

Abstract
Pathological pain is a common and debilitating condition that is often poorly managed. Central sensitization is an important mechanism underlying pathological pain. However, candidate molecules involved in central sensitization remain unclear. Store-operated calcium channels (SOCs) mediate important calcium signals in nonexcitable and excitable cells. SOCs have been implicated in a wide variety of human pathophysiological conditions, including immunodeficiency, occlusive vascular diseases, and cancer. However, the role of SOCs in CNS disorders has been relatively unexplored. Orai1, a key component of SOCs, is expressed in the human and rodent spinal cord dorsal horn, but its functional significance in dorsal horn neurons is poorly understood. Here we sought to explore a potential role of Orai1 in the modulation of neuronal excitability and A-type potassium channels involved in pain plasticity. Using both male and female Orai1 knock-out mice, we found that activation of Orai1 increased neuronal excitability and reduced A-type potassium channels via the protein kinase C–extracellular signal-regulated protein kinase (PKC–ERK) pathway in dorsal horn neurons. Orai1 deficiency significantly decreased acute pain induced by noxious stimuli, nearly eliminated the second phase of formalin-induced nociceptive response, markedly attenuated carrageenan-induced ipsilateral pain hypersensitivity and abolished carrageenan-induced contralateral mechanical allodynia. Consistently, carrageenan-induced increase in neuronal excitability was abolished in the dorsal horn from Orai1 mutant mice. These findings uncover a novel signaling pathway involved in the pain process and central sensitization. Our study also reveals a novel link among Orai1, ERK, A-type potassium channels, and neuronal excitability.
SIGNIFICANCE STATEMENT Orai1 is a key component of store-operated calcium channels (SOCs) in many cell types. It has been implicated in such pathological conditions as immunodeficiency, autoimmunity, and cancer. However, the role of Orai1 in CNS disorders remains poorly understood. The functional significance of Orai1 in neurons is elusive. Here we demonstrate that activation of Orai1 modulates neuronal excitability and Kv4-containing A-type potassium channels via the protein kinase C–extracellular signal-regulated protein kinase (PKC–ERK) pathway. Genetic knock-out of Orai1 nearly eliminates the second phase of formalin-induced pain and markedly attenuates carrageenan-induced pain hypersensitivity and neuronal excitability. These findings reveal a novel link between Orai1 and neuronal excitability and advance our understanding of central sensitization.
Keywords: A-type potassium channels, ERK, Orai1, pain, spinal cord dorsal horn, store-operated calcium channels
Introduction
Store-operated calcium channels (SOCs) are highly Ca2+-selective cation channels activated by depletion of calcium stores from the endoplasmic reticulum (ER; Smyth et al., 2010). SOCs underlie sustained Ca2+ influx, which is required for Ca2+-dependent cellular functions, such as secretion, proliferation, neurotransmitter release, and enzymatic activity (Emptage et al., 2001; Chang et al., 2006; Abdullaev et al., 2008; Prakriya, 2009). SOCs are composed of the pore-forming Orai proteins Orai1–Orai3 [often referred to as calcium release-activated calcium (CRAC) channels] and are coupled to ER Ca2+ levels by the Ca2+-sensing stromal interaction molecules STIM1 and STIM2 (Liou et al., 2005; Roos et al., 2005; Feske et al., 2006). SOC entry (SOCE) has been extensively studied and its importance is well recognized in non-neuronal cells (Takezawa et al., 2006; Muik et al., 2012). SOCE has been shown in dorsal root ganglion neurons (Gemes et al., 2011), hippocampal neurons (Narayanan et al., 2010), and cortical neurons (Berna-Erro et al., 2009; Gruszczynska-Biegala and Kuznicki, 2013). STIM1 and STIM2 have been implicated in brain functions and pathological conditions (Berna-Erro et al., 2009; Hartmann et al., 2014). However, the role of Orai1 in the nervous system remains elusive. A previous study has reported Orai1 expression in the dorsal horn of the human spinal cord (Guzman et al., 2014). We recently demonstrated that SOCs are functional in dorsal horn neurons. Using pharmacological and siRNA knockdown approaches, we have demonstrated that Orai1 is responsible for SOCE in dorsal horn neurons (Xia et al., 2014). However, its functional significance in dorsal horn neurons remains elusive. Here, we show that Orai1-mediated SOCE modulates neuronal excitability and A-type currents in dorsal horn neurons. These effects are mediated by the protein kinase C–extracellular signal-regulated protein kinase (PKC–ERK) cascade. We also demonstrate that Orai1 deficiency reduces acute pain and almost eliminates the second phase of formalin-induced nociceptive behavior through the ERK signaling pathway, while it markedly attenuates carrageenan-induced pain hypersensitivity and neuronal excitability. These findings reveal a novel link of Orai1 to ERK, A-type channels, neuronal excitability, and pain, and provide insights into the molecular mechanisms that underlie central sensitization associated with inflammatory pain.
Materials and Methods
Animals.
All experiments were performed in accordance with the guidelines of the National Institutes of Health and with the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain and were approved by the Animal Care and Use Committee of Drexel University College of Medicine. Pregnant mice were purchased from Taconic or Charles River and individually housed in standard cages and maintained in a room with a 12 h light/dark cycle. Both sexes were used for all in vitro and in vivo experiments. Neonatal CD1 mice were used for cell cultures and adult CD1 mice were used for slice preparations (Xia et al., 2014). Orai1 mutant mice were developed in the laboratory of Dr. Jean-Pierre Kinet by insertion of a β-Geo cassette in the first intron of the Orai1 gene (Vig et al., 2008), and purchased from the Mutant Mouse Regional Resource Centers. The original Orai1 mutant mice came from a mixed C57BL/6 and 129P2/OlaHsd background. These mice had been backcrossed to CD1 for >7 generations.
Genotyping.
Mice were genotyped by PCR of DNA extracted from tail clips. The following primers were used: for β-Geo: 5′-CAAATGGCGATTACCGTTGA (F), 5′-TGCCCAGTCATAGCCGAATA (R); for Tcrd: 5′-CAAATGTTGCTTGTCTGGTG (F), 5′-GTCAGTCGAGTGCACAGTTT (R). Genotypes were further determined by the copy number analysis using a TaqMan Genotyping Master Mix kit (Applied Biosystems) following the manufacturer's instructions. Briefly, the TaqMan copy number assay (detecting the β-Geo) was run simultaneously with a TaqMan copy number reference assay [detecting the telomerase reverse transcriptase (Tert)] in a duplex real-time PCR. Real-time quantitative PCR was performed in a 7900HT fast real-time PCR system (Applied Biosystems) using the following amplification conditions: 10 min of initial denaturation at 95°C, then 40 cycles at 95°C for 15 s, and at 60°C for 1 min. The Applied Biosystems CopyCaller software was used for post-PCR data analysis of copy number quantitation.
Real-time PCR analysis of Orai1 mRNA expression.
Real-time PCR was performed according to our previous study (Gao et al., 2016). Total RNA was extracted from adult spinal cords and acutely dissociated neurons using TRIzol Reagent (Molecular Research Center). RNA concentration was determined by optical density at 260 nm. Total RNA was reverse-transcribed into cDNA for each sample using a Fermentas maxima first-strand cDNA synthesis kit (Thermo Fisher Scientific) following the manufacturer's instructions. Primers specific for mouse Orai1 (Mm00774349_m1) and GAPDH were purchased from Applied Biosystems. Real-time quantitative PCR was performed in a 7900HT fast real-time PCR system (Applied Biosystems) using the following conditions: 5 min of initial denaturation at 96°C, then 35 cycles at 96°C for 30 s, at 55°C for 30 s, and at 72°C for 1.5 min. The threshold cycle for each gene was determined and analyzed using the relative quantitation software (Applied Biosystems). The relative expression of Orai1 was calculated using the 2−ΔΔCt method (Livak and Schmittgen, 2001). The mRNA levels of Orai1 were normalized to the housekeeping gene GAPDH.
Cell culture and slice preparation.
Primary cultures of spinal cord superficial dorsal horn neurons were prepared from postnatal day 1 or 2 mice as previously described (Hu et al., 2003). Briefly, neonatal mice were decapitated after inducing hypothermia on ice. A laminectomy was performed and the spinal cord superficial dorsal horn was carefully dissected in the dorsal side of the spinal cord (approximately lamina III) with a surgical blade. The superficial dorsal horn strips were then incubated for 30 min at 37°C in HBSS (Invitrogen; in mm: 137 NaCl, 5.4 KCl, 0.4 KH2PO4, 1 CaCl2, 0.5 MgCl2, 0.4 MgSO4, 4.2 NaHCO3, 0.3 Na2HPO4, and 5.6 glucose) containing papain (15 U/ml; Worthington Biochemical), rinsed three times with HBSS, and placed in culture medium containing Neurobasal A (Invitrogen), 2% fetal calf serum (Invitrogen), 2% heat-inactivated horse serum (Invitrogen), 2% B-27 (Invitrogen), 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 2 mm GlutaMAX-1 (Invitrogen). The fragments were mechanically dissociated by gently triturating with a fire-polished Pasteur pipette until the solution turned cloudy. The dispersed cells were plated at a density of 3000 cells per well onto 12 mm coverslips coated with poly-d-lysine and laminin (Sigma-Aldrich). Neurons were cultured in a humidified atmosphere containing 5% CO2 at 37°C for 1–2 d for recording of A-type currents.
Lumbar spinal cord slices (300–350 μm) were prepared from CD-1 mice as we described previously (Xia et al., 2014). Briefly, after decapitation, the vertebral column and surrounding tissue were isolated and immersed in ice-cold oxygenated N-methyl-d-glucamine (NMDG) artificial CSF (ACSF) containing the following (in mm): 93 NMDG, 93 HCl, 30 NaHCO3, 20 HEPES, 2.5 KCl, 1.2 NaH2PO4, 10 MgCl2, 0.5 CaCl2, 25 glucose, 3 myo-inositol, 1 sodium-L-ascorbate, 5 ethyl pyruvate (osmolality, ∼305–310 mmol/kg). The lumbar enlargement of the spinal cord was removed and glued onto the cutting platform with the adhesive Loctite 404 (Loctite). Transverse spinal cord slices were cut in NMDG ACSF with a Vibratome 3000 (Vibratome). Slices were incubated at 32–34°C for 15 min and then maintained at room temperature (20–24°C) for 1 h before electrophysiological recording in the continuously oxygenated recovery solution containing the following (in mm): 26 NaHCO3, 3 myo-inositol, 120 NaCl, 5 KCl, 1.25 NaH2PO4, 1 MgCl2, 1 CaCl2, 12.5 glucose (osmolality, ∼305–310 mmol/kg).
Transfection.
For transfection of Orai1-CFP (inserted in the pIRESneo plasmid, a generous gift from Dr. Donald Gill, Penn State College of Medicine, Hershey, Pennsylvania), neurons were electroporated using mouse primary cell Nucleofector kit according to the manufacturer's instructions (Lonza Group). Briefly, neurons were transfected with Orai1-CFP (2.5 μg) per 1 × 106 cells and were seeded on 12 mm glass coverslips. Calcium imaging and patch clamp were performed 48–72 h after transfection.
Calcium imaging.
Calcium imaging was performed using fura-2-based microfluorimetry and imaging analysis as we previously described (Xia et al., 2014). Neurons were loaded with 4 μm fura-2AM (Life Technologies) for 30 min at room temperature in HBSS, washed, and further incubated in normal bath solution (Tyrode's) containing the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 5.6 glucose for 20 min. Coverslips were mounted in a small perfusion chamber (Warner Instruments, Model RC-25) and continuously perfused at 5–7 ml/min with Tyrode's solution. Images were acquired at 3 s intervals at room temperature using an Olympus inverted microscope equipped with a CCD camera (Hamamatsu, ORCA-03G). The 340:380 ratios were recorded and analyzed using the software MetaFluor 7.7.9 (Molecular Devices). Only one recording was made from each coverslip.
Electrophysiological recording.
Standard whole-cell recordings were performed at room temperature using EPC 10 amplifier and PatchMaster software (HEKA Elektronik) as we described previously (Hu et al., 2003). Electrode resistances were ∼3–6 MΩ with series resistances of 6–15 MΩ and were compensated to the maximal current amplitude. Resting membrane potentials were −56.7 ± 0.4 mV (n = 110). Only neurons with a resting membrane potential more hyperpolarized than −50 mV were used. For recording A-type currents in cultured neurons, the bath solution contained (in mm) 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 5.6 glucose with 500 nm tetrodotoxin (TTX) to block voltage-gated Na+ currents. The electrode solution contained (in mm) 140 KCl, 1 MgCl2, 10 HEPES, 1 EGTA, 3 Na2ATP, and 0.3 Na2GTP, pH adjusted to 7.4 with KOH. The membrane voltage was held at −80 mV and transient potassium currents (A-type currents) were isolated by a two-step voltage protocol as previously described (Hu and Gereau, 2011). To determine the voltage dependence of activation, voltage steps of 150 ms were applied at 5 s intervals in +10 mV increments from −90 mV to a maximum of +70 mV. To determine the voltage dependence of inactivation, conditioning prepulses ranging from −100 to +40 mV were applied in +10 mV increments for 150 ms followed by a voltage step to +40 mV for 150 ms. For current-clamp recording in slices, the bath solution was ACSF bubbled with 95% O2 and 5% CO2. The intracellular solution contained the following (in mm): 140 KMeSO4, 2 MgCl2, 1 EGTA, 10 HEPES, 3 Na2ATP, and 0.3 Na2GTP, pH 7.4. Action potentials were generated by current injection from a holding potential of −75 mV. Throughout the recording, the holding potential was maintained by current injection. The rheobase (the minimal current to evoke an action potential) was first determined for every neuron by depolarization pulses (1 s duration) ranging from 10 to 100 pA in 10 or 5 pA increments. Modulation of action potentials was assessed every 15 s using a small constant depolarization pulse (1 s, 1.1–1.3 times rheobase). The current amplitude that evoked 3–6 action potentials during the baseline period was selected for evaluating effects of compounds on action potentials and remained constant throughout the recording. The first spike-latency was measured as the time between the stimulus onset and the first spike of the response. The spike frequency was measured by counting the number of spikes within a depolarizing pulse (1 s duration; Hu et al., 2006).
Western blot analysis.
Mice were killed after an overdose of isoflurane anesthesia. The lumbar section of the spinal cord was dissected and homogenized using a Dounce homogenizer in an ice-cold radio immunoprecipitation assay (RIPA) buffer containing 50 mm Tris HCl, 150 mm NaCl, 0.2 mm EDTA, 1% Triton X-100, 2% SDS, 1% deoxycholate, 0.1 mm PMSF, and protease inhibitor mixtures (Thermo Fisher Scientific). For cultures, neurons were washed in ice-cold PBS and lysed in RIPA buffer. The lysed tissues and neurons were sonicated at a constant intensity of 2.5 for 10 s, and centrifuged at 18 000 × g at 4°C for 5 min. The concentrations of total protein were determined using a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific) following the manufacturer's instructions. Protein samples were heated at 95°C for 5 min, electrophoresed in 10% SDS polyacrylamide gel, and transferred onto nitrocellulose membranes (Bio-Rad). The blots were blocked with Odyssey blocking buffer (TBS) for 1 h at room temperature and probed with rabbit anti-Orai1 (1:500; ProSci), anti-STIM1 (1:8000; Cell Signaling Technology), anti-STIM2 (1:4000; ProSci), anti-Orai2 (1:500; ProSci), anti-Orai3 (1:500; ProSci), anti-p-ERK (1:1000; Cell Signaling Technology), anti-ERK (1:10,000; Cell Signaling Technology), mouse anti-GAPDH (1:20,000; Cell Signaling Technology), and mouse anti-Actin (1:20,000; Sigma-Aldrich) primary antibodies at 4°C overnight. The blots were washed and incubated for 1 h at room temperature with IRDye donkey anti-rabbit/mouse secondary antibodies (1:10,000; LI-COR). The bands were quantified using Odyssey Image Studio Software (LI-COR).
Immunofluorescent staining.
CD-1 mice were injected subcutaneously in the right hindpaw with 15 μl of PBS or 2% formalin solution. After 8 min, animals were deeply anesthetized with ketamine and xylazine and perfused intracardially with saline followed by 4% cold buffered paraformaldehyde (PFA)/0.1 m phosphate buffer (PB) solution. The lumbar spinal cord (L4–L5) was removed then postfixed overnight in 4% PFA/0.1 m PB, followed by cryoprotection in 30% sucrose in 0.1 m PB for 2 d. Spinal cords were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek), and sections were cut into 30-μm-thick slices and blocked with PBS containing 5% normal goat serum and 0.3% Triton X-100 for 1 h. Primary antibody for p-ERK (1:1000; Cell Signaling Technology) was incubated overnight at 4°C in blocking solution. After three washes in PBS, slices were incubated with secondary antibody (Thermo Fisher Scientific, Alexa Fluor) for 1 h in the blocking solution. Spinal cord slices were then mounted on glass slides and coverslips were applied using mounting media (Southern Biotech). Images of the superficial dorsal horn were captured using the Olympus Fluoview FV1000 confocal microscope equipped with a 10× objective. Imaging capture parameters, such as gain, offset, high voltage (HV), and laser intensity, were kept constant during the acquisition of images for valid comparisons of p-ERK intensity. Blind quantitative analysis was performed by using ImageJ to count the number of cells positive for p-ERK fluorescence in the dorsal horn from ≥3 randomly chosen spinal cord slices from each animal.
Behavioral studies.
Orai1 knock-out mice (Orai1−/−) and wild-type (Orai1+/+) littermate mice were used. All behavioral tests were conducted in 8–10-week-old mice and were performed blind to genotypes.
Motor function.
Mice were tested for motor function using the accelerating rotarod (5–30 rpm; Ugo Basile). Motor coordination of mice was analyzed by measuring latency to falling off a rod rotated with increasing velocity. All mice were trained the day before testing. The time spent on the rotarod (drop latency) was recorded.
The open-field test.
The spontaneous activity of mice was tested using an open-field system (Med Associates), which automatically detects the free movements of mice. Activity monitor software was used to record and quantify behaviors, including the distance traveled. Animals were acclimatized to the equipment 30 min before the test. All mice were tested for 30 min.
Tail-flick test.
The tail-flick test was performed by applying focused, radiant heat to the tail as described previously (Li et al., 2016). Mice were loosely held with soft tissues for 1 min each time (>3 times) before testing. The withdrawal response was detected using the tail-flick test analgesia meter (Columbus Instruments). A light beam was focused onto the caudal end (2.5 cm from the tip) of the tail. The intensity of heat was adjusted to evoke a tail-flick baseline latency of ∼5–6 s. A cutoff time was set at 10 s to avoid tissue damage. The latencies were averaged over three trials, separated by 30 min intervals.
Tail-pressure test.
The tail-pressure test was conducted using the Basile Analgesy-Meter (Ugo Basile), and the response (struggle/vocalization) threshold was measured as previously described (Hu et al., 2006). Animals were placed in a restrainer and left to settle for a few minutes. Force was applied on the dorsal aspect of the tail at two adjacent localities approximately two-thirds the length of the tail from the tail base.
Hot-plate test.
The hot-plate test was conducted using the Hot Plate Analgesia Meter (IITC Life Science). Hot-plate latencies were measured as the time taken for a mouse to lick, shake its hindpaw, or jump at hot-plate temperatures of either 52°C or 56°C as previously described (Hu et al., 2006).
Formalin-induced spontaneous nociceptive behavior.
The formalin test was performed as previously described (Gao et al., 2013). Briefly, CD1 mice were habituated for 1 h in a transparent Plexiglas test box (5 × 5 ×10 inches) before any injections. Fifteen microliters of 2% formalin in PBS solution was injected subcutaneously into the plantar surface of the right hindpaw, and the mouse was returned to the test box immediately. The spontaneous nociceptive behavior was quantified by recording the total time that the mouse spent licking or lifting the injected paw with stopwatch in 5 min intervals for 1 h.
Carrageenan-induced pain hypersensitivity.
Carrageenan-induced inflammatory pain was induced in the right hindpaw by intraplantar injection of 15 μl of carrageenan (2% in PBS solution). Mechanical sensitivity was measured before injection of carrageenan and 3 and 24 h after injection of carrageenan in both injected and uninjected paws using von Frey filaments (North Coast Medical). A series of von Frey filaments were applied to both right and left paws. The 50% hindpaws withdrawal threshold was determined using the Dixon's up–down method (Dixon, 1980). Thermal sensitivity was also measured before injection of carrageenan and 3 and 24 h after injection of carrageenan using a different group of mice and the Hargreaves' method as previously described (Gao et al., 2015). The paw withdrawal latency was set to ∼10 s with a maximum of 20 s as the cutoff to prevent potential injury. The latencies were averaged over three trials, separated by 30 min intervals.
Weight distribution on each hindlimb was measured before carrageenan injection and 5 and 24 h after carrageenan injection (after the von Frey filaments test) using an incapacitance analgesia meter (IITC Life Science). Mice were placed into an angled Plexiglas chamber of the incapacitance meter with their hindpaws on separate sensors. Each data point is the average of three readings. Data were expressed as ipsilateral/contralateral. A value of 1.0 represented equal weight distribution across ipsilateral and contralateral hindlimbs.
Paw thickness was measured at the mid-plantar level before carrageenan injection and 4 and 24 h after carrageenan injection (after the Hargreaves' test) using an electronic outside micrometer (World Precision Instruments), as described previously (Boettger et al., 2007). Each paw was measured twice and an average was calculated. Paw thickness was measured after thermal latencies were determined. Paw edema was determined by subtraction of preinjection from postinjection thickness (mm).
Measurement of IL-1β and prostaglandin E2 in the injected paw.
Mice were anesthetized with isoflurane 24 h after carrageenan or PBS injection. The entire injected paw was removed using a scalpel. Multiple incisions were made on both dorsal and plantar surfaces. The paw was placed in a 12-well plate with 1.5 ml of PBS containing 20 μg/ml indomethacin and gently shaken at 100 rpm for 1 h. The exudate was collected after being centrifuged at a speed of 10,000 rpm for 10 min. The IL-1β and prostaglandin E2 (PGE2) levels in the supernatant were measured with ELISA kits according to the manufacturer's instructions (R & D Systems).
Drug application.
Thapsigargin (TG), TTX, formalin, carrageenan, indomethacin, and 4-aminopyridine (4-AP) were purchased from Sigma-Aldrich. YM-58483 (N-[4-[3,5-bis(trifluoromethyl)-1H-pyrazol-1-yl] phenyl]-4-methyl-1,2,3-thiadiazole-5-carboxamide), U0126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene), PD98059 [2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one], and GF109203X (GF) were purchased from Tocris Bioscience. They were dissolved in Milli-Q water or dimethyl sulfoxide (DMSO) as stock solutions and further diluted to final concentrations in 0.1% DMSO.
Electrophysiological data analysis.
Off-line data analysis was processed using PatchMaster (HEKA) and Origin 8.1 software (OriginLab). The voltage dependency of activation and inactivation of A-type currents were fitted with the Boltzmann function. For activation, peak currents were converted to conductance (G) by the formula G = I/(Vm − Vrev), where Vm is the membrane voltage of depolarization pulses and Vrev is the calculated potassium reversal potential (−84 mV). The function G/Gmax = 1/(1 + exp[(V1/2 − V)/k]) was used to normalize conductance, where Gmax is the maximal conductance obtained with a depolarizing pulse to +70 mV, V1/2 is the half maximal voltage, and k is the slope factor. For inactivation, I/Imax = 1/(1 + exp[(V1/2 − V)/k]) was used, where Imax is the maximal current obtained with a −100 mV prepulse (Hu et al., 2003). Current density was calculated as the peak currents divided by the capacitance (pA/pF).
Statistical analyses.
Data are expressed as original traces or as mean ± SEM. Treatment effects in vitro or behavioral data were statistically analyzed with paired, two-sample Student's t tests, one-way ANOVA, or two-way ANOVA followed by the Bonferroni's multiple-comparison test. Error probabilities of p < 0.05 were considered statistically significant. Statistical analyses were performed using Origin 8.1 or GraphPad Prism 7.0.
Results
Depletion of ER calcium stores increases neuronal excitability
We have demonstrated that SOCs are expressed and function in dorsal horn neurons (Xia et al., 2014). To explore the functional significance of SOCs in dorsal horn neurons, we first assessed the effect of ER Ca2+ store depletion on neuronal excitability. Current-clamp recordings were performed in substantia gelatinosa (SG) neurons of adult mouse spinal cord slices. Action potentials were evoked in response to current injection from a holding potential of −75 mV. The firing rate was increased upon increase in current injection (Fig. 1A). Bath application of TG, an inhibitor of the ER Ca2+-ATPase, slightly induced a depolarization (from −74.6 ± 0.5 mV to −71.4 ± 0.8 mV, df = 9, t = 4.49, p < 0.01), increased the firing rate under the same current injection (Fig. 1A), and significantly reduced the rheobase compared with the vehicle (0.1%DMSO, Pre-TG) in ∼85% neurons (Fig. 1B). To assess the effects of TG on the first-spike latency and spike frequency, action potentials were evoked by injection of a current amplitude between 1.1 and 1.3 times the rheobase. TG significantly decreased the first-spike latency and increased spike frequency (Fig. 1B), but steady-state input resistance was not significantly changed after TG application (data not shown). To confirm whether TG-induced modulation of action potentials is mediated by SOCE, we bath-applied 3 μm YM58483 (a SOC inhibitor) for 5 min before TG addition. YM58483 had no significant effect on action potentials alone (data not shown), but blocked TG-induced changes of the first-spike latency and spike frequency (Fig. 1C). These results indicate that activation of SOCs increases neuronal excitability.
Figure 1.
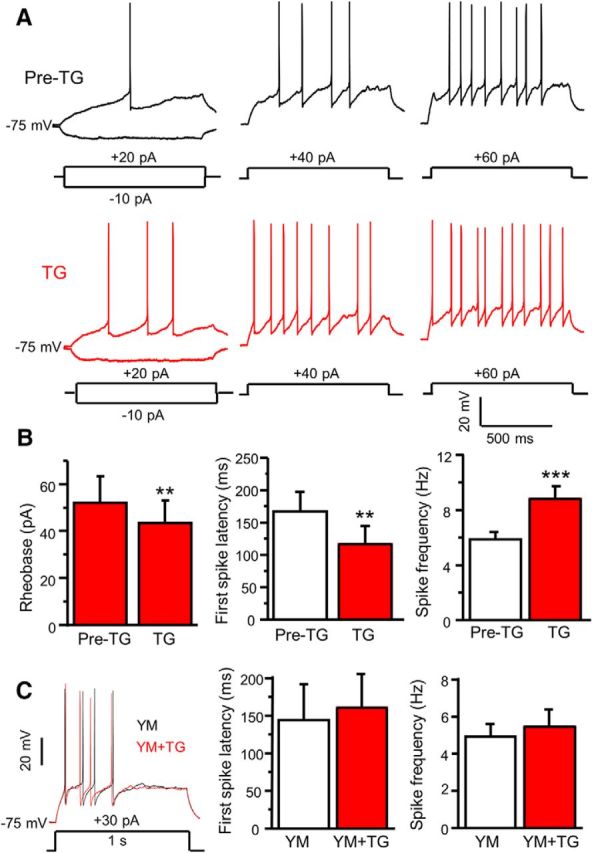
Depletion of ER Ca2+ stores modulates neuronal excitability of SG neurons in spinal cord slices. A, Representative action potentials generated by increasing series of current injections recorded before (Pre-TG) and 5 min after application of 2 μm TG. B, Summary of changes in the rheobase (df = 6, t = 4.044, p = 0.007), the first-spike latency (df = 15, t = 3.922, p = 0.0014), spike frequency (df = 15, t = 4.935, p = 0.0001) induced by TG in dorsal horn neurons. C, The effect of 3 μm YM 58483 (YM) on TG-induced modulation of the first-spike latency (df = 4, t = 1.195, p = 0.298) and spike frequency (df = 6, t = 0.764, p = 0.474). Values represent mean ± SEM; n = 7–16 neurons; **p < 0.01, ***p < 0.001 compared with pre-TG by the paired Student's t test.
Depletion of ER calcium stores decreases A-type currents
It is well established that A-type currents contribute to the onset of action potential generation and the rate of firing. To determine whether TG modulates Kv4-containing A-type currents, which contribute to pain plasticity (Hu et al., 2006), we used the whole-cell patch-clamp technique in cultured dorsal horn neurons. Outward potassium currents were evoked by a step depolarization from a holding potential of −80 mV to +40 mV. The A-type current was dissected away from the sustained current by the voltage protocol as described in our previous report (Hu and Gereau, 2011). Bath application of TG significantly reduced A-type currents (Fig. 2A). This time-dependent effect reached maximal effect at 5 min and lasted ≥10 min (Fig. 2B). Sustained currents were not significantly reduced by TG treatment (Fig. 2C). Steady-state inactivation and activation curves showed no shift in response to TG application (Fig. 2D). To confirm depletion of ER Ca2+ stores-induced modulation of A-type currents, we also tested a structurally distinct ER Ca2+-ATPase inhibitor, cyclopiazonic acid (CPA). Similarly, CPA significantly decreased A-type currents with no effect on sustained currents (Fig. 2E,F). These results demonstrate that depletion of ER Ca2+ stores inhibits A-type currents in dorsal horn neurons.
Figure 2.

Depletion of ER Ca2+ stores reduces A-type currents in spinal cord dorsal horn neurons. A, Representative traces of A-type currents recorded before (Pre-TG) and 5 min after application of 2 μm TG. B, Time course of TG's effect on A-type current peak amplitude (n = 9 neurons). C, Summary of the effects of TG on A-type and sustained currents (n = 15–17 neurons, df = 12, t = 7.4, p = 0.0001). D, Steady-state activation and inactivation curves before (Pre-TG) and 5 min after bath application of 2 μm TG (n = 13 neurons). E, Representative traces of A-type and sustained currents recorded before (Pre-CPA) and 5 min after application of 20 μm CPA. F, Summary of the effects of CPA on A-type and sustained currents (n = 6 neurons, df = 8, t = 6.7, p = 0.0002). Values are means ± SEM; ***p < 0.001 compared with predrug administration by the paired Student's t test.
TG-induced increase in neuronal excitability is mediated by modulation of A-type channels
To determine whether blockage of A-type channels occludes TG-induced modulation of A-type currents and neuronal excitability, we performed patch-clamp recordings and tested TG-induced modulation of A currents and neuronal excitability in the presence of 4-AP, a blocker of A-type channels. We found that 4-AP reduced A-type currents in a concentration-dependent manner (Fig. 3A,B). At a higher concentration, 4-AP eliminated A-type currents. Four-AP pretreatment abolished TG-induced modulation of A-type currents in cultured neurons. Consistently, 4-AP markedly reduced the rheobase and the first-spike latency, and increased spike frequency in SG neurons of spinal cord slices (Fig. 3C,D), suggesting that 4-AP increases neuronal excitability. Importantly, blockage of A-type channels by 4-AP occluded TG-induced modulation of neuronal excitability.
Figure 3.

Blockage of A-type channels occludes TG-induced modulation of A-type currents and neuronal excitability. A, Representative traces of A-type currents recorded before treatment (control), 3 min after 4-AP treatment, and 5 min after application of 2 μm TG in the presence of 4-AP. B, Summary of the effects of 4-AP and TG + 4-AP on A-type currents (n = 5–6, F = 10.57618, p = 0.0005. C, Representative action potentials generated by current injection recorded before treatment (control), 3 min after 0.5 mm 4-AP treatment, and 5 min after application of 2 μm TG in the presence of 4-AP. D, Summary of changes in the first-spike latency, spike frequency, and rheobase (n = 6). For the rheobase, F = 12.67, p = 0.007. For the first-spike latency, F = 9.34745, p = 0.003. For spike frequency, F = 5.739, p = 0.009. Values are means ± SEM, ns, not significant. **p < 0.01, ***p < 0.001 compared with control by one-way ANOVA.
Orai1 deficiency impairs SOCE in dorsal horn neurons
Using the siRNA knockdown approach, we have demonstrated that Orai1 is a key component of SOCs in dorsal horn neurons (Xia et al., 2014). To confirm whether Orai1 is required for SOCE, we took advantage of the Orai1 mutant mouse line, which has been characterized previously (Vig et al., 2008; Davis et al., 2015). We backcrossed these mice (C57BL/6 background) to CD-1 for ≥7 generations to improve their growth and survival rate. To identify genotypes of the offspring from Orai1 heterozygous (Orai1+/−) breeding pairs, we first performed PCR of DNA extracted from tail clips of litters. Orai1 knock-out (Orai1−/−) and heterozygous (Orai1+/−) mice showed β-Geo expression, which was undetectable in wild-type (Orai1+/+) mice (Fig. 4A). The genotypes of Orai1+/− and Orai1−/− mice were further assessed by the copy numbers of β-Geo. Orai1−/− mice, compared with Orai1+/− mice, had twice the number of copies of β-Geo. In addition, Orai1 mRNA was absent in the spinal cord of Orai1−/− mice while it was reduced to 50% in Orai1+/− mice compared with Orai1+/+ mice (Fig. 4B). Western blot analysis showed that the Orai1 band at ∼50 kDa (glycosylated Orai1) was not detectable in spinal cord tissues from Orai1−/− mice (Fig. 4B, inset). Consistent with a previous study showing that the mRNA levels of Orai2, Orai3, STIM1, or STIM2 are not altered in Orai1−/− mice (Davis et al., 2015), we did not observe compensatory changes in protein levels of Orai2, Orai3, STIM1, or STIM2 using specific antibodies that we have confirmed (Xia et al., 2014; data not shown). Importantly, TG-induced SOCE was eliminated in Orai1−/− neurons, while it was reduced in Orai1+/− neurons when compared with neurons from Orai1+/+ littermates (Fig. 4C). To determine whether lack of SOCE observed in Orai1−/− dorsal horn neurons is due to loss of Orai1 expression, we transfected Orai1−/− dorsal horn neurons with wild-type Orai1-CFP. Neurons transfected with Orai1-CFP showed robust SOCE, while SOCE was absent in nontransfected neurons (from the same coverslips; Fig. 4D).
Figure 4.
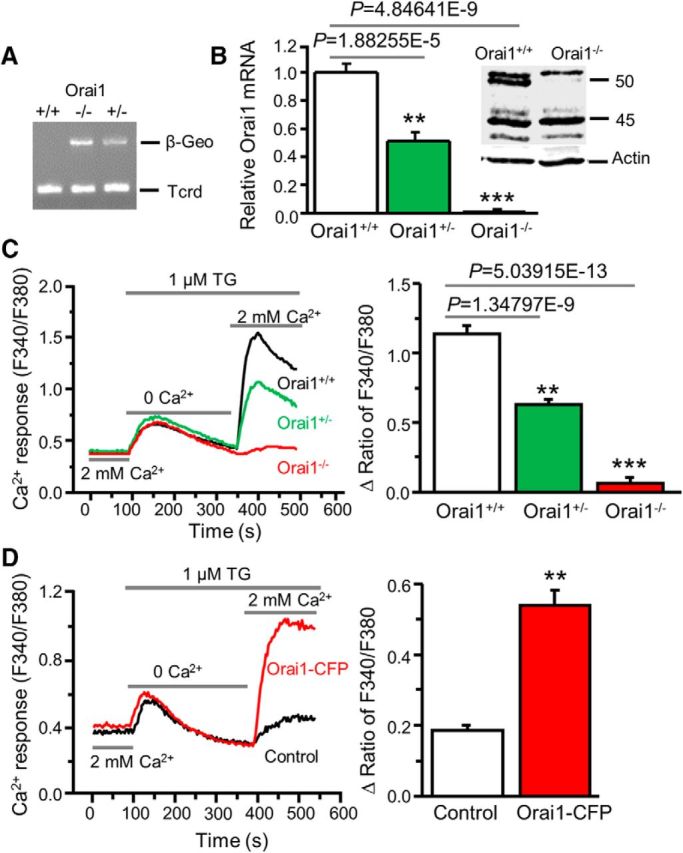
Orai1 deficiency abolishes SOCE in dorsal horn neurons. A, β-Geo expression in Orai1 knock-out (Orai1−/−), heterozygous (Orai1+/−), and wild-type (Orai1+/+) mice. T-cell receptor delta chain (Tcrd) is a control for presence of DNA. B, Orai1 mRNA expression (relative to GAPDH and normalized to Orai1+/+) in Orai1+/+, Orai1+/−, and Orai1−/− mice (n = 5–6 mice, F = 94.5, p values indicated in graph). Inset, Western blot of Orai1 protein extracted from spinal cord tissues of Orai1+/+ and Orai1−/− mice. C, TG-induced Ca2+ entry in Orai1+/+, Orai1+/−, and Orai1−/− dorsal horn neurons (n = 26–34 neurons, F = 44.1, p values indicated in graph). D, TG-induced Ca2+ entry in Orai1−/− neurons transfected with Orai1+/+ Orai1-CFP or GFP (as control; n = 17–19 neurons, df = 16, t = 7.58176, p = 1.10506E-6). Values represent mean ± SEM; **p < 0.01, ***p < 0.001 compared with Orai1+/+ or control neurons by one-way ANOVA or the paired t test.
Orai1 is required for TG-induced modulation of A-type currents and neuronal excitability in the spinal cord dorsal horn
To determine whether Orai1 modulates A-type channels, we recorded outward potassium currents in Orai1−/− mice and their littermate Orai1+/+ mice. The peak of total potassium currents, A-type currents, and sustained currents were measured and compared between Orai1−/− and Orai1+/+ neurons. They were indistinguishable between the two groups (data not shown), suggesting that Orai1 does not change the function of A-type channels at the basal condition. However, TG-induced modulation of A-type currents was robust in dorsal horn neurons from Orai1+/+ littermates (Fig. 5A) and absent in Orai1−/− neurons (Fig. 5B). To determine whether wild-type Orai1 rescues TG-induced modulation of A-type currents in Orai1−/− neurons, we transfected wild-type Orai1-CFP plasmid to Orai1−/− neurons. As expected, TG-induced modulation of A-type currents was completely rescued in these neurons (Fig. 5C). These results demonstrated that Orai1 is necessary for TG-induced modulation of A-type channels.
Figure 5.
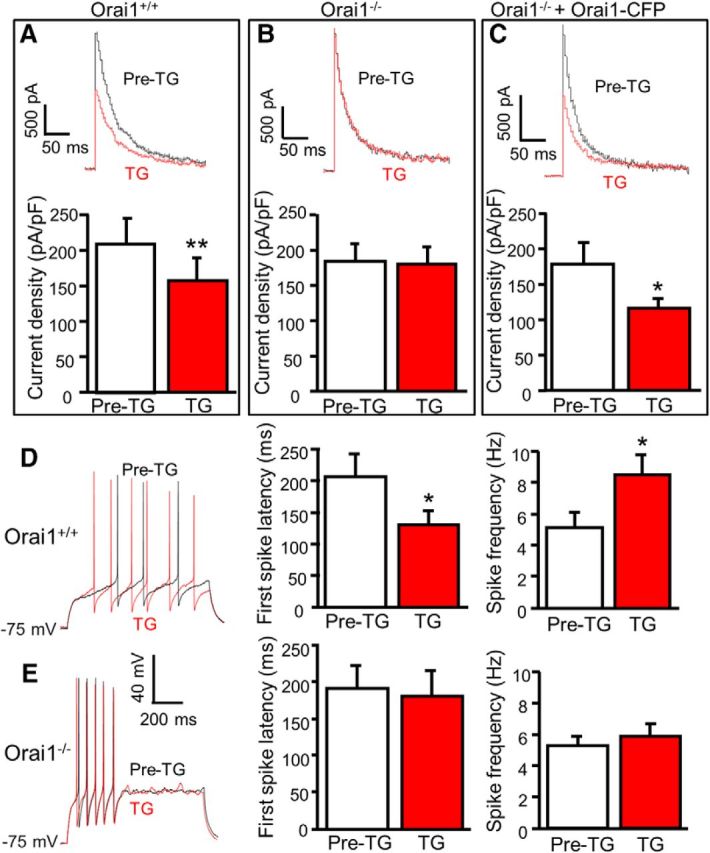
Orai1 is required for TG-induced modulation of A-type currents and neuronal excitability. A–C, TG-induced modulation of A-type currents before (Pre-TG) and 5 min after application of 2 μm TG in Orai1+/+ neurons (A; n = 6, df = 5, t = 5.020, p = 0.004), Orai1−/− neurons (B; n = 11, df = 10, t = 0.461, p = 0.655), and Orai1−/− neurons transfected with Orai1+/+ Orai1-CFP (C; n = 8, df = 7, t = 3.457, p = 0.011). D, TG-induced modulation of action potentials before (Pre-TG) and 5 min after application of 2 μm TG in SG neurons from Orai1+/+ mice (n = 7). For the first-spike latency, df = 6, t = 2.905, p = 0.027; for spike frequency, df = 6, t = 3.243, p = 0.018. E, TG-induced modulation of action potentials before (Pre-TG) and 5 min after application of 2 μm TG in SG neurons from Orai1−/− mice (n = 15). For the first-spike latency, df = 14, t = 0.140, p = 0.891; for spike frequency, df = 14, t = 1.468, p = 0.164. Values are means ± SEM; *p < 0.05, **p < 0.01 compared with pre-TG by the paired t test.
To determine whether Orai1 contributes to neuronal excitability, we performed current-clamp recordings in SG neurons of adult mouse slices prepared from Orai1−/− and Orai1+/+ littermates. The resting membrane potential, rheobase, and input resistance were indistinguishable between Orai1−/− neurons and Orai1+/+ neurons (data not shown). However, TG-induced modulation of action potential properties was absent in dorsal horn neurons from Orai1−/− mice while TG-induced modulation of neuronal excitability was robust in SG neurons from Orai1+/+ littermates (Fig. 5D,E). These findings suggest that functional expression of Orai1 is not essential for basal neuronal excitability, but it is required for SOC activation-induced modulation of neuronal excitability.
ERK signaling mediates TG-induced modulation of A-type currents and neuronal excitability
We and others have reported that ERK activation downregulated A-type currents in dorsal horn and hippocampal neurons (Adams et al., 2000; Hu et al., 2003). It has been shown that SOC activation leads to ERK activation in T cells and neutrophils (Denys et al., 2004; Kim et al., 2015). To determine whether depletion of Ca2+ stores leads to ERK activation in dorsal horn neurons, we performed Western blot analysis using an antibody specific for phosphorylated (active) ERK1/2 (p44/42). Dorsal horn neurons were cultured in six-well plates for 24 h and TG was added to cultured neurons for 1, 5, 10, 30, 60 min. Phosphorylation of ERK1/2 (p-ERKs) was drastically increased after 5 min, which lasted for ≥60 min (Fig. 6A). To determine whether TG-induced ERK activation is mediated by Orai1, we cultured Orai1+/+ and Orai1−/− neurons from the same litter. Orai1+/+ neurons showed a significant increase in p-ERKs after TG treatment, while TG did not induce ERK activation in Orai1−/− neurons (Fig. 6B). These results indicate that Orai1 is required for TG-induced ERK activation and that ERKs are downstream in the signaling cascade of SOC activation.
Figure 6.
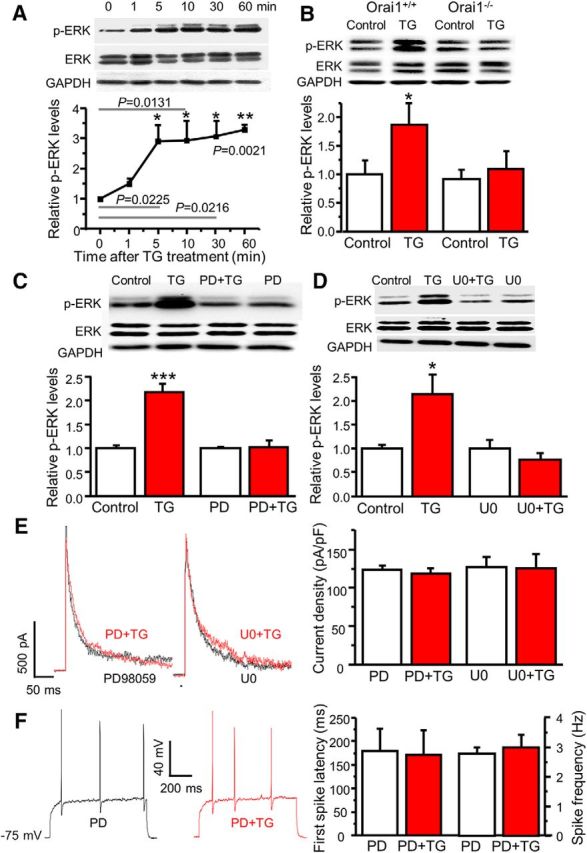
SOCs mediate modulation of A-type currents via ERKs. A, The time course of TG-induced ERK activation (n = 4, F = 67.7, p values indicated in the graph). B, TG-induced ERK activation in cultured dorsal horn neurons from Orai1+/+ and Orai1−/− littermates. For Orai1+/+ mice, n = 7 samples, df = 6, t = 3.25629, p = 0.0173; for Orai1−/− mice, n = 7 samples, df = 6, t = 0.990, p = 0.360. C, The effects of PD98059 (PD) on TG-induced ERK activation. For vehicle treatment, n = 10 samples, df = 9, t = 6.43439, p = 0.00012; for PD treatment, n = 8 samples, df = 7, t = 0.072, p = 0.944. D, The effects of U0126 (U0) on TG-induced ERK activation. For vehicle treatment, n = 4 samples, df = 3, t = 4.42385, p = 0.02145; for U0 treatment, n = 4 samples, df = 3, t = 2.415, p = 0.0946. E, The effects of PD and U0 on TG-induced modulation of A-type currents. For PD treatment, n = 9 neurons, df = 8, t = 1.289, p = 0.233; for U0 treatment, n = 7 neurons, df = 6, t = 0.206, p = 0.843. F, The effects of PD on TG-induced modulation of action potentials. For the first-spike latency, n = 8 neurons, df = 7, t = 0.760, p = 0.472. For spike frequency, n = 8 neurons, df = 7, t = 1.000, p = 0.351. Values are means ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001 compared with control by the two-sample or paired-sample Student's t test.
To determine whether TG-induced modulation of A-type currents is mediated by the activation of ERKs, we used PD98059 and U0126, inhibitors of MEK (an upstream regulator of ERKs). To confirm whether 20 μm PD98059 and 0.5 μm U0126 are effective in blocking TG-induced ERK activation, we pretreated neurons with PD98059 or U0126 for 10 min. Both PD98059 and U0126 did not change the basal p-ERKs, but TG-induced ERK activation was eliminated in the presence of PD98059 or U0126 (Fig. 6C,D). Patch-clamp results showed that while PD98059 and U0126 had no significant effect on A-type currents, TG-induced modulation of A-type currents was abolished by PD98059 or U0126 (Fig. 6E). Consistently, TG-induced changes in the first-spike latency and spike frequency were absent in the presence of PD98059 (Fig. 6F). These results suggest that SOCE-induced modulation of A-type currents and neuronal excitability is mediated by the ERK signaling cascade.
PKC is involved in the SOC–ERK pathway
Since Ca2+ cannot directly phosphorylate ERK, what mediates SOCE-induced ERK activation? It is well known that PKC can be activated by Ca2+ and plays an important role in various signal-transduction pathways. We have previously demonstrated that PKC is an upstream activator of ERKs in dorsal horn neurons (Hu and Gereau, 2003). To determine whether PKC is involved in the SOC–ERK pathway, we used a potent PKC inhibitor, GF. When cultured neurons were pretreated with 0.5 μm GF for 5 min, TG-induced ERK activation was drastically reduced (Fig. 7A). To rule out the possibility that GF has a direct effect on SOCE, we performed calcium imaging recordings. SOCE was intact in the presence of GF (Fig. 7B,C). Application of 0.5 μm GF slightly increased A-type currents by 10%. Importantly, SOCE-mediated modulation of A-type currents was eliminated (Fig. 7D,E). These results suggest that PKC is downstream of SOCE and upstream of ERKs.
Figure 7.

PKC is involved in the SOC–ERK pathway. A, The effect of GF109203X (GF) on TG-induced ERK activation (n = 8 samples, F = 13.3, p values indicated in the graph. B, Representative TG-induced Ca2+ responses recorded in neurons pretreated with vehicle (Control) or GF. C, Summary of the effect of GF on TG-induced SOCE presented as the peak amplitude (n = 9–11 neurons, df = 19, t = 0.368, p = 0.717). D, The effect of GF on TG-induced modulation of A-type currents. E, Summary of the effects of GF on TG-induced modulation of A-type currents. For vehicle treatment, n = 5 neurons, df = 4, t = 5.868, p = 0.0042; for GF treatment, n = 6 neurons, df = 5, t = 0.941, p = 0.390. Values are means ± SEM, **p < 0.01, ***p < 0.001 compared with the control group by one-way ANOVA or the Student's t test; ##p < 0.01 compared with the TG group.
Orai1 deficiency attenuates painful stimuli-induced nociception
To determine whether Orai1 deficiency alters pain sensitivity, we first evaluated locomotor activity and motor function in Orai1−/− mice and their Orai1+/+ littermates (at the age of 8–10 weeks) using the open-field and accelerating-rotarod tests. The distance traveled during a 30 min trial was indistinguishable between Orai1−/− and Orai1+/+ mice (Fig. 8A). In the rotarod test, drop latency varied in both Orai1−/− and Orai1+/+ mice, and there was no difference between the two groups (Fig. 8B). We then measured the basal sensitivity to mechanical and mild thermal stimuli using von Frey filaments and the Hargreaves' method. Paw withdrawal threshold and latency were not altered in Orai1−/− mice (Fig. 8C,D). However, Orai1 mutant mice showed decreased sensitivity to noxious mechanical and thermal stimuli compared with their Orai1+/+ littermates in the tail-pressure, tail-flick, and hot-plate tests (Fig. 8E–G). These results suggest that Orai1 plays an important role in nociception.
Figure 8.

Orai1 deficiency attenuates physical painful stimuli-induced nociception. A, Spontaneous activity (n = 16–17 mice, df = 31, t = 0.143, p = 0.887). B, Locomotor activity (n = 13–18 mice, df = 29, t = 0.073, p = 0.523). C, Basal mechanical sensitivity (n = 16–17 mice, df = 32, t = 0.645, p = 0.887). D, Basal thermal sensitivity (n = 16–17 mice, df = 32, t = 0.315, p = 0.754). E, Pain threshold to noxious mechanical stimulation (n = 8 mice each, df = 14, t = 3.533, p = 0.0033). F, Tail-flick threshold to noxious heat stimulation (n = 6–7 mice, df = 11, t = 5.035, p = 0.00038). G, Heat-induced pain response (n = 6–8 mice). For 52°C, df = 12, t = 2.883, p = 0.0137; for 56°C, df = 12, t = 4.227, p = 0.0011. Values are means ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 compared with Orai1+/+ mice by the Student's t test.
Orai1 deficiency attenuates formalin-induced nociceptive behavior
The ERK signaling cascade plays a key role in pain plasticity (Ji et al., 2009; Alter et al., 2010; Otsubo et al., 2012). Our data above showed that Orai1-mediated modulation of A-type currents and neuronal excitability is ERK-dependent in dorsal horn neurons. We hypothesize that Orai1 plays a role in ERK-dependent pain behavior. We performed the formalin test because the second phase of formalin-induced nocifensive behavior is ERK-dependent (Ji et al., 1999; Karim et al., 2001). Intraplantar injection of 15 μl of formalin (2%) subcutaneously resulted in intensive spontaneous licking, lifting, flinching/shaking of the injected paw with a classic biphasic nociception in littermate Orai1+/+ mice. The second phase of nociception was almost eliminated in Orai1−/− mice. Interestingly, the first phase was not significantly reduced in these mice (Fig. 9A,B). To confirm that reduction of formalin-induced nociceptive behavior in Orai1−/− mice is mediated by ERKs, we performed immunostaining using p-ERK antibody. Formalin induced robust activation of ERKs (increased number of p-ERK-positive cells) in the ipsilateral dorsal horn (Fig. 9C,D). However, formalin-induced ERK activation was markedly decreased in Orai1−/− mice (Fig. 9C,D). To confirm this result, we also performed Western blot analysis. As expected, formalin injection increased p-ERK in the ipsilateral side of the spinal cord (L4–L5 segments) from Orai1+/+ mice, which was not observed in Orai1−/− mice (Fig. 9E,F). These results demonstrated that Orai1 plays a critical role in nociceptive behavior through the ERK cascade.
Figure 9.

Orai1 deficiency attenuates formalin-induced nociceptive behavior. A, Formalin-induced spontaneous nociceptive behavior in Orai1+/+ and Orai1−/− mice. Two-way ANOVA revealed significant differences between genotypes (F(1,20) = 26.4, p = 0.0001). The adjusted p values are indicated in the graph. B, Summary of the first (5–10 min) and second (15–60 min) phases of formalin-induced nociceptive behavior in Orai1+/+ and Orai1−/− mice (n = 10–12 mice; for the first phase, df = 20, t = 1.752, p = 0.095; for the second phase, df = 20, t = 6.388, p = 3.11837E-6). C, Confocal images of p-ERKs in the spinal cord dorsal horn at 8 min after formalin injection. D, The number of p-ERK-positive cells after formalin injection (n = 5–6 mice; F = 82.08247, p = 1.07806E-10). The Bonferroni-adjusted p values for multiple comparisons are indicated in the graph. E, Western blots of p-ERKs and ERKs in the spinal cord (L4–L5 segments) at 8 min after PBS or formalin injection. F, Summary of formalin-induced ERK activation (n = 4–5 mice). The p values are indicated in the graph. Values are means ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 compared with Orai1+/+ mice; #p < 0.05, ###p < 0.001 compared with the contralateral paw by two-way repeated-measures ANOVA or the Student's t test.
Orai1 deficiency diminishes carrageenan-induced pain hypersensitivity
Our study and other studies have shown that carrageenan-induced pain hypersensitivity is associated with ERK activation and A-type channel function in the spinal cord dorsal horn (Galan et al., 2002; Hu et al., 2006). To determine whether Orai1 is involved in carrageenan-induced inflammatory pain, we injected 2% carrageenan (15 μl) into the plantar surface of the right hindpaw of Orai1+/+ and Orai1−/− mice. The Orai1+/+ mice developed robust hypersensitivity to mechanical stimuli in both the ipsilateral and contralateral paws at 3 and 24 h after injection of carrageenan, while the mechanical hypersensitivity was markedly reduced in the ipsilateral paw and abolished in the contralateral paw of Orai1−/− mice (Fig. 10A,B). Thermal hypersensitivity was observed in the ipsilateral paw of both Orai1+/+ and Orai1−/− mice. However, thermal hypersensitivity in Orai1−/− mice was less profound (Fig. 10C). We also measured weight bearing, an alternative nonreflex readout. Weight distribution in hindlimbs was significantly impaired in carrageenan-injected Orai1+/+ mice at 5 and 24 h time points, while Orai1−/− mice showed less impairment at 5 h and normal distribution after 24 h (Fig. 10D). To determine whether the reduction of pain hypersensitivity in Orai1−/− mice was due to a lack of ERK-dependent modulation in the dorsal horn, we measured p-ERK levels in the dorsal horn after behavioral tests (24 h after carrageenan injection). ERK activation was observed only in the ipsilateral paw of Orai1+/+ mice, but not in that of Orai1−/− mice (Fig. 10E). Interestingly, carrageenan-induced inflammation and inflammatory mediator production in Orai1−/− mice did not differ from those in Orai1+/+ mice (Fig. 10F–H). These results suggest that Orai1 is crucial for inflammatory pain, but not for inflammation.
Figure 10.
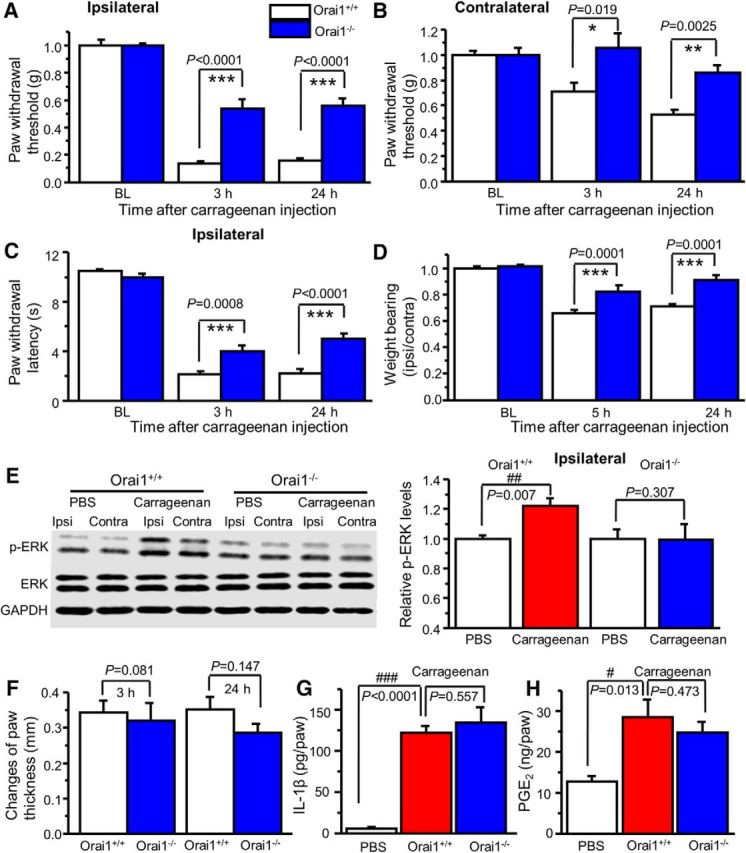
Orai1 deficiency attenuates carrageenan-induced pain hypersensitivity. A, B, Carrageenan-induced mechanical hypersensitivity in the ipsilateral paw (A) and the contralateral paw (B) from Orai1+/+ and Orai1−/− mice. Two-way ANOVA revealed significant differences between genotypes in the ipsilateral paw (F(1,13) = 35.0, p < 0.0001) and the contralateral paw (F(1,13) = 16.7, p = 0.0013). C, Carrageenan-induced thermal hypersensitivity in the ipsilateral paw. Two-way ANOVA revealed significant differences between genotypes (F(1,13) = 16.4, p = 0.0014). D, Weight distribution in Orai1+/+ and Orai1−/− mice. Two-way ANOVA revealed significant differences between genotypes (F(1,13) = 49.7, p < 0.0001). E, Carrageenan-induced ERK activation in the ipsilateral dorsal horn from Orai1+/+ mice, but not from Orai1−/− mice (p values are indicated in the graph). Ipsi, Ipsilateral; Contra, Contralateral. F, Paw thickness changes induced by intraplantar injection of carrageenan in Orai1+/+ and Orai1−/− mice. G, H, Carrageenan-induced IL-1β (G) and PGE2 (H) production in the injected paw from Orai1+/+ and Orai1−/− mice. Values are means ± SEM (n = 6–9 mice; p values indicated in the graph). *p < 0.05, **p < 0.01, ***p < 0.001 compared with Orai1+/+ mice; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with the PBS group by the Student's t test or one-way ANOVA.
Orai1 is responsible for carrageenan-induced neuronal excitability
As shown above, Orai1 activation increased p-ERK and neuronal excitability in vitro. In addition, Orai1 is critical for formalin-induced and carrageenan-induced ERK activation in the ipsilateral side of the spinal cord. To determine whether carrageenan induces a change of neuronal excitability and Orai1 is necessary for this change, we performed current-clamp recordings in ipsilateral SG neurons of spinal cord slices from adult Orai1−/− and Orai1+/+ mice 24 h after intraplantar injection of carrageenan. There was no significant difference of the resting membrane potential between neurons from carrageenan-injected and PBS-injected mice (data not shown). However, we observed that the same amount of current injections caused more action potentials in SG neurons from carrageenan-injected Orai1+/+ mice compared with those from PBS-injected mice (Fig. 11A). Correspondingly, the rheobase was significantly reduced, and the current–frequency relationship curve was shifted upward in neurons from carrageenan-injected Orai1+/+ mice, but not carrageenan-injected Orai1−/− mice, compared with their counterpart PBS-injected mice (Fig. 11B). This finding suggests that Orai1 is critical for carrageenan-induced neuronal excitability.
Figure 11.
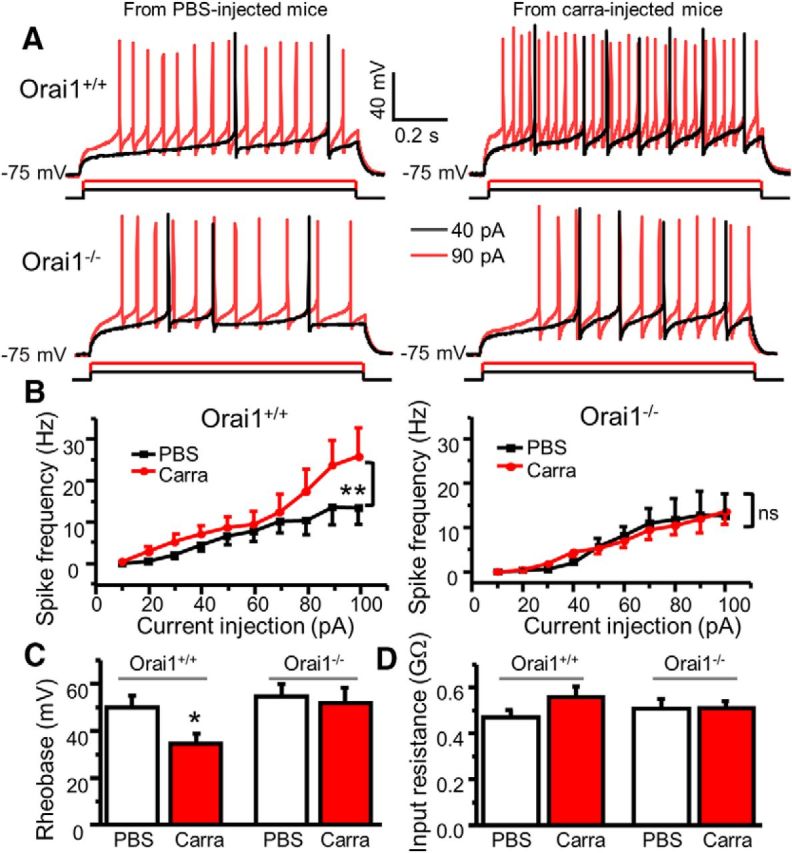
Orai1 is responsible for carrageenan-induced increase in neuronal excitability. A, Representative action potentials generated by 40 and 90 pA current injections recorded in SG neurons from PBS-injected and carrageenan-injected Orai1+/+ and Orai1−/− mice. B, The current–spike frequency curves from PBS-injected and carrageenan-injected Orai1+/+ and Orai1−/− mice. For Orai1+/+ mice, two-way ANOVA revealed significant differences between PBS-injected and carrageenan-injected groups (F(1,234) = 11.4, p = 0.001). For Orai1−/− mice, F(1,389) = 0.046, p = 0.831. C, Summary of changes in rheobase. For Orai1+/+ mice, n = 16–27, df = 41, t = 2.38645, p = 0.0217. For Orai1−/− mice, n = 19–20, df = 37, t = 0.34836, p = 0.729. D, Summary of changes in input resistance. For Orai1+/+ mice, n = 17–33, df = 48, t = 1.34738, p = 0.18418. For Orai1−/− mice, n = 27–33, df = 58, t = −0.05781, p = 0.95407. Carra, Carrageenan. Values are means ± SEM, *p < 0.05, **p < 0.01 compared with the PBS group by two-way ANOVA or the Student's t test.
Discussion
Findings from the present study provide the first evidence that Orai1 plays a critical role in central sensitization and modulates neuronal excitability and A-type currents through the ERK signaling pathway. Our results reveal a functional link between Orai1 and neuronal excitability at both cellular and behavioral levels and support a model in which activation of CRAC channel Orai1 leads to an increase in p-ERKs via PKC, subsequently downregulating A-type channels and increasing neuronal excitability in dorsal horn neurons and pain sensitivity.
The present study demonstrates that TG decreases the first-spike latency and increases spike frequency, suggesting that SOC activation increases neuronal excitability in the spinal cord dorsal horn. A-type channels are believed to be important in controlling the rate of action potential generation and delaying the onset of firing. Indeed, the electrophysiological results show that activation of SOCs by TG or CPA caused downregulation of A-type currents, which are consistent with the crucial role of A-type currents in neuronal activity. We also confirmed that TG-induced increase in neuronal excitability is mediated by modulating A-type channels since blockade of A-type channels by 4-AP abolished TG-induced modulation of A-type currents in cultured neurons and neuronal excitability in spinal cord slices. In line with our hypothesis, Orai1 deficiency abolished TG-induced modulation of A-type currents and neuronal excitability. Together, these data indicate that activation of Orai1 increases neuronal excitability via modulation of A-type channels.
Our findings show that total outward potassium currents and A-type currents were not altered in Orai1−/− neurons under the basal conditions, demonstrating that Orai1 does not regulate A-type channels when it is not activated. What then is the mediator of this modulation? A-type currents are generated by the Kv4 subfamily of voltage-gated potassium channels in several types of neurons, including dorsal horn neurons (Maletic-Savatic et al., 1995; Guo et al., 2005; Hu et al., 2006). We have previously demonstrated that the Kv4 mediates most A-type currents in dorsal horn neurons (Hu et al., 2006). We and others have shown that Kv4.2 is a downstream target of ERKs (Hu et al., 2006; Schrader et al., 2006). It has also been reported that SOCE induces ERK activation in nonexcitable cells (Denys et al., 2004; Kim et al., 2015). Thus, we hypothesized that ERKs mediate interaction between Orai1 and A-type channels. In agreement with our hypothesis, Western blot analysis revealed that activation of Orai1 leads an increase in p-ERKs. Additionally, SOCE-induced modulation of A-type currents and neuronal excitability is mediated by ERK activation. We further showed that PKC is a downstream of SOCE and involved in the SOC-ERK signaling pathway. At the behavioral level, we used the formalin model, where ERKs and Kv4.2-containing A-type channels are required in the second phase (Karim et al., 2001). We found that the formalin-induced second phase of spontaneous nociceptive behavior was nearly eliminated in Orai1−/− mice, which is consistent with our previous finding from Kv4.2 knock-out mice (Hu et al., 2006). There was no significant difference in the first phase between Orai1+/+ and Orai1−/− mice, suggesting that Orai1 may not be involved in formalin-induced acute nociception. The decreased second phase was mediated by ERKs since formalin-induced ERK activation was markedly reduced in Orai1−/− mice. In the carrageenan model, we also observed that Orai1−/− mice showed a marked reduction of ipsilateral pain hypersensitivity. Most interestingly, secondary (contralateral) pain hypersensitivity was abolished in Orai1−/− mice. These findings indicate that Orai1 plays a crucial role in development of central sensitization. Further, to determine whether this role is mediated by modulation of neuronal excitability, we conducted current-clamp recordings from ipsilateral SG neurons in PBS-injected and carrageenan-injected Orai1+/+ and Orai1−/− mice. There were no differences in the resting membrane potential among the groups (data not shown), which is consistent with a previous report (Kurihara et al., 2014). However, we observed a carrageenan-induced increase in neuronal excitability in Orai1+/+ mice, but not in Orai1−/− mice, suggesting that Orai1 is essential to increase neuronal excitability induced by inflammatory pain. This result provides evidence that Orai1 plays a role in central sensitization by modulating neuronal excitability. It is worth pointing out that we found a slight, but not significant increase in p-ERK in the contralateral side of the spinal cord at 24 h after carrageenan injection (data not shown). It is possible that ERK activation may occur in very small populations of contralateral dorsal horn neurons. In addition, activation of ERK was significantly, but not robustly increased in the ipsilateral side of the spinal cord at the 24 h time point. It has been reported that carrageenan-induced maximal ERK activation in the spinal cord was observed at the 1 h time point (Galan et al., 2002). Therefore, we may have missed a significant increase in p-ERK in contralateral spinal cord tissue due to our chosen time point.
We wondered whether Orai1 contributes carrageenan-induced peripheral inflammation since Orai1 plays a role in cytokine production and inflammatory reactions in immune cells (Gwack et al., 2007; Yang et al., 2012; Gaida et al., 2015). Surprisingly, carrageenan-induced edema and cytokine production were intact in Orai1−/− mice. Carrageenan is a nonspecific inflammatory irritant from the cell walls of red algae. Carrageenan is known to stimulate macrophages, leading them to secrete PGE2 and cytokines and thus cause inflammation (Tsuji et al., 2003; Min et al., 2011). A recent report has demonstrated that SOCE is not required for macrophages and dendritic cells to function (Vaeth et al., 2015). This at least partially explains why an Orai1 deficit did not affect carrageenan-induced inflammation.
In summary, we discovered that the activation of CRAC-channel Orai1 increases neuronal excitability by modulating A-type channels in the spinal cord dorsal horn via the PKC–ERK signaling cascade. Orai1 plays an essential role in ERK-dependent pain behavior. Our findings advance our understanding of the mechanisms underlying central sensitization and establish a novel link among SOCs, PKC, ERKs, A-type channels, neuronal excitability, and pain.
Footnotes
This work was supported by National Institutes of Health Grants R21NS077330 and R01NS087033 (to H.H.). We thank Dr. Donald Gill (Penn State College of Medicine) for providing Orai1-CFP.
The authors declare no competing financial interests.
References
- Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M (2008) Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res 103:1289–1299. 10.1161/01.RES.0000338496.95579.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD (2000) The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem 75:2277–2287. [DOI] [PubMed] [Google Scholar]
- Alter BJ, Zhao C, Karim F, Landreth GE, Gereau RW 4th (2010) Genetic targeting of ERK1 suggests a predominant role for ERK2 in murine pain models. J Neurosci 30:11537–11547. 10.1523/JNEUROSCI.6103-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G, Nieswandt B (2009) STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Science Signal 2:ra67. 10.1126/scisignal.2000522 [DOI] [PubMed] [Google Scholar]
- Boettger MK, Uceyler N, Zelenka M, Schmitt A, Reif A, Chen Y, Sommer C (2007) Differences in inflammatory pain in nNOS-, iNOS- and eNOS-deficient mice. Eur J Pain 11:810–818. 10.1016/j.ejpain.2006.12.008 [DOI] [PubMed] [Google Scholar]
- Chang WC, Nelson C, Parekh AB (2006) Ca2+ influx through CRAC channels activates cytosolic phospholipase A2, leukotriene C4 secretion, and expression of c-fos through ERK-dependent and -independent pathways in mast cells. FASEB J 20:2381–2383. 10.1096/fj.06-6016fje [DOI] [PubMed] [Google Scholar]
- Davis FM, Janoshazi A, Janardhan KS, Steinckwich N, D'Agostin DM, Petranka JG, Desai PN, Roberts-Thomson SJ, Bird GS, Tucker DK, Fenton SE, Feske S, Monteith GR, Putney JW Jr (2015) Essential role of Orai1 store-operated calcium channels in lactation. Proc Natl Acad Sci U S A 112:5827–5832. 10.1073/pnas.1502264112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denys A, Aires V, Hichami A, Khan NA (2004) Thapsigargin-stimulated MAP kinase phosphorylation via CRAC channels and PLD activation: inhibitory action of docosahexaenoic acid. FEBS Lett 564:177–182. 10.1016/S0014-5793(04)00361-8 [DOI] [PubMed] [Google Scholar]
- Dixon WJ. (1980) Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20:441–462. 10.1146/annurev.pa.20.040180.002301 [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A (2001) Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron 29:197–208. 10.1016/S0896-6273(01)00190-8 [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441:179–185. 10.1038/nature04702 [DOI] [PubMed] [Google Scholar]
- Gaida K, Salimi-Moosavi H, Subramanian R, Almon V, Knize A, Zhang M, Lin FF, Nguyen HQ, Zhou L, Sullivan JK, Wong M, McBride HJ (2015) Inhibition of CRAC with a human anti-ORAI1 monoclonal antibody inhibits T-cell-derived cytokine production but fails to inhibit a T-cell-dependent antibody response in the cynomolgus monkey. J Immunotoxicol 12:164–173. 10.3109/1547691X.2014.915897 [DOI] [PubMed] [Google Scholar]
- Galan A, Lopez-Garcia JA, Cervero F, Laird JM (2002) Activation of spinal extracellular signaling-regulated kinase-1 and -2 by intraplantar carrageenan in rodents. Neurosci Lett 322:37–40. 10.1016/S0304-3940(02)00078-2 [DOI] [PubMed] [Google Scholar]
- Gao R, Gao X, Xia J, Tian Y, Barrett JE, Dai Y, Hu H (2013) Potent analgesic effects of a store-operated calcium channel inhibitor. Pain 154:2034–2044. 10.1016/j.pain.2013.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XH, Gao R, Tian YZ, McGonigle P, Barrett JE, Dai Y, Hu H (2015) A store-operated calcium channel inhibitor attenuates collagen-induced arthritis. Br J Pharmacol 172:2991–3002. 10.1111/bph.13104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Xia J, Munoz FM, Manners MT, Pan R, Meucci O, Dai Y, Hu H (2016) STIMs and Orai1 regulate cytokine production in spinal astrocytes. J Neuroinflammation 13:126. 10.1186/s12974-016-0594-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH (2011) Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci 31:3536–3549. 10.1523/JNEUROSCI.5053-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszczynska-Biegala J, Kuznicki J (2013) Native STIM2 and ORAI1 proteins form a calcium-sensitive and thapsigargin-insensitive complex in cortical neurons. J Neurochem 126:727–738. 10.1111/jnc.12320 [DOI] [PubMed] [Google Scholar]
- Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM (2005) Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res 97:1342–1350. 10.1161/01.RES.0000196559.63223.aa [DOI] [PubMed] [Google Scholar]
- Guzman R, Valente EG, Pretorius J, Pacheco E, Qi M, Bennett BD, Fong DH, Lin FF, Bi V, McBride HJ (2014) Expression of ORAII, a plasma membrane resident subunit of the CRAC channel, in rodent and non-rodent species. J Histochem Cytochem 62:864–878. 10.1369/0022155414554926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, Rao A (2007) Biochemical and functional characterization of Orai proteins. J Biol Chem 282:16232–16243. 10.1074/jbc.M609630200 [DOI] [PubMed] [Google Scholar]
- Hartmann J, Karl RM, Alexander RP, Adelsberger H, Brill MS, Rühlmann C, Ansel A, Sakimura K, Baba Y, Kurosaki T, Misgeld T, Konnerth A (2014) STIM1 controls neuronal Ca(2) (+) signaling, mGluR1-dependent synaptic transmission, and cerebellar motor behavior. Neuron 82:635–644. 10.1016/j.neuron.2014.03.027 [DOI] [PubMed] [Google Scholar]
- Hu HJ, Gereau RW 4th (2003) ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol 90:1680–1688. 10.1152/jn.00341.2003 [DOI] [PubMed] [Google Scholar]
- Hu HJ, Gereau RW 4th (2011) Metabotropic glutamate receptor 5 regulates excitability and Kv4.2-containing K(+) channels primarily in excitatory neurons of the spinal dorsal horn. J Neurophysiol 105:3010–3021. 10.1152/jn.01050.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Glauner KS, Gereau RW 4th (2003) ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I. Modulation of A-type K+ currents. J Neurophysiol 90:1671–1679. 10.1152/jn.00340.2003 [DOI] [PubMed] [Google Scholar]
- Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW 4th (2006) The kv4.2 potassium channel subunit is required for pain plasticity. Neuron 50:89–100. 10.1016/j.neuron.2006.03.010 [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ (1999) Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci 2:1114–1119. 10.1038/16040 [DOI] [PubMed] [Google Scholar]
- Ji RR, Gereau RW 4th, Malcangio M, Strichartz GR (2009) MAP kinase and pain. Brain Res Rev 60:135–148. 10.1016/j.brainresrev.2008.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW 4th (2001) Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci 21:3771–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Li J, Tseng A, Andrews RK, Cho J (2015) NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood 126:1952–1964. 10.1182/blood-2014-10-605261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara T, Sakurai E, Toyomoto M, Kii I, Kawamoto D, Asada T, Tanabe T, Yoshimura M, Hagiwara M, Miyata A (2014) Alleviation of behavioral hypersensitivity in mouse models of inflammatory pain with two structurally different casein kinase 1 (CK1) inhibitors. Mol Pain 10:17. 10.1186/1744-8069-10-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Han ZL, Wang ZL, Xing YH, Sun YL, Li XH, Song JJ, Zhang T, Zhang R, Zhang MN, Xu B, Fang Q, Wang R (2016) BN-9, a chimeric peptide with mixed opioid and neuropeptide FF receptor agonistic properties, produces nontolerance-forming antinociception in mice. Br J Pharmacol 173:1864–1880. 10.1111/bph.13489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE Jr, Meyer T (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15:1235–1241. 10.1016/j.cub.2005.05.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Lenn NJ, Trimmer JS (1995) Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci 15:3840–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Park YJ, Kim DH (2011) Kakkalide and its metabolite irisolidone ameliorate carrageenan-induced inflammation in mice by inhibiting NF-kappaB pathway. Inflammation 34:344–351. 10.1007/s10753-010-9240-1 [DOI] [PubMed] [Google Scholar]
- Muik M, Schindl R, Fahrner M, Romanin C (2012) Ca(2+) release-activated Ca(2+) (CRAC) current, structure, and function. Cell Mol Life Sci 69:4163–4176. 10.1007/s00018-012-1072-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Dougherty KJ, Johnston D (2010) Calcium store depletion induces persistent perisomatic increases in the functional density of h channels in hippocampal pyramidal neurons. Neuron 68:921–935. 10.1016/j.neuron.2010.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsubo Y, Satoh Y, Kodama M, Araki Y, Satomoto M, Sakamoto E, Pagès G, Pouysségur J, Endo S, Kazama T (2012) Mechanical allodynia but not thermal hyperalgesia is impaired in mice deficient for ERK2 in the central nervous system. Pain 153:2241–2252. 10.1016/j.pain.2012.07.020 [DOI] [PubMed] [Google Scholar]
- Prakriya M. (2009) The molecular physiology of CRAC channels. Immunol Rev 231:88–98. 10.1111/j.1600-065X.2009.00820.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Veliçelebi G, Stauderman KA (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169:435–445. 10.1083/jcb.200502019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader LA, Birnbaum SG, Nadin BM, Ren Y, Bui D, Anderson AE, Sweatt JD (2006) ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am J Physiol Cell Physiol 290:C852–C861. 10.1152/aipcell.00358.2005 [DOI] [PubMed] [Google Scholar]
- Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW (2010) Activation and regulation of store-operated calcium entry. J Cell Mol Med 14:2337–2349. 10.1111/j.1582-4934.2010.01168.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa R, Cheng H, Beck A, Ishikawa J, Launay P, Kubota H, Kinet JP, Fleig A, Yamada T, Penner R (2006) A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol Pharmacol 69:1413–1420. 10.1124/mol.105.021154 [DOI] [PubMed] [Google Scholar]
- Tsuji RF, Hoshino K, Noro Y, Tsuji NM, Kurokawa T, Masuda T, Akira S, Nowak B (2003) Suppression of allergic reaction by lambda-carrageenan: toll-like receptor 4/MyD88-dependent and -independent modulation of immunity. Clin Exp Allergy 33:249–258. 10.1046/j.1365-2222.2003.01575.x [DOI] [PubMed] [Google Scholar]
- Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, Zahra A, Kozhaya L, Weidinger C, Philips J, Unutmaz D, Feske S (2015) Ca2+ signaling but not store-operated Ca2+ entry is required for the function of macrophages and dendritic cells. J Immunol 195:1202–1217. 10.4049/jimmunol.1403013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP (2008) Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol 9:89–96. 10.1038/ni1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Pan R, Gao X, Meucci O, Hu H (2014) Native store-operated calcium channels are functionally expressed in mouse spinal cord dorsal horn neurons and regulate resting calcium homeostasis. J Physiol 592:3443–3461. 10.1113/jphysiol.2014.275065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Mo X, Lv J, Liu X, Yuan M, Dong M, Li L, Luo X, Fan X, Jin Z, Liu Z, Liu J (2012) Lipopolysaccharide enhances FcepsilonRI-mediated mast cell degranulation by increasing Ca2+ entry through store-operated Ca2+ channels: implications for lipopolysaccharide exacerbating allergic asthma. Exp Physiol 97:1315–1327. 10.1113/expphysiol.2012.065854 [DOI] [PubMed] [Google Scholar]