

Inherited photoreceptor degeneration (IPD): The human retina is a highly specialised tissue that enables the perception of light across a range of intensities and colours. It covers about 65% of the inner surface of the eye and contains three layers of cells: the outer nuclear layer (ONL) containing the cell bodies and nuclei of the light-sensitive rod and cone photoreceptors whose photopigment-containing outer segments form the photoreceptor layer; the inner nuclear layer (INL) containing bipolar, horizontal and amacrine cells; and the ganglion cell layer (GCL) from which the optic nerve arises. There are two layers of synaptic connections between these three layers: the photoreceptors synapse with second order neurons, mainly bipolar cells, in the outer plexiform layer (OPL), while in turn the bipolar cells form connections in the inner plexiform layer (IPL) with ganglion cells. The retinal pigment epithelium (RPE) lies directly behind the photoreceptor layer, is heavily pigmented to reduce scattering of light, and is essential for the nourishment, maintenance and metabolism of photoreceptors.
IPD, usually referred to as monogenic retinal disease, is characterized by the dysfunction or death of photoreceptor cells (Bramall et al., 2010). IPD has a large degree of genotypic and phenotypic heterogeneity. Currently, 261 genes and 298 loci have been reported to be associated with IPD (https://sph.uth.edu/RetNet). IPD associated genes function in a wide range of biological processes, including photoreceptor cell development and maintenance, phototransduction, ciliary trafficking, phagocytosis, protein quality control, and metabolism (Bramall et al., 2010). Clinical features in IPD can be classified into rod dystrophy, cone dystrophy, and rod-and-cone dystrophy, depending on which type of photoreceptor cells are initially affected. Photoreceptor cell death in IPD is mainly through caspase-dependent and caspase-independent apoptosis, though necrosis is also reported to be involved (Bramall et al., 2010). Both oxidative stress and inflammation play a critical role in the death of photoreceptor cells.
Oxidative stress in inherited photoreceptor degeneration: Oxidative stress reflects an imbalance between the production of free radicals and their counteraction by the antioxidant defence system. Photoreceptor cells consume oxygen at possibly the highest rate in the body, producing extremely large quantities of reactive oxygen and nitrogen species (RONS) and subsequently causing oxidative damage to macromolecules such as nucleic acids, proteins and lipids (Zhang et al., 2017). Previously we investigated oxidative stress in the retinae of four IPD mouse models (Rho–/–, Prph2rds/rds, Pde6brd1/rd1 and Pde6batrd1/atrd1) and found that they had significantly decreased activities of mitochondrial complex I compared to that of wildtype retinae, an effect that would have been compounded by the consequent increased production of reactive oxygen species (ROS) by the mitochondria. Rho–/– and Pde6brd1/rd1 mouse retinae also had markedly lower levels of the reduced form of the antioxidant glutathione (GSH) (Vlachantoni et al., 2011). Patients with retinitis pigmentosa, a common IPD, exhibited ocular oxidative damage with significantly reduced total antioxidant capacity and a reduced ratio of GSH to GSSG (the oxidised form of glutathione) in the aqueous humour compared to that of controls (Campochiaro et al., 2015). A mix of antioxidants (α-tocopherol, ascorbic acid, α-lipoic acid and Mn(III)tetrakis (4-benzoic acid) porphyrin) significantly reduced cone death and preserved cone function in an IPD mouse model (rd1), possibly through inhibiting oxidative damage (Komeima et al., 2006). Co-expression of catalase and superoxide dismutase (SOD)2 in the photoreceptor cells of an IPD mouse model (rd10) significantly reduced superoxide radicals and carbonyl contents, decreased cone cell death, and preserved cone function (Usui et al., 2009).
Light exposure also generates RONS and this makes photoreceptor cells vulnerable to oxidative damage. Accumulated evidence demonstrates that light damage to photoreceptors is due to the accumulation of all-trans retinal released from photo-activated rhodopsin, particularly when the recycling of all-trans retinal to 11-cis retinal is blocked due to genetic mutations. All-trans retinal molecules can form toxic bis-retinoids, such as N-retinylidene-N-retinylethanolamine (A2E), which accumulate in the RPE cells through phagocytosis (Organisciak and Vaughan, 2010). Most components of bis-retinoids are able to produce RONS when undergoing photo-oxidation. Synthetic antioxidants such as dimethylthiourea have been shown to protect photoreceptors from light-induced damage, functioning as a quencher of free radicals. Natural products also demonstrate a protective role against light-induced retinal damage: some substances act directly as antioxidants, while others are involved in up-regulation of antioxidant gene expression.
Microglia l activation in inherited photoreceptor degeneration: Microglia are a type of macrophage resident in the nervous system including the brain and retina. In the developing retina, microglia precursors may move from retinal vascular vessels and the ciliary body into the retina. After entering the retina, these precursors move along the axon fascicles to their final location in the retina. Microglia ultimately settle in the plexiform layers of the retina where their development completes. They exhibit a ramified form, constantly moving to surveille the retinal neurons. The cellular markers and receptors of microglia indicate the status of microglia (i.e., inactive or active form). These cell surface receptors also provide microglia with special immune functions. Microglia have a phagocytic role in engulfing presynaptic inputs. This function depends on microglia-specific phagocytic signalling pathway, complement receptor 3 (CR3)/C3. Under healthy conditions, microglia fulfil specialized immune tasks by releasing cytokines and neurotrophic factors. A typical interaction between microglia and neurons is the FKN/CX2CR1 system (fractalkine or CX3CL1 and its G protein-coupled receptor, CX3CR1). CX3CL1 is a chemokine in the form of a membrane-bound protein, which can bind to CX3CR1 on microglia and which modulates the activation of microglia (Vecino et al., 2016). CX3CR1 signalling in retinal microglia plays an important role in controlling motility of the thin microglial process that continuously monitor the microenvironment in the healthy retina. Following retinal damage or a change in its condition (such as inflammation, light injury or genetic defects), microglia become activated and change their morphology, with expression of surface markers such as complement receptor, major histocompatibility complex (MHC)-II, and CD68. Subsequently, activated microglia exhibit amoeboid movement and enter the photoreceptor layer to carry out phagocytosis (Karlstetter et al., 2015).
Activated microglia may play a pathological role in the degenerating photoreceptor layer by releasing neurotoxic factors, such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β). In IPD, evidence that the dying rods may attract the resting microglia to mediate phagocytosis in retinitis pigmentosa patients was first presented by Gupta et al. (Gupta et al., 2003). Activated microglia were found in the region of dying rods and it was proposed that the initial rod cell death further triggers the onset of cone death. In the widely used rd1 mouse model which contains nonsense mutation in PDE6b gene, activated microglia were detected from postnatal day 12 in the outer nuclear layer with releasing cytokines (such as TNF-α) and chemokines C-C motif ligand 2 (CCL2), C-C motif ligand 5 (CCL5) and monocyte chemoattractant protein 1 (MCP1) (Karlstetter et al., 2015). Similarly, in rd10 mouse, the rod degeneration is accompanied by infiltration of microglia. The dying rods exhibited the “eat me” signal PtdSer on the cell surface to induce the invasion of activated microglia into the ONL. Genetic ablation of retinal microglia significantly reduced rod cell death in rd10 mice (Zhao et al., 2015). Inhibition of microglia activation by minocycline also protected photoreceptor cells from apoptosis in IPD mouse models rds and rd10 (Bramall et al., 2010; Karlstetter et al., 2015). Given this evidence, activated microglia-mediated phagocytosis appears to be the underlying cell death mechanism contributing to retinal degeneration in IPD.
Coenzyme Q10 (CoQ10) protects retinal cells from oxidative stress and inhibits retinal microglia activation: CoQ10 is a vitamin-like molecule that is endogenously synthesized in all aerobic organisms, existing as oxidized (ubiquinone), partially reduced (semiquinone) and reduced form (ubiquinol) within the body. The lipophilic molecule plays a central role in cellular growth and metabolism through the electron transport chain where it functions as an electron acceptor, collecting electrons from mitochondrial respiratory chain complex I and complex II, then shuttling them to complex III. Additionally, CoQ10 plays a role as an antioxidant, acting as a free radical scavenger and so protecting cells from oxidative damage. It is particularly abundant in tissues that are highly oxygen-demanding, such as cardiac muscle. Although present in the retina, levels are comparatively low relative to other tissues dependent on oxidative metabolism. Following a peak in the third decade of life, the level of CoQ10 declines with age due to decreased synthesis and increased degradation. CoQ10 supplementation has been shown to slow the progression of age-related neurodegenerative diseases, such as Parkinson's disease and age-related macular degeneration (Zhang et al., 2017).
Previous work demonstrated that CoQ10 treatment protected RGC-5 cells (a rat ganglion cell line) from oxidative damage caused by exposure to H2O2, antimycin (mitochondria complex III inhibitor), radiation, or serum starvation (Zhang et al., 2017). We have also treated human retinal pigment epithelium cells (ARPE-19) with H2O2 only or with H2O2 and CoQ10, and found that cell viability was significantly increased in ARPE-19 cell exposure to CoQ10 and H2O2 when compared to cells exposed to H2O2 alone (data not shown). In addition, inflammatory cytokines such as IL-1β were markedly decreased and expression of antioxidant genes (e.g., SOD1) was notably increased in ARPE-19 cells co-exposed to CoQ10 and H2O2 when compared to cells treated with H2O2 only (data not shown).
Senescence accelerated P8 (SAMP8) mice have high levels of protein oxidation and lipid peroxidation in examined tissues, demonstrate some pathological features similar to those presented in age-related neurodegenerative diseases, and have been widely used as a model for oxidative stress and ageing diseases (Morley et al., 2012). To investigate the effect of CoQ10 on retinal microglial activation, we treated SAMP8 mice with CoQ10 via oral administration (500 mg/kg, daily) over a two-month period. We found that microglia migrated into the inner portion of the ONL in untreated SAMP8 mice, while microglia were settled in the OPL in CoQ10-treated SAMP8 mice. We also observed that expression of inflammatory cytokine IL-1β in the OPL was noticeably increased in untreated SAMP8 mice when compared to CoQ10-treated mice (Figure 1).
Figure 1.
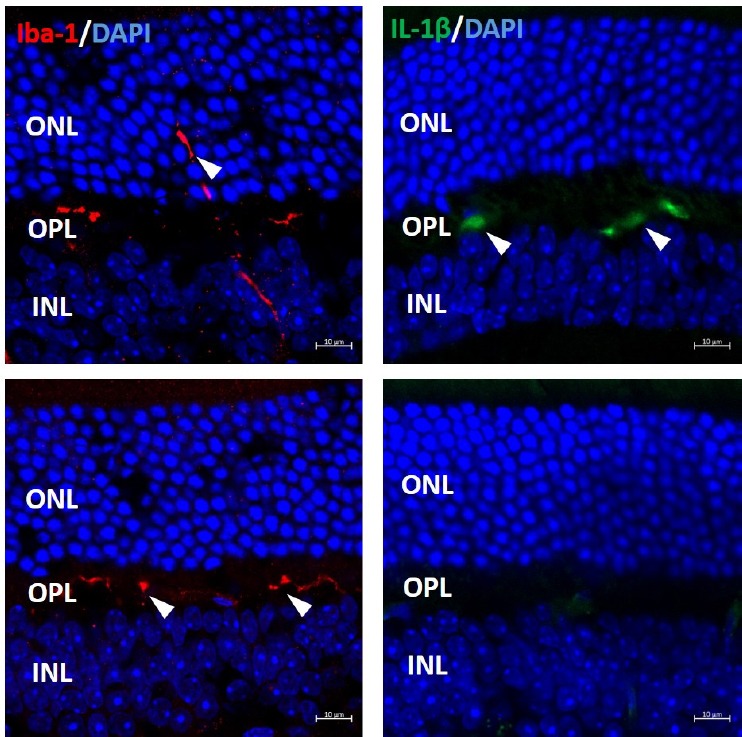
Coenzyme Q10 (CoQ10) treatment blocked microglial migration to outer nuclear layer (ONL) and suppressed inflammation.
Untreated and CoQ10-treated senescence accelerated P8 (SAMP8) mouse eyes were subjected to cryosection and immunostaining with anti-Iba-1 antibody (labelling microglia) and anti-IL-1β antibody. In untreated (No Q10) SAMP8 mouse retina, microglia (arrowheads in left-hand boxes) migrated into the inner portion of ONL; in CoQ10 treated (Q10) SAMP8 retina, microglia stayed in the outer plexiform layer (OPL). Expression of inflammatory cytokine IL-1β (arrowheads in right-hand boxes), was noticeably higher in OPL of untreated SAMP8 retina, but not in CoQ10 treated retina. INL: Inner nuclear layer; Iba-1: ionized calcium bindingadaptor molecule-1; IL-1β: interleukin-1 beta.
Future directions: Although CoQ10 supplement treatment shows protective effects in age-related neurodegenerative diseases and inhibits oxidative damage, inflammation and microglial activation in retina cell lines and SAMP8 retinae, more pre-clinical studies in IPD animal models are needed before it can be used effectively in a therapeutic setting. Mutations in the RPGRIP1 gene can cause Leber congenital amaurosis (LCA), juvenile retinitis pigmentosa (RP) and cone-rod dystrophy (common types of IPD) in humans. Recently, we characterized a zebrafish IPD model that carries a nonsense mutation in the rpgrip1 gene (unpublished). Rpgrip1 mutant zebrafish did not develop photoreceptor outer segments and displayed mislocalization of rhodopsin, early rod degeneration and subsequently cone degeneration (Figure 2) (unpublished), similar to clinical features presented in LCA and juvenile RP patients. The potential for delaying photoreceptor cell death by CoQ10 can be evaluated in this IPD zebrafish. More evaluation may be needed in other mouse IPD models. Based on the preclinical data, clinical trial can be carried out in IPD patients. In conclusion, CoQ10 has the capacity to inhibit oxidative stress, decrease inflammation and suppress microglia activation, which will offer potential therapy for IPD patients.
Figure 2.
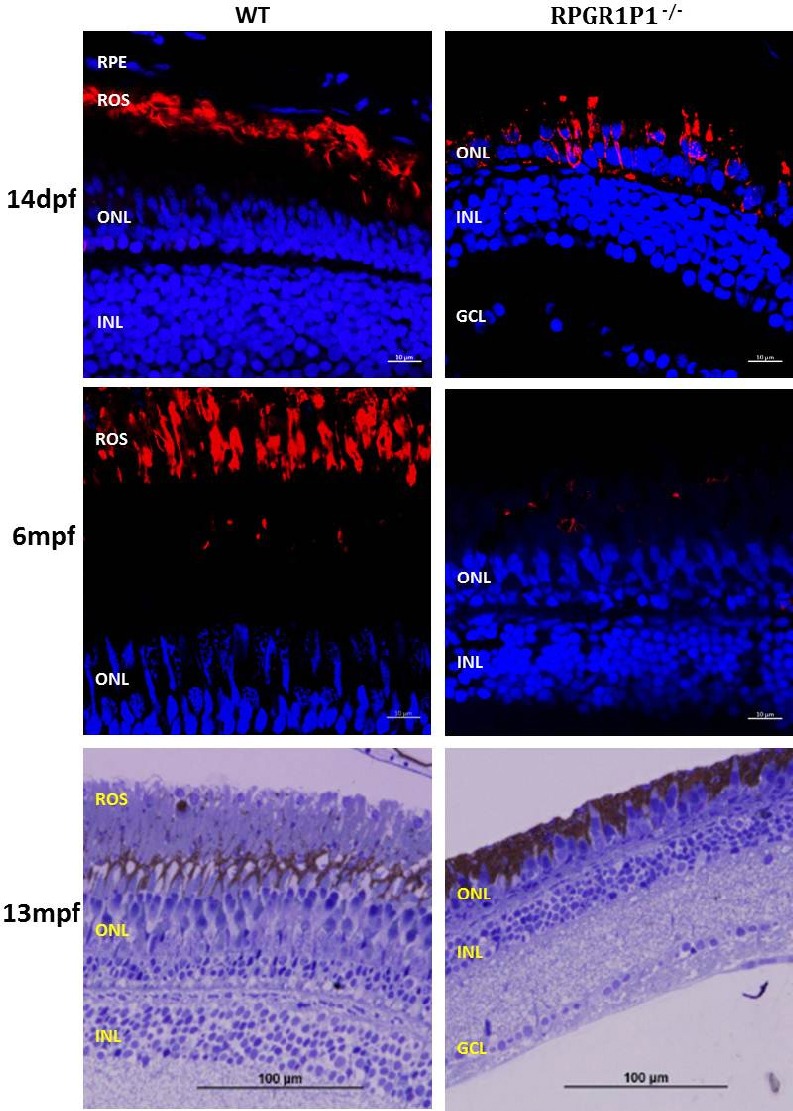
RPGRIP1–/– zebrafish displayed mislocalization of rhodopsin, early rod degeneration and subsequent cone degeneration.
Eyes from both wildtype and RPGRIP1 mutant zebrafish at 14 days (14dpf) and 6 months old (6mpf) were subjected to cryosection and immunostaining using Rho 4D2 antibody (labelling rod outer segments, ROS), mislocalization of rhodopsin was seen in mutant retina at 14dpf, while rod cells showed early signs of degeneration. All rod cells in mutant retina were degenerated at 6 mpf. Zebrafish have long cones (red and green cones) and short cones (blue and UV cones), methylene blue staining showed that only short cones remained in the retina of 13mpf RPGRIP1 mutant zebrafish. RPGRIP1: Retinitis pigmentosa GTPase regulator interacting protein 1; GCL: ganglion cell layer; INL: inner nuclear layer; ONL: outer nuclear layer; RPE: retinal pigment epithelium.
Work in Dr. Shu's lab was supported by the Rosetrees Trust (No. M160 and M160-F1), the Fight for Sight, the Glasgow Children's Hospital Charity (No. YRSS/PSG/2014) and the Visual Research Trust (No. VR2014).
Footnotes
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Jenny R. Lenkowski, Goucher College, USA.
References
- Bramall AN, Wright AF, Jacobson SG, McInnes RR. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu Rev Neurosci. 2010;33:441–472. doi: 10.1146/annurev-neuro-060909-153227. [DOI] [PubMed] [Google Scholar]
- Campochiaro PA, Strauss RW, Lu L, Hafiz G, Wolfson Y, Shah SM, Sophie R, Mir TA, Scholl HP. Is there excess oxidative stress and damage in eyes of patients with retinitis pigmentosa? Antioxid Redox Signal. 2015;23:643–648. doi: 10.1089/ars.2015.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471. doi: 10.1016/s0014-4835(02)00332-9. [DOI] [PubMed] [Google Scholar]
- Karlstetter M, Scholz R, Rutar M, Wong WT, Provis JM, Langmann T. Retinal microglia: just bystander or target for therapy? Prog Retin Eye Res. 2015;45:30–57. doi: 10.1016/j.preteyeres.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Komeima K, Rogers BS, Lu L, Campochiaro PA. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc Natl Acad Sci U S A. 2006;103:11300–11305. doi: 10.1073/pnas.0604056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JE, Armbrecht HJ, Farr SA, Kumar VB. The senescence accelerated mouse (SAMP8) as a model for oxidative stress and Alzheimer's disease. Biochim Biophys Acta. 2012;1822:650–656. doi: 10.1016/j.bbadis.2011.11.015. [DOI] [PubMed] [Google Scholar]
- Organisciak DT, Vaughan DK. Retinal light damage: mechanisms and protection. Prog Retin Eye Res. 2010;29:113–134. doi: 10.1016/j.preteyeres.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui S, Komeima K, Lee SY, Jo YJ, Ueno S, Rogers BS, Wu Z, Shen J, Lu L, Oveson BC, Rabinovitch PS, Campochiaro PA. Increased expression of catalase and superoxide dismutase 2 reduces cone cell death in retinitis pigmentosa. Mol Ther. 2009;17:778–786. doi: 10.1038/mt.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. Glia-neuron interactions in the mammalian retina. Prog Retin Eye Res. 2016;51:1–40. doi: 10.1016/j.preteyeres.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Vlachantoni D, Bramall AN, Murphy MP, Taylor RW, Shu X, Tulloch B, Van Veen T, Turnbull DM, McInnes RR, Wright AF. Evidence of severe mitochondrial oxidative stress and a protective effect of low oxygen in mouse models of inherited photoreceptor degeneration. Hum Mol Genet. 2011;20:322–335. doi: 10.1093/hmg/ddq467. [DOI] [PubMed] [Google Scholar]
- Zhang X, Tohari AM, Marcheggiani F, Zhou X, Reilly J, Tiano L, Shu X. Therapeutic potential of co-enzyme Q10 in retinal diseases. Curr Med Chem. 2017 doi: 10.2174/0929867324666170801100516. doi: 10.2174/0929867324666170801100516. [DOI] [PubMed] [Google Scholar]
- Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, Qian H, Parkhurst CN, Gan WB, Wong WT. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med. 2015;7:1179–1197. doi: 10.15252/emmm.201505298. [DOI] [PMC free article] [PubMed] [Google Scholar]