

Abstract
BACKGROUND
Fibrosis is the pathological consequence of stress-induced tissue remodeling and matrix accumulation. Increased levels of plasminogen activator inhibitor type I (PAI-1) have been shown to promote fibrosis in multiple organ systems. Paradoxically, homozygous genetic deficiency of PAI-1 is associated with spontaneous age-dependent, cardiac-selective fibrosis in mice. We have identified a novel PAI-1-dependent mechanism that regulates cardiomyocyte-derived fibrogenic signals and cardiac transcriptional pathways during injury.
METHODS
Cardiac fibrosis in subjects with homozygous mutation in SERPINE-1 was evaluated with late gadolinium-enhanced cardiac magnetic resonance imaging. A murine cardiac injury model was performed by subcutaneous infusion of either saline or Angiotensin II by osmotic minipumps. We evaluated blood pressure, cardiac function (by echocardiography), fibrosis (with Masson Trichrome staining), and apoptosis (with TUNEL staining), and we performed transcriptome analysis (with RNA sequencing). We further evaluated fibrotic signaling in isolated murine primary ventricular myocytes.
RESULTS
Cardiac fibrosis was detected in 2 otherwise healthy humans with complete PAI-1 deficiency because of a homozygous frameshift mutation in SERPINE-1. In addition to its suppressive role during spontaneous cardiac fibrosis in multiple species, we hypothesized that PAI-1 also regulates fibrosis during cardiac injury. Treatment of young PAI-1−/− mice with Angiotensin II induced extensive hypertrophy and fibrotic cardiomyopathy, with increased cardiac apoptosis and both reactive and replacement fibrosis. Although Angiotensin II-induced hypertension was blunted in PAI-1−/− mice, cardiac hypertrophy was accelerated. Furthermore, ventricular myocytes were found to be an important source of cardiac transforming growth factor-β (TGF-β) and PAI-1 regulated TGF-β synthesis by cardiomyocytes in vitro as well as in vivo during cardiac injury. Transcriptome analysis of ventricular RNA after Angiotensin II treatment confirmed that PAI-1 deficiency significantly enhanced multiple TGF-β signaling elements and transcriptional targets, including genes for extracellular matrix components, mediators of extracellular matrix remodeling, matricellular proteins, and cardiac integrins compared with wild-type mice.
CONCLUSIONS
PAI-1 is an essential repressor of cardiac fibrosis in mammals. We define a novel cardiomyocyte-specific regulatory mechanism for TGF-β production by PAI-1, which explains the paradoxical effect of PAI-1 deficiency in promoting cardiac-selective fibrosis. Thus, PAI-1 is a molecular switch that controls the cardiac TGF-β axis and its early transcriptional effects that lead to myocardial fibrosis.
Keywords: cardiac remodeling, cardiomyopathy, protease inhibitor
Cardiac fibrosis is a common pathologic feature of a broad spectrum of cardiovascular diseases, including pulmonary arterial hypertension, hypertensive heart disease, dilated cardiomyopathy, and myocardial infarction.1–4 Excessive cardiac remodeling after both acute and chronic injury leads excessive deposition of fibrotic extracellular matrix (ECM), including collagen. Scar disrupts normal tissue architecture and adversely affects biochemical and mechanical properties of the myocardium, leading to defects in filling and contractility.5 Heart failure currently represents the leading healthcare expenditure in the United States, and the estimated total lifetime risk for cardiovascular disease is estimated to be 35.3%.6 The presence and extent of clinically detectable cardiac fibrosis is a major independent predictive factor of adverse cardiac outcomes in multiple clinical trials.7 Various molecular mechanisms for cardiac fibrosis have been proposed, yet limited availability of targeted pharmacotherapy highlights the need for further investigation. Currently, organ transplantation represents the only effective cure for end-stage fibrotic disease.
Plasminogen activator inhibitor type I (PAI-1) is a member of the serine protease inhibitor superfamily, inhibits the plasminogen system, negatively regulates the activation of matrix metalloproteinases (MMPs), and is important for tissue homeostasis.8 In organs such as the liver, skin, lung, and kidney, PAI-1 exerts a profibrotic effect by its inhibition of the plasmin-MMP system, which is responsible for the clearance of collagen and fibrin.9,10 Paradoxically, 2 previous studies established that genetic deficiency of PAI-1 promotes spontaneous cardiac-selective fibrosis during aging in mice.3,11 In this study, we report 2 cases of cardiac fibrosis because of complete PAI-1 deficiency in humans, validating our observations in murine models of cardiac fibrosis and broadening the significance of PAI-1 in mammalian cardiovascular disease.
The cardiac-specific effect of PAI-1 deficiency on fibrosis has been attributed to enhanced vascular permeability, MMP-2 and MMP-9 activity, cardiac transforming growth factor-β (TGF-β) content, and endothelial to mesenchymal transition.3,11,12 It has been established that TGF-β induces PAI-1 expression in a variety of cell types,10 and that PAI-1 transcription is inducible by both canonical and noncanonical TGF-β-dependent signaling mechanisms.13,14 Although PAI-1 is a well-defined downstream target of TGF-β, the in vivo and in vitro models described in this study highlight an upstream regulatory role of PAI-1 in TGF-β expression in cardiomyocytes. Because cardiomyocytes are central to cardiac fibrosis through fibroblast-myocyte communication, direct expression and release of TGF-β and TβR signaling,15–17 we provide a molecular explanation for this unique form of fibrotic cardiomyopathy.
METHODS
Mice
Male and female C57BL/6 and PAI-1−/− mice on C57BL/6 background (Jackson Laboratories) were used for the Angiotensin II (AngII) injury model and primary ventricular myocyte isolation. All protocols were approved by the Animal Care and Use Committee and Northwestern University, and all animal procedures were performed in accordance with Northwestern’s guidelines.
Human Participants
Plasma TGF-β1 was measured as described in the online-only Data Supplement. Cine and late gadolinium-enhanced cardiac magnetic resonance imaging were performed on otherwise healthy individuals with a homozygous genetic mutation in SERPINE-1. The institutional review boards at the Indiana Center for Hemophilia and Thrombosis (St. Vincent Hospital) and Northwestern University approved the study protocols. The study participants provided written informed consent. The participants were genotyped for the c.699_700dupTA frameshift mutation in SERPINE1 at the University of Michigan with a preidentified primer.
AngII Model
Wild-type mice and PAI-1−/− mice were infused with AngII (1 μg/kg/min; Sigma-Aldrich) or a vehicle control (PBS) using an osmotic minipump (Alzet, model 1002) as previously described18; 9 to 10 male and female mice (16–20 weeks old) were used per group. Pumps remained implanted for 7 or 14 days. An additional group of 6 PAI-1−/− mice were infused with AngII as described earlier but also received injections of 300 μg/kg recombinant human BMP-7 in PBS (R&D Systems) every other day starting the day of minipump implantation. Arterial pressures were measured using the Coda2 noninvasive cuff system (Kent Scientific) for ≥8 consecutive measurements. Heart and tibia were extracted at day 7 or 28 after initiation of AngII. Transthoracic 2-dimensional M-mode echocardiography was performed using the Vevo770 (VisualSonics) equipped with a 30-MHz transducer. Mice were lightly anesthetized using a mixture of 1.5% isofluorane and oxygen (2 L/min) to ensure heart rates >400 to 450 bpm. Change in left ventricular area during the cardiac cycle was determined by endocardial border tracings to determine ejection fraction (average of parasternal long and short axis measurements). M-mode tracings were used to measure left ventricular systolic and diastolic chamber dimensions. The mean value from 3 beats was determined for each parameter.
Cardiac and Aortic Fibrosis
Mice were anesthetized as previously described and perfused by ventricular puncture with 20 cc of PBS. Hearts and aortas were harvested, fixed in 10% formalin, and paraffin embedded. Cardiac short-axis sections at the mid-ventricle are shown. The levels of collagen deposition were determined by Masson trichrome staining. Photographs were taken with an Olympus DP71 camera. To quantify the extent of fibrosis, stained sections (n=9–10 per group) were analyzed using Image J. Percent fibrotic area represents the number of blue pixels in each section divided by the total number of pixels taken up by myocardium or aorta.
Immunohistochemistry
Briefly, cardiac sections were deparrafinized and rehydrated, and antigens were retrieved using Retreivit 6 (InnoGennex). Sections were treated with 2% Sudan Black (Sigma-Aldrich) in 70% ethanol to quench autofluorescence and blocked with 3% BSA (Sigma-Aldrich). Slides were incubated with primary antibodies against TGF-β 1/2/3 (sc-7892, Santa Cruz Biotechnology), TGF-β (ab66043, Abcam), Troponin I (ab188877, Abcam), Integrin β3 (ab75872, Abcam), and Laminin (ab11575, Abcam) in PBS with 1% BSA. AlexaFluor-labeled secondary antibodies with ‒488 or ‒555 conjugates were used (Thermo Fisher Scientific). Images were obtained from 5 fields of 4 separate hearts for each staining condition.
Adult Mouse Ventricular Myocyte and Cardiac Fibroblast Isolation
Mice were anesthetized and euthanized by cervical dislocation. Hearts were collected and cardiomyocytes/fibroblasts prepared as described in full detail in the online-only Data Supplement.
Western Blot
Cardiac tissue or cardiomyocyte lysates were prepared using ice-cold RIPA buffer (Thermo Fisher Scientific) containing Complete Protease Inhibitor Cocktail and PhosSTOP (Roche). Lysates or culture medium were cleared by centrifugation, reduced, subjected to Western blot using a NuPage system (Invitrogen), and probed with antibodies against TGF-β 1/2/3 (Santa Cruz Biotechnology), TGF-β (Abcam), Troponin I (Abcam), Smad6 (ab13727, Abcam), phospho-Smad1/5 (Thermo Fisher), and phospho-Smad2/3 (sc-11769, Santa Cruz Biotechnology).
RNA Sequencing
Mouse whole ventricular RNA was isolated with an RNeasy fibrous tissue midi kit per the manufacturer’s instructions (Qiagen). RNA sequencing and statistical analysis were performed as described in the online-only Data Supplement.
Statistics
Values represent mean±SD or SEM as indicated in each figure legend. All group comparisons (except for transcriptome analysis ‒ supplemental methods) were performed by 1-way analysis of variance or nonpaired 2-tailed Student’s t test. Sample sizes and results are provided in the figures and tables.
RESULTS
Homozygous Loss-of-Function Mutation in SERPINE-1 Is Associated With Spontaneous Cardiac Fibrosis in Humans
A rare frameshift mutation in SERPINE-1 (c.699_700dupTA) was originally described in the Old Order Amish community in Indiana, and this mutation has been shown to abolish circulating PAI-1 levels in homozygous individuals.19,20 We have previously observed that aged PAI-1−/− mice developed spontaneous cardiac-specific fibrosis.3 We sought to confirm our observations in the murine model by evaluating the myocardium of homozygous affected individuals for cardiac fibrosis. Two otherwise healthy nonfirst-degree relatives (a 20-year-old homozygous male [subject 1] and a 22-year-old homozygous female [subject 2]) volunteered to undergo cardiac magnetic resonance imaging with contrast. Late gadolinium-enhanced images demonstrate multiple foci of scar in a noncoronary distribution, suggesting nonischemic etiology (Figure 1). The epicardial and midmyocardial patterns of enhancement are fibrotic patterns that we later observe in a murine model of cardiac injury. It is interesting to note that circulating plasma levels of TGF-β1 are elevated in individuals with homozygous mutation in SERPINE-1, compared with age-matched controls with at least 1 functional allele (Figure I in the online-only Data Supplement), suggesting that cardiac fibrosis in these individuals may involve the TGF-β axis. Thus, the role of PAI-1 in the regulation of cardiac fibrosis in humans is a novel finding and reinforces our previous observations that PAI-1 regulates cardiac-specific age-dependent fibrosis in mice.
Figure 1. Spontaneous cardiac fibrosis in 2 human subjects with homozygous deficiency of plasminogen activator inhibitor type I (PAI-1).

Late gadolinium-enhanced cardiac magnetic resonance imaging short-axis views (base, mid, and apex). Multiple bright foci in the left ventricle represent cardiac scar. Normal myocardium appears black. Subepicardial enhancement (single arrowhead) and a midmyocardial stripe (double arrowhead) are evident. This pattern of fibrosis is consistent with a noncoronary distribution.
PAI-1 Deficiency Accelerates AngII-Induced Cardiac Fibrosis and Leads to Systolic Dysfunction
Although several groups have characterized the spontaneous cardiac fibrosis that occurs in PAI-1−/− mice, it remains unclear whether the accelerated age-dependent profibrotic phenotype can be recapitulated in shorter time frames after cardiac injury. We chose an AngII infusion model because of its known effects of cardiomyocyte hypertrophy, fibroblast proliferation, myofibroblast activation, and interstitial cardiac fibrosis.21,22 Although previous studies demonstrated that pharmacological inhibition of PAI-1 may exacerbate AngII-mediated cardiac fibrosis,23 the mechanistic effect of genetic PAI-1 deficiency was not characterized in these studies. Using a chronic AngII infusion model, we investigated whether young 16- to 20-week-old PAI-1−/− mice exhibit accelerated cardiac fibrosis compared with age-matched wild-type (WT) controls, and whether this could be prevented by coadministration of bone morphogenetic protein (BMP)-7, a potent inhibitor of TGF-β signaling. Mice were implanted with osmotic minipumps that would deliver either 14 days of PBS or AngII (1000 ng/kg/min). An additional group of PAI-1−/− mice received intraperitoneal BMP-7 (300μg/kg every other day, a dose shown to inhibit cardiac fibrosis in vivo24 to inhibit AngII-mediated cardiac fibrosis). The timeline for treatment and measured cardiovascular parameters is summarized in Figure 2A. At baseline, no difference in blood pressure, heart weight, or systolic function was observed between PBS-treated WT and PAI-1−/− mice. AngII increased systolic blood pressure in both WT and PAI-1−/− mice compared with PBS (Figure 2B). However, PAI-1 deficiency was protective against AngII-mediated hypertension possibly because of a protective effect of PAI-1 deficiency on arteriosclerosis (reduced aortic fibrosis compared with WT; Figure II in the online-only Data Supplement). Despite decreased hemodynamic load, PAI-1 deficiency did not blunt AngII-mediated hypertrophy compared with WT as measured by cardiac mass (Figure 2C) and left ventricular wall thickness (Table I in the online-only Data Supplement). Notably, only PAI-1−/− mice treated with AngII alone developed moderate to severe systolic dysfunction compared with WT (ejection fraction 0.40 versus 0.57 in WT, P<0.005; Figure 2D and fractional shortening 0.22 versus 0.40 in WT, P=0.003; Table I in the online-only Data Supplement). Representative M-mode tracings (Figure 2E) illustrate that AngII induces cardiomyopathy in PAI-1−/− mice. Furthermore, BMP-7 was able to inhibit AngII-mediated cardiac dysfunction in PAI-1−/− mice (Figure 2D and 2E). There was no observed difference in fibrosis by Masson Trichrome staining between PBS-treated WT and PAI-1−/− hearts (Figure 3A). However, 2-week infusion of AngII induced significantly more fibrosis in PAI-1−/− mice compared with age-matched WT controls, and fibrosis was partially but significantly reduced by BMP-7. PAI-1 deficiency promoted a dramatic increase in the total area of AngII-induced collagen deposition (P<0.00005; Figure 3C), with both reactive and replacement fibrosis observed in the PAI-1−/− myocardium. At this dose, AngII induced fibrosis in WT hearts characterized by mild perivascular and interstitial fibrosis (Figure 3A and 3B). In contrast, fibrosis in the young PAI-1−/− mice is biventricular and involves all 3 layers of the myocardium, including the epicardium, midmyocardium, and endocardium (Figure 3A and 3B). In addition, a distinct midwall stripe pattern of collagen deposition was seen. The areas of replacement fibrosis observed in the epicardium, midmyocardium, and right ventricular insertion sites suggest cardiomyocyte loss and only occurred in PAI-1−/− mice treated with AngII alone. We further demonstrated that fibrosis is dramatically attenuated by the coadministration of BMP-7, suggesting that the fibrotic mechanism is TGF-β dependent (Figure 3A and 3C). Collectively, these findings suggest a specific protective role of PAI-1 against stress-induced cardiac fibrosis, which in PAI-1−/− mice results in a combined hypertrophic and fibrotic cardiomyopathy independent of pressure overload.
Figure 2. Angiotensin II (AngII)-mediated cardiovascular effects in wild-type (WT) and plasminogen activator inhibitor type I (PAI-1)−/− mice.
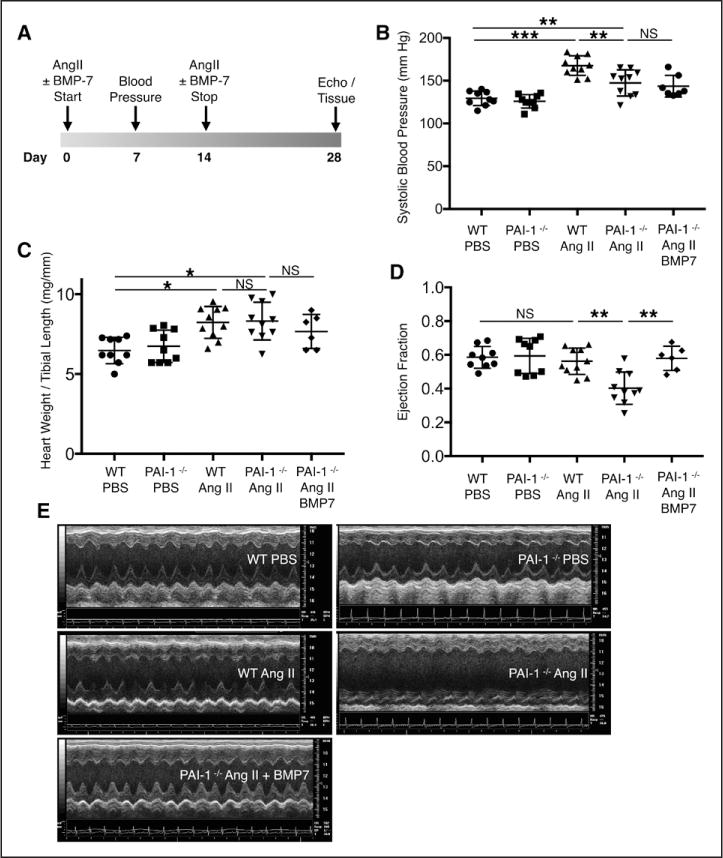
A, Mice were implanted with osmotic minipumps loaded with PBS or AngII (1000 ng/kg/min) and were studied for 28 days. One group of PAI-1−/− mice also received intraperitoneal injections of BMP-7 (300 μg/kg). B, Changes in systolic blood pressure after 1 week of treatment. AngII increases blood pressure in both WT and PAI-1−/− mice compared with saline. PAI-1 deficiency confers partial protection from AngII-mediated hypertension. C, Changes in heart weight to tibial length ratio. Cardiac mass increases to a similar degree in both WT and PAI-1−/− 28 days after initiation of AngII. D, EF was quantified using 2-dimensional (2D) echocardiography. AngII treatment induced moderate to severe systolic dysfunction in PAI-1 −/− mice but did not affect the EF of WT mice. Coadministration of BMP-7 restored systolic function in PAI-1−/− mice. E, Representative short-axis M-mode images from each group. Images were obtained at similar heart rates. B through E, n=6 to 10 per group. Error bars represent SD. All statistical analyses were performed using 1-way analysis of variance followed by Tukey’s HSD test. EF indicates ejection fraction; and NS, not significant. *P<0.05; **P<0.005; ***P<0.0005.
Figure 3. Homozygous deficiency of plasminogen activator inhibitor type I (PAI-1) accelerates Angiotensin II (AngII)-mediated cardiac fibrosis, which is inhibited by exogenous BMP-7.
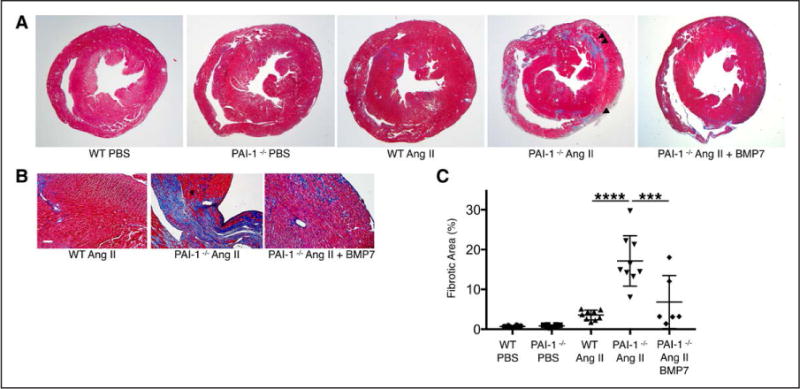
A, Short-axis sections through the ventricles of wild-type (WT) and PAI-1−/− mice at the level of the papillary muscles. Representative images with Masson’s Trichrome stain demonstrate a prominent increase in collagen deposition (blue stain) in PAI-1−/− hearts compared with WT. Hearts of WT and PAI-1−/− mice both develop interstitial and perivascular fibrosis in response to AngII compared with PBS. In addition, PAI-1−/− hearts contain areas of replacement fibrosis in the epicardial (single arrowhead) and midmyocardial (double arrowhead) regions. Coadministration of BMP-7 with AngII significantly reduces fibrotic deposition. B, Higher magnification images from AngII-treated WT and PAI-1−/− hearts stained with Masson’s Trichrome. Interstitial and perivascular collagen deposition occurs in both WT and PAI-1−/− hearts. Replacement fibrosis only occurs in the PAI-1−/− myocardium treated with AngII alone. Bar=20 μm. C, Quantification of percent fibrotic area was performed using Image J (n=6 to 10 per group). Error bars represent SD. Statistical analysis was performed using 1-way analysis of variance followed by Tukey’s HSD test. *** P<0.0001; ****P<0.00001.
PAI-1 Deficiency Stimulates AngII-Mediated Cardiomyocyte TGF-β Expression in Vivo
The profibrotic cytokine TGF-β is a downstream mediator of AngII-induced hypertrophy and matrix deposition in the heart.21,25,26 Persistent TGF-β signaling induces fibroblast-to-myofibroblast transition, and myofibroblasts are the primary source of collagen and other ECM proteins during fibrotic remodeling.27–29 Furthermore, AngII has been shown to stimulate mRNA and protein expression of TGF-β in both fibroblasts and cardiomyocytes.16,22,30 Cardiac sections from either PBS- or AngII-treated WT or PAI-1−/− mice were immunostained with antibodies against either TGF-β alone or TGF-β and troponin (Figure 4A through 4C). High magnification fluorescence images from PBS-treated WT and PAI-1−/− hearts reveals that TGF-β is not detectable in the absence of stimulation (negative controls; Figure 4A). In contrast, AngII induces the production of TGF-β in both WT and PAI-1-deficient myocardium (red). Costaining of TGF-β (red) positive cells with troponin (green) indicates that this cytokine is produced in ventricular myocytes. In addition, we observed that stressed PAI-1−/− myocardium exhibited an augmentation in staining intensity with anti-TGF-β Ab and contained abundant positive myocytes, which were sparse in WT hearts. Figure 4B shows negative controls for both the red and green channels for the experiment in Figure 4A, indicating low background levels. Enhanced TGF-β production in the knockout is better visualized on lower magnification images (Figure 4C), where TGF-β (now green) is localized to the epicardium and perivascular regions in WT hearts. In contrast, TGF-β is expressed in a more diffuse pattern in PAI-1−/− myocardium, and several waves of cardiomyocyte TGF-β production are evident. It is important to note that the cytoplasmic immunostaining pattern on confocal z-stack imaging suggests increased cardiomyocyte TGF-β synthesis rather than increased binding to cell surface receptors (Figure III in the online-only Data Supplement). Taken together, the immunohistochemical findings establish that PAI-1 is a potent regulator of stress-induced cardiomyocyte TGF-β expression.
Figure 4. Homozygous deficiency of plasminogen activator inhibitor type I (PAI-1) potentiates Angiotensin II (AngII)-induced transforming growth factor-β (TGF-β) production by ventricular myocytes in vivo.
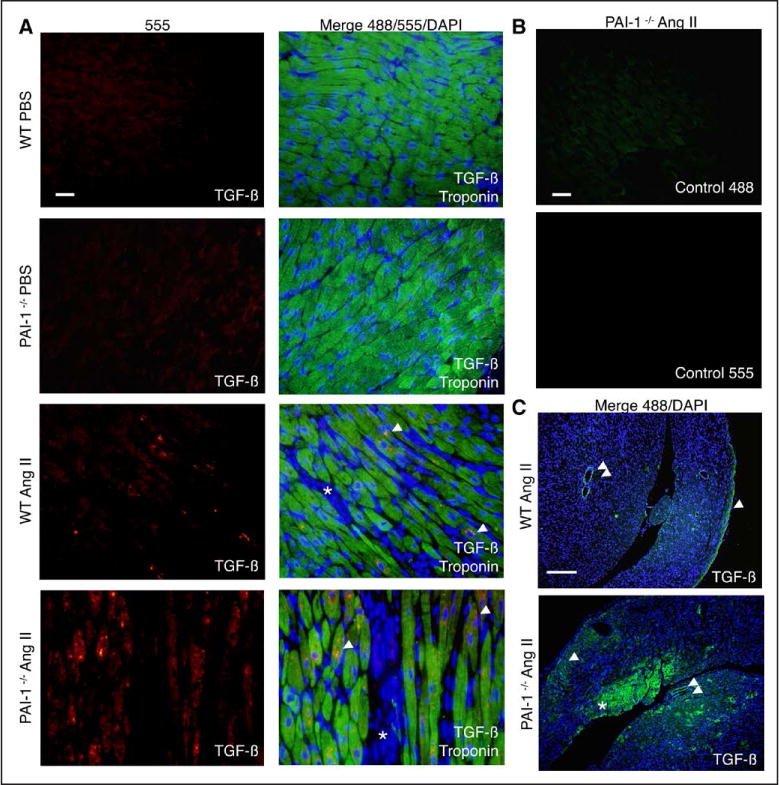
A, Fluorescence microscopy images of mouse cardiac sections immunostained for TGF-β (red/555), troponin (green/488), and with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Left, TGF-β staining. Right, Staining merged with troponin and DAPI for improved tissue visualization. Hearts of wild-type (WT) and PAI-1−/− mice treated with PBS do not stain for TGF-β. AngII induces the production of TGF-β in ventricular myocytes (troponin positive, single arrowhead). Compared with WT myocardium, PAI-1−/− sections stain more intensely for TGF-β, with more positive myocytes. Interstitial areas that are troponin negative are marked with an asterisk. Images were taken with identical exposure times for each channel. Representative images from 4 experiments (4 different hearts per group). Bar=10 μm. B, Sections from PAI-1−/− hearts were treated with secondary antibody alone and imaged with identical exposure times to sections from A. Both the red/555 and green/488 controls are shown. Representative images from 4 experiments (4 different hearts per group). Bar=10 μm. C, Fluorescence microscopy images from AngII-treated mouse cardiac sections immunostained for TGF-β (green/488) and with DAPI (blue). Merged images are shown. WT myocardium stains most intensely for TGF-β in the epicardium (single arrowhead) and perivascular areas (double arrowhead). Compared with WT, PAI-1−/− myocardium more diffusely expresses TGF-β, and several waves of involvement are evident: epicardial (single arrowhead), subendocardial (asterisk), and perivascular (double arrowhead). Representative images from 4 experiments (4 different hearts per group). Bar=200 μm.
PAI-1 Deficiency Enhances TGF-β Expression and Secretion by Cardiomyocytes
We confirmed that AngII stimulates cardiomyocyte TGF-β, and that PAI-1 deficiency upregulates its expression in vivo. PAI-1 is known to exert many of its biological effects by its inhibition of the plasmin-MMP system, which regulates ECM proteolysis, or by inhibiting cell migration through its interaction with vitronectin.9 In the AngII model, it remained unclear whether the effect of PAI-1 was cardiomyocyte specific or whether PAI-1 deficiency alters cardiomyocyte interactions with the ECM or other cardiac cell types. To answer this question, we isolated adult ventricular myocytes from 10-week-old WT and PAI-1−/− mice and examined whether 24-hour exposure to high-dose TGF-β (5 ng/mL) or AngII (1 μM) could recapitulate the enhanced TGF-β expression by PAI-1−/− cardiomyocytes that we observed in vivo. An increase in TGF-β is detectable by Western blot in both WT and PAI-1−/− cardiomyocytes that were stimulated with either TGF-β or AngII (Figure 5A). Agonist-mediated Smad-2/3 phosphorylation indicates that the expressed TGF-β is functional and capable of canonical autocrine stimulation. Troponin serves as loading control. Lysates from PAI-1−/− ventricular myocytes demonstrated significantly more intracellular TGF-β (Figure 5B). Examination of the culture medium for cardiomyocyte-secreted TGF-β reveals that PAI-1−/− myocytes secrete more TGF-β than do WT myocytes when stimulated with either TGF-β or AngII (Figure 5C and 5D). The nonspecific band reflects loading control, and medium that was not incubated with cells does not specifically react for TGF-β at the observed molecular weight, despite containing 5 ng/mL exogenous TGF-β. A typical preparation of isolated primary ventricular myocytes is shown in Figure 5E. It is important to note that lentiviral overexpression of PAI-1 in human inducible pluripotent stem cell-derived cardiomyocytes is sufficient to inhibit autocrine TGF-β production (Figure IVA through IVC in the online-only Data Supplement), direct evidence that human cardiomyocytes synthesize TGF-β and PAI-1 is an essential upstream negative regulator of this process. In contrast, we show that PAI-1 deficiency is associated with reduced TGF-β in primary cardiac fibroblasts compared with WT, and that TGF-β production did not significantly change in response to a 24-hour treatment with TGF-β or AngII despite expected increases in collagen (Figure VA and VD in the online-only Data Supplement). In addition, cardiac fibroblasts do not secrete detectable levels of TGF-β under conditions shown to increase expression in ventricular myocytes (Figure VB in the online-only Data Supplement). Collectively, these data demonstrate that the regulation of TGF-β by PAI-1 is likely cardiomyocyte-specific and helps explain the cardiac-selective phenotype in PAI-1-deficient mice and humans.
Figure 5. Homozygous deficiency of plasminogen activator inhibitor type I (PAI-1) enhances transforming growth factor-β (TGF-β) production in cardiomyocytes.
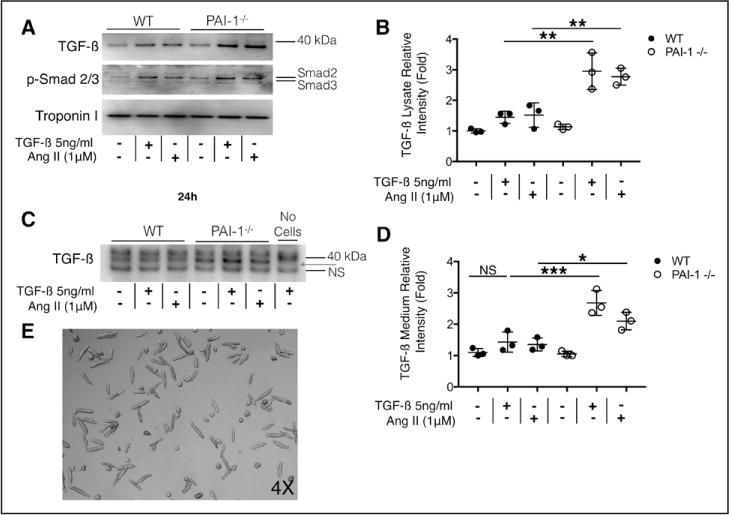
A, Production of TGF-β by cardiomyocytes. Wild-type (WT) and PAI-1−/− adult mouse ventricular myocytes were isolated and cultured for 24 hours in the presence or absence of TGF-β or Angiotensin II (AngII). Cells were rinsed and solubilized in radioimmunoprecipitation assay (RIPA) buffer. Lysates were analyzed by Western blot for TGF-β, phospho-SMAD2/3, and troponin (loading control). TGF-β or AngII-induced TGF-β production in WT and PAI-1−/− cardiomyocytes. Intracellular TGF-β is increased in treated PAI-1−/− cardiomyocytes compared with WT, suggesting increased susceptibility to either TGF-β or AngII stimulation. SMAD2/3 phosphorylation suggests that TGF-β produced by the myocytes is functionally active. Representative Western blot from 3 separate experiments. B, Quantification of normalized relative intensity in A. Each sample was normalized to troponin intensity. C, Culture medium from cardiomyocytes in A. Equal volume of medium was loaded in each lane and analyzed by Western blot for TGF-β. The nonspecific (NS) band reflects loading. Medium in lane 7 contains 5ng/mL TGF-β but did not incubate with cardiomyocytes and did not react with TGF-β Ab. PAI-1−/− myocytes secrete more TGF-β into the medium than WT cardiomyocytes (red arrow). Representative Western blot from 3 separate experiments. D, Quantification of normalized relative intensity in C. Each sample was normalized to troponin intensity. E, Representative image of adult mouse ventricular myocyte preparations. All statistical analyses were performed using 1-way analysis of variance followed by Tukey’s HSD test. NS indicates not significant. *P<0.05; **P<0.005; ***P<0.0005.
PAI-1 Deficiency Accelerates Cardiac Hypertrophy and Upregulates Multiple Early AngII-Induced Cardiac Transcriptional Pathways
High-throughput techniques have provided significant insight into the global molecular events associated with acute infusion of AngII.31 To better understand the effect of PAI-1 on the complex molecular pathways induced by AngII, we performed whole transcriptome analysis on ventricular RNA (Figure 6A). RNA was purified from the ventricles of WT and PAI-1−/− mice treated with either PBS or AngII, and the total transcriptome was sequenced as described in Methods. A short treatment of 7 days was chosen, predicting that significant divergence of early signaling elements would occur at this time point. In Figure 2C, we showed that chronic infusion of AngII increased cardiac mass to a similar extent in WT and PAI-1−/− when evaluated at 28 days. It is interesting to note that subacute treatment with AngII accelerated increase in cardiac mass in the PAI-1−/− compared with WT (P<0.005; Figure 6B). Only 26 genes were differentially expressed in the hearts of young WT and PAI-1-deficient mice in the absence of injury (Figure 6C). Genes that were different (up- or down regulated) at least ≈50% are summarized in Table II in the online-only Data Supplement. The most notably upregulated gene in the PBS-treated PAI-1−/− myocardium is Wdfy1, which encodes a critical mediator of the Toll-like receptor signaling pathway.32 Also of interest, 2 genes encoding proteins that have been implicated in the negative regulation of the TGF-β signaling pathway, nuclear receptor 4a133 and Smad6,34 are downregulated in the unstimulated PAI-1−/− ventricle. Subacute treatment with AngII affected significantly more cardiac transcripts in the PAI-1−/− versus WT (6005 versus 551). When comparing AngII-treated groups, PAI-1 deficiency was associated with significant alteration of 4168 transcripts. To examine the characteristics of differentially expressed genes, functional classification of up- and downregulated transcripts was performed using the pathway analysis (Metascape). A heat map of differentially expressed pathways among the 4 groups is depicted in Figure 6D. Maximal log 10-fold change (≈20) in a given pathway is depicted as dark red. AngII upregulated multiple pathways involved in inflammation, cellular activation, ECM remodeling, and wound healing. When compared with AngII-treated WT hearts, pathways increased by a log 10-fold change of 10 to 20 in the PAI-1−/− myocardium, serving as biochemical confirmation of the robust effect of PAI-1 deficiency on cardiac fibrosis. Pathway analysis reveals that the cardiac transcriptomes in young PBS-treated WT and PAI-1−/− mice are nearly indistinguishable.
Figure 6. Plasminogen activator inhibitor type I (PAI-1) regulates early Angiotensin II (AngII)-mediated cardiac hypertrophy and transcriptional events.
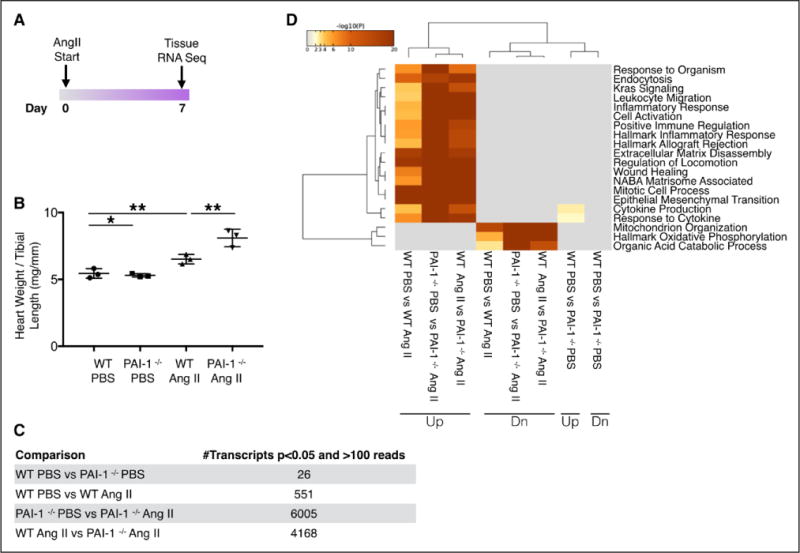
A, Mice were implanted with osmotic minipumps loaded with PBS or AngII (1000 ng/kg/min) for 7 days before ventricular RNA isolation. B, Changes in heart weight to tibial length ratio. Cardiac hypertrophy is accelerated in PAI-1−/− mice treated with AngII compared with wild type (WT) (n=3 mice per group). Error bars represent SD. Statistical analysis was performed using 1-way analysis of variance followed by Tukey’s HSD test. *P<0.05; **P<0.005. C, RNA sequencing results. Number of significantly different genes (P<0.05) with ≥100 reads. Hearts from WT and PAI-1−/− mice treated with PBS were transcriptionally similar with the exception of 26 genes. In contrast, treatment with 7 days of AngII induced 4168 differentially expressed genes in the PAI-1−/−compared with WT (n=3 per group). Statistical analysis was performed using DESeq2 with adjustment for multiple comparisons as described in the online-only Data Supplement (false discovery rate [FDR]-adjusted P value). D, Computational analysis reveals that numerous pathways involving inflammation, matrix remodeling, and wound healing are markedly upregulated in the AngII-treated PAI-1−/− heart compared with WT (n=3 hearts per group). Pathway analysis was performed using Metascape.
PAI-1 Deficiency Enhances Early AngII-Induced Transcription of Genes Encoding TGF-β and ECM-Associated Proteins
Although PAI-1 is a known downstream transcriptional target of TGF-β, we showed that PAI-1 deficiency upregulates TGF-β at the protein level. RNA sequencing provided us with the opportunity to comprehensively characterize the effect of PAI-1 on the induction of TGF-β-related transcripts during AngII-mediated cardiac injury (Table). Although the different TGF-β isoforms have similar effects in vitro, they carry out diverse biological functions, and deletion of individual isoforms in mice results in distinct phenotypes.35 Ubiquitously, transcripts for all isoforms of TGF-β were increased in the PAI-1−/− heart, most prominently, Tgfb2, Tgfb3, and Tgfbi rose >100% (log2-fold change >1) more than in AngII-treated WT myocardium. The RNA sequencing results further expand our understanding of specific functional effects of TGF-β induction on the myocardial matrix. Accordingly, we observed significant elevation in transcripts for 12 collagen subunits in the AngII-treated PAI-1−/− heart compared with WT (Table). Isoform-specific collagen RNA content was increased 150% to 550% (log2-fold change 1.3–2.7) after 7 days of AngII treatment. Although collagen is the main constituent of fibrotic scar, PAI-1 deficiency results in a 330% relative rise in fibronectin RNA, as well as increases in transcripts associated with matrix elastic fibers (Table III in the online-only Data Supplement). Genes involved in matrix remodeling were dramatically upregulated in the PAI-1−/− heart, including Timp1, which is amplified 550% compared with WT (Table). Another group of functionally related transcripts enhanced by PAI-1 deficiency are genes encoding matricellular proteins. Nearly every member of the matricellular protein family has been shown to play a role in cardiac fibrosis in response to injury.36 Compared with WT, transcriptome analysis of PAI-1-deficient ventricular RNA demonstrates robust upregulation of matricellular genes in response to AngII, including TnC, Spp1, Thbs1, Thbs2, Thbs 4, Ctgf, and Sparc (Table). In fact, TnC (tenascin C) is the most highly upregulated transcript in the PAI-1−/− heart, increasing by 880%. The striking effect of PAI-1 deficiency on the genetic regulation of cardiac ECM composition is also associated with an altered expression pattern of adhesion receptors, most prominently integrins. Numerous integrin receptor subunits have been reported to play specialized roles in the heart and are centrally involved in the development of cardiac fibrosis and injury.37 AngII failed to significantly induce changes in cardiac integrin expression in WT mice. In contrast, AngII stimulation in PAI-1-deficient mice promotes significant increases in cardiac integrin RNAs (Table). The most substantial elevation was observed in cardiac Itgb3 (integrin-β3), which increased 120% relative to WT. Collectively, the RNA sequencing results provide nonbiased confirmatory evidence for the upstream regulatory role of PAI-1 on cardiac expression of TGF-β, as well as multiple TGF-β gene targets known to be centrally involved in ECM homeostasis and fibrosis. Furthermore, cardiac sections from PBS-treated WT and PAI-1−/− mice demonstrate normal laminin architecture, whereas β3-integrin expression is localized to vascular structures as expected38 (Figure VIA in the online-only Data Supplement). Although WT mice demonstrate conserved laminin architecture and β3-integrin expression pattern after AngII infusion, PAI-1 deficiency is associated with marked laminin disorganization and cardiomyocyte β3-integrin expression (Figure VIA in the online-only Data Supplement). Colocalization with troponin confirms cardiomyocyte β3-integrin expression in the PAI-1−/− hearts (Figure VIB in the online-only Data Supplement). These data further confirm at the protein level that PAI-1 conserves the integrity of cardiac ECM and cardiomyocyte integrin-matrix interactions, in addition to regulating cardiac TGF-β.
Table.
Differentially Expressed Genes in Angiotensin II–Treated Wild-Type Versus Plasminogen Activator Inhibitor Type I−/− Hearts
| Gene | Transcript Protein Product | Log 2-Fold Change | FDR-Adjusted P Value | Function |
|---|---|---|---|---|
| Smad9 | SMAD family member 9 | −0.9 | 0.0034 | TGF-β signaling and regulation |
| Smad6 | SMAD family member 6 | −0.6 | 0.0018 | |
| Ltbp2 | Latent TGF-β binding protein 2 | 1.9 | 2.42E-06 | |
| Tgfb1 | Transforming growth factor, β1 | 0.6 | 0.0016 | |
| Tgfb2 | Transforming growth factor, β2 | 1.5 | 3.93E-25 | |
| Tgfb3 | Transforming growth factor, β3 | 1.2 | 5.32E-12 | |
| Tgfbi | Transforming growth factor, β induced | 1.4 | 0.00044 | |
| Tgfbr1 | Transforming growth factor, β receptor I | 0.6 | 7.13E-05 | |
| Smad1 | SMAD family member 1 | 0.6 | 6.34E-05 | |
| Col1a1 | Collagen, type I, alpha 1 | 2.6 | 3.75E-17 | Collagen |
| Col1a2 | Collagen, type I, alpha 2 | 2.3 | 2.60E-14 | |
| Col3a1 | Collagen, type III, alpha 1 | 1.9 | 2.45E-10 | |
| Col5a1 | Collagen, type V, alpha 1 | 1.8 | 1.60E-14 | |
| Col8a1 | Collagen, type VIII, alpha 1 | 1.2 | 6.28E-08 | |
| Col8a2 | Collagen, type VIII, alpha 2 | 2.1 | 1.05E-11 | |
| Col12a1 | Collagen, type XII, alpha 1 | 2.7 | 1.17E-13 | |
| Col14a1 | Collagen, type XIV, alpha 1 | 1.2 | 0.00053 | |
| Col16a1 | Collagen, type XVI, alpha 1 | 1.7 | 4.92E-13 | |
| Col18a1 | Collagen, type XVIII, alpha 1 | 1.2 | 0.00062 | |
| Col27a1 | Collagen, type XXVII, alpha 1 | 1.3 | 2.51E-07 | |
| Timp1 | Tissue inhibitor of metalloproteinase 1 | 2.7 | 1.05E-11 | Matrix remodeling |
| Adam12 | A disintegrin and metallopeptidase domain 12 (meltrin alpha) | 2.2 | 8.68E-08 | |
| Adamts4 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 4 | 1.8 | 7.27E-08 | |
| Mmp14 | Matrix metallopeptidase 14 (membrane-inserted) | 1.7 | 2.05E-08 | |
| Mmp23 | Matrix metallopeptidase 23 | 1.3 | 8.27E-08 | |
| Adamts12 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 12 | 1.1 | 6.08E-08 | |
| Adamts2 | A disintegrin-like and metallopeptidase (reprolysin type) with thrombospondin type 1 motif, 2 | 1.1 | 1.82E-11 | |
| Tnc | Tenascin C | 3.3 | 9.33E-17 | Matricellular proteins |
| Spp1 | Secreted phosphoprotein 1 (osteopontin) | 2.3 | 6.43E-10 | |
| Thbs1 | Thrombospondin 1 | 1.8 | 3.74E-06 | |
| Ctgf | Connective tissue growth factor (CCN2) | 1.7 | 1.18E-06 | |
| Sparc | Secreted acidic cysteine rich glycoprotein | 1.3 | 1.41E-09 | |
| Thbs2 | Thrombospondin 2 | 1.1 | 2.85E-08 | |
| Thbs4 | Thrombospondin 4 | 1.1 | 0.00094 | |
| Itgb3 | Integrin β3 | 1.7 | 1.71E-07 | Integrins |
| Itga5 | Integrin alpha 5 (fibronectin receptor alpha) | 1.6 | 2.83E-16 | |
| Itgb2 | Integrin β2 | 1.3 | 0.00018 | |
| Itgam | Integrin alpha M | 1.1 | 0.0023 |
PAI-1 indicates plasminogen activator inhibitor type I.
Inhibitory Smad6 Is Reduced in the PAI-1−/− Heart, and BMP-7 Inhibits PAI-1−/− Cardiomyocyte TGF-β Signaling and Expression
It has been previously reported that Smad6 is activated by BMP and inhibits signaling by the TGF-β superfamily.34 Having observed that levels of cardiac Smad6 RNA are reduced in young PAI-1−/− mice (Table II in the online-only Data Supplement), we investigated whether protein levels of inhibitory Smad6 were also reduced. Indeed, the level of Smad6 detectable by Western blot in cardiac lysates is also significantly reduced in PAI-1−/− mice compared with WT, confirming the RNA sequencing results (Figure 7A and 7B). Troponin serves as loading control. We also demonstrate that BMP-7-induced Smad6 protein expression rapidly occurs within 3 hours in primary adult mouse ventricular myocytes (Figure VII in the online-only Data Supplement). In Figures 2 and 3, we showed that BMP-7 inhibits AngII-mediated cardiac fibrosis and cardiomyopathy in PAI-1−/− mice. Therefore, we further investigated whether BMP-7 could specifically inhibit cardiomyocyte TGF-β production and autocrine signaling. As previously shown, PAI-1−/− ventricular myocytes highly express and secrete TGF-β when incubated with 5ng/mL TGF-β or 1 μM AngII (Figure 7C through 7F). However, simultaneous incubation with BMP-7 (1000 ng/mL) inhibits Smad2/3 phosphorylation and abrogates TGF-β production (Figure 7C and 7D) as well as secretion (Figure 7E and 7F). These data suggest that BMP-7 inhibits cardiac fibrosis in the PAI-1−/− mice by opposing TGF-β signaling and thus reducing autocrine cardiomyocyte TGF-β production and secretion.
Figure 7. Homozygous deficiency of plasminogen activator inhibitor type I (PAI-1) is associated with reduced cardiac SMAD6, and BMP-7 inhibits cardiomyocyte transforming growth factor-β (TGF-β) expression and release.
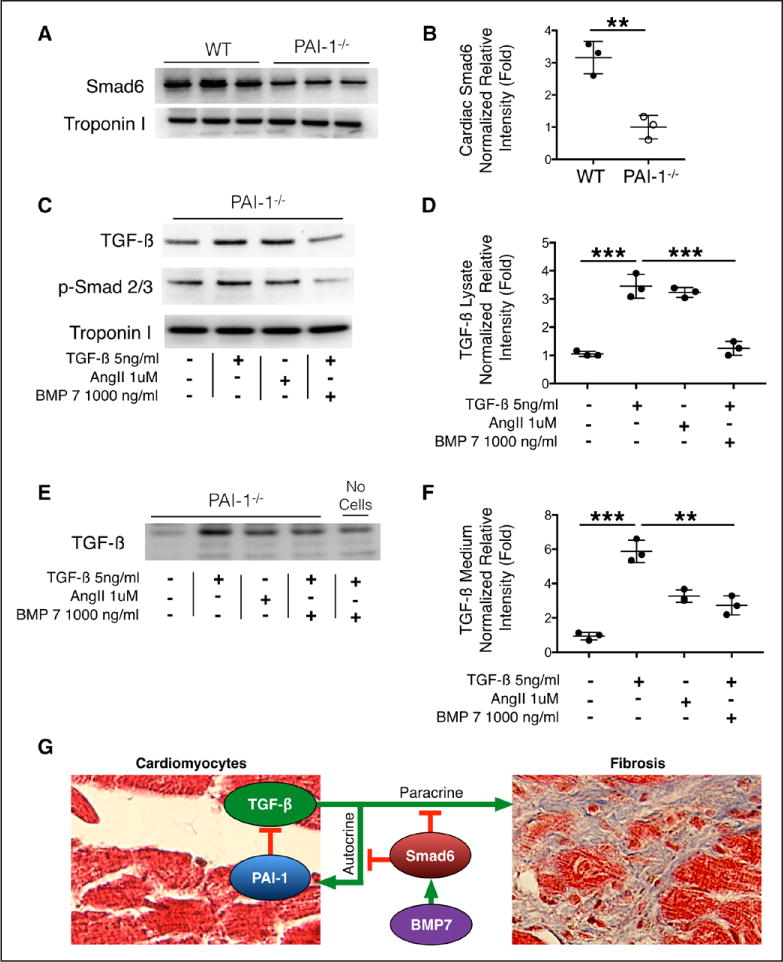
A, Cardiac SMAD6 content. Wild-type (WT) and PAI-1−/− ventricular tissue was solubilized in RIPA buffer. Lysates were analyzed by Western blot for SMAD6 and troponin (loading control). Lysates from 3 different hearts per group are shown. B, Quantification of relative intensity in A. Statistical analysis performed by nonpaired Student’s t test. C, PAI-1−/− adult mouse ventricular myocytes were isolated and treated for 24 hours with either TGF-β, Angiotensin II (AngII), or TGF-β+BMP-7. Cells were rinsed and solubilized in RIPA buffer. Lysates were analyzed by Western blot for TGF-β, phospho-SMAD2/3, and troponin (loading control). TGF-β or AngII induced TGF-β production. Intracellular TGF-β and phospho-SMAD 2/3 are decreased in BMP-7-treated PAI-1−/− cardiomyocytes, suggesting decreased susceptibility to autocrine stimulation. Representative Western blot from 3 separate experiments. D, Quantification of normalized relative intensity in C. E, Culture medium from cardiomyocytes in C. Equal volume of medium was loaded in each lane and analyzed by Western blot for TGF-β. Medium in lane 5 contains 5ng/mL TGF-β but did not incubate with cardiomyocytes. F, Quantification of normalized relative intensity in E. Values from each sample were normalized to troponin intensity, and statistical analyses in D and F were performed using 1-way analysis of variance followed by Tukey’s HSD test. **P<0.005; ***P<0.0005. G, Model for the role of cardiomyocyte PAI-1 in TGF-β-mediated cardiac fibrosis. PAI-1 is required for feedback inhibition of early autocrine TGF-β expression and secretion by cardiomyocytes. BMP-7-induced Smad6 activation suppresses both early autocrine and late paracrine TGF-β-mediated fibrogenesis.
DISCUSSION
Fibrosis is a common pathological manifestation of numerous diseases and is known to affect every major organ system. Normal wound healing is initiated by inflammation or cellular stress, but prolonged insult triggers the release of damage-associated molecular patterns from injured cells.39 Sustained exposure to cytokines, including TGF-β, has been implicated in the activation of resident fibroblasts to myofibroblasts and the transition of endothelial and epithelial cells to matrix secreting mesenchymal cells,12,18,40,41 resulting in ECM deposition. In addition, the balance of the rates of synthesis and proteolytic degradation determines ECM accumulation. The enabling effect of PAI-1 excess in liver, skin, kidney, and lung fibrosis is likely derived from its inhibition of fibrotic matrix clearance by the plasmin-MMP system.9 It recently became evident that PAI-1 paradoxically protects against cardiac fibrosis,3,11 suggesting that PAI-1 plays a fundamentally unique role in the myocardium. It has been reported that targeting the ability of cardiomyocytes to participate in TGF-β signaling disrupts the pathological response to cardiac injury.17 Thus, cardiomyocytes are key integrators of the TGF-β-fibrotic axis in the heart and regulate damage-associated molecular patterns required for ECM deposition in response to injury. In the present study, we report that PAI-1 controls cardiomyocyte expression of TGF-β, and that PAI-1 deficiency augments cardiac TGF-β and its associated transcriptional pathways, leading to enhanced cardiac fibrosis. Our studies further reveal a novel paradigm for the role of PAI-1 as an upstream regulator of TGF-β in the heart because PAI-1 has previously been designated as a bona fide downstream transcriptional target of TGF-β.
Data from the AngII model indicate that PAI-1 deficiency accelerates maladaptive responses to cardiac injury despite reducing hemodynamic stress on the heart, including both reactive and reparative fibrogenesis. We also observed areas of replacement fibrosis in the PAI-1-deficient myocardium, possibly related to enhanced AngII-mediated apoptosis (Figure VIII in the online-only Data Supplement). Cardiac hypertrophy occurred earlier, and ECM deposition was exaggerated in the PAI-1−/− heart despite blunted increase in systolic blood pressure compared with WT. The observed protection from AngII-mediated hypertension is possibly caused by reduced arteriosclerosis and consistent with our previous findings that PAI-1 deficiency or pharmacological inhibition of PAI-1 attenuate Nω-nitro-L-arginine methyl ester (L-NAME) and AngII-induced hypertension.42 Another beneficial effect of reduced or absent PAI-1 is a decline in vascular senescence, leading to decreased vascular fibrosis and arterial stiffness.42 Despite this finding, PAI-1 deficiency results in one of the more extensive forms of fibrotic cardiomyopathy that has been described in the literature. Notably, 1 of the AngII-treated PAI-1−/− mice exhibited collagen deposition in >50% of the myocardial area and suffered from right ventricular rupture after 9 days of treatment (Figure IX in the online-only Data Supplement).
We further show that effects of PAI-1 deficiency on cardiac hypertrophy and reactive fibrosis are pathophysiologic consequences of enhanced myocyte TGF-β synthesis and release. Previous studies have described that cardiac injury induces expression and secretion of TGF-β from ventricular myocytes and is dependent on autocrine cardiomyocyte TGF-β signaling through noncanonical TβR-II-dependent mechanisms.16,17 In the present study, we demonstrate that AngII indeed is capable of inducing TGF-β in both WT and PAI-1−/− murine cardiomyocytes, as well as human inducible pluripotent stem cell -derived cardiomyocytes, and is detectable by Western blot, suggesting that this response is substantial and conserved in mammals. We observed increased TGF-β protein and RNA in the PAI-1−/− heart and were able to reproduce this in vitro by stimulating increased levels of intracellular and secreted TGF-β from primary PAI-1−/− ventricular myocytes. Furthermore, overexpression of PAI-1 in human inducible pluripotent stem cell -derived cardiomyocytes blunted TGF-β production. Using 2 different antibodies, we detect the same 35-to 40-kDa isoform of TGF-β in primary cell lysates and in the culture medium, consistent with the molecular weight of unprocessed pre-TGF-β.43,44 Although the structural and biochemical modifications leading to activation of the TGF-β latent complex are well characterized,43,45 further studies using in vivo models are required to elucidate the signals that trigger formation of 14-kDa mature active TGF-β during cardiac injury.
Published data suggest reciprocal regulation between the BMP and TGF-β pathway during cardiac injury. TGF-β1 signaling by cardiomyocyte TβRII is known to suppress cardiac BMP-7 expression, and BMP7 is the dominant upregulated gene in TβRII−/− hearts subject to pressure overload.17 Conversely, upregulation of BMP-7 opposes TGF-β signaling and inhibits cardiac fibrosis.24 In addition, BMP-2-dependent activation of Smad6 has been shown to antagonize TGF-β-mediated signaling and attenuate cardiac fibrosis in a model of pressure overload.46 To date, several groups have shown that the inhibitory Smad6 is capable of regulating TGF-β signaling, and forced expression of Smad6 opposes TGF-β, mimicking the protective effects of BMP-7.34,46,47 We now present evidence that levels of Smad6 RNA and protein are both uniformly decreased in the PAI-1−/− heart, and Smad6 is activated by BMP-7 in adult murine ventricular myocytes. We further present direct evidence that BMP-7 antagonizes TGF-β signaling and production in cardiomyocytes and can suppress AngII-mediated cardiac fibrosis and apoptosis (Figure VIII in the online-only Data Supplement) in PAI-1−/− mice in vivo, which suggests potential therapeutic utility for BMP-7 and related compounds in the treatment of TGF-β-mediated fibrotic cardiomyopathies.
TGF-β is critical regulator of cardiac ECM during wound healing and fibrosis.36,45,48 Transcriptome sequencing of PAI-1−/− ventricular tissue confirms the early brisk activation of multiple gene networks that are independently validated downstream targets of TGF-β during cardiac injury. In addition to structural matrix components, the most highly upregulated genes in the PAI-1−/− heart encode matricellular proteins. Matricellular proteins dynamically integrate environmental signals by modulating cellular-ECM interactions. Although present at low levels in normal myocardium, they are expressed during cardiac injury and development.36 Furthermore, several members of the matricellular protein family are capable of inducing or activating TGF-β, and TGF-β has also been shown to induce genes encoding matricellular proteins in a feed-forward manner.36 The most highly upregulated transcript in the PAI-1−/− heart encodes Tenascin C, which is transiently expressed in fibrotic myocardium after multiple modes of injury, and has been studied as a circulating biomarker for heart failure and remodeling in humans.49,50
In conclusion, we propose a novel upstream regulatory role of PAI-1 in the TGF-β-dependent fibrotic axis in the heart, which is demonstrated in Figure 7G. In this model, PAI-1 inhibits early autocrine expression and secretion of TGF-β by ventricular myocytes. Furthermore, BMP-7 activates Smad6, which inhibits both the early autocrine cardiomyocyte-dependent pathway as well as the late TGF-β paracrine pathways involved in mesenchymal cell matrix production.
Supplementary Material
Clinical Perspective.
What Is New?
Otherwise healthy human subjects with complete plasminogen activator inhibitor type I (PAI-1) deficiency exhibit spontaneous cardiac fibrosis, and PAI-1-deficient mice develop accelerated fibrotic cardiomyopathy after cardiac injury.
We define a novel upstream regulatory mechanism for PAI-1 in transforming growth factor-β (TGF-β)-mediated cardiac-selective fibrogenesis.
We provide direct evidence that cardiomyocytes are a source of cardiac TGF-β, and myocyte PAI-1 expression modulates TGF-β synthesis and provides feedback inhibition of early autocrine signaling.
Furthermore, PAI-1 deficiency is associated with reduced cardiac levels of the inhibitory Smad6, and pharmacological treatment with BMP-7 ameliorates injury-mediated cardiac fibrosis and cardiomyocyte TGF-β autocrine stimulation.
What Are the Clinical Implications?
Tissue fibrosis is the leading cause of organ failure but persists as one of the most pressing global health problems because of the lack of effective pharmacotherapies.
The profibrotic cytokine TGF-β is broadly implicated in nearly every form of fibrotic cardiomyopathy, and the burden of cardiac scar is closely related to decline in systolic and diastolic function, arrhythmia, and patient outcomes.
Supported by clinical and functional data, our work provides exciting new evidence that modulation of the cardiomyocyte TGF-β axis represents a unique therapeutic strategy to abrogate fibrotic signaling and cardiac fibrosis.
Acknowledgments
The authors thank our Amish liaisons, the Indiana Hemophilia and Thrombosis Center Amish Research staff and Feinberg Cardiovascular Research Institute volunteers. The authors also thank Northwestern University Pathology Core Facility, Chrissy Kamide, Veronica Ramirez, Elizabeth Lux, Ting Liu, and Guillermo Oliver Laboratory for help with echocardiography and histology. The authors thank Dr Matthew Schipma and Northwestern Genomics Core for providing transcriptome sequencing. The authors Alexander Mackie, John Wasserstrom, David Barefield, and William Marszalec for helping optimize the cardiomyocyte protocol.
SOURCES OF FUNDING
This work was supported by grants from the National Institutes of Health to Dr Vaughan (R01 HL051387), to Dr Flevaris (F32 HL131301-01), and to Dr Khan (F32 HL129695-01), and from the American Heart Association to Dr. Ghosh (16GRNT31130010).
Footnotes
DISCLOSURES
None.
FOOTNOTES
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.117.028145/-/DC1.(/p)(p)Circulation is available at http://circ.ahajournals.org.
References
- 1.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–575. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellmers LJ, Scott NJ, Medicherla S, Pilbrow AP, Bridgman PG, Yandle TG, Richards AM, Protter AA, Cameron VA. Transforming growth factor-beta blockade down-regulates the renin-angiotensin system and modifies cardiac remodeling after myocardial infarction. Endocrinology. 2008;149:5828–5834. doi: 10.1210/en.2008-0165. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh AK, Bradham WS, Gleaves LA, De Taeye B, Murphy SB, Covington JW, Vaughan DE. Genetic deficiency of plasminogen activator inhibitor-1 promotes cardiac fibrosis in aged mice: involvement of constitutive transforming growth factor-beta signaling and endothelial-to-mesenchymal transition. Circulation. 2010;122:1200–1209. doi: 10.1161/CIRCULATIONAHA.110.955245. [DOI] [PubMed] [Google Scholar]
- 4.Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, Sheppard MN, Poole-Wilson PA, Pennell DJ. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1977–1985. doi: 10.1016/j.jacc.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 5.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007;87:1285–1342. doi: 10.1152/physrev.00012.2007. [DOI] [PubMed] [Google Scholar]
- 6.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB, American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mewton N, Liu CY, Croisille P, Bluemke D, Lima JA. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J Am Coll Cardiol. 2011;57:891–903. doi: 10.1016/j.jacc.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 9.Flevaris P, Vaughan D. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin Thromb Hemost. 2017;43:169–177. doi: 10.1055/s-0036-1586228. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Castellino FJ, Ploplis VA. Plasminogen activator inhibitor-1 (PAI-1) is cardioprotective in mice by maintaining microvascular integrity and cardiac architecture. Blood. 2010;115:2038–2047. doi: 10.1182/blood-2009-09-244962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Nagpal V, Covington JW, Michaels MA, Vaughan DE. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal. 2012;24:1031–1036. doi: 10.1016/j.cellsig.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samarakoon R, Higgins PJ. Integration of non-SMAD and SMAD signaling in TGF-beta1-induced plasminogen activator inhibitor type-1 gene expression in vascular smooth muscle cells. Thromb Haemost. 2008;100:976–983. [PMC free article] [PubMed] [Google Scholar]
- 15.Kakkar R, Lee RT. Intramyocardial fibroblast myocyte communication. Circ Res. 2010;106:47–57. doi: 10.1161/CIRCRESAHA.109.207456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taimor G, Schlüter KD, Frischkopf K, Flesch M, Rosenkranz S, Piper HM. Autocrine regulation of TGF beta expression in adult cardiomyocytes. J Mol Cell Cardiol. 1999;31:2127–2136. doi: 10.1006/jmcc.1999.1055. [DOI] [PubMed] [Google Scholar]
- 17.Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA. Pivotal role of cardiomyocyte TGF-β signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–2312. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, Vaughan DE. MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation. 2016;133:291–301. doi: 10.1161/CIRCULATIONAHA.115.018174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fay WP, Shapiro AD, Shih JL, Schleef RR, Ginsburg D. Brief report: complete deficiency of plasminogen-activator inhibitor type 1 due to a frame-shift mutation. N Engl J Med. 1992;327:1729–1733. doi: 10.1056/NEJM199212103272406. [DOI] [PubMed] [Google Scholar]
- 20.Fay WP, Parker AC, Condrey LR, Shapiro AD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. 1997;90:204–208. [PubMed] [Google Scholar]
- 21.Schnee JM, Hsueh WA. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc Res. 2000;46:264–268. doi: 10.1016/s0008-6363(00)00044-4. [DOI] [PubMed] [Google Scholar]
- 22.Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997;29:1947–1958. doi: 10.1006/jmcc.1997.0435. [DOI] [PubMed] [Google Scholar]
- 23.Weisberg AD, Albornoz F, Griffin JP, Crandall DL, Elokdah H, Fogo AB, Vaughan DE, Brown NJ. Pharmacological inhibition and genetic deficiency of plasminogen activator inhibitor-1 attenuates angiotensin II/salt-induced aortic remodeling. Arterioscler Thromb Vasc Biol. 2005;25:365–371. doi: 10.1161/01.ATV.0000152356.85791.52. [DOI] [PubMed] [Google Scholar]
- 24.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 25.Brand T, Schneider MD. The TGF beta superfamily in myocardium: ligands, receptors, transduction, and function. J Mol Cell Cardiol. 1995;27:5–18. doi: 10.1016/s0022-2828(08)80003-x. [DOI] [PubMed] [Google Scholar]
- 26.Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. 2007;74:184–195. doi: 10.1016/j.cardiores.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh AK, Quaggin SE, Vaughan DE. Molecular basis of organ fibrosis: potential therapeutic approaches. Exp Biol Med (Maywood) 2013;238:461–481. doi: 10.1177/1535370213489441. [DOI] [PubMed] [Google Scholar]
- 28.Ghosh AK, Vaughan DE. Fibrosis: is it a coactivator disease? Front Biosci (Elite Ed) 2012;4:1556–1570. doi: 10.2741/e480. [DOI] [PubMed] [Google Scholar]
- 29.Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. 1985;82:8681–8685. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gray MO, Long CS, Kalinyak JE, Li HT, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40:352–363. doi: 10.1016/s0008-6363(98)00121-7. [DOI] [PubMed] [Google Scholar]
- 31.Li TT, Li XY, Jia LX, Zhang J, Zhang WM, Li YL, Qi YF, Du J. Whole transcriptome analysis of hypertension induced cardiac injury using deep sequencing. Cell Physiol Biochem. 2016;38:670–682. doi: 10.1159/000438659. [DOI] [PubMed] [Google Scholar]
- 32.Hu YH, Zhang Y, Jiang LQ, Wang S, Lei CQ, Sun MS, Shu HB, Liu Y. WDFY1 mediates TLR3/4 signaling by recruiting TRIF. EMBO Rep. 2015;16:447–455. doi: 10.15252/embr.201439637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palumbo-Zerr K, Zerr P, Distler A, Fliehr J, Mancuso R, Huang J, Mielenz D, Tomcik M, Fürnrohr BG, Scholtysek C, Dees C, Beyer C, Krönke G, Metzger D, Distler O, Schett G, Distler JH. Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat Med. 2015;21:150–158. doi: 10.1038/nm.3777. [DOI] [PubMed] [Google Scholar]
- 34.Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- 35.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 36.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. doi: 10.1152/phys-rev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Israeli-Rosenberg S, Manso AM, Okada H, Ross RS. Integrins and integrin-associated proteins in the cardiac myocyte. Circ Res. 2014;114:572–586. doi: 10.1161/CIRCRESAHA.114.301275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88:1112–1119. doi: 10.1161/hh1101.091862. [DOI] [PubMed] [Google Scholar]
- 39.Turner NA. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs) J Mol Cell Cardiol. 2016;94:189–200. doi: 10.1016/j.yjmcc.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SB, Kalluri R. Mechanistic connection between inflammation and fibrosis. Kidney Int Suppl. 2010;119:S22–S26. doi: 10.1038/ki.2010.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boe AE, Eren M, Murphy SB, Kamide CE, Ichimura A, Terry D, McAnally D, Smith LH, Miyata T, Vaughan DE. Plasminogen activator inhibitor-1 antagonist TM5441 attenuates Nω-nitro-L-arginine methyl ester-induced hypertension and vascular senescence. Circulation. 2013;128:2318–2324. doi: 10.1161/CIRCULATIONAHA.113.003192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gentry LE, Lioubin MN, Purchio AF, Marquardt H. Molecular events in the processing of recombinant type 1 pre-pro-transforming growth factor beta to the mature polypeptide. Mol Cell Biol. 1988;8:4162–4168. doi: 10.1128/mcb.8.10.4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116(Pt 2):217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 45.Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S, Sun A, Li L, Zhao G, Jia J, Wang K, Ge J, Zou Y. Up-regulation of BMP-2 antagonizes TGF-β1/ROCK-enhanced cardiac fibrotic signalling through activation of Smurf1/Smad6 complex. J Cell Mol Med. 2012;16:2301–2310. doi: 10.1111/j.1582-4934.2012.01538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang S, Hirschberg R. Bone morphogenetic protein-7 signals opposing transforming growth factor beta in mesangial cells. J Biol Chem. 2004;279:23200–23206. doi: 10.1074/jbc.M311998200. [DOI] [PubMed] [Google Scholar]
- 48.Schiller M, Javelaud D, Mauviel A. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci. 2004;35:83–92. doi: 10.1016/j.jdermsci.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 49.Terasaki F, Okamoto H, Onishi K, Sato A, Shimomura H, Tsukada B, Imanaka-Yoshida K, Hiroe M, Yoshida T, Kitaura Y, Kitabatake A, Study Group for Intractable Diseases by a Grant from the Ministry of Health, Labor and Welfare of Japan Higher serum tenascin-C levels reflect the severity of heart failure, left ventricular dysfunction and remodeling in patients with dilated cardiomyopathy. Circ J. 2007;71:327–330. doi: 10.1253/circj.71.327. [DOI] [PubMed] [Google Scholar]
- 50.Yokokawa T, Sugano Y, Nakayama T, Nagai T, Matsuyama TA, Ohta-Ogo K, Ikeda Y, Ishibashi-Ueda H, Nakatani T, Yasuda S, Takeishi Y, Ogawa H, Anzai T. Significance of myocardial tenascin-C expression in left ventricular remodelling and long-term outcome in patients with dilated cardiomyopathy. Eur J Heart Fail. 2016;18:375–385. doi: 10.1002/ejhf.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.