

Abstract
Vitamin D serves as a precursor to the potent steroid hormone calcitriol, which has widespread actions throughout the body. Calcitriol regulates numerous cellular pathways that could have a role in determining cancer risk and prognosis. Low Vitamin D levels have been implicated in numerous disease processes including fracture risk, falls, cardiovascular disease, hypertension, diabetes mellitus, and cancers. Metabolite of 1, 25-dihydroxyvitamin D3 (1,25[OH]2D3) regulates numerous genes that control gut physiology and homeostasis. 1,25(OH)2D3 serves various functions such as maintaining the integrity of epithelial barrier and absorption of calcium and phosphate, and the host's defense against pathogens, and the inflammatory response by several types of secretory and immune cells. Although epidemiological data remain inconsistent, and randomized control trials in humans do not yet exist to conclusively support a beneficial role for Vitamin D, results from some correlating studies strongly suggest that Vitamin D deficiency increases the risk of developing cancer and that avoiding deficiency and adding Vitamin D supplements might be an economical and safe way to reduce cancer incidence and improve cancer prognosis and outcome. The present review highlights the role of Vitamin D in cancer of the gastrointestinal tract including esophagus, gastric (stomach), liver, pancreas, and colon.
Keywords: Cancer, Vitamin D receptor, Vitamin D
Introduction
Vitamin D is a pluripotent fat-soluble secosteroid that is essential for a variety of physiological and pathological processes in the human body. Vitamin D has classic effect on calcium homeostasis as well as shows multiple biological actions in different tissues, including the breast, colon, and pancreas.[1,2]
In particular, Vitamin D regulates numerous cellular pathways that affect cell proliferation, differentiation, and apoptosis; henceforth, Vitamin D has been correlated with cancer incidence, prognosis, and mortality.[2,3] The major source of Vitamin D for most people is endogenous exposure to ultraviolet B radiation which converts 7-dehydrocholesterol in skin to Vitamin D, which is then hydroxylated to 25-hydroxyvitamin D (25[OH] D) that belongs to the secosteroid hormone family, and later, the enzyme 1-α-hydroxylase converts 25(OH) D to 1, 25-dihydroxyvitamin D (1,25[OH] 2D) the most active metabolite of Vitamin D. Vitamin D is antiproliferative in cells of the skin, colon, breast, and prostate, among others, and limits proinflammatory stresses.[3,4]
The traditional role of Vitamin D has been centered on the control of calcium and bone metabolism but however recent studies emphasis the role of Vitamin D in the development and progression of various cancers. Because Vitamin D is known to participate in cell cycle regulation, cellular proliferation, apoptosis, angiogenesis and molecular cell signaling, it stands to reason that this metabolite is involved in the tumorigenic activity.[5]
It has been found that low serum Vitamin D levels are associated with increased cancer risk of breast, colon, and prostate, and thus, lack of VDR or with severe Vitamin D deficiency results in increased tumorigenesis.[6]
Vitamin D and Vitamin D Receptor in Gastrointestinal Stromal Tumor Cancer
Vitamin D has an important role in regulating body levels of calcium and mineralization. The hormonal activity of Vitamin D is mediated through its binding with Vitamin D receptor (VDR within the cell nuclei). VDR is expressed in all body tissues, so it also shows extraskeletal effects. VDR is involved in the activation and regulation of Vitamin D and calcium, insulin-like growth factor signaling, inflammation, and estrogen-related pathways. The involvement of VDR in multiple pathways and points of convergence within these pathways indicate the potential importance of VDR in the etiology of cancer.
VDR belongs to the superfamily of transacting transcriptional regulatory factors, which includes the steroid and thyroid hormone receptors as well as the retinoid-X receptors and retinoic acid receptors.[7,8] VDR is encoded by a large gene (>100 kb) located on the chromosome 12q12. The VDR gene encompasses two promoter regions, eight protein-coding exons and six untranslated exons (1a-1f).[6]
It has a promoter region capable of generating multiple tissue-specific transcripts. Vitamin D response elements are further known to control various cell cycle-related processes such as cellular growth, differentiation, apoptosis, invasion, and metastasis of tumor cells involving different genes like p21/WAF1, cyclin A and cyclin E, human nm23.H1 human c-fms, c-fos, c-jun, and c-myc. Therefore, it can be assumed that VDR-mediated signaling pathways and VDR gene polymorphisms may be of importance for cancer.[9]
Vitamin D enters the cell through membrane proteins and binds to the VDR. Vitamin D and VDR form a heterodimer with the retinoid X receptor (RXR) and bind to the Vitamin D responsive element on the respective responsive gene. After binding, transcription and translation occur leading to protein formation, for example, the formation of the calcium-binding protein or osteocalcin. For example, in intestinal cells, Vitamin D binds to VDR synthesizing the calcium-binding protein. Although VDR is predominantly a nuclear protein but its traces have also been found in the cytoplasm of Vitamin D target cells. The interaction between RXR and VDR is essential for VDR transcriptional activity. VDR elements (VDREs) play a major role in initiating gene transcription. The RXR-VDR complex recruits specific coactivator molecules such as steroid receptor coactivators, histone acetyltransferases, and the mediator complex subunit 1. The VDR-RXR complex translocates to the nucleus binding to VDREs that allows for promotion or suppression of specific cellular events, including tumorigenesis.[10]
Esophageal cancer
Esophageal cancer is a relatively rare form of cancer characterized by two subtypes:
Adenocarcinoma and
Squamous cell carcinoma.
It has been shown that heavy smoking or alcohol use increases the incidence of squamous cell esophageal cancer.[11] The diagnosis, treatment, and outcome for patients afflicted with esophageal cancers remain difficult and are diagnosed mainly at the advanced stage with symptoms of dysphagia. The 5-year survival rate is approximately 15%, though, most patients die by the 1st year after diagnosis.[12] Some studies suggest that Vitamin D and VDR may allow for earlier detection or alternative therapeutic strategies; other than this, there are no reliable markers for the diagnosis of esophageal cancer till date. The majority of studies have been inconclusive and controversial. For example, in a study by Trowbridge et al., the authors found that in human esophageal adenocarcinomas, VDR expression declined with tumor dedifferentiation. It was also found that VDR translocated out to the cytoplasm during neoplastic transformation.[13]
The primary precursor to esophageal adenocarcinoma is Barrett's Esophagus, and it has been found that VDR expression is unregulated in Barrett's mucosa when compared to the normal squamous epithelium of the esophagus although no conclusive evidence regarding the definite role of Vitamin D could be demonstrated.[10]
Esophageal adenocarcinoma arises from a background of gastroesophageal reflux disease, and in Barrett's esophagus, the normal stratified squamous epithelium is replaced by condition defined by metaplastic columnar epithelium that predisposes to cancer development.
In Barrett's esophagus, the metaplastic changes within the epithelium occur due to alterations in gene expression. Vitamin D may regulate mechanisms that contribute to an environment associated with metaplastic and neoplastic change in the esophagus.[14]
Refluxed acid and bile can initiate an inflammatory response that stimulates esophageal keratinocytes to release proinflammatory molecules including substance P, platelet-activating factor, and Interleukin-8, which recruit neutrophils and other blood leukocytes and stimulate these cells to produce H2O2, nitric oxide, reactive oxygen species (ROS), and hypochlorous acid. These inflammatory processes have the potential to modify genetic phenotypes causing metaplasia or initiating carcinogenesis. Bile acids are capable of inducing COX-2, a key enzyme that is upregulated in the early stages of inflammation and prostaglandin receptor expression in human esophageal adenocarcinoma cells. A single-nucleotide polymorphism in the promoter of COX-2 may increase the risk of developing esophageal adenocarcinoma.[14]
1,25(OH)2D may alter the progression of metaplasia and neoplasia by mitigating the disease-promoting effects of COX-2 and the genotoxicity of ROS. 1,25(OH)2D may play a role in the direct metabolism of bile acids. Lithocholic acid (LCA), a toxic secondary bile acid, is partially catabolized by CYP3A. Both lithocholic acid and 1,25(OH)2D interact with VDR to upregulate transcription of CYP3A, an enzyme involved in bile acid detoxification thus reducing the risk of initiating carcinogenesis.[14]
Gastric cancer
Gastric cancer arises from any part of the stomach. It is the fourth most common cancer worldwide and incurs poor prognosis, as being diagnosed at an advanced stage. The majority of gastric cancers are due to Helicobacter pylori infection, but dietary factors may also play an important contribution. Smoking as well as alcohol consumption significantly increases the risk of gastric cancer.[10] The most common malignancy of the stomach is adenocarcinoma arising from gastric epithelium. Gastric adenocarcinoma is often asymptomatic, but some nonspecific symptoms reported include indigestion, abdominal discomfort, and appetite loss. Later in the disease phase, there is bleeding which leads to anemia.[15]
Vitamin D plays an important role during tumorigenesis. Increased levels of serum Vitamin D reduce the risk of gastric cancer.[16] Paricalcitol (an analog to calcitriol) suppresses the growth of gastric cancer cells by regulating cell cycle, apoptosis, and inflammation without inducing the hypercalcemia effects. Bao et al.[17] found that direct usage of 1,25(OH)2D3 induces cellular apoptosis in gastric cancer cells and also increased the expression of VDR and CYP24A1 further supporting the antitumoral role that Vitamin D may activate in gastric cancer. Vitamin D acts through the hedgehog signaling pathway and decreases cell viability by the inhibition of the expression of numerous hedgehog signaling target genes including patched1 and Gli1 in gastric cancer cells.[10]
Functional VDR elements have been identified in the promoter of phosphatase and tensin homolog (PTEN), suggesting that Vitamin D may play a role in the regulation of PTEN expression.
Vitamin D substantially promotes apoptosis in undifferentiated gastric malignant cells, specifically HCG-27.[16]
Vitamin D may prevent gastric cancers from progressing by modulating the extracellular microenvironment, as Vitamin D has been shown to alter the expression of multiple genes in the extracellular matrix remodeling. Vitamin D can inhibit Wnt signaling by interrupting the crosstalk between tumor epithelial cells and its microenvironment. Functional VDR elements have been identified in the promoter of PTEN, suggesting that Vitamin D may play a role in the regulation of PTEN expression. In conclusion, Vitamin D level is a significant independent prognostic factor in gastric cancer patients, and Vitamin D deficiency may be associated with poor prognosis.[16]
Liver cancer
Liver cancers are metastatic tumors derived from other organs including breast, colon, lung, and kidney. The two primary cancers that arise from cells within the liver are hepatocellular carcinoma (HCC) (arising from hepatocytes, HCC) and cholangiocarcinoma (CCH, derived from cholangiocytes, cells that line the bile ducts). HCC is a primary tumor of the liver that typically results from viral hepatitis infections or cirrhosis. CCH typically is the result of bile duct damage from diseases such as primary sclerosing cholangitis.[10,18] Numerous studies have shown that both HCC and CCH express high levels of CYP24A1, which leads to lower level of Vitamin D thereby allowing for tumor growth. In these studies, treatment with Vitamin D decreased the proliferative rate in numerous HCC and CCH cell lines.
Vitamin D Regulating Multiple Genes/Proteins in Hepatocellular Cancer
The transforming growth factor beta (TGF-β) signaling pathway is aberrant in fibrosis as well as in liver and gastrointestinal (GI) cancers, with a complex context-dependent role, promoting epithelial–mesenchymal transition, to suppressing and prompting oncogenesis. It is often considered a driving pathway for these specific tumors. TGF-β mediates its effects through type I and type II serine-threonine receptor kinases.[19] The ligand-activated TGF-β receptor complex phosphorylates and activates Smads, specifically the receptor-regulated Smad2 and Smad3 which then form a complex with Smad4 and translocate into the nucleus. Activated Smad complexes additionally recruit transcriptional coactivators and corepressors that regulate a multitude of target genes, leading to complex outcomes that include connective tissue deposition, cell cycle arrest in G1/S phase, induction of apoptosis, immune suppression, as well as tumorigenesis.[19]
Vitamin D is most likely to be an antifibrotic agent. Vitamin D possesses antifibrosis effect both in bone marrow mesenchymal stem cells and hepatic cells, with the increasing expression of VDR caused by Vitamin D, which inhibits the proliferation of hepatic stellate cells and the expression of cyclin D1, tissue inhibitor of metalloproteinase 1, and collagen.[20]
Vitamin D metabolites suppress TGF-β-mediated fibrosis through modulating multiple profibrotic proteins, for instance, lowering collagen I and III expression and raising expression levels of matrix metallopeptidase 8, a metalloproteinase that degrades collagen. Vitamin D and synthetic analogs are also potent cytostatic or apoptotic agents in hepatic malignant cells that express VDR.
Vitamin D treatment restores levels of TGF-β pathway members and suppresses β-catenin in patients with cirrhosis or hepatocellular cancer.[19]
Pancreatic cancer
Pancreatic cancer is a life-threatening disease, with the highest mortality rate among all major cancers. It is a malignant neoplasm that arises from transformed cells of the pancreas. Adenocarcinoma arises in exocrine component of the pancreas is the most common type of pancreatic cancer with poor prognosis. Smoking is a confirmed risk factor.
Abundant research has been done regarding Vitamin D and pancreatic tumor progression. It has been demonstrated that the ductal cells of the pancreas in both normal and adenocarcinoma tissues express the enzyme that catalyzes the conversion of 25(OH) D to 1, 25-dihydroxy Vitamin D. The VDR has been detected in numerous pancreatic cell lines and its expression is greatly decreased when compared to normal pancreatic cells.[10]
VDRs are expressed in pancreatic cancer cell lines as well as in tumor tissue. A set of experiments by Hummel and colleagues found a dysregulation of the Vitamin D system in the islets and the transformed ducts in pancreatic cancer.
Based on genetic analysis, Jones et al. determined a core set of 12 cellular signaling pathways involved in the development of pancreatic cancer, and further, on these sites, calcitriol analogs can have their antitumor effects [Figure 1].[21]
Figure 1.
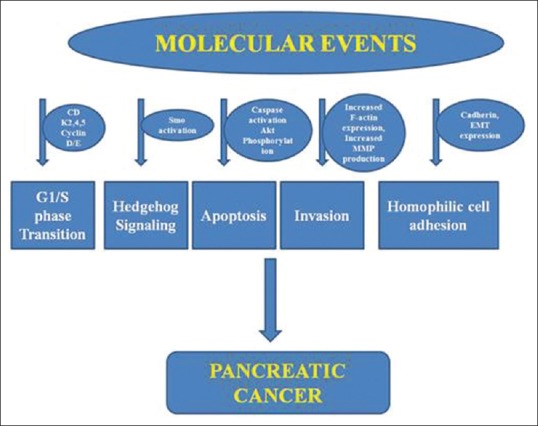
A schematic representation of different potential sites involved in pancreatic cancer
Calcitriol analogs inhibit tumor cell proliferation and their growth (inducing cell cycle arrest at G1/S point), induce apoptosis, and repress migration and invasion of pancreatic cancer cells in cell lines, human pancreatic cancer tissue, and pancreatic xenografts. These analogs affect the Akt pathway resulting in an upregulation of cyclin-dependent kinase inhibitors p21 and p27 and downregulation of cyclin D3 and CDK 4 and 5. Other potential pathways include inhibition of cell growth by inactivation of Smo and the downstream hedgehog pathway, blocking the cadherin switch, repressing F-actin synthesis, and decreasing secretion of matrix metalloproteinases 2 and 9. The calcitriol analog 19-nor-2α-(3-hydroxypropyl)-1α,25(OH)2D3 was able to inhibit epithelial to mesenchymal transition through downregulation of Snail, Slug, and Vimentin expressions. The VDR acts as a master genomic regulator of the pancreatic stellate cell activation program in cells from human pancreatic cancer stroma.[22]
The Vitamin D system is altered in pancreatic cancer. Pancreatic cancer tissue expresses VDRs, but the calcitriol analogs may affect pancreatic cancer tissue by mechanisms that do not involve interaction with its receptors.
Colorectal adenoma and cancer
Colorectal cancer (CRC) is defined as cancer that arises from the colon or rectum (parts of the large intestine) or in the appendix. Most of the CRCs are concerned with lifestyle habit and increasing age with only a minority of cases associated with underlying genetic disorders. The CRC principally starts in the lining of the bowel and if not treated can grow into the muscle layers underneath and the bowel wall. The carcinogenesis can be potentiated by the interactions of environmental (chemicals, infectious agents, radiation) and genetic (mutations, immune system, and hormone dysfunction) factors.[23,24] Symptoms of CRC include increased worsening constipation, bloody stool, weight loss, and loss of appetite. These symptoms increase with increasing age. It has been analyzed that during tumor progression, VDR regulates the effect of Vitamin D on CRC. The regulating enzymes that metabolize Vitamin D are under epigenetic regulation, and Vitamin D (via VDR) regulates proliferation, differentiation, and apoptosis in an autocrine fashion within colonic epithelium. VDR overexpression in CRC is associated with PI3K-AKT pathway and KRAS mutations. Like other GI cancers, treatment with Vitamin D for CRC should be monitored closely and perhaps even personalized to the individual's genetic makeup.[10]
Thus, the polymorphisms of the VDR gene may be possible risk factors for colorectal carcinogenesis.[25]
Further, Vitamin D is associated with the severity of intestinal injury. Vitamin D deficiency is common in inflammatory bowel diseases (IBD) and colon cancer patients. 1,25(OH)2D3 induces the expression of antimicrobial peptide gene cathelicidin and β2-defensins in colon cancer cell lines, bone marrow-derived macrophages, and fresh bone marrow cells. Oral 1, 25(OH)2D3 treatment shifts the composition of the gut bacterial microflora and protects against IBD.[26]
Conclusion
In conclusion, Vitamin D plays a pivotal role in GI malignancies mediated by VDR. Vitamin D affects several cellular mechanisms such as inhibiting differentiation, proliferation, metastasis and invasion, angiogenesis, inducing cell cycle arrest, and apoptosis, thus supporting its anticancer role. Serum levels of Vitamin D may serve as an important biomarker of gastrointestinal malignancies. The exact knowledge of molecular mechanisms involved in various gastrointestinal malignancies further aids in treating these malignancies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Norman AW. From Vitamin D to hormone D: Fundamentals of the Vitamin D endocrine system essential for good health. Am J Clin Nutr. 2008;88:491S–9S. doi: 10.1093/ajcn/88.2.491S. [DOI] [PubMed] [Google Scholar]
- 2.Rosen CJ, Adams JS, Bikle DD, Black DM, Demay MB, Manson JE, et al. The nonskeletal effects of Vitamin D: An Endocrine Society scientific statement. Endocr Rev. 2012;33:456–92. doi: 10.1210/er.2012-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleet JC. Molecular actions of Vitamin D contributing to cancer prevention. Mol Aspects Med. 2008;29:388–96. doi: 10.1016/j.mam.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore C, Murphy MM, Keast DR, Holick MF. Vitamin D intake in the United States. J Am Diet Assoc. 2004;104:980–3. doi: 10.1016/j.jada.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 5.Fleet JC, DeSmet M, Johnson R, Li Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem J. 2012;441:61–76. doi: 10.1042/BJ20110744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llor X, Jacoby RF, Teng BB, Davidson NO, Sitrin MD, Brasitus TA. K-ras mutations in 1,2-dimethylhydrazine-induced colonic tumors: Effects of supplemental dietary calcium and Vitamin D deficiency. Cancer Res. 1991;51:4305–9. [PubMed] [Google Scholar]
- 7.Ingles SA, Ross RK, Yu MC, Irvine RA, La Pera G, Haile RW, et al. Association of prostate cancer risk with genetic polymorphisms in Vitamin D receptor and androgen receptor. J Natl Cancer Inst. 1997;89:166–70. doi: 10.1093/jnci/89.2.166. [DOI] [PubMed] [Google Scholar]
- 8.Haussler MR, Mangelsdorf DJ, Komm BS, Terpening CM, Yamaoka K, Allegretto EA, et al. Molecular biology of the Vitamin D hormone. Recent Prog Horm Res. 1988;44:263–305. doi: 10.1016/b978-0-12-571144-9.50013-2. [DOI] [PubMed] [Google Scholar]
- 9.Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and Vitamin D3 signalling. Nature. 1992;355:446–9. doi: 10.1038/355446a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hargrove L, Francis T, Francis H. Vitamin D and GI cancers: Shedding some light on dark diseases. Ann Transl Med. 2014;2:9. doi: 10.3978/j.issn.2305-5839.2013.03.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349:2241–52. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 12.Polednak AP. Trends in survival for both histologic types of esophageal cancer in US surveillance, epidemiology and end results areas. Int J Cancer. 2003;105:98–100. doi: 10.1002/ijc.11029. [DOI] [PubMed] [Google Scholar]
- 13.Trowbridge R, Sharma P, Hunter WJ, Agrawal DK. Vitamin D receptor expression and neoadjuvant therapy in esophageal adenocarcinoma. Exp Mol Pathol. 2012;93:147–53. doi: 10.1016/j.yexmp.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trowbridge R, Kizer RT, Mittal SK, Agrawal DK. 1,25-dihydroxyvitamin D in the pathogenesis of Barrett's esophagus and esophageal adenocarcinoma. Expert Rev Clin Immunol. 2013;9:517–33. doi: 10.1586/eci.13.38. [DOI] [PubMed] [Google Scholar]
- 15.Vyas N, Companioni RC, Tiba M, Alkhawam H, Catalano C, Sogomonian R, et al. Association between serum Vitamin D levels and gastric cancer: A retrospective chart analysis. World J Gastrointest Oncol. 2016;8:688–94. doi: 10.4251/wjgo.v8.i9.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ren C, Qiu MZ, Wang DS, Luo HY, Zhang DS, Wang ZQ, et al. Prognostic effects of 25-hydroxyvitamin D levels in gastric cancer. J Transl Med. 2012;10:16. doi: 10.1186/1479-5876-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bao A, Li Y, Tong Y, Zheng H, Wu W, Wei C. Tumor-suppressive effects of 1, 25-dihydroxyvitamin D3 in gastric cancer cells. Hepatogastroenterology. 2013;60:943–8. doi: 10.5754/hge121003. [DOI] [PubMed] [Google Scholar]
- 18.Sirica AE. Cholangiocarcinoma: Molecular targeting strategies for chemoprevention and therapy. Hepatology. 2005;41:5–15. doi: 10.1002/hep.20537. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Katz LH, Muñoz NM, Gu S, Shin JH, Jogunoori WS, et al. Vitamin D deficiency promotes liver tumor growth in transforming growth factor-ß/Smad3-deficient mice through WNT and Toll-like receptor 7 pathway modulation. Sci Rep. 2016;6:30217. doi: 10.1038/srep30217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen EQ, Shi Y, Tang H. New insight of Vitamin D in chronic liver diseases. Hepatobiliary Pancreat Dis Int. 2014;13:580–5. doi: 10.1016/s1499-3872(14)60295-2. [DOI] [PubMed] [Google Scholar]
- 21.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barreto SG, Neale RE. Vitamin D and pancreatic cancer. Cancer Lett. 2015;368:1–6. doi: 10.1016/j.canlet.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 23.Heidelbaugh JJ, Tortorello M. The adult well male examination. Am Fam Physician. 2012;85:964–71. [PubMed] [Google Scholar]
- 24.Whitlock EP, Lin JS, Liles E, Beil TL, Fu R. Screening for colorectal cancer: A targeted, updated systematic review for the U.S. Preventive Services Task Force. Ann Intern Med. 2008;149:638–58. doi: 10.7326/0003-4819-149-9-200811040-00245. [DOI] [PubMed] [Google Scholar]
- 25.Gündüz M, Cacina C, Toptas B, Yaylim-Eraltan I, Tekand Y, Isbir T. Association of Vitamin D receptor gene polymorphisms with colon cancer. Genet Test Mol Biomarkers. 2012;16:1058–61. doi: 10.1089/gtmb.2012.0044. [DOI] [PubMed] [Google Scholar]
- 26.Shang M, Sun J. Vitamin D/VDR, probiotics, and gastrointestinal diseases. Curr Med Chem. 2017;24:876–87. doi: 10.2174/0929867323666161202150008. [DOI] [PMC free article] [PubMed] [Google Scholar]