotype with loss of TH+ neurons, striatal DA depletion, and motor coordination defects (Pickrell et al., 2011a).
ParkinKO mice were generated by deleting exon 3 of PARK2 (Goldberg et al., 2003). They are viable and fertile without obvious abnormalities, have normal brain morphology, and have no significant alterations in their general behavior. The only disruption detected in the nigrostriatal system of ParkinKO mice is an elevated level (20%) of extracellular DA in the striatum due to defective reuptake (Jiang et al., 2004; Oyama et al., 2010).
We crossed the PD-mito-PstI with ParkinKO animals to obtain ParkinKO-PD-mito-PstI. In the PD-mito-PstI and ParkinKO-PD-mito-PstI mice, mito-PstI expression was continuously induced from birth. Both mouse models were born at expected Mendelian ratios and appeared normal at birth, apart from a small but significant decrease in body weight as previously reported (Kim et al., 2011). ParkinKO-PD-mito-PstI mice had reduced body weight, starting from 2 months of age and persisting until 12 months of age (Fig. 1A). The life span of PD-mito-PstI mice was not altered by the lack of Parkin up to 24 months.
Figure 1.

Locomotive phenotypes in ParkinKO PD-mito-PstI mice. A, Weight measurements (grams) taken at 2, 4, 8, and 12 months of age (n = 15/group). Error bars ± SEM (statistics are detailed in Fig. 1-2). B, Locomotive activity of 2-month-old animals measured by the number of beam breaks that occurred in an activity cage during nocturnal hours (12 h; n = 6–8/group). Error bars ± SEM. *Cntrl/ParkKO-PD-mitoPstI, #Cntrl/PD-mitoPstI, $PD-mitoPstI/ParkKO-PD-mitoPstI, &ParkKO/ParkKO-PD-mitoPstI (statistics are detailed in Fig. 1-3). C–F, Coordination tests performed on 2-month-old animals: pole test (n = 14–23/group; C); Rotarod (n = 7–9/group; D); open field, measurement of total distance traveled on a 15 min period (n = 7–10/group; E); and open field, measurement of rearing activity (n = 7–10; F; statistics are detailed in Fig. 1-4). *p < 0.05, **p < 0.01, ***p < 0.001. Figure 1-1 describes the same behavioral analyses in older mice (statistics are detailed in Fig. 1-5).
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Coordination tests performed on older animals: A: Pole test performed on 4 months old animals (n=8-19/group), B: Activity cage performed on 4 months old animals (n=7-10/group), C: Rotarod performed on 12 months old animals (5-11/group), D: Open field, measurement of total distance travelled on a 15 minute period by 12 months old animals (n=5-12/group), E: Open field, measurement of 12 months old animals rearing activity on a 15 minutes period (n=5-12/group). * = p < 0.05, ** = p < 0.01, *** = p < 0.001. Download Figure 1-1, EPS file (1.1MB, eps)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for weight measurements taken at 2, 4, 8, and 12 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-2, EPS file (1.3MB, eps)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for activity cage measurements taken at 2 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-3, EPS file (10.6MB, eps)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for motor tests measurements taken at 2 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-4, PDF file (26.6KB, pdf)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for motor tests measurements taken at 12 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-5, PDF file (37.4KB, pdf)
The lack of Parkin anticipates but does not worsen motor behavior in PD-mito-PstI mice
We performed behavioral testing (Meredith and Kang, 2006) to determine whether the lack of Parkin combined with damaged mtDNA worsened the motor defects in the PD-mito-PstI mice. We used an ambulatory activity cage to measure spontaneous horizontal activity and exploration time for a period of 12 h. At 2 months of age, we observed that ParkinKO-PD-mito-PstI mice moved significantly less in the cage compared with PD-mito-PstI mice (Fig. 1B), but this difference between the two groups was not significant at later time points (4, 8, and >12 months of age; Fig. 1-1), suggesting that the loss of Parkin anticipates the onset but does not worsen the motor impairment in this model. When we tested 2-month-old mice for coordination (pole test, Rotarod) and with other motor tests (open field), we found that ParkinKO-PD-mito-PstI mice had some degree of motor impairment compared with control mice (Fig. 1D,F), but we did not detect any difference when we compared them to PD-mito-PstI mice (Fig. 1C–F), indicating that the lack of Parkin did not worsen the motor phenotype already present in the PD-mito-PstI mice at 2 months of age. We tested the mice for the same motor skills at later time points (4, 8, and >12 months of age), but we did not detect any significant difference between PD-mito-PstI mice with or without the expression of Parkin (Fig. 1-1).
Loss of endogenous Parkin anticipates neuropathological signs in PD-mito-PstI mice
We previously reported that the earliest time point when we observed a significant loss of TH+ cells in the SN of PD-mito-PstI mice was 8 months (Pickrell et al., 2011a; Fig. 2E,F). We tested whether the absence of Parkin worsened the dopaminergic neurodegeneration present in the PD-mito-PstI mice. We found that TH+ neurons were decreased in the SN (A9) of the ParkinKO-PD-mito-PstI mice already at 4 months of age (Fig. 2A,B; p = 0.009). We noticed that ParkinKO-PD-mito-PstI mice showed a decreased number of TH+ neurons also in the VTA (A10) at 4 months (Fig. 2A,B; p = 0.0026) and at 8 months (Fig. 2E,F; p = 0.0016). These data suggest that the lack of Parkin exacerbated the neurodegenerative process.
Figure 2.

Degeneration of dopaminergic neurons in substantia nigra and loss of striatal axonal projections. A, E, Representative images of the midbrain sections immunostained to identify TH+ neurons for PD-mito-PstI and ParkinKO-PD-mito-PstI mice at 4 (A) and 8 (E) months of age. Scale bar, 500 μm. B, F, Graph representing the stereological quantification A9 and A10 TH+ cells (n = 4–5/group) in 4-month-old (B) and 8-month-old (F) mice. C, D, Western blots showing levels of TH and DAT in 4-month-old mice striata (C) and relative quantification (D). G, H, Western blots showing levels of TH and DAT in 8-month-old mice striata (G) and relative quantification (H). Molecular weights are indicated on the left of the gels. Error bars ± SEM. n = 5/group. *p < 0.05, **p < 0.01.
The SN neurons project their axons to the striatum. The degeneration of these dopaminergic axons and the consequent depletion of dopamine in the striatum is the cause of the motor impairments in PD. We previously showed that in PD-mito-PstI mice the striatal content of TH and DAT (indicative of the presence of dopaminergic axons) was reduced already at 4 months, before the degeneration of the cell body in the substantia nigra (Pickrell et al., 2011a). We performed immunostaining and Western blot assays using anti-TH and anti-DAT antibodies in the ParkinKO-PD-mito-PstI mice, and, although TH levels were reduced in the striatum at both 4 months (Fig. 2D; p = 0.021) and 8 months (Fig. 2H; p = 0.0028), we did not detect significant changes compared with the PD-mito-PstI mice. The absence of Parkin also did not affect the levels of DAT in the striatum (Fig. 2C,D,G,H).
We then quantified the amount of dopamine and its downstream metabolites dihydroxyphenylacetic acid (DOPAC), 3-methoxytyramine (3-MT), and homovanillic acid (HVA) in freshly isolated striatum samples from animals of different ages. Although DA levels were markedly reduced in ParkinKO-PD-mito-PstI mice compared with control animals already at 4 months of age (Fig. 3A), the absence of Parkin had no effect at 4, 8, or 12 months of age (Fig. 3A and Fig. 3-1), which is consistent with our finding that there are similar amounts of innervating fibers in these mice. However, we noticed higher levels of 3-MT and HVA both in the absolute quantification and after normalization for dopamine levels (Fig. 3B; p < 0.0001) in ParkinKO-PD-mito-PstI compared with PD-mito-PstI mice. This increase in dopamine metabolites despite equal amounts of striatal DA suggests that the loss of Parkin affects the turnover rate of DA in the surviving TH+ neurons.
Figure 3.

Striatal alterations of dopamine and its metabolites. A, Quantification of DA normalized for milligrams of proteins in striata of 4-month-old animals (n = 3–5/group). B, Quantification of degradation metabolites normalized for dopamine content (3-MT/DA, HVA/DA, and DOPAC/DA) in striata of 4-month-old animals (n = 3–5/group). C, Representation of a dopaminergic synapses and cartooned scheme of enzymes involved in the degradation of dopamine. D, E, Western blots showing levels of MAO-B and COMT protein in striata of 4-month-old mice (D) and relative quantification (E). Soluble (sol) and membrane bound (mb) are two forms of COMT. Molecular weights are indicated on the left of the gels. *p < 0.05, **p < 0.01, ***p < 0.001. Figure 3-1 describes the same dopaminergic metabolites analyses in older mice. Figure 3-2 describes detailed statistical analyses.
Striatal alterations of dopamine and its metabolites. A: Quantification of dopamine (DA) normalized for mg of proteins in 8 months old animals striata (n=4-7/group). B: Quantification of degradation metabolites normalized for dopamine content (3-MT/DA, HVA/DA, and DOPAC/DA) in 8 months old animals’ striata (n=4-7/group). C: Quantification of dopamine (DA) normalized for mg of proteins in 12 months old animals striata (n=3-6/group). D: Quantification of degradation metabolites normalized for dopamine content (3-MT/DA, HVA/DA, and DOPAC/DA) in 8 months old animals’ striata (n=3-6/group). E: Western blots showing levels of MAO-B and COMT protein in 8 months old mice striata and (F), relative quantification. * = p < 0.05, ** = p < 0.01, *** = p < 0.001. Download Figure 3-1, PDF file (25.4KB, pdf)
Striatal alterations of dopamine and its metabolites. Statistical analyses for dopamine and dopamine metabolites at 4, 8 and 12 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 3-2, PDF file (25.6KB, pdf)
Monoamine oxidase (MAO) and catechol O-methyltransferase (COMT) are the two main enzymes involved in the turnover of DA (Fig. 3C). After the release in the synaptic cleft, the extracellular DA has to be removed, and it is either recycled after reuptake by dopaminergic neurons or degraded after uptake by glial cells (Vaughan and Foster, 2013). In the dopaminergic neurons, DA is degraded to DOPAC by MAO. In the glial cells, DA is degraded by MAO and also by COMT to HVA (Meiser et al., 2013). To analyze whether the increase of DA metabolites in the striata of ParkinKO mice was due to increased activity of these enzymes, we performed a Western blot probing for anti-MAO-B and anti-COMT. We did find a small but significant increase in the concentration of COMT in the striatum of 4 months old ParkinKO and ParkinKO-PD-mito-PstI mice (Fig. 3D,E; p = 0.019) that could explain the accelerated metabolism of DA in the absence of Parkin. COMT is prevalently expressed by glial cells (Myöhänen et al., 2010). Since in our mice Parkin is knocked out ubiquitously, we believe that this effect is due to the lack of Parkin in striatal glial cells. At later time points (8 and 12 months), we also found an increase in MAO-B (Fig. 2-1E,F).
MtDNA levels are increased in mice lacking endogenous Parkin
Parkin was shown to participate in the clearance of mitochondria with damaged mtDNA in cultured cells (Suen et al., 2010). Mito-PstI promotes DSB in two mtDNA sites, causing mainly mtDNA depletion and, to a smaller extent, the generation of recombined mtDNA. To analyze how the absence of Parkin affects mitochondria with damaged mtDNA, we measured the levels of different types of mtDNA alterations (depletion, deletions, and oxidative lesions) in ParkinKO-PD-mito-PstI mice and relative controls.
To measure relative mtDNA levels in the cell bodies of dopaminergic neurons, we isolated TH+ cells by laser capture microdissection (LCM) after staining consecutive midbrain slices. We extracted the DNA from the collected cells and measured the mtDNA copy number using quantitative real-time qPCR. To determine the relative levels of total mtDNA in each sample, we used a primer set that amplifies an mtDNA region between one of the PstI sites and the D-loop (Pickrell et al., 2011a; Fig. 4A, COXI region). As expected, mtDNA levels were reduced in the PD-mito-PstI mice compared with controls, but we detected higher levels of total mtDNA in ParkinKO-PD-mito-PstI mice compared with PD-mito-PstI mice (Fig. 4B). Likewise, ParkinKO TH+ neurons also had more mtDNA than in wild-type mice (Fig. 4B).
Figure 4.
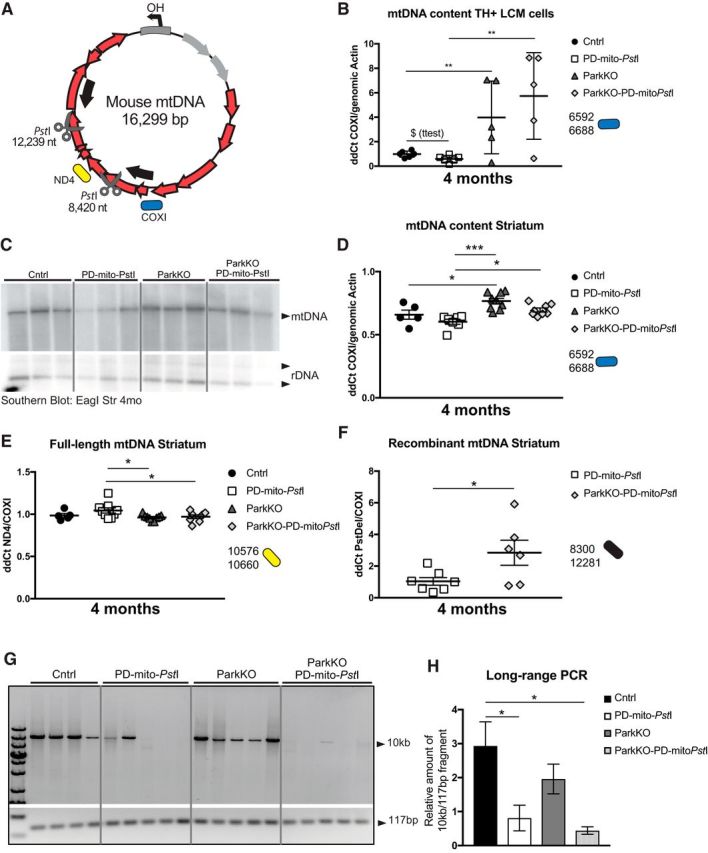
mtDNA deletions and mutations increase when endogenous Parkin is lost in striata of PD-mito-PstI mice. A, Schematic representation of mouse mtDNA. Scissors indicate the PstI cleavage sites. Red arrows represent encoded proteins. Gray arrows represent mt-tRNAs. Color-coded representation of the position of the amplicons used for the qPCRs is maintained in the graphs. B, mtDNA levels measured by qPCR of LCM-collected SN TH+ neurons (n = 5/group; T = p value after t test). C, D, Southern blot probing striatal DNA with mtDNA and nuclear DNA probes (C) and mtDNA content measured by qPCR (D) in 4-month-old animals. E, Quantification by qPCR of full-length mtDNA content in striata of 4-month-old animals. F, Recombination events detected by qPCR using primers flanking the PstI sites in striatal samples. G, H, Long-range PCR amplification of a 10 kb and a 117 bp fragment from striatal DNA (G) and relative quantification of a 10 kb fragment normalized to the 117 bp fragment (H). Boxes in graphs B, D, and F represent one individual animal. *p < 0.05, **p < 0.01, ***p < 0.001. Figure 4-1 describes mtDNA levels in older mice and different brain regions. Figure 4-2 and Figure 4-3 describe detailed statistical analyses.
MtDNA deletions and mutations in older mice in midbrain and striata. A: Schematic representation of mouse mtDNA. Color-coded representation of the position of the amplicons used for the qPCRs is maintained in the graphs (B-G). B-C:qPCR in dissected midbrain of 4 months old animals measuring (B) total mtDNA and (C) full-length mtDNA. D: qPCR in dissected midbrain of 12 and 19 months old animals measuring total mtDNA and full-length mtDNA.
E: Recombination events detected by qPCR using primers flanking the PstI sites in dissected midbrain of 12 and 19 months old animals. F: qPCR in strata of 12 and 19 months old animals measuring total mtDNA and full-length mtDNA. G: Recombination events detected by qPCR using primers flanking the PstI sites in striata of 12 and 19 months old animals. Color coded and numbers in the graphs represent the position of the primers on the mtDNA. H: Southern blots probing (top) DNA from dissected midbrains with mtDNA and nDNA probes in 4 months old animals and (bottom) striatal DNA with mtDNA and nDNA probes in 12 months old animals. I: Representative western blots probing for mitochondrial markers in 12 months old animals’ striata. J: Representative western blots probing for autophagy markers (p62 and LC3b) and for mitochondrial fusion markers (OPA1 and Mfn2) in 12 months old animals’ striata. * = p < 0.05, ** = p < 0.01, *** =p<0.001. Download Figure 4-1, PDF file (21.5KB, pdf)
MtDNA deletions and mutations increase when endogenous Parkin is lost in PD-mito-PstI mice striata. Statistical analyses for qPCR performed on DNA after LCM and on DNA from striatum at 4 months. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 4-2, PDF file (20.8KB, pdf)
MtDNA deletions and mutations increase when endogenous Parkin is lost in PD-mito-PstI mice striata. Statistical analyses for qPCR performed on DNA from striatum and SN at 12 and 19 months. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 4-3, PDF file (26.4KB, pdf)
The axons of dopaminergic neurons project to the striatum. By using qPCR and Southern blot analysis, we assessed the mtDNA levels in striatal homogenates. It is important to note that, while we used LCM for dissecting dopaminergic cell bodies, we used homogenized striatal tissue for the analysis of the axons. The manually isolated striatal region is enriched in dopaminergic terminals but also contains different cell populations and different neuronal terminals. Also in striata homogenates, higher levels of total mtDNA were detected in ParkinKO-PD-mito-PstI mice compared with PD-mito-PstI mice (Fig. 4C,D). After being cleaved by mito-PstI, mtDNA undergoes depletion and, albeit at low levels, recombination between the two PstI sites (Pickrell et al., 2011a). Because Parkin is supposed to target damaged mitochondria to mitophagy, we expected mice lacking Parkin to accumulate defective mitochondria with deleted mtDNA molecules. We designed primers flanking the PstI sites to specifically amplify the breakpoint region (Fig. 4A, arrows). A qPCR of DNA from striata of the different groups of mice showed higher levels of recombinant mtDNA in the ParkinKO-PD-mito-PstI mice compared with PD-mito-PstI mice (Fig. 4F) and lower levels of full-length mtDNA (Fig. 4E).
To analyze whether the mtDNA in ParkinKO-PD-mito-PstI striata mice accumulated oxidative damage, we performed a previously characterized quantitative long-range PCR approach (see Materials and Methods). This assay is based on the observation that different types of damage (e.g., single-strand breaks, oxidative modifications, abasic sites, and bulky adducts) slow down or halt TaqDNA polymerase progression, resulting in reduced amplification yield (Santos et al., 2006; Maslov et al., 2013). We used two pairs of primers in separate reactions, one amplifying a 10 kb fragment, which is susceptible to impaired amplification, and a second, which amplifies a 117 bp internal control fragment, not expected to be significantly affected by these modifications. (Furda et al., 2012). When we analyzed mtDNA from striata of 4-month-old mice, we found that mtDNA from PD-mito-PstI and ParkinKO-PD-mito-PstI mice showed reduced amplifications when compared with controls, indicating increased mtDNA damage associated with the presence of mito-PstI. There were no significant changes between PD-mito-PstI and ParkinKO-PD-mito-PstI mice, suggesting either that the accumulation of base modifications is not exacerbated by the absence of Parkin or that the method is not sensitive enough to differentiate already markedly impaired amplifications (Fig. 4G,H).
We performed the same analysis in the mtDNA at later time points (12 and 19 months) by analyzing both striatum and dissected midbrain, but we did not detect any changes in the mtDNA, probably because of the depletion of dopaminergic axons and neurons at these ages (Fig. 4-1).
Mito-PstI and lack of Parkin did not lead to A10 cortical axon degeneration
As part of the meso-cortico-limbic pathway, part of VTA dopaminergic neurons project their axons to the cortex, specifically to the prefrontal and cingulate cortex. In PD, the VTA neurons (A10) are relatively spared. In our PD-mito-PstI mice, we did not detect a depletion of A10 neurons, while in ParkinKO-PD-mito-PstI mice there is already a 50% decrease of A10 cells early in the course of the disease (Fig. 2B). To investigate whether the degenerating cells were the ones projecting to the cortex, we analyzed cortices in 12-month-old animals. The content of cortical TH was much lower compared with striatal TH, but the expression of mito-PstI did not cause any further TH depletion in this region (Fig. 5A). We then analyzed the mtDNA depletion and recombination by Southern blot and found an accumulation of recombinant mtDNA molecules in cortices of 12-month-old animals (Fig. 5B,C,G). The mtDNA accumulation was not accompanied by an increase in Porin, indicating that also in this region, mitochondrial mass was not affected.
Figure 5.

Alterations of dopaminergic terminals and mtDNA in cortical region of ParkKO-PD-mitoPstI mice. A, Quantification of cortical TH in 12-month-old animals and representative Western blot. CTX, cortex; STR, striatum. B, Southern blot probing cortical DNA with mtDNA and nuclear DNA (nDNA) probes and relative Western blot probing for Porin in 12-month-old animals. Molecular weights are indicated on the left of the gels. C, Southern blot probing cortical DNA with mtDNA and nDNA probes in 19-month-old animals. D, Schematic representation of mouse mtDNA and qPCR analysis of mtDNA in cortices of 12- and 19-month-old mice. Color-coded representation of the position of the amplicons used for the qPCRs is maintained in the graphs. E, Total mtDNA levels measured by qPCR of cortex from 12- and 19-month-old animals. F, Quantification by qPCR of full-length mtDNA content in cortices of 12- and 19-month-old animals. G, Recombination events detected by qPCR using primers flanking the PstI sites in cortical samples from 12- and 19-month-old animals. *p < 0.05, **p < 0.01. Figure 5-1 describes detailed statistical analyses.
Alterations of dopaminergic terminals and mtDNA in cortical region of ParkKO-PD-mitoPstI mice. Statistical analyses for qPCR performed on DNA from cortex at 12 and 19 months. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 5-1, PDF file (28.1KB, pdf)
Full-length mtDNA was slightly decreased in cortex of mito-PstI-expressing mice. The absence of Parkin resulted in even higher levels of deleted mtDNA (Fig. 5E–G). It is interesting to note that mito-PstI-generated deletions are usually present at very low levels, detectable only by qPCR. However, in this case the deleted mtDNA was clearly detected by Southern blot.
Lack of Parkin modified mitochondrial morphology but not total mitochondrial mass
The fact that the recombinant mtDNA was increased in the mice lacking Parkin is compatible with the suggested role of Parkin in the clearance of mitochondria with damaged mtDNA by mitophagy. To study the state of mitochondria and the activation of mitophagy in our mice, we analyzed mitochondrial and mitophagy markers.
Porin, Tim23, and several OXPHOS proteins (ATP5a, UQRC2, mtCO1, SDHB, and NDUFB8) were not changed in 4-month-old mice lacking Parkin (Fig. 6A), suggesting that mitochondrial mass was not altered by the lack of Parkin. Likewise, we did not observe a change in the levels of PARIS, a repressor of PGC-1α (Fig. 6A). When we measured p62 and LC3b (autophagy markers), we also did not detect significant changes (Fig. 6B). We performed the same quantification in older mice (12 months old) obtaining the same results (Fig. 4-1I,J).
Figure 6.

Mitochondrial morphology in striatal and nigral dopaminergic axons. A, Representative Western blots probing for mitochondrial markers Porin, Tim23, OXPHOS cocktail proteins, and PARIS in striata of 4-month-old animals. Actin was used as a housekeeping gene for normalization (n = 3–4/group). B, Representative Western blots probing for autophagy markers p62 and LC3b in striata of 4-month-old animals. Actin and tubulin were used as housekeeping genes for normalization (n = 3–4/group). Molecular weights are indicated on the left of the gels. C, Quantification of the mitochondrial total number in dopaminergic cell axons, normalized for axon length. D, Representative black and white images of axonal mitochondria in mice expressing mito-eYFP. E, Quantification of axonal mitochondria divided by size. *p < 0.05, **p < 0.01, ***p < 0.001.
Because mtDNA was increased but total mitochondrial mass was not, we analyzed mitochondrial morphology of mice lacking Parkin and controls. To do so, we crossed our mice with TRE-mito-eYFP mice that express eYFP targeted to the mitochondria (Chandrasekaran et al., 2006). Using Fiji software (see Materials and Methods), we were able to quantify axonal mitochondria in dopaminergic neurons and separate them into four groups depending on their size. We analyzed midbrain slices from 4-month-old animals, and we observed a small but significant decrease in mitochondrial number only in the dopaminergic axons of ParkinKO-PD-mito-PstImito-eYFP mice (Fig. 6C). When we analyzed mitochondrial size, we observed that the lack of Parkin led to the accumulation of larger mitochondria (Fig. 6D,E). We also examined the steady-state levels of the following two proteins associated with mitochondrial dynamics: Mfn2 and OPA1, which is involved in the fusion of the outer and the inner mitochondrial membrane, respectively. Although we did not detect changes in Mfn2 levels, there was a slight decrease in OPA1 (long form) levels in striatal homogenates from 4- and 12-month-old animals (Fig. 6B and Fig. 4-1J).
Discussion
Park2 is one of the confirmed genes mutated in monogenic forms of Parkinson's disease, but the loss of Parkin does not severely affect the nigrostriatal pathway in mice (Goldberg et al., 2003; Von Coelln et al., 2004; Perez and Palmiter, 2005). A few possible explanations have been proposed for this observation. Mitochondrial defects accumulate with aging and play a role in the pathology of PD. It is possible that mitochondrial dysfunctions in mice do not reach thresholds high enough for the loss of Parkin to have any detrimental effect (LaFrance et al., 2005), but this hypothesis is conflicting with the clearly severe effects that Parkin loss has in even more short-lived organisms such as zebrafish and Drosophila (Hattori et al., 1998; Greene et al., 2003; Flinn et al., 2009). Another hypothesis is that other pathways such as unselective macroautophagy or the recently described Rheb mitophagic pathway, which controls basal levels of mitochondria in normal conditions, compensate for the loss of Parkin (Hara et al., 2006; Melser et al., 2013). Finally, knocking out Parkin in the mouse germline could also induce compensatory mechanisms, and the fact that deleting the Parkin gene after development does cause dopaminergic neurodegeneration corroborates this hypothesis (Shin et al., 2011; Stevens et al., 2015).
Other groups have crossed mouse models of mitochondrial dysfunction with the ParkinKO mouse and, depending on the type of mitochondrial insult, different results have been reported. In TFAM KO-ParkinKO mice, where TFAM is specifically knocked out in dopaminergic neurons, there was no worsening of dopaminergic neurodegeneration (Sterky et al., 2011). In this case, it has been proposed that the rapid depletion of mtDNA and neurodegeneration may not allow enough time for it to be partially compensated by endogenous Parkin. In addition, there are reports that the loss of Parkin did not worsen motor phenotypes in two mouse models affecting mitochondrial fission and fusion [Purkinje cell-specific dynamin-related protein 1 (Drp-1) KO mice; Lee et al., 2012] and dopaminergic neuron-specific Mfn2 KO mice (Kageyama et al., 2012; Lee et al., 2012). However, it has been shown that the loss of Drp-1 or Mfn2 in a cell culture does not cause sufficient mitochondrial dysfunction to recruit Parkin (Narendra et al., 2008; Tanaka et al., 2010). On the other hand, two recent publications showed that the lack of Parkin exacerbates neurodegeneration in two mouse models of PD obtained by increasing mtDNA mutations. In the Mutator mouse, the lack of Parkin combined with the accumulation of mtDNA mutations was associated with neurodegeneration, striatal DA depletion and parkinsonian-like phenotypes, which were absent in the Mutator mouse alone (Pickrell et al., 2015). In the Twinkle-TG mouse, in which mtDNA deletions are increased specifically in substantia nigra, the absence of Parkin led to higher mtDNA deletion levels, lower mitochondrial function and membrane potential, and severe neurobehavioral deficits at 19 months (Song et al., 2017).
Our results showed that the lack of Parkin mildly accelerated, but overall did not worsen, the motor defects and the dopaminergic neurodegeneration present in the PD-mito-PstI mice. A curious observation was the fact that dopamine metabolism was affected by the lack of Parkin: we detected an increase in COMT and later an increase in MAO, enzymes involved in the metabolism of dopamine. Parkin overexpression was shown to reduce MAO expression in cultured cells (Jiang et al., 2006). Because COMT is present mostly in glial cells, we suggest that the COMT increase was due to the lack of Parkin in the glial cells rather than in the dopaminergic striatal axons.
Another novel observation obtained from our analysis is that the lack of Parkin resulted in an increase of mtDNA levels in midbrain (Fig. 4-1B, homogenates and Fig. 4B, LCM microdissected neurons) and striata (Fig. 4D) of 4-month-old mice. These changes were not observed at later time points, likely because of neuronal (and axonal) loss.
Recent work showed that the loss-of-function mutations in Parkin and PINK1 in Drosophila lead to the accumulation of dysfunctional mitochondria due to defective mitophagy halting mitochondrial protein turnover (Burman et al., 2012; Vincow et al., 2013). Despite our results being compatible with the previously suggested mechanistic mitophagy model, it is important to analyze them without a preconceived notion. In fact, although the levels of mtDNA were increased, we could not detect a corresponding increase in mitochondrial proteins in striatal samples, and markers of mitophagy (LC3b and p62) were also unchanged.
Interestingly, when we analyzed axonal mitochondrial number and morphology in the midbrain of ParkinKO-PD-mito-PstI mice, we observed a clear shift from small-sized to enlarged organelles. The number of total mitochondria was decreased. This would suggest that the fusion/fission process in defective mitochondria is affected by the absence of Parkin. This phenomenon could be explained by the fact that, in pathological conditions, Parkin inhibits the fusion of defective mitochondrial by the ubiquitination of Mfn2 (Poole et al., 2010; Ziviani et al., 2010). When we analyzed Mfn2 levels in our mice, though, we detected no changes. We also analyzed the levels of OPA1, which is involved in the mitochondrial inner membrane fusion, and again we detected no change (in midbrain; data not shown) or only a mild decrease (in striatal samples; Fig. 6B,E,F and Fig. 4-1J).
Another possible explanation for the accumulation of mtDNA without an increase in mitochondria biogenesis, is that a mtDNA pool is increased in the striatum of ParkinKO mice. Parkin has not been shown to interact with the mtDNA replicative processes, but the full repertoire of Parkin ubiquitination substrates is still being defined. Rothfuss et al. (2009) showed that Parkin can associate with mtDNA and protect it against oxidative stress. Sarraf et al. (2013) showed how the Parkin-dependent ubiquitylome includes not only proteins of the mitochondrial outer membrane but also proteins expressed in the mitochondrial matrix, such as the mtDNA single-strand binding protein 1 (SSBP1), implying the possibility that Parkin could have a role in the mtDNA maintenance. In fact, the increased levels of deleted mtDNA could be due to increased replication, which could favor smaller molecules (Diaz et al., 2002).
Mitochondrial morphology, which was impaired in our mice, is intrinsically associated with mtDNA replication: lack of mitofusins leads to a severe mtDNA depletion (Chen et al., 2010), and OPA1 appears to regulate mtDNA copy number (Elachouri et al., 2011). Moreover, Parkin seems to regulate OPA1 through NEMO (nuclear factor-κB essential modulator; Müller-Rischart et al., 2013), and a recent study reported the occurrence of syndromic parkinsonism with an OPA1-dominant mutation associated with accumulation of mtDNA deletions (Carelli et al., 2015). In our model, however, the increase in mtDNA was not accompanied by an increase in OPA1; therefore, we could not confirm this effect in our system.
Finally, the observed increase in mtDNA levels in our mice may not be a direct consequence of the lack of Parkin but a compensatory protective mechanism of dopaminergic neurons to partially rescue the nigrostriatal integrity (Perier et al., 2013). Accordingly, it was recently described that mtDNA content is increased in dopaminergic neurons of aged healthy individuals, suggesting the presence of a mechanism to maintain the pool of wild-type mtDNA population despite accumulating deletions. This process appears to fail in PD, resulting in the loss of wild-type mtDNA, suggesting that the dysregulation of mtDNA homeostasis is a key process in the pathogenesis of neuronal loss in Parkinson disease (Dölle et al., 2016).
In conclusion, we showed that the absence of Parkin, in a mouse model where mtDNA undergoes double-strand breaks and recombination in dopaminergic cells, does not lead to a worsening of the PD phenotype. However, lack of Parkin was associated with an increase in both wild-type and recombinant mtDNA. The simplest explanation for these observations would be that mitochondrial dynamics was altered, impairing mitophagy and mtDNA degradation. However, because markers of mitophagy were not changed, our observations suggest that Parkin has a role (direct or indirect) in mtDNA turnover or maintenance.
Footnotes
This work was supported in part by National Institutes of Health (NIH) Grants 1R01-AG-036871, 1R01-NS-079965, and 5R01-EY-010804 (to C.T.M.), Parkinson's Disease Foundation Fellowship PDF-FBS-1316 (to M.P.), and American Heart Association Fellowship 16PRE30480009 and Lois Pope LIFE Fellows Program (to N.N.). Support was also received by National Eye Institute Center Grant P30-EY-014801 and from the NIH.
The authors declare no competing financial interests.
References
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. 10.1038/ng1769 [DOI] [PubMed] [Google Scholar]
- Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res 318:121–134. 10.1007/s00441-004-0956-9 [DOI] [PubMed] [Google Scholar]
- Bratic A, Larsson NG (2013) The role of mitochondria in aging. J Clin Invest 123:951–957. 10.1172/JCI64125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burman JL, Yu S, Poole AC, Decal RB, Pallanck L (2012) Analysis of neural subtypes reveals selective mitochondrial dysfunction in dopaminergic neurons from parkin mutants. Proc Natl Acad Sci U S A 109:10438–10443. 10.1073/pnas.1120688109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagniard B, Beeler JA, Britt JP, McGehee DS, Marinelli M, Zhuang X (2006) Dopamine scales performance in the absence of new learning. Neuron 51:541–547. 10.1016/j.neuron.2006.07.026 [DOI] [PubMed] [Google Scholar]
- Carelli V, Musumeci O, Caporali L, Zanna C, La Morgia C, Del Dotto V, Porcelli AM, Rugolo M, Valentino ML, Iommarini L, Maresca A, Barboni P, Carbonelli M, Trombetta C, Valente EM, Patergnani S, Giorgi C, Pinton P, Rizzo G, Tonon C, et al. (2015) Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol 78:21–38. 10.1002/ana.24410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran K, Hazelton JL, Wang Y, Fiskum G, Kristian T (2006) Neuron-specific conditional expression of a mitochondrially targeted fluorescent protein in mice. J Neurosci 26:13123–13127. 10.1523/JNEUROSCI.4191-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC (2010) Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141:280–289. 10.1016/j.cell.2010.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano M, Gautier CA, Bulteau AL, Ferrando-Miguel R, Gouarne C, Paoli MG, Pruss R, Auchère F, L'Hermitte-Stead C, Bouillaud F, Brice A, Corti O, Lombès A (2014) Tissue- and cell-specific mitochondrial defect in Parkin-deficient mice. PLoS One 9:e99898. 10.1371/journal.pone.0099898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909. 10.1016/S0896-6273(03)00568-3 [DOI] [PubMed] [Google Scholar]
- Diaz F, Bayona-Bafaluy MP, Rana M, Mora M, Hao H, Moraes CT (2002) Human mitochondrial DNA with large deletions repopulates organelles faster than full-length genomes under relaxed copy number control. Nucleic Acids Res 30:4626–4633. 10.1093/nar/gkf602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dölle C, Flønes I, Nido GS, Miletic H, Osuagwu N, Kristoffersen S, Lilleng PK, Larsen JP, Tysnes OB, Haugarvoll K, Bindoff LA, Tzoulis C (2016) Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 7:13548. 10.1038/ncomms13548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elachouri G, Vidoni S, Zanna C, Pattyn A, Boukhaddaoui H, Gaget K, Yu-Wai-Man P, Gasparre G, Sarzi E, Delettre C, Olichon A, Loiseau D, Reynier P, Chinnery PF, Rotig A, Carelli V, Hamel CP, Rugolo M, Lenaers G (2011) OPA1 links human mitochondrial genome maintenance to mtDNA replication and distribution. Genome Res 21:12–20. 10.1101/gr.108696.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinn L, Mortiboys H, Volkmann K, Köster RW, Ingham PW, Bandmann O (2009) Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 132:1613–1623. 10.1093/brain/awp108 [DOI] [PubMed] [Google Scholar]
- Fukui H, Moraes CT (2009) Mechanisms of formation and accumulation of mitochondrial DNA deletions in aging neurons. Hum Mol Genet 18:1028–1036. 10.1093/hmg/ddn437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furda AM, Bess AS, Meyer JN, Van Houten B (2012) Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol Biol 920:111–132. 10.1007/978-1-61779-998-3_9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaweda-Walerych K, Zekanowski C (2013) Integrated pathways of parkin control over mitochondrial maintenance-relevance to Parkinson's disease pathogenesis. Acta Neurobiol Exp (Wars) 73:199–224. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J (2003) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278:43628–43635. 10.1074/jbc.M308947200 [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 100:4078–4083. 10.1073/pnas.0737556100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM (2016) Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann Neurol 79:366–378. 10.1002/ana.24571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889. 10.1038/nature04724 [DOI] [PubMed] [Google Scholar]
- Hattori N, Kitada T, Matsumine H, Asakawa S, Yamamura Y, Yoshino H, Kobayashi T, Yokochi M, Wang M, Yoritaka A, Kondo T, Kuzuhara S, Nakamura S, Shimizu N, Mizuno Y (1998) Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann Neurol 44:935–941. 10.1002/ana.410440612 [DOI] [PubMed] [Google Scholar]
- Jiang H, Jiang Q, Feng J (2004) Parkin increases dopamine uptake by enhancing the cell surface expression of dopamine transporter. J Biol Chem 279:54380–54386. 10.1074/jbc.M409282200 [DOI] [PubMed] [Google Scholar]
- Jiang H, Jiang Q, Liu W, Feng J (2006) Parkin suppresses the expression of monoamine oxidases. J Biol Chem 281:8591–8599. 10.1074/jbc.m510926200 [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Zhang Z, Roda R, Fukaya M, Wakabayashi J, Wakabayashi N, Kensler TW, Reddy PH, Iijima M, Sesaki H (2012) Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol 197:535–551. 10.1083/jcb.201110034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Stevens MV, Akter MH, Rusk SE, Huang RJ, Cohen A, Noguchi A, Springer D, Bocharov AV, Eggerman TL, Suen DF, Youle RJ, Amar M, Remaley AT, Sack MN (2011) Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J Clin Invest 121:3701–3712. 10.1172/JCI44736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. 10.1038/33416 [DOI] [PubMed] [Google Scholar]
- Kitada T, Tong Y, Gautier CA, Shen J (2009) Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J Neurochem 111:696–702. 10.1111/j.1471-4159.2009.06350.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh H, Chung J (2010) PINK1 and Parkin to control mitochondria remodeling. Anat Cell Biol 43:179–184. 10.5115/acb.2010.43.3.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38:518–520. 10.1038/ng1778 [DOI] [PubMed] [Google Scholar]
- LaFrance R, Brustovetsky N, Sherburne C, Delong D, Dubinsky JM (2005) Age-related changes in regional brain mitochondria from Fischer 344 rats. Aging Cell 4:139–145. 10.1111/j.1474-9726.2005.00156.x [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ (2005) Dopamine covalently modifies and functionally inactivates parkin. Nat Med 11:1214–1221. 10.1038/nm1314 [DOI] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG (2012) Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet 21:4827–4835. 10.1093/hmg/dds352 [DOI] [PubMed] [Google Scholar]
- Lee Y, Stevens DA, Kang SU, Jiang H, Lee YI, Ko HS, Scarffe LA, Umanah GE, Kang H, Ham S, Kam TI, Allen K, Brahmachari S, Kim JW, Neifert S, Yun SP, Fiesel FC, Springer W, Dawson VL, Shin JH, et al. (2017) PINK1 primes Parkin-mediated ubiquitination of PARIS in dopaminergic neuronal survival. Cell Rep 18:918–932. 10.1016/j.celrep.2016.12.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov AY, Ganapathi S, Westerhof M, Quispe-Tintaya W, White RR, Van Houten B, Reiling E, Dollé ME, van Steeg H, Hasty P, Hoeijmakers JH, Vijg J (2013) DNA damage in normally and prematurely aged mice. Aging Cell 12:467–477. 10.1111/acel.12071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Kabuto H, Makino H, Ogawa N (1997) Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J Neurosci Methods 73:45–48. 10.1016/S0165-0270(96)02211-X [DOI] [PubMed] [Google Scholar]
- Meiser J, Weindl D, Hiller K (2013) Complexity of dopamine metabolism. Cell Commun Signal 11:34. 10.1186/1478-811X-11-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, Goorden S, Priault M, Elgersma Y, Rezvani HR, Rossignol R, Bénard G (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 17:719–730. 10.1016/j.cmet.2013.03.014 [DOI] [PubMed] [Google Scholar]
- Meredith GE, Kang UJ (2006) Behavioral models of Parkinson's disease in rodents: a new look at an old problem. Mov Disord 21:1595–1606. 10.1002/mds.21010 [DOI] [PubMed] [Google Scholar]
- Müller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn PH, Lichtenthaler SF, Motori E, Hrelia S, Wurst W, Trümbach D, Langer T, Krappmann D, Dittmar G, Tatzelt J, Winklhofer KF (2013) The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol Cell 49:908–921. 10.1016/j.molcel.2013.01.036 [DOI] [PubMed] [Google Scholar]
- Myöhänen TT, Schendzielorz N, Männistö PT (2010) Distribution of catechol-O-methyltransferase (COMT) proteins and enzymatic activities in wild-type and soluble COMT deficient mice. J Neurochem 113:1632–1643. 10.1111/j.1471-4159.2010.06723.x [DOI] [PubMed] [Google Scholar]
- Narendra DP, Youle RJ (2011) Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal 14:1929–1938. 10.1089/ars.2010.3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyama G, Yoshimi K, Natori S, Chikaoka Y, Ren YR, Funayama M, Shimo Y, Takahashi R, Nakazato T, Kitazawa S, Hattori N (2010) Impaired in vivo dopamine release in parkin knockout mice. Brain Res 1352:214–222. 10.1016/j.brainres.2010.06.065 [DOI] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD (2005) Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A 102:2174–2179. 10.1073/pnas.0409598102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier C, Bender A, García-Arumí E, Melià MJ, Bové J, Laub C, Klopstock T, Elstner M, Mounsey RB, Teismann P, Prolla T, Andreu AL, Vila M (2013) Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain 136:2369–2378. 10.1093/brain/awt196 [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85:257–273. 10.1016/j.neuron.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Pinto M, Hida A, Moraes CT (2011a) Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci 31:17649–17658. 10.1523/JNEUROSCI.4871-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Fukui H, Wang X, Pinto M, Moraes CT (2011b) The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J Neurosci 31:9895–9904. 10.1523/JNEUROSCI.6223-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ (2015) Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87:371–381. 10.1016/j.neuron.2015.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5:e10054. 10.1371/journal.pone.0010054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve A, Meagher M, Lax N, Simcox E, Hepplewhite P, Jaros E, Turnbull D (2013) The impact of pathogenic mitochondrial DNA mutations on substantia nigra neurons. J Neurosci 33:10790–10801. 10.1523/JNEUROSCI.3525-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T, Patenge N (2009) Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet 18:3832–3850. 10.1093/hmg/ddp327 [DOI] [PubMed] [Google Scholar]
- Santos JH, Meyer JN, Mandavilli BS, Van Houten B (2006) Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol 314:183–199. 10.1385/1-59259-973-7:183 [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496:372–376. 10.1038/nature12043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990a) Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54:823–827. 10.1111/j.1471-4159.1990.tb02325.x [DOI] [PubMed] [Google Scholar]
- Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD (1990b) Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson's disease. J Neurochem 55:2142–2145. 10.1111/j.1471-4159.1990.tb05809.x [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM (2011) PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 144:689–702. 10.1016/j.cell.2011.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, McMackin M, Nguyen A, Cortopassi G (2017) Parkin deficiency accelerates consequences of mitochondrial DNA deletions and Parkinsonism. Neurobiol Dis 100:30–38. 10.1016/j.nbd.2016.12.024 [DOI] [PubMed] [Google Scholar]
- Srivastava S, Moraes CT (2005) Double-strand breaks of mouse muscle mtDNA promote large deletions similar to multiple mtDNA deletions in humans. Hum Mol Genet 14:893–902. 10.1093/hmg/ddi082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterky FH, Lee S, Wibom R, Olson L, Larsson NG (2011) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A 108:12937–12942. 10.1073/pnas.1103295108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, Shin JH, Dawson TM (2015) Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci U S A 112:11696–11701. 10.1073/pnas.1500624112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ (2010) Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A 107:11835–11840. 10.1073/pnas.0914569107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380. 10.1083/jcb.201007013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan RA, Foster JD (2013) Mechanisms of dopamine transporter regulation in normal and disease states. Trends Pharmacol Sci 34:489–496. 10.1016/j.tips.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ (2013) The PINK1-Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci U S A 110:6400–6405. 10.1073/pnas.1221132110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM (2004) Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A 101:10744–10749. 10.1073/pnas.0401297101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906. 10.1016/j.cell.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A 107:5018–5023. 10.1073/pnas.0913485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Coordination tests performed on older animals: A: Pole test performed on 4 months old animals (n=8-19/group), B: Activity cage performed on 4 months old animals (n=7-10/group), C: Rotarod performed on 12 months old animals (5-11/group), D: Open field, measurement of total distance travelled on a 15 minute period by 12 months old animals (n=5-12/group), E: Open field, measurement of 12 months old animals rearing activity on a 15 minutes period (n=5-12/group). * = p < 0.05, ** = p < 0.01, *** = p < 0.001. Download Figure 1-1, EPS file (1.1MB, eps)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for weight measurements taken at 2, 4, 8, and 12 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-2, EPS file (1.3MB, eps)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for activity cage measurements taken at 2 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-3, EPS file (10.6MB, eps)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for motor tests measurements taken at 2 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-4, PDF file (26.6KB, pdf)
Locomotive phenotypes in ParkinKO PD-mito-PstI mice. Statistical analyses for motor tests measurements taken at 12 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 1-5, PDF file (37.4KB, pdf)
Striatal alterations of dopamine and its metabolites. A: Quantification of dopamine (DA) normalized for mg of proteins in 8 months old animals striata (n=4-7/group). B: Quantification of degradation metabolites normalized for dopamine content (3-MT/DA, HVA/DA, and DOPAC/DA) in 8 months old animals’ striata (n=4-7/group). C: Quantification of dopamine (DA) normalized for mg of proteins in 12 months old animals striata (n=3-6/group). D: Quantification of degradation metabolites normalized for dopamine content (3-MT/DA, HVA/DA, and DOPAC/DA) in 8 months old animals’ striata (n=3-6/group). E: Western blots showing levels of MAO-B and COMT protein in 8 months old mice striata and (F), relative quantification. * = p < 0.05, ** = p < 0.01, *** = p < 0.001. Download Figure 3-1, PDF file (25.4KB, pdf)
Striatal alterations of dopamine and its metabolites. Statistical analyses for dopamine and dopamine metabolites at 4, 8 and 12 months of age. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 3-2, PDF file (25.6KB, pdf)
MtDNA deletions and mutations in older mice in midbrain and striata. A: Schematic representation of mouse mtDNA. Color-coded representation of the position of the amplicons used for the qPCRs is maintained in the graphs (B-G). B-C:qPCR in dissected midbrain of 4 months old animals measuring (B) total mtDNA and (C) full-length mtDNA. D: qPCR in dissected midbrain of 12 and 19 months old animals measuring total mtDNA and full-length mtDNA.
E: Recombination events detected by qPCR using primers flanking the PstI sites in dissected midbrain of 12 and 19 months old animals. F: qPCR in strata of 12 and 19 months old animals measuring total mtDNA and full-length mtDNA. G: Recombination events detected by qPCR using primers flanking the PstI sites in striata of 12 and 19 months old animals. Color coded and numbers in the graphs represent the position of the primers on the mtDNA. H: Southern blots probing (top) DNA from dissected midbrains with mtDNA and nDNA probes in 4 months old animals and (bottom) striatal DNA with mtDNA and nDNA probes in 12 months old animals. I: Representative western blots probing for mitochondrial markers in 12 months old animals’ striata. J: Representative western blots probing for autophagy markers (p62 and LC3b) and for mitochondrial fusion markers (OPA1 and Mfn2) in 12 months old animals’ striata. * = p < 0.05, ** = p < 0.01, *** =p<0.001. Download Figure 4-1, PDF file (21.5KB, pdf)
MtDNA deletions and mutations increase when endogenous Parkin is lost in PD-mito-PstI mice striata. Statistical analyses for qPCR performed on DNA after LCM and on DNA from striatum at 4 months. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 4-2, PDF file (20.8KB, pdf)
MtDNA deletions and mutations increase when endogenous Parkin is lost in PD-mito-PstI mice striata. Statistical analyses for qPCR performed on DNA from striatum and SN at 12 and 19 months. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 4-3, PDF file (26.4KB, pdf)
Alterations of dopaminergic terminals and mtDNA in cortical region of ParkKO-PD-mitoPstI mice. Statistical analyses for qPCR performed on DNA from cortex at 12 and 19 months. p value and Degree of freedom (DF) after one-way ANOVA analysis and Tukey’s multiple comparison test. Download Figure 5-1, PDF file (28.1KB, pdf)