

Abstract
Mammalian species differ dramatically in telomere biology. Species larger than 5–10 kg repress somatic telomerase activity and have shorter telomeres, leading to replicative senescence. It has been proposed that evolution of replicative senescence in large-bodied species is an anti-tumour mechanism counteracting increased risk of cancer due to increased cell numbers. By contrast, small-bodied species express high telomerase activity and have longer telomeres. To counteract cancer risk due to longer lifespan, long-lived small-bodied species evolved additional telomere-independent tumour suppressor mechanisms. Here, we tested the connection between telomere biology and tumorigenesis by analysing the propensity of fibroblasts from 18 rodent species to form tumours. We found a negative correlation between species lifespan and anchorage-independent growth. Small-bodied species required inactivation of Rb and/or p53 and expression of oncogenic H-Ras to form tumours. Large-bodied species displayed a continuum of phenotypes requiring additional genetic ‘hits’ for malignant transformation. Based on these data we refine the model of the evolution of tumour suppressor mechanisms and telomeres. We propose that two different strategies evolved in small and large species because small-bodied species cannot tolerate small tumours that form prior to activation of the telomere barrier, and must instead use telomere-independent strategies that act earlier, at the hyperplasia stage.
This article is part of the theme issue ‘Understanding diversity in telomere dynamics’.
Keywords: telomeres, ageing, lifespan, cancer, evolution, mammals
1. Introduction
(a). Evolution of telomere maintenance mechanisms in mammals
(i). Telomerase activity levels correlate with species body mass
Mammalian species differ dramatically in telomere biology, ranging from humans with very short telomeres and limited telomerase activity in the soma to mice with extremely long telomeres and active telomerase in multiple tissues. Research conducted in the past 10 years has revealed important discoveries in the evolution of telomere maintenance mechanisms.
To obtain insights into the evolutionary forces that shape telomere maintenance mechanisms, we quantified telomerase activity across multiple organs in a collection of rodent species (figure 1). Rodents were chosen because this mammalian order contains the greatest number of species. Furthermore, rodent species differ dramatically in maximum lifespan (MLS) and body mass, which aids in distinguishing the effects driven by either lifespan or size. Importantly, both slow ageing and large body size have evolved multiple times in rodents, which also allows parsing out of the effects of shared ancestry. Telomerase activity measured by the TRAP assay [4] across seven organs showed a very strong negative correlation with species body mass, but not with MLS [5] (figure 2). Thus, larger species had lower levels of telomerase activity across tissues. Among small-bodied species with active telomerase, the members of the family Sciuridae (chipmunk, grey squirrel and woodchuck) stood out, as they had extremely high telomerase activity in every tissue tested. However, within this family telomerase activity still showed a linear negative correlation to body mass. The largest rodents such as beaver and capybara had completely repressed telomerase in the soma, similar to human organs. Remarkably, other large mammals, such as cows and sheep, also show repressed telomerase activity in the soma [6,7]. The collection of species was further expanded by the study of Gomes et al. [8], which analysed telomerase expression in fibroblasts from 60 mammalian species. The analysis showed a negative correlation between body mass and telomerase activity in primary fibroblasts. Thus, it appears a universal trend exists among mammals where species with larger body masses evolved repression of telomerase activity. Interestingly, aside from telomerase inhibition, extremely large mammals such as elephants [8] evolved duplications of the p53 gene as an additional mechanism to enhance their tumour suppressor function [9,10].
Figure 1.

Phylogeny and life-history traits for 18 rodent species used in the study. The tree topology and estimated divergence times were derived from Fabre et al. [1] and Meredith et al. [2]. The data for maximum lifespan and adult body mass were obtained from the Animal Ageing and Longevity Database [3].
Figure 2.

Telomerase activity across somatic tissues shows a strong negative correlation with body mass. Telomerase is more active in tissues of small rodents and repressed in the tissues of large rodents [5]. NMR, naked mole rat.
Whether the ancestral state for mammals represents active or repressed telomerase is a subject of debate. Gomes et al. [8] concluded from phylogenetic analysis that in the ancestral mammal telomerase was repressed and, consequently, telomeres were short. However, this is difficult to reconcile with the fact that telomerase is active in the soma of most invertebrates and vertebrates. In addition, the ancestral mammal is believed to have been relatively small, suggesting the presence of active telomerase based on the trends observed.
(ii). Telomere length
Telomere lengths among mammals range from 8 to 12 kb in humans to over 100 kb in laboratory mice. In humans, telomeres shorten with age, and short telomeres are associated with higher mortality in the elderly [11,12], posing the question of whether a relationship exists between telomere length and lifespan across mammalian species.
In rodent species that were examined for telomerase activity, telomere lengths could be classified into three groups: (i) shorter telomeres, observed in large-bodied species with repressed telomerase activity, which are similar to the human telomere length of 10–15 kb; (ii) long telomeres, seen in the majority of small-bodied species, averaging 20–50 kb; and (iii) extremely long telomeres, as observed in laboratory mice and rats, ranging from 50 to 150 kb. Naked mole rats and deer mice, both small, were outliers having shorter telomeres, similar to human. Overall, there was no significant correlation between telomere length and either MLS or body mass among rodents, but a trend could be seen for shorter telomeres in longer-lived species. The analysis of fibroblasts from 60 mammalian species found a significant negative correlation between telomere length and MLS [8]. Importantly, it has been reported that this negative correlation is independent of other lifespan-correlative factors such as body mass and the GC content of mitochondrial DNA [13]. Thus, different relationships between telomere length and lifespan exist within species and across species. While shorter telomeres may be associated with higher mortality within species, on the evolutionary scale, longer-lived species evolved shorter telomeres.
(iii). Replicative senescence
Active telomerase and long telomeres offer multiple benefits related to enhanced cell proliferation, which may contribute to faster wound healing and stronger immune function. Therefore, for repressed telomerase and shorter telomeres to evolve in large-bodied species, there must be a selective pressure acting on these species that outweighs the benefits of long telomeres and active telomerase. It was hypothesized that such a selective pressure is the increased risk of cancer resulting from the large number of cells. The so-called Peto's paradox posits that large-bodied species have statistically higher risk of developing cancer, but in reality are less cancer prone than small-bodied species [14]. Therefore, large species must have evolved additional tumour suppressor mechanisms.
Human fibroblasts have short telomeres and repressed telomerase. As a result, human cells undergo permanent growth arrest, or replicative senescence, triggered by critically short telomeres upon serial passaging in culture [15,16]. Replicative senescence is an important barrier in the way of tumour progression, as malignant tumours must reactivate telomerase or use the alternative lengthening of telomeres mechanism to acquire unlimited proliferation potential [4]. Interestingly, Tacutu et al. found that the genes involved in cellular senescence show a similar pattern of evolutionary conservation to cancer-related genes, suggesting that the genes that control cellular senescence and cancer resistance may have coevolved [17].
Similar to human cells, fibroblasts from large-bodied rodents (figure 3) [18] and a wide range of large mammals [7] undergo replicative senescence in culture. It was proposed that repression of telomerase activity in large-bodied species evolved as a tumour suppressor to counteract the increased risk of cancer resulting from larger body size. Thus, replicative senescence may be the solution to Peto's paradox.
Figure 3.
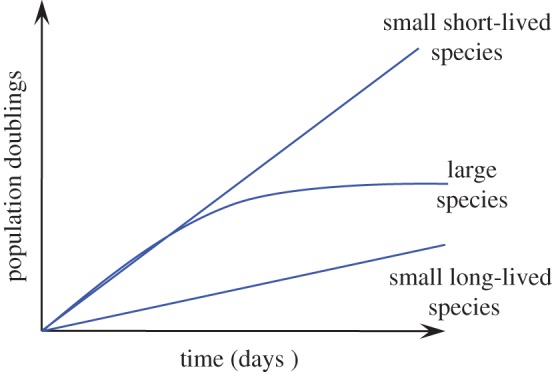
Typical growth curves for fibroblasts from large and small rodent species. Cells from large (greater than 5–10 kg) animals display replicative senescence. Cells from smaller animals are immortal and proliferate continuously, with cells from short-lived small rodents displaying rapid proliferation, and cells from long-lived small rodents displaying slow proliferation in culture. (Online version in colour.)
Where exactly is the body mass threshold where replicative senescence emerges, and is it a sharp transition or is there a gradient of phenotypes? Based on the species examined thus far, the threshold for the evolution of replicative senescence lies around a body mass of 5–10 kg. Species with a body mass in the 5–10 kg range may display intermediate phenotypes with regard to telomerase repression. For example, the paca, a medium-sized South American rodent, has an average telomere length of 12 kb and represses telomerase, but its telomerase becomes spontaneously reactivated when cells approach growth arrest after passaging in culture. Although the selective pressure imposed by cancer offers an elegant explanation for the evolution of replicative senescence in large species, the relationship between telomere maintenance mechanisms and propensity to form tumours has not been experimentally investigated in these animals.
(iv). Telomere-independent anti-cancer mechanisms in small-bodied species
Large body mass increases the risk of malignancy owing to the larger number of cells. However, longer MLS is also expected to increase the risk of cancer owing to a greater number of cell divisions over time. Surprisingly, small-bodied species do not evolve repression of telomerase activity and replicative senescence [5,18]. Fibroblasts from rodents with body mass smaller than 5–10 kg display an unlimited proliferative capacity in culture (figure 3). However, there is a marked difference in proliferation rates between short- and long-lived species. Fibroblasts from short-lived species proliferate much faster, while fibroblasts from long-lived small species proliferate slowly in culture [18]. This slow proliferation likely reflects more stringent cell cycle checkpoint mechanisms in these species that provide additional levels of tumour suppression. Detailed analyses of some of the long-lived small-bodied species revealed unique telomere-independent mechanisms of tumour suppression. These include early contact inhibition due to unique properties of the extracellular matrix in the longest-lived rodent, the naked mole rat, and interferon-mediated cell death in another very long-lived small rodent, the blind mole rat. Cells of the eastern grey squirrel proliferate very slowly in culture and do not display either early contact inhibition or interferon-mediated response, suggesting the presence of a third unique anti-cancer adaptation in this long-lived species. These mechanisms appear to be unique to each species and target overproliferation of cells at the early stages of tumour progression.
(b). The impact of telomere maintenance mechanisms on tumorigenesis
Previous studies suggested that the evolution of telomere maintenance mechanisms is driven by the selective pressure imposed by cancer. However, the cancer rates and the types of mutations driving cancer for the majority of mammalian species have not been studied. Furthermore, it is unknown whether small- and large-bodied species require different numbers of ‘hits’ for malignant transformation. The minimal requirements for malignant transformation have been experimentally compared in human, mouse and naked mole rat cells [19–22]. Mouse fibroblasts require only two ‘hits’ for malignant transformation: inactivation of p53 or Rb and an activating mutation of H-Ras. By contrast, human fibroblasts require five ‘hits’: inactivation of both Rb and p53, inactivation of PP2A, an activating mutation of H-Ras and activation of telomerase. Naked mole rat fibroblasts are unique in that synthesis or accumulation of high molecular weight hyaluronan must be abolished, in addition to inactivation of both Rb and p53 and activation of H-Ras, to achieve malignant transformation. Here, we set out to investigate the requirements for malignant transformation in a collection of 18 rodent species that differ in their telomere maintenance strategies. The collection includes small species with active telomerase such as mouse, rat, hamster, gerbil, deer mouse, chipmunk, squirrels, woodchuck, guinea pig, naked mole rat and blind mole rat, as well as large-bodied species with repressed telomerase such as beaver, porcupine, capybara and paca. In addition to diverse body sizes, species in this collection differ in their MLS from 4 to 32 years (figure 1), which allowed us to examine a relationship between cancer resistance and longevity.
2. Material and methods
(a). Animal samples and cell culture
Rodent animals used in this study were the same as previously described [5,18]. Primary skin fibroblasts were isolated from under-arm skin samples. Cells were recovered from liquid nitrogen and passaged twice before being used for experiments. Cells were grown in EMEM medium (ATCC) supplemented with 15% fetal bovine serum (FBS; Gibco) and 1× penicillin–streptomycin (Corning). Cells were cultured at 37°C in 5% CO2 and 3% O2 except for naked mole rat cells, which were cultured at 32°C in 5% CO2 and 3% O2.
(b). Plasmids
Complementary DNAs (cDNAs) encoding SV40 large T (LT), LT K1, LTΔ434–444, SV40 small T (ST) + LT, H-Ras V12 and hTERT were PCR amplified from corresponding plasmids (Addgene) and subcloned into piggyBac (pPB) expression vector under the CAG promoter. The puromycin selection marker was replaced with the hygromycin resistance gene for the H-Ras V12 plasmid, and with the neomycin resistance gene for the hTERT plasmid to allow for respective selection after integration into the genome.
(c). Transfections
Rodent cells were plated at 5 × 105 cells per 10 cm plate 2 days before transfection, except for naked mole rat cells which were seeded at 2 × 105 cells per 10 cm plate 7 days before transfection. Cells were harvested, resuspended in NHDF transfection solution (Amaxa) and transfected with corresponding plasmid DNA using an Amaxa Nucleofector II on programme U-20, except for porcupine cells, which were transfected using the A-30 programme because of excessive cell death after U-20 transfection.
(d). Soft agar assays
A measure of 106 exponentially growing skin fibroblast cells were transfected with a mixture of 5 µg pPB-LT plasmid, 5 µg pPB-H-Ras V12 plasmid, 1 µg pPB-GFP plasmid and 5 µg plasmid expressing piggyBac transposase (pBase). After transfection, cells were replated and allowed to recover for 1 day before harvesting for soft agar assays. The following day, 1% autoclaved Difco Agar Novel (BD Biosciences) was mixed with 2× MEM medium (Gibco) supplemented with FBS and penicillin–streptomycin at a 1 : 1 ratio. A 3 ml mixture was added into 6 cm plates and allowed to solidify. Live cells were harvested, counted and titrated into 5.0 × 104 cell aliquots. Cells were then resuspended in 1.5 ml 2× complete MEM medium before 1.5 ml of 0.8% agar liquid was added. The mixture was inverted to mix and was immediately poured on top of the solidified 0.5% medium agar. The top layer was allowed to solidify at room temperature and then transferred into incubators; 1 ml 1× complete EMEM medium was added into each plate, which was refreshed every week. The frequency of GFP+ cells was measured in leftover cells, not used for soft agar assay, as an indication of transfection efficiency using a FACS Canto machine (BD Biosciences).
After incubation for four weeks, cells and colonies were fixed by adding 5 ml 70% cold ethanol directly into each agar plate and waiting for 1 h. Ethanol was removed and plates were washed with phosphate-buffered saline (PBS) for three times before adding 2 ml 5 μg ml−1 ethidium bromide staining solution. After staining for 2 h, photos were taken with a Bio-Rad Gel Doc imager under UV light. The experiments were repeated with at least another one unrelated animal for each species.
(e). Colony quantification
The soft agar colonies were quantified using the Quantify One program (Bio-Rad) when there were a lot of colonies grown. Plates with fewer colonies were counted by hand. Oncogenic transformation for each species was calculated by the following formula:
 |
(f). Phylogenetically independent contrast analysis
Phylogenetically independent contrast analysis was performed using the PDAP package which was run in the Mesquite program [23]. Phylogeny information and divergence time of 18 rodents were obtained from Fabre et al. [1] and Meredith et al. [2].
(g). Tumorigenicity assays
NIH-III nude mice (Crl:NIH-Lystbg−J Foxn1nuBtkxid) were used in xenograft studies. Cells from different rodents were transfected with a mixture of plasmids expressing the following different combinations: LT + H-Ras V12 + pBase, STLT + H-Ras V12 + pBase (only for beaver and human cells), LT K1 + H-Ras V12 + pBase, LTΔ434–444 + H-Ras V12 + pBase. Stable integrants were selected with puromycin and hygromycin for about three weeks. Cell colonies were pooled and expanded for xenograft experiments. For paca, capybara, porcupine, beaver and human cells, the hTERT gene was integrated by co-transfection with pPB-hTERT and pBase plasmids and selection with G418. For injection, cells were harvested and resuspended into 4 × 106 cells per 50 µl in PBS, followed by adding the same volume of ice-cold Matrigel (Corning). One hundred microlitres of cell–Matrigel mixture was injected subcutaneously into the flank of the mice close to the hind legs using a 22-G needle. The mice were sacrificed and the tumours were excised and analysed when the following endpoints were reached: tumour sizes exceeding 20 mm in the longest dimension, mice losing greater than 15% of their body weight or mice displaying severe distress. If there were no signs of tumour growth for at least six weeks, the mice were euthanized and dissected to verify the absence of tumours.
3. Results
(a). Cancer resistance coevolves with longevity
To characterize the susceptibility of the animal species to malignant transformation, we analysed the frequency of colony formation in soft agar. Anchorage-independent growth in soft agar is a good indication of tumorigenicity in vivo. Primary skin fibroblasts of 18 rodent species (figure 1) were transfected with LT and H-Ras V12, and anchorage-independent growth was measured after four weeks of growth in soft agar. Transformed cells from different species produced dramatically different numbers of anchorage-independent colonies (electronic supplementary material, figure S1). In general, colonies from telomerase-repressed species, which include paca, capybara, porcupine and beaver, are fewer and smaller than those from telomerase-active species. Interestingly, species within either group also showed a large diversity of susceptibility to oncogenic transformation. Notably, naked mole rat and beaver cells formed very few colonies.
Colonies were quantified and the data subjected to regression analyses against lifespan and body mass of the species. Considering that telomerase suppression presents an additional barrier to tumour formation in large species, we only included small-bodied species in such analyses. The frequency of anchorage-independent colonies in such species showed a significant negative correlation with species MLS (r² = 0.5489, p = 0.0024; figure 4a), but not with body mass (r² = 0.0198, p = 0.63; figure 4b). The correlation between the frequency of anchorage-independent colonies and lifespan remained significant after phylogenetic independent contrast analysis, which was used to correct for phylogenetic non-independence [24] (lifespan: r² = 0.3058, p = 0.040; body mass: r² = 0.0020, p = 0.88). These results demonstrate that longer-lived species are more resistant to oncogenic transformation than shorter-lived species.
Figure 4.

The efficiency of oncogenic transformation negatively correlates with maximum lifespan. Skin fibroblasts from 14 rodent species with active telomerase (electronic supplementary material, figure S1) were transfected with SV40 large T (LT) and H-Ras V12 expressing plasmids together with a GFP plasmid. The anchorage-independent growth was measured in soft agar plates. The efficiency of oncogenic transformation was calculated as follows: no. colonies/(no. seeded cells × frequency of GFP+ cells). Experiments were repeated at least three times and included cell lines from at least two animals of each species. p-values for the uncorrected data were determined by Student's t-test for Pearson's correlation; p-values for phylogenetically corrected data are shown in the text. BMR, blind mole rat; NMR, naked mole rat.
(b). Large-bodied species evolved differential levels of tumour suppression
We next tested the tumorigenic growth of the transformed cells. Skin fibroblasts stably expressing LT and H-Ras V12 (for telomerase-active species) or LT, H-Ras V12 and human telomerase (hTERT) (for telomerase-repressed species) were injected into immunodeficient mice. Tumour growth was analysed upon reaching the endpoints or at least six weeks after injection if there were no signs of tumour growth. All LT + Ras V12 transformed cells gave rise to tumours except cells from naked mole rat, beaver and human (figure 5a; electronic supplementary material, S2a). Tumour growth rate, for the species that formed tumours, showed negative correlation with species MLS and body mass (figure 5b). After phylogenetic correction, the correlation between tumour growth and MLS was borderline significant (r² = 0.2312, p = 0.059), and the correlation between tumour growth and body mass lost significance (r² = 0.0015, p = 0.887).
Figure 5.

Factors required for tumour formation in immunodeficient mice by cells from different species. (a) Rodent cells stably expressing the indicated combinations of oncogenes were assayed for tumorigenicity in immunodeficient mice. The number of tumours formed per number of xenografts injected is shown in parentheses. Representative images of the tumours are shown in electronic supplementary material, figure S2 and figure S4. Asterisk (*) indicates the result from a previous publication [25] that had not been repeated in the current study. 1Low population doubling (PD) cells were used for injection (see electronic supplementary material, figure S3). 2High PD cells were used for injection (see electronic supplementary material, figure S3). (b) Tumour growth rate shows negative correlation with maximum lifespan and body mass. Tumour growth rate was calculated as tumour weight in grams at the endpoint divided by the number of days to the endpoint. The endpoints for the tumour study were defined as either six weeks post injection or the tumours reaching 20 mm in the longest dimension or the mice exhibiting distress in accordance with the guidelines of the University of Rochester Institutional Animal Care and Use Committee.
It was previously demonstrated that a combination of SV40 small T (ST), LT, hTERT and H-Ras V12 is required to fully transform human cells [21]. We found that the same combination of oncogenes also suffices to transform beaver cells (figure 5a; electronic supplementary material, S2a). ST is known to perturb protein phosphatase 2A (PP2A), which regulates multiple mitogenic signalling pathways, including PI3K and Wnt pathways [26]. The requirement for ST in transforming beaver cells suggests that beavers evolved additional tumour suppression mechanisms compared with other shorter-lived telomerase-repressed rodent species, which include pacas, capybaras and porcupines.
It was previously shown that telomerase is required for tumorigenic growth of human cells transformed with ST, LT and oncogenic Ras [27]. Without telomerase expression, human cells expressing LT or a combination of LT and Ras V12 undergo crisis. Surprisingly, paca, porcupine and capybara cells transformed with LT and H-Ras V12 were capable of forming tumours (figure 5a; electronic supplementary material, S2a), suggesting that tumour initiation in these three species does not require telomerase. However, similar to human cells, beaver, capybara and paca cells transformed with LT and H-Ras V12 eventually entered crisis and exhibited massive cell death (electronic supplementary material, figure S3). Importantly, paca and capybara cells in pre-crisis stage failed to form tumours two months after injection (electronic supplementary material, figure S2a), suggesting that telomerase is required for malignant tumour growth in these two species. Interestingly, LT and Ras transformed porcupine cells slowed down their growth and exhibited some cell death at around day 120, but the remaining cells resumed fast proliferation (electronic supplementary material, figure S3).
When the tumours from the telomerase-repressed species were dissected, the tumours formed by capybara and paca cells were hollow inside (electronic supplementary material, figure S2b), suggesting that these tumours were benign and the cells inside the tumour were possibly dying due to telomere-mediated crisis. This is consistent with the crisis exhibited by these cells during in vitro passaging. Thus, telomerase-repressed larger rodents display a continuum of phenotypes requiring increasing numbers of ‘hits’ (porcupine < paca < capybara < beaver) for malignant transformation.
(c). Oncogene cooperation in different species
Wild-type LT binds and inactivates both p53 and pRb tumour suppressor proteins. Inhibition of either p53 or pRb combined with oncogenic Ras suffices to transform mouse cells [22,28]. We next set out to determine the cooperative transforming ability between Ras and the disruption of either of these two tumour suppressors in different rodent cells. The LTK1 mutant inactivates only p53, while the LTΔ434–444 mutant inactivates only pRb and its family members including p107 and p130 [21]. We generated rodent cells co-expressing H-Ras V12 and either LTK1 or LTΔ434–444, and tumorigenic growth of these cells was tested in immunodeficient mice. Similar to mouse cells, cells from species with MLS ≤ 10 years, which include hamster, rat, gerbil, deer mouse, chipmunk and red squirrel, were readily transformed and formed tumours with either oncogene combination (figure 5a; electronic supplementary material, S4). Unexpectedly, the grey squirrel, which is a long-lived rodent species with a MLS of 24 years, was susceptible to oncogenic transformation induced by either oncogene combination (figure 5a; electronic supplementary material, S4). We previously found that primary eastern grey squirrel cells have a very slow proliferation rate [18], suggesting that the grey squirrel may not rely on mechanisms that resist tumour growth, but evolved tight cell-cycle regulation mechanisms to prevent mutation accumulations that initiate tumour formation. Tumours formed with either LTK1 or LTΔ434–444 tended to be smaller than the tumours formed with the wild-type LT, suggesting that inactivation of both p53 and Rb tumour suppressors provides growth advantage.
Interestingly, woodchuck, fox squirrel and paca cells were resistant to one oncogene combination, but not the other one (figure 5a; electronic supplementary material, S4). This result indicates that pRb and p53 have differential cooperative functions with oncogenic Ras in these three species.
Guinea pig, chinchilla, blind mole rat, capybara and porcupine cells could not be fully transformed with either oncoprotein combination (figure 5a; electronic supplementary material, S4), suggesting that pRb or p53 that is not inhibited by LTK1 or LTΔ434–444 is sufficient to suppress tumour growth induced by oncogenic Ras.
4. Discussion
Here, we examined the connection between telomere maintenance, life-history traits, and tumour suppression. Tumorigenesis was induced by introducing viral oncoprotein LT, oncogenic Ras and hTERT into primary fibroblasts of 18 rodent species. Then malignant phenotype of the cells was assayed in vitro in soft agar and in vivo in immunodeficient mice.
In vitro assays in soft agar demonstrated that the combination of LT and oncogenic Ras was sufficient to confer anchorage independence to fibroblasts from all small-bodied species that express telomerase, with the exception of naked mole rat. Cells of the larger, telomerase-inactive species formed either no or rare abortive small colonies. This result demonstrates that repression of telomerase increases resistance to malignant transformation.
Within the small-bodied species group, there was a significant negative correlation between the MLS and the frequency of colonies formed in soft agar. This shows that although LT and oncogenic Ras are sufficient for anchorage-independent growth, longer-lived species evolved additional mechanisms that limit proliferation of pre-malignant cells.
The naked mole rat, with its MLS of 32 years, is an extreme example of a small and long-lived mammal that evolved additional mechanisms of tumour suppression. Naked mole rat fibroblasts could not be transformed by the combination of LT and oncogenic Ras, and our previous studies showed that malignant transformation of naked mole rat cells requires abrogation of high molecular weight hyaluronan [20]. Cells from two other long-lived small rodents, grey squirrel and blind mole rat, whose MLSs are over 20 years, formed colonies, albeit at somewhat lower frequency than the shorter-lived species. The absence of a robust protection from transformation with LT + Ras V12 suggests that the tumour suppressors that evolved in these species may be acting through different mechanisms. Indeed, blind mole rat cells display concerted cell death due to interferon release at the early stages of hyperplasia [29]. In vivo, such a mechanism would be activated prior to the loss of Rb and p53. In the case of grey squirrel, the specific mechanism that evolved in this species is not yet understood. Grey squirrel cells display very slow proliferation, which may be controlled by Rb and/or p53, as LT abrogates this protective mechanism.
In vivo experiments showed that cells from small-bodied telomerase-active species (except naked mole rat) expressing LT + Ras V12 formed tumours in immunodeficient mice. There was a trend towards greater tumour latency in longer-lived species. This result is consistent with anchorage-independent growth in soft agar.
Fibroblasts from telomerase-inactive rodents showed a range of phenotypes upon tumour tests in immunodeficient mice. Porcupine cells formed tumours in mice with just LT and Ras V12; however, these tumours took twice as long to grow as the tumours from most long-lived telomerase-active species. Interestingly, upon serial passaging, porcupine cells transfected with LT + Ras V12 entered crisis, but immortal clones rapidly emerged. This suggests that telomerase can readily become reactivated in porcupine cells. By contrast, no immortal cells emerged in capybara and paca cells undergoing crisis. Consequently, the tumours formed by early passage capybara and paca cells were benign. These tumours were hollow inside and capybara tumours took a very long time to grow. Importantly, late passage capybara and paca transformed cells failed to form tumours. This result indicates that in vivo the tumours from paca and capybara entered crisis due to telomere shortening.
Beaver cells did not form any tumours when transfected with LT + Ras V12. Beaver cells could only be transformed with a combination of LT, ST, Ras V12 and telomerase, which are the same five ‘hits’ required for transformation of human fibroblasts. Interestingly, beaver MLS is almost double that of paca and capybara. Based on these observations, large-bodied telomerase-inactive species examined in our study can be arranged with regard to tumour resistance in the following order: porcupine < paca < capybara < beaver = human. Cumulatively these results indicate that telomerase repression serves as an effective barrier against tumour formation. However, species differ in the ease of telomerase reactivation and evolve additional anti-cancer barriers, such as the requirement for PP2A inactivation in beaver and human.
The results of this study allowed us to test and refine the model for the evolution of tumour suppressor mechanisms [18] (figure 6a). Large-bodied species evolve repression of telomerase activity [5]. The tumorigenesis experiments confirm that repressed telomerase activity provides an added layer of protection against malignant transformation. Therefore, we conclude that repression of telomerase activity and replicative senescence evolved as a tumour suppressor mechanism in response to an increased risk of cancer due to the greater number of cells in the lager-bodied species. Based on the available data, the threshold for the evolution of replicative senescence appears to be between 5 and 10 kg of body mass, with the species close to this threshold having intermediate phenotypes. For instance, the North American porcupine (body mass 8.6 kg) possesses repressed telomerase that can be reactivated in LT + Ras V12 transfected cells.
Figure 6.

A model for the evolution of telomere maintenance and tumour suppressor mechanisms. (a) Body size and lifespan shape the evolution of telomere maintenance and tumour suppressor mechanisms. As species evolved large body mass, their cancer risk increased owing to the greater number of cells in the body. To counteract this risk, large-bodied species evolved repression of somatic telomerase activity and replicative senescence as an additional tumour suppressor mechanism. Replicative senescence represents a late-acting barrier for tumour progression, because it still allows the formation of small tumours prior to the activation of the telomere checkpoint. A long lifespan also increases the risk of cancer, and small, long-lived species, which cannot tolerate the formation of small tumours, evolved telomere-independent tumour suppressor mechanisms. These mechanisms target hyperplasia and manifest in slow cell proliferation in vitro. (b) Small- and large-bodied animals are expected to have different tolerance to tumour size. Mouse and capybara are drawn to scale with a 3 g tumour. Such a tumour would likely affect fitness of a 30 g mouse but would be insignificant in a 55 kg capybara. (Online version in colour.)
Small, short-lived species such as house mouse, hamster and rat require fewer tumour suppressor mechanisms to reach their reproductive age without developing cancer. Fibroblasts from these species express telomerase activity, have longer telomeres, proliferate rapidly and readily form tumours upon transfection with LT + Ras V12. Small long-lived species do not repress telomerase but instead evolve a variety of mechanisms that slow cell proliferation [18,20,29]. These mechanisms counteract the increased risk of malignant transformation due to the greater number of cell divisions their cells undergo over the course of the animal lifespan. Soft agar experiments indicate that the frequency of anchorage-independent colonies negatively correlates with species MLS for the small-bodied animals.
Why have the large and small species used two different strategies for tumour suppression? Why did the small-bodied long-lived animals not evolve telomerase repression? Our in vivo tumorigenesis experiments provide clues to this puzzle. In paca and capybara, the combination of LT and Ras V12 led to formation of benign tumours that eventually would arrest proliferation owing to the telomere checkpoint. However, in a small mouse-sized animal, a 1–3 g tumour would affect fitness and survival. Under experimental conditions, mice with tumours of this size had to be euthanized for humane reasons. However, for a larger animal, a small benign tumour would not have a significant effect on fitness (figure 6b). Therefore, we propose that the telomere barrier does not evolve in small-bodied species because a benign tumour that continues growing prior to activation of the telomere checkpoint would be detrimental for an animal of small size. Therefore, the anti-cancer mechanisms that evolve in long-lived small-bodied animals prevent hyperplasia which occurs at the earlier stages of tumour development.
Active telomerase confers multiple advantages such as faster cell renewal, wound healing and a more efficient immune response. However, the risk of cancer associated with active telomerase outweighs these benefits for large-bodied species. Humans are a typical example of a large-bodied species with repressed somatic telomerase. Multiple age-related pathologies, especially cardiovascular disease [30–33], may be attributed to telomere shortening in humans. However, activating telomerase increases the risk of malignancy. Once we understand the anti-hyperplastic mechanisms that evolved in small long-lived species, it may be possible to activate these mechanisms in humans in combination with telomerase activation to alleviate age-related pathologies due to telomere shortening and mitigate the risk of cancer.
5. Conclusion
Large and small mammals evolved two different types of telomere maintenance. Large mammals with body mass greater than 5–10 kg evolved repression of somatic telomerase activity and replicative senescence. Small mammals are characterized by active telomerase in somatic cells, longer telomeres and lack of replicative senescence. These two telomere maintenance strategies were likely shaped by differential cancer risks across small and large animals. Large species face a greater risk of cancer, and replicative senescence evolved in these animals as an additional anti-cancer mechanism to mitigate this risk. Small species do not rely on replicative senescence, possibly because they cannot tolerate small tumours that form prior to the activation of the telomere barrier. Instead, small species evolved telomere-independent anti-cancer mechanisms that act to slow down cell proliferation and prevent pre-malignant hyperplasia. Species in the 5–10 kg body mass range display intermediate phenotypes with regard to telomere maintenance. Some may repress telomerase, but telomerase could be readily reactivated upon oncogenic insult.
Supplementary Material
Acknowledgements
We thank Apolline Jungels and Taylor Guinan for their help with mouse xenograft experiments and Matthew Simon for critically reading the manuscript.
Ethics
All animal experiments were approved by the University of Rochester Institutional Animal Care and Use Committee, and conform to the legal requirements of the USA.
Data accessibility
This article has no additional data.
Authors' contributions
X.T. designed research; X.T., K.D., R.P. and A.N. performed xenograft experiments and grew cells for PD curves; X.T., A.C.R.Q. and C.H. performed soft agar experiments. A.S. and V.G. designed and supervised research. X.T., A.S. and V.G. analysed results and wrote the manuscript with input from all authors.
Competing interests
We declare we have no competing interests.
Funding
V.G. and A.S. were supported by grants from the US National Institutes of Health (P01AG047200) and Life Extension Foundation.
References
- 1.Fabre PH, Hautier L, Dimitrov D, Douzery EJ. 2012. A glimpse on the pattern of rodent diversification: a phylogenetic approach. BMC Evol. Biol. 12, 88 ( 10.1186/1471-2148-12-88) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meredith RW, et al. 2011. Impacts of the Cretaceous terrestrial revolution and KPg extinction on mammal diversification. Science 334, 521–524. ( 10.1126/science.1211028) [DOI] [PubMed] [Google Scholar]
- 3.Tacutu R, Craig T, Budovsky A, Wuttke D, Lehmann G, Taranukha D, Costa J, Fraifeld VE, de Magalhães JP. 2013. Human ageing genomic resources: integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Res. 41, D1027–D1033. ( 10.1093/nar/gks1155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim NW, et al. 1994. Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. ( 10.1126/science.7605428) [DOI] [PubMed] [Google Scholar]
- 5.Seluanov A, Chen Z, Hine C, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. 2007. Telomerase activity coevolves with body mass not lifespan. Aging Cell 6, 45–52. ( 10.1111/j.1474-9726.2006.00262.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorbunova V, Seluanov A. 2009. Coevolution of telomerase activity and body mass in mammals: from mice to beavers. Mech. Ageing Dev. 130, 3–9. ( 10.1016/j.mad.2008.02.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis T, Kipling D. 2005. Telomeres and telomerase biology in vertebrates: progress towards a non-human model for replicative senescence and ageing. Biogerontology 6, 371–385. ( 10.1007/s10522-005-4901-4) [DOI] [PubMed] [Google Scholar]
- 8.Gomes NMV, et al. 2011. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell 10, 761–768. ( 10.1111/j.1474-9726.2011.00718.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sulak M, Fong L, Mika K, Chigurupati S, Yon L, Mongan NP, Emes RD, Lynch VJ. 2016. TP53 copy number expansion is associated with the evolution of increased body size and an enhanced DNA damage response in elephants. eLife 5, e11994 ( 10.7554/eLife.11994) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schiffman JD, Schmitt DL, Maley CC. 2016. TP53 gene and cancer resistance in elephants–reply. JAMA 315, 1790–1791. ( 10.1001/jama.2016.0457) [DOI] [PubMed] [Google Scholar]
- 11.Cawthon RM, Smith KR, O'Brien E, Sivatchenko A, Kerber RA. 2003. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 361, 393–395. ( 10.1016/S0140-6736(03)12384-7) [DOI] [PubMed] [Google Scholar]
- 12.Steenstrup T, et al. 2017. Telomeres and the natural lifespan limit in humans. Aging 9, 1130–1142. ( 10.18632/aging.101216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lehmann G, Muradian KK, Fraifeld VE. 2013. Telomere length and body temperature-independent determinants of mammalian longevity? Front. Genet. 4, 111 ( 10.3389/fgene.2013.00111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peto R, Roe FJ, Lee PN, Levy L, Clack J. 1975. Cancer and ageing in mice and men. Br. J. Cancer 32, 411–426. ( 10.1038/bjc.1975.242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayflick L, Moorhead PS. 1961. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621. ( 10.1016/0014-4827(61)90192-6) [DOI] [PubMed] [Google Scholar]
- 16.Harley CB, Futcher AB, Greider CW. 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460. ( 10.1038/345458a0) [DOI] [PubMed] [Google Scholar]
- 17.Tacutu R, Budovsky A, Yanai H, Fraifeld VE. 2011. Molecular links between cellular senescence, longevity and age-related diseases - a systems biology perspective. Aging 3, 1178–1191. ( 10.18632/aging.100413) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seluanov A, Hine C, Bozzella M, Hall A, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. 2008. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell 7, 813–823. ( 10.1111/j.1474-9726.2008.00431.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seluanov A, Hine C, Azpurua J, Feigenson M, Bozzella M, Mao Z, Catania KC, Gorbunova V. 2009. Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole-rat. Proc. Natl Acad. Sci. USA 106, 19 352–19 357. ( 10.1073/pnas.0905252106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian X, et al. 2013. High-molecular-mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499, 346–349. ( 10.1038/nature12234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA. 2002. Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 22, 2111–2123. ( 10.1128/MCB.22.7.2111-2123.2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Serrano M, Lee HW, Chin L, CordonCardo C, Beach D, DePinho RA. 1996. Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37. ( 10.1016/S0092-8674(00)81079-X) [DOI] [PubMed] [Google Scholar]
- 23.Garland T, Dickerman AW, Janis CM, Jones JA. 1993. Phylogenetic analysis of covariance by computer-simulation. Syst. Biol. 42, 265–292. ( 10.1093/sysbio/42.3.265) [DOI] [Google Scholar]
- 24.Felsenstein J. 1985. Phylogenies and the comparative method. Am. Nat. 125, 1–15. ( 10.1086/284325) [DOI] [Google Scholar]
- 25.Liang S, Mele J, Wu Y, Buffenstein R, Hornsby PJ. 2010. Resistance to experimental tumorigenesis in cells of a long-lived mammal, the naked mole-rat (Heterocephalus glaber). Aging Cell 9, 626–635. ( 10.1111/j.1474-9726.2010.00588.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westermarck J, Hahn WC. 2008. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 14, 152–160. ( 10.1016/j.molmed.2008.02.001) [DOI] [PubMed] [Google Scholar]
- 27.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. 1999. Creation of human tumour cells with defined genetic elements. Nature 400, 464–468. ( 10.1038/22780) [DOI] [PubMed] [Google Scholar]
- 28.Tanaka N, et al. 1994. Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor Irf-1. Cell 77, 829–839. ( 10.1016/0092-8674(94)90132-5) [DOI] [PubMed] [Google Scholar]
- 29.Gorbunova V, Hine C, Tian X, Ablaeva J, Gudkov AV, Nevo E, Seluanov A. 2012. Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc. Natl Acad. Sci. USA 109, 19 392–19 396. ( 10.1073/pnas.1217211109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fyhrquist F, Saijonmaa O, Strandberg T. 2013. The roles of senescence and telomere shortening in cardiovascular disease. Nat. Rev. Cardiol. 10, 274–283. ( 10.1038/nrcardio.2013.30) [DOI] [PubMed] [Google Scholar]
- 31.Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. 2001. Telomere shortening in atherosclerosis. Lancet 358, 472–473. ( 10.1016/S0140-6736(01)05633-1) [DOI] [PubMed] [Google Scholar]
- 32.Ogami M, et al. 2004. Telomere shortening in human coronary artery diseases. Arterioscler. Thromb. Vasc. Biol. 24, 546–550. ( 10.1161/01.ATV.0000117200.46938.e7) [DOI] [PubMed] [Google Scholar]
- 33.Stone RC, Horvath K, Kark JD, Susser E, Tishkoff SA, Aviv A. 2016. Telomere length and the cancer–atherosclerosis trade-off. PLoS Genet. 12, e1006144 ( 10.1371/journal.pgen.1006144) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This article has no additional data.