

Abstract
Cystic fibrosis (CF) is a lethal genetic disorder caused by mutation of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Despite recent groundbreaking approval of genotype-specific small-molecule drugs, a significant portion of CF patients still lack effective therapeutic options that address the underlying cause of the disease. Through a phenotypic high-throughput screen of approximately 54,000 small molecules, we identified a novel class of CFTR modulators called amplifiers. The identified compound, the characteristics of which are represented here by PTI-CH, selectively increases the expression of immature CFTR protein across different CFTR mutations, including F508del-CFTR, by targeting the inefficiencies of early CFTR biosynthesis. PTI-CH also augments the activity of other CFTR modulators and was found to possess novel characteristics that distinguish it from CFTR potentiator and corrector moieties. The PTI-CH–mediated increase in F508del-CFTR did not elicit cytosolic or endoplasmic reticulum–associated cellular stress responses. Based on these data, amplifiers represent a promising new class of CFTR modulators for the treatment of CF that can be used synergistically with other CFTR modulators.
Keywords: amplifiers, high-throughput screening, CFTR modulator, PTI-CH, phenotypic assays
賦活薬および矯正薬との官能基の相乗効果を示す新たな薬効分類の低分子薬の増幅装置を特定するハイスループット表現型スクリーニング戦略の活用
嚢胞性線維症(CF)は、嚢胞性線維症膜コンダクタンス制御因子(CFTR)遺伝子の変異によって引き起こされる致死性の遺伝病である。近年になり遺伝子型特異的低分子薬が承認され始めたものの、多くのCF患者にとっては、疾患の根本原因に対処できる有効な治療選択肢が依然として不足している。我々は、約54,000の低分子にハイスループット表現型スクリーニングを行い、「増幅装置」といわれる新規クラスのCFTR修飾薬を特定した。特定されたこれらの化合物は、その特徴についてはここにPTI-CHが示すとおりであるが、初期のCFTR生合成の非効率性をターゲットとすることにより、F508del-CFTRを含めさまざまなCFTR変異において未熟なCFTRタンパク質の発現を選択的に増強させる。ほかのCFTR修飾薬の活性も高めるPTI-CHは、CFTR賦活薬およびCFTR矯正薬の成分を識別する新たな特性を備えていることが明らかにされている。PTI-CHを介して増強されたF508del-CFTRが細胞基質または細胞質網状構造に伴う細胞性ストレス応答を誘発することはなかった。これらのデータに基づくと、増幅装置とされるものはCFの治療に向けた有望な新規クラスのCFTR修飾薬であり、ほかのCFTR修飾薬と相乗的に利用できる。
고속 대량 표현형 스크리닝 전략을 사용하여 증강제 및 보정제와의 기능적 시너지 효과를 나타내는 소분자의 새로운 약리학적 분류인 증폭제 식별
낭포성 섬유증(CF)은 췌장 섬유증 막 관통 전도 조절제(CFTR) 유전자의 돌연변이로 인한 치명적인 유전 질환이다. 최근 유전자형 특이적 소분자 의약품의 획기적인 승인에도 불구하고 상당 부분의 CF 환자는 여전히 근본적인 원인을 다루는 효과적인 치료법이 부족하다. 대략 54,000개의 소분자에 대한 표현형 고속 대량 스크리닝을 통해, 우리는 증폭제라 불리는 새로운 종류의 CFTR 조절제를 식별했다. 식별된 화합물은 여기에서 PFT-CH로 나타내는 특성으로, 초기 CFTR 생합성의 비효율성을 타겟으로 하여 F508del-CFTR을 비롯한 다양한 CFTR 돌연변이를 통해 미성숙 CFTR 단백질의 발현을 선택적으로 증가시킨다. PTI-CH는 또한 다른 CFTR 조절제의 활성을 증가시키고 CFTR 증강제 및 보정제 부분과 구별되는 새로운 특징을 갖는 것으로 밝혀졌다. F508del-CFTR의 PTI-CH 매개 증가는 세포질 또는 소포체 관련 세포 스트레스 반응을 유도하지 않았다. 이러한 데이터를 바탕으로, 증강제는 다른 CFTR 조절제와 함께 상승 작용할 수 있는 유망한 새로운 종류의 CF 치료용 CFTR 조절제이다.
采用高通量表型筛选策略来甄别催化因子(Amplifiers)—一类与增效因子和修正因子在功能上协同作用的新型药理学小分子。
囊性纤维性变(CF)是由囊性纤维化跨膜传导调节因子(CFTR)基因突变诱发的致命性基因缺陷疾病。当前,尽管在基因型特效小分子药物的获批上取得突破性进展,但是仍然有很大一部分CF患者无法获得有效的根治手段。通过表型高通量筛选大约54,000个小分子,我们发现了一类新型CFTR调节因子,叫做催化因子(Amplifiers)。在此,我们用PTI-CH来代表这一类已经确认的合成物的特性,这类化合物能选择性地增加未成熟CFTR蛋白质的表达,这一催化作用可见于多种CFTR变异中,包括F508del-CFTR,其作用机理是攻击早期CFTR生物合成中的低效现象。PTI-CH同时也能增强其他CFTR调节因子的活性,我们也发现了它的一些新奇特性,使得它有别于CFTR增强因子和校正因子。PTI-CH调节的F508del-CFTR增长并不会诱发与细胞溶质或细胞内质网有关的细胞应激反应。基于这些数据,催化因子代表一类新的CFTR调节因子,有希望被应用于CF的治疗,并与其他CFTR调节因子协同作用。
Introduction
Cystic fibrosis (CF) is an autosomal recessive, multisystem disease caused by defects in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which codes for an ion channel that mediates chloride transport across epithelial cell membranes.1 CFTR mutations include those that reduce levels of CFTR messenger RNA (mRNA) or prevent biosynthesis of CFTR protein, as well as those that encode misfolded, unstable, gating-defective, chloride conductance-defective, or otherwise nonfunctional protein.1,2
Currently, two types of small-molecule CFTR modulators have been developed that target the underlying defective mutant CFTR protein: potentiators and correctors. To date, ivacaftor (VX-770), a potentiator, and lumacaftor/ivacaftor, a corrector/potentiator combination, are the only two approved mutation-specific drug options for CF patients. Ivacaftor is approved for the treatment of CF patients with the G551D gating defect mutation, which affects approximately 3% to 4% of the CF population, as well as a handful of additional mutations that cause a similar deficit either in gating or in chloride conductance.3–8 The utility of ivacaftor by itself is therefore limited to select genotype-specific patients.3–6
The F508del mutation is present in approximately 90% of CF patients, and a beneficial response to the lumacaftor/ivacaftor combination was observed in patients homozygous for F508del, whereas no benefit was seen in compound heterozygous patients in which the second allele is not F508del.7,9,10 The effectiveness of this combination in compound heterozygous patients may be limited by reduced mutant CFTR protein substrates.9 It is possible that increasing the available mutant CFTR substrate may enhance therapeutic benefit. Effective future modulators will need to treat the full range of CFTR genotypes with the goal of providing a meaningful therapeutic benefit for all patients. CF mutations interfere with the quantity or function of CFTR protein biosynthesis, a process dictated by a complex cellular protein quality control system that reduces its efficiency.2,11 Current CFTR modulators are only as effective as the number of available substrates produced during biosynthesis.9 New strategies are therefore needed to address this underlying substrate deficiency.
In this study, we report the results of a high-throughput screen (HTS) aimed at identifying novel first-in-class small molecules that exhibit functional synergy in combination with ivacaftor and lumacaftor (VX-809) and do not necessarily function as potentiators or correctors themselves. One of the active series identified, represented by PTI-CH, was shown to possess novel characteristics relative to known modulators. This newly identified class of CFTR modulators, which we have termed amplifiers, may advance our understanding of CF biology and, more important, be a promising candidate as a therapy for patients with CF.
Methods
HTS
A phenotypic HTS was used to identify novel modulators of CFTR function ( Fig. 1 ). An immortalized cystic fibrosis bronchial epithelial cell line, CFBE41o¯,12,13 which stably expresses a previously described halide-sensitive (H128Q/I152L variant) yellow fluorescent protein (YFP) reporter,14–16 was used. This line is also stably transfected with a construct exogenously expressing F508del-CFTR complementary DNA (cDNA) under the control of a cytomegalovirus (CMV) promoter.13,16,17 We refer to this cell line as CFBE41-YFP. The assay involves the addition of sodium iodide to the cells and kinetically monitoring the fluorescence signal following iodide addition.18,19 If functional CFTR is present at the plasma membrane, iodide influx is accelerated and will quench fluorescence of the halide-sensitive reporter protein. This allows quantitation of CFTR function based on the forskolin- and genistein-dependent fluorescent quenching in response to modulators of CFTR function. Compounds were considered hits if they provided F508del-CFTR–mediated YFP quenching at 25% or greater than the positive control.
Figure 1.

Screening strategy, compound triage, and potency and maximum efficacy improvements during optimization of PTI-CH. (A) The screening funnel showing the steps used to confirm and characterize compound hits from the small-molecule library. (B) The number of compounds at the beginning and end of each of the triage steps in the screening process. The common pharmacophore for PTI-CA and PTI-CH is shown, with R1-3 and Ra side groups as described.20 (C) The yellow fluorescent protein (YFP) fluorescence quenching in response to sodium iodide (NaI) addition is shown for the DMSO control wells from one plate (±5 standard deviations) in comparison to the three indicated reference compounds that were included as test compounds in single wells in the compound library. These same reference compounds were also present on each plate in control wells (see text). CFTR, cystic fibrosis transmembrane conductance regulator; HBE, human bronchial epithelial; HTS, high-throughput screen.
CFBE41-YFP cell culture was scaled up in multilayer hyperflasks (cat. 10024; Corning, Corning, NY) inoculated at a density of 75,000,000 cells per hyperflask. MEM alpha (cat. 12571071; Thermo-fisher, Waltham, MA) was used to maintain the cells. For every 500 mL MEM alpha, the following components were added: 50 mL Gibco (Gaithersburg, MD) fetal bovine serum (FBS), 7.5 mL 50 mg/mL G418, 5 mL 10,000 µg/10,000 U/mL penicillin/streptomycin, and 100 µL of 10 mg/mL puromycin. After 3 days in the hyperflask, cells were seeded onto 384-well clear bottom assay plates (cat. 3962; BD Falcon, Tewksbury, MA). Cells were plated (12,500 cells per well) in 80 µL complete MEM alpha using a Titertek Multidrop dispenser (Titertek-Berthold, Pforzheim, Germany). Plates were incubated for 2 days at 37 °C and 5% CO2 prior to treatment with compound.
On day 2, 384-well plates containing DMSO compound stocks (at a concentration of ~10 mM and stored at −20 °C) were thawed at room temperature. Control compounds (96 samples per microplate) were added to the top four empty rows using a Beckman-Coulter Biomek FX liquid handler (cat. 4101500; Beckman-Coulter, Brea, CA). The 96 samples of control “compounds” consisted of 24 wells each of DMSO only (negative control), 3 µM lumacaftor (positive control), 10 µM tezacaftor (VX-661)21 (corrector mechanism secondary positive control), and 3 µM suberoylanilide hydroxamic acid (SAHA, proteostasis mechanism secondary positive control). The medium in the 384-well plates seeded 2 days prior was aspirated down from 80 µL to 25 µL/well with a 384-well aspirator (Tecan Falcon 25 Model PW384; Tecan, Morrisville, NC). The Biomek FX liquid handling program was used to treat cells with 25 µL of 2× compound at a final DMSO concentration of 0.2% and a final compound concentration of ~10 µM. Cells were then incubated for 24 h at 37 °C and 5% CO2.
On day 3, the 384-well plates were aspirated to 6 µL/well. Using a Multidrop dispenser (Model 832; Titertek-Berthold), 95 µL modified Dulbecco’s phosphate-buffered saline (DPBS) was added to each well. The washing step was repeated by aspirating to 6 µL per well and replacing 95 µL/well DPBS. The plates were again aspirated back down to 6 µL/well, and 25 µL of (1.25×) 12.5 µM forskolin (cat. F6886; Sigma-Aldrich, St. Louis, MO) and 62.5 µM genistein (cat. G6649; Sigma-Aldrich) in modified DPBS were added to a final concentration of 10 µM forskolin and 50 µM genistein. Plates were incubated at room temperature for 1 h. Before reading plates on a FDSS/µCell instrument (model A11529-01; Hamamatsu, Hamamatsu City, Japan), the internal Ligand A reservoir was filled with NaI buffer (0.7 mM CaCl2, 2.7 mM KCl, 1.5 mM KH2PO4, 1.1 mM MgCl2, 145 mM NaI, 8.1 mM NaHPO4, pH 7.4). Plates were read individually, collecting a 10-s baseline signal before injection with NaI buffer (30 µL delivered 4 mm above the bottom of the plate within 1 s), followed by a reading every 500 ms for 300 reads or a total of 2 min and 40 s. In between each plate, the in-instrument wash cycle was used to flush tips with water five times. After the reads were complete, data were analyzed to derive the YFP quenching rate the percent quench at 30 s after NaI addition as reporters for CFTR chloride channel activity.
Primary Human Bronchial Epithelial Cell Culturing and Differentiation
Human bronchial epithelial (HBE) cells from non-CF and CF donors of the indicated genotypes were provided by the Tissue Procurement and Cell Culture Core of the Cystic Fibrosis and Pulmonary Diseases Research and Treatment Center of the University of North Carolina Marsico Lung Institute (Chapel Hill, NC). HBEs were cultured essentially as described22 and were differentiated in an air-liquid interface essentially as described.23 HBEs were differentiated with medium replacement three times a week for a minimum of 4 weeks prior to electrophysiology (Ussing chamber) or other experiments.
Measuring CFTR Activity in Ussing Chambers
Primary HBEs with CF-causing mutations were differentiated for a minimum of 4 weeks in an air-liquid interface on SnapWell filter plates (cat. 3801; Corning Costar, Corning, NY) prior to Ussing measurements. Cells were apically mucus-washed for 30 min prior to treatment with compounds. The basolateral medium was removed and replaced with medium containing the compound of interest diluted to its final concentration from DMSO stocks. Treated cells were incubated at 37 °C and 5% CO2 for 24 h. At the end of the treatment period, the cells on filters were transferred to the Ussing chamber and equilibrated for 20 min in assay buffer (Coon’s F12 without sodium bicarbonate [cat. F6636; Sigma-Aldrich], 10 mM HEPES, pH 7.4). The short-circuit current (Isc) was measured in voltage clamp mode (Vhold = 0 mV), and the entire assay was conducted at 36.0 °C to 36.5 °C. Once the voltages had stabilized, the chambers were clamped and were recorded with readings every 10 s. Following baseline current stabilization, the following were added, and changes in the current and resistance of the cells were monitored: (1) 10 µM benzamil to the apical chamber to inhibit the ENaC sodium channel,24 (2) 10 µM forskolin to both chambers to activate CFTR by phosphorylation, (3) 1 µM ivacaftor to both chambers to potentiate CFTR channel opening, and (4) 20 µM CFTRinh-172 to the apical chamber to inhibit CFTR Cl– conductance.25 The inhibitable current (the current that is blocked by CFTRinh-172) is reported as the CFTR activity, and increased activity in response to compound beyond that observed in vehicle-treated samples is the improvement of F508del-CFTR function imparted by the compound tested.
Approach to Determining Synergy versus Additivity with Other CFTR Modulators
To evaluate whether the combination of PTI-CH with other modulators was additive or synergistic in the study, the DMSO CFTR activity was subtracted from the 3-µM PTI-CH CFTR activity, and this difference was then added to the “without PTI-CH” treatments for ivacaftor, lumacaftor, and the dual combination of ivacaftor + lumacaftor. This “expected” additivity was then compared to the observed activity for the respective treatments “with PTI-CH,” and the lumacaftor and ivacaftor + lumacaftor combinations were significantly greater (by two-way analysis of variance [ANOVA]) than the activity predicted for an additive interaction.
CFTR Immunoblotting
Cells were apically mucus-washed for 30 min prior to treatment with compounds. The basolateral medium was removed and replaced with medium containing the compound of interest diluted to its final concentration from DMSO stocks. Treated cells were incubated at 37 °C and 5% CO2 for 24 h. Cells were lysed and supernatants collected as follows: cells were washed with cold phosphate-buffered saline (PBS) three times and then lysed with 100 µL IP lysis buffer (cat. PI-87788; Thermo-Fisher Scientific, Grand Island, NY) containing protease inhibitors (cat. 11697498001, cOmplete tablets; Sigma-Aldrich). Lysates were placed on ice for 20 min and then centrifuged at 100,000 × g for 30 min at 4 °C. Supernatants were collected and protein assay performed using Lowry reagent from Bio-Rad (Hercules, CA). Protein samples (20–40 µg) were loaded onto a 3% to 8% Tris-Tricine gel (cat. 345-0129; Bio-Rad). After electrophoresis, proteins were transferred onto nitrocellulose membrane. The nitrocellulose membrane was then blocked in Tris-buffered saline + 0.1% Tween-20 (TBST) containing 20% goat serum for 1 h at room temperature with gentle agitation. Primary CFTR antibody (anti-CFTR; UNC-596) was prepared at a 1:1000 dilution in TBST, added to the membrane, and incubated overnight with gentle agitation at 4 °C. The following day, the membrane was washed three times (5 min each) with TBST at room temperature with gentle agitation and then incubated with goat anti-mouse IgG horseradish peroxidase (HRP)–labeled secondary antibody (sc-2005; Santa Cruz, Santa Cruz, CA) at a 1:10,000 dilution in TBST containing 10% goat serum for 1 to 3 h at room temperature with gentle agitation. Subsequently, the membrane was washed three times (5 min each) in TBST at room temperature with gentle agitation, and SuperSignal Femto solution (cat. PI-34096; Thermo-Fisher Scientific) was used to develop the HRP signal. Following detection, the membrane was stripped (cat. 21059, Restore Western Blot Stripping Buffer; Thermo-Fisher Scientific), blocked, and incubated with anti–Na+/K+ ATPase antibody as a loading control (cat. 3010S; Cell Signaling Technologies, Danvers, MA) at a 1:2000 dilution overnight at 4 °C with gentle agitation. Pierce ECL 2 plus substrate (cat. PI80196; Thermo-Fisher Scientific) was used the following day to develop the HRP signal. Band B/C quantification of protein amount was performed using an AlphaImager, Protein Simple, San Jose, CA densitometry software.
Surface and Total Protein Expression
The CFTR Mutant Surface and Total Protein Expression Assay and the P-gp Mutant Surface and Total Protein Expression Assay (Sharp Edge Labs, Pittsburgh, PA) were used to evaluate the effects of compounds on surface and total protein levels of F508del-CFTR and G268V–P-gp, respectively, in HEK293 cells. The assays make use of a fluorogen-activating protein fused to F508del-CFTR or G268V–P-gp that is detected through the use of a dye, which only fluoresces when bound to the protein.26 HEK293 cells stably transfected with constructs containing the peptide fused to F508del-CFTR or G268V–P-gp were incubated at 37 °C for 24 h with PTI-CH or one of two corrector compounds (VRT-325 or lumacaftor). Flow cytometry was then used to quantitate the levels of surface and total F508del-CFTR or G268V–P-gp protein using this system. A cell-impermeable dye was used to detect surface protein, while a cell-permeable dye detected total protein. Propidium iodide–positive cells were excluded from quantification.
RNA Isolation
Total RNA was isolated from nasal epithelial cells using the RNeasy Plus Universal Mini kit (cat. 73404; Qiagen, Oslo, Norway). Nasal epithelial lysate was thawed at room temperature. The lysate was agitated by running the Eppendorf tube across the top of a plastic Eppendorf tube rack twice. Samples were vortexed briefly and pipetted up and down five times with a P1000 pipette. Tubes were spun briefly in a benchtop centrifuge to allow the lysate to settle. The liquid was then transferred to a clean RNase-free Eppendorf tube, leaving behind the brush. An equivalent volume (about 350 µL) of 70% ethanol was added to the lysate, and the sample was mixed by pipetting up and down 7 to 10 times and vortexed briefly. The entire mixture was transferred, including all precipitate, to an RNeasy spin column in a 2-mL collection tube. The sample was centrifuged for 1 min at 10,000 × g in a benchtop centrifuge, and the flow-through was discarded; this step was repeated for the remainder of the sample, if necessary. The column filter was washed by adding 700 µL RWT wash buffer to the RNeasy spin column and spinning for 30 s at 10,000 × g. Buffer RPE (500 µL) was then added to the RNeasy spin column, the sample was centrifuged for 30 s at 10,000 × g, and flow-through was discarded. Then, 500 µL Buffer RPE was added to the RNeasy spin column, and the sample was centrifuged for 2 min at 10,000 × g. The RNeasy spin column was then transferred to a new collection tube and centrifuged for 1 min at 16,000 × g to dry the membrane. The RNeasy spin column was then placed in a new 1.5-mL RNase-free Eppendorf tube, and 35 µL of 37 °C RNase-free water was added directly to the spin column membrane and allowed to incubate for 5 min at room temperature. The tube was centrifuged for 1 min at 16,000 × g to elute the RNA, and the RNA concentration was measured.
Results
A High-Throughput Phenotypic Screening Strategy Identifies a Novel CFTR Modulator
A highly curated library of approximately 54,000 small molecules selected for novelty and drug-like properties was screened via HTS using a phenotypic assay in CFBE41-YFP cells to detect F508del-CFTR function. The assay performance for the HTS was monitored by negative control wells and three different positive control compounds, present on every plate. The DMSO-only negative control had a coefficient of variance of 4%, and the average Z′ across the plates was 0.5 for SAHA and tezacaftor and 0.7 for lumacaftor. The screening strategy shown in Figure 1A was used to confirm and validate hits as follows. From the library, 370 primary hits were identified and retested to determine the concentration dependence of their activity ( Fig. 1B ). The three control compounds mentioned above were also added to their own single empty well position on a single plate within the library to see if they would be identified as a hit. Figure 1C shows the quenching data from the primary HTS for these “library well” reference compounds that were indeed identified as hits in the screen and went on to show a concentration dependence to their activity. These were excluded from further characterization.
Of the 128 confirmed compounds that showed a concentration-dependent response, 27 were selected for further testing of their ability to improve endogenously expressed CFTR electrophysiological function in HBE cells from patients homozygous for the F508del mutation (F508del/F508del HBE cells). Five series of compounds were identified with activity in both the F508del CFBE41-YFP cell line and in F508del/F508del HBE cells. Of the five series of compounds that showed activity in F508del/F508del HBE cells, one of the compounds, PTI-CA, was found to increase F508del-CFTR levels in these cells. Based on the drug-like properties of PTI-CA and its observed activity in HBE cells, we pursued an optimization campaign to improve the efficacy and potency of the series. Among the optimized compounds, PTI-CH was selected as a representative of the chemical class for further characterization. PTI-CH has superior potency and in vitro efficacy compared to its parent compound in both the F508del CFBE41-YFP cell line assay ( Fig. 2A ) and F508del/F508del HBE cells ( Fig. 2B ). While the relative potency and maximal effect for PTI-CH are greater than PTI-CA in both the cell line and the primary cells, approximately 10 times more compound is needed in the overexpression F508del CFBE41-YFP cell line assay than in the primary F508del/F508del HBE cells for both compounds. This difference may be due to the increased abundance in CFTR in the cell line system relative to the endogenously expressing primary cells. Potency and maximal effective concentration differences between an overexpression cell line and F508del/F508del HBE cells have also been shown for lumacaftor.27
Figure 2.
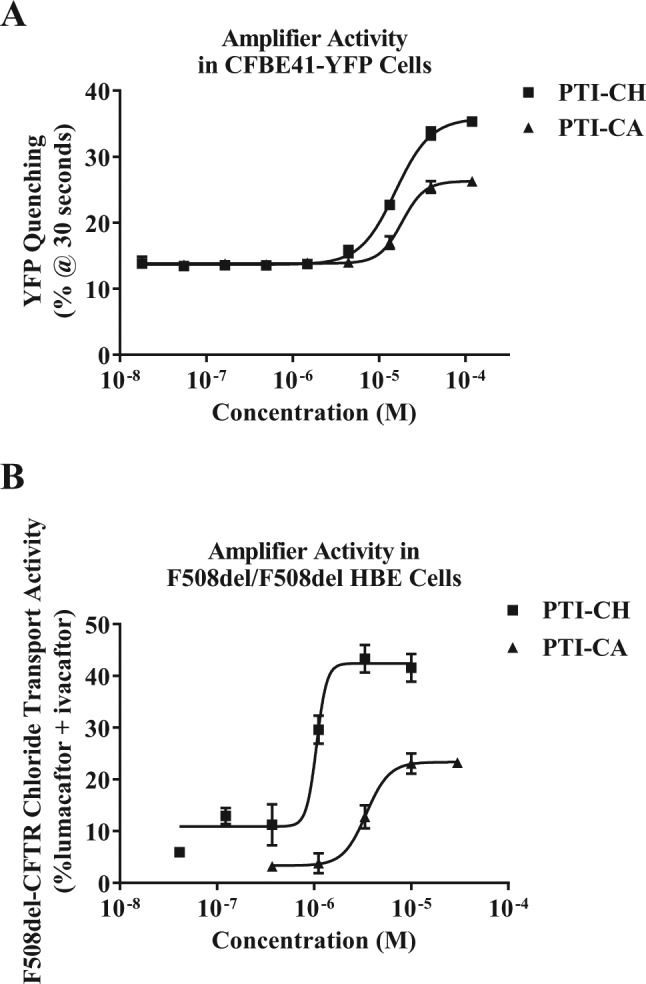
Potency and maximum efficacy improvements during optimization of PTI-CH. (A) Comparison of relative potency and activity of original hit compound PTI-CA with the optimized compound PTI-CH in the original CFBE41-YFP F508del–cystic fibrosis transmembrane conductance regulator (CFTR) functional assay. (B) Comparison of the relative potency and activity of PTI-CA with PTI-CH in the F508del/F508del human bronchial epithelial (HBE) CFTR functional assay. YFP, yellow fluorescent protein.
PTI-CH Increases F508del-CFTR Function in Primary HBE Cells and Augments the Activity of Other CFTR Modulators
Electrophysiological analysis was used to evaluate CFTR function in response to PTI-CH in comparison to and in combination with the CFTR modulators ivacaftor and lumacaftor in F508del/F508del HBE cells. The Ussing chamber was used to detect the short-circuit current across HBE monolayers, with the difference in short-circuit currents upon treatment with various compounds being measured to indicate their activity. The concentration-dependent activity of PTI-CH alone in HBE cells was recapitulated when incubated in combination with the corrector lumacaftor and acutely potentiated with ivacaftor ( Fig. 3A ). The forskolin- and ivacaftor-induced increases in the short-circuit current seen in vehicle control-treated cells were enhanced in cells treated with lumacaftor ( Fig. 3B ). Coadministration of PTI-CH with lumacaftor further enhanced the levels of both the forskolin-responsive and ivacaftor-potentiated short-circuit currents, nearly doubling the current in response to both molecules during the assay. As shown in Figure 3C , measurement of the CFTRinh172-sensitive current demonstrated a 1.5- to 2-fold increase in F508del-CFTR function upon treatment with PTI-CH alone, in combination with ivacaftor, in combination with lumacaftor, and in combination with both lumacaftor and ivacaftor. The synergy of the combination of all three CFTR modulators provided the greatest activity, indicating that all three molecules act nonredundantly to provide increased CFTR chloride transport activity.
Figure 3.

PTI-CH improves the functional rescue of F508del–cystic fibrosis transmembrane conductance regulator (CFTR) conferred by approved CFTR modulators. (A) PTI-CH shows a concentration-dependent increase in the activity of the combination of ivacaftor and lumacaftor in Ussing chamber short-circuit current measurements. (B) Representative Ussing chamber Isc traces are shown for the indicated treatments. Acute additions (10 µM forskolin, 1 µM ivacaftor, 20 µM CFTRinh172) during the Ussing assay are shown with arrows indicating the time of addition. All other compound incubations (3 µM lumacaftor with or without 3 µM PTI-CH) were for 24 h prior to the assay. Not shown is the 10 µM benzamil addition at the beginning of the assay to inhibit ENaC activity. (C) Summary of PTI-CH–mediated increases in F508del-CFTR CFTRinh172-sensitive current relative to other treatments. Concentrations as in panel B. Data are presented relative to the ivacaftor and lumacaftor combination. Solid bars represent means and black error bars represent the standard error of the mean (SEM) of three biological replicates. Open bars represent calculated values for expected additivity of PTI-CH with the indicated treatment, and gray error bars are drawn from the experimentally observed combinations. EXP, expected activity if PTI-CH were additive to the without PTI-CH condition; OBS, observed functional activity. See Methods for details. ns, non-significant. *p < 0.05, ****p < 0.0001 by two-way ANOVA with Tukey’s multiple comparisons test.
The results shown in Figure 3 indicate that the addition of PTI-CH nearly doubles the activity of a corrector (lumacaftor) and potentiator (ivacaftor) in combination when each compound is used at a concentration greater than its EC90.27,28 Although the observed synergy, in particular in the triple combination, suggests that PTI-CH functions through a mechanism different from that of lumacaftor or ivacaftor, the possibility remained that PTI-CH functions as a corrector or potentiator with a distinct and complementary mechanism.
PTI-CH Is Neither a Potentiator nor a Corrector of CFTR but Possesses Novel Characteristics
To investigate whether PTI-CH acts as a potentiator, we assessed its ability to acutely increase forskolin-dependent F508del-CFTR activity in the Ussing chamber assay ( Fig. 4A , B ). In contrast to ivacaftor, PTI-CH induced no effect on CFTR current upon acute addition, consistent with a lack of potentiator activity ( Fig. 4A ). Figure 4B contains the summary of the mean responses to the acutely added CFTR modulators shown in the Figure 4A traces.
Figure 4.

PTI-CH is distinct from the potentiator and corrector class of cystic fibrosis transmembrane conductance regulator (CFTR) modulators. (A) Average Isc traces of the indicated treatments are shown for F508del/F508del human bronchial epithelial (HBE) cells that had been incubated for 24 h with 3 µM lumacaftor. (B) Quantitation of the three chambers assayed for each treatment shown in panel A relative to the 1-µM ivacaftor acute addition. Bars represent means, and error bars represent the standard error of the mean (SEM) of three biological replicates. (C) Immunoblot of F508del/F508del HBE cells showing the levels of immature (band B) and mature (band C) of F508del-CFTR protein. NaATPase was used as a loading control. Lumacaftor and PTI-CH were both used at 3 µM, and F508del-CFTR was detected with MAB-596. (D) Surface and total expression of F508del-CFTR and G268V–P-gp were measured in the Sharp Edge Labs Mutant Surface and Total Protein Expression Assays in HEK293 cells. All compounds were used at 10 µM. Bars represent means, and error bars represent the SEM of six biological replicates. (E) CFTR messenger RNA (mRNA) levels were quantitated in F508del/F508del or non-CF HBE cells incubated for 24 h with 3 µM PTI-CH or DMSO prior to CFTR mRNA quantitation. Bars represent means, and error bars represent the SEM of three biological replicates. (F) F508del-CFTR and G268V–P-gp mRNA levels were measured from the Sharp Edge Labs stable transfectant HEK293 cells used in panel D. All compounds were used at 10 µM. Bars represent means, and error bars represent the SEM of four biological replicates.
To determine whether PTI-CH acts as a corrector, we next examined its effect on F508del-CFTR maturation ( Fig. 4C ). The degree of CFTR maturation was determined by immunoblot analysis, measuring the conversion of the lower molecular weight, immature, core-glycosylated form of F508del-CFTR (the 150-kDa band, band B) into the higher molecular weight, mature, fully glycosylated form (the 250-kDa band, band C).29,30 Treatment of F508del/F508del HBE cells with lumacaftor increased only the levels of the slower-migrating mature form of F508del-CFTR ( Fig. 4C ), consistent with its previously described activity as a corrector.27 In contrast, PTI-CH increased both the immature and mature forms of F508del-CFTR, consistent with an increase in total CFTR protein and consistent with PTI-CH acting through a mechanism distinct from direct F508del-CFTR correction. Interestingly, the combined treatment with PTI-CH and lumacaftor led to an enhanced increase in F508del-CFTR protein greater than that observed with either modulator alone, underscoring the synergistic effects observed in the functional assay (see Fig. 3 ).
HEK293 cells stably transfected with reporter constructs for F508del-CFTR or a related mutant ATP-binding cassette (ABC) transporter protein, G268V–P-gp, fused to a fluorogen-activating protein, were subjected to flow cytometry to quantitate cell surface and total protein levels. The G268V–P-gp was used as a control for specificity. The effect of PTI-CH compared with two distinct and established correctors, lumacaftor (previously shown to be selective for F508del-CFTR correction27) and VRT-325 (previously shown to also be highly active in correcting G268V–P-gp27), is shown in Figure 4D . While both correctors caused a modest increase in the cell surface levels of F508del-CFTR and a decrease in total protein, PTI-CH increased total F508del-CFTR levels to nearly 300% of vehicle-treated cells. This effect was selective, as PTI-CH did not increase total G268V–P-gp levels, indicating that PTI-CH does not increase the levels of other mutant membrane proteins and likely has a mechanism of action with some specificity toward CFTR. VRT-325 produced a 400% increase in surface expression of G268V–P-gp, with no increase in the total protein expressed ( Fig. 4D ).
The increase in the levels of total F508del-CFTR protein in response to PTI-CH could be explained by increasing the levels of CFTR mRNA. Primary bronchial epithelial cells were treated for 24 h with PTI-CH, and RNA was isolated and subjected to quantitative reverse transcription (RT)–PCR to measure the levels of CFTR mRNA. Figure 4E shows that PTI-CH indeed increased CFTR mRNA levels by ~1.5- to 2-fold in these cells, similar to the magnitude of its effect in functional assays (see Fig. 3C ). Importantly, the increase in CFTR mRNA due to PTI-CH was not dependent on the F508del mutation, as an increase was seen in HBE cells derived from a donor with the F508del/F508del genotype, as well as in HBE cells from a non-CF donor ( Fig. 4E ). While this manuscript was in revision, a separate study was published that showed that PTI-CH acts posttranscriptionally to increase endogenously expressed CFTR that had been engineered to have another CF-causative mutation, c.3700 A>G.31
In addition, CFTR mRNA was increased in the CFBE41-YFP cells where CFTR cDNA was expressed under the control of an exogenous promoter (data not shown) and in the HEK293 surface expression reporter system (see Fig. 4D ) where only the open reading frame (ORF) of F508del-CFTR was present ( Fig. 4F ). The same HEK293 reporter system expressing the G268V–P-gp ORF did not respond to PTI-CH. The ability of PTI-CH to selectively increase CFTR mRNA across different expression contexts is a novel aspect to its mechanism of action that will require further studies.
The PTI-CH–Mediated Increase in Misfolded CFTR Does Not Trigger a Cellular Stress Response
Since F508del-CFTR is a misfolded membrane protein, it was important to eliminate the possibility that an increase in expression of the mutant protein in response to PTI-CH triggers cellular stress responses, such as the cytosolic heat shock response or the endoplasmic reticulum–based unfolded protein response (UPR) ( Fig. 5 ). F508del/F508del HBE cells were treated either with PTI-CH, the CFTR potentiator ivacaftor, or an F508del-CFTR corrector, tezacaftor. Following 24 h of incubation with these modulators, gene expression of stress response constituents was measured using quantitative PCR (qPCR). For the cytosolic stress response, we selected genes that play induced or constitutive roles in this response: HSPA1A (Hsp70A1),32 HSPA8 (Hsc70),33 HSP90AA1 (Hsp90),34 and HSP90B1 (GRP94).35 No increase in mRNA levels of these stress genes was detected in response to the PTI-CH–induced increase in F508del-CFTR expression ( Fig. 5A ). Possible activation of the UPR was assessed by measuring the transcript levels of the following genes, which are transcriptional targets of the three arms of the UPR signaling pathway: HSPA5 (BiP),36 DNAJB9 (ERdj4),37 DNAJC3 (p58-IPK),38 EDEM,37 ATF4,36 DDIT3 (CHOP),36,37 and both total and spliced XBP1.36 In response to treatment with PTI-CH, none of the UPR genes exhibited an increase in transcript levels ( Fig. 5B ), indicating that the UPR is not activated by the PTI-CH–mediated increase in F508del-CFTR expression. The other two modulators, ivacaftor and tezacaftor, both resulted in increases in CHOP, suggesting those modulators activate some signaling through the UPR in vitro in primary HBE cells derived from F508del/F508del donors. In summary, an increase in F508del-CFTR expression in response to PTI-CH activated neither the cytosolic stress response nor the UPR, consistent with this increase not leading to an induction of cellular stress.
Figure 5.

The increase in F508del–cystic fibrosis transmembrane conductance regulator (CFTR) does not result in the induction of cytosolic stress or endoplasmic reticulum (ER)–associated unfolded protein response pathways. (A) Quantitative PCR (qPCR) results showing the effect of incubation with PTI-CH for 24 h at the indicated concentrations on the indicated HSF1-regulated messenger RNAs (mRNAs) in F508del/F508del human bronchial epithelial (HBE) cells. (B) qPCR results showing the effect of incubation with PTI-CH for 24 h at the indicated concentrations on the indicated UPR-regulated mRNAs in F508del/F508del HBE cells. Bars represent means, and error bars represent the standard error of the mean (SEM) of three biological replicates.
Discussion
As CFTR is a complex, multidomain, membrane-spanning protein, it undergoes tightly regulated steps during its biosynthesis to achieve a correctly localized and functional mature protein within epithelial cells.2 Mutations in CFTR affect the function or quantity of the CFTR channel at the cell surface.1,2 Current CFTR modulators are only as effective as the amount of available substrate protein.9 New strategies are therefore needed to address this underlying substrate limitation. While the recent regulatory approval of ivacaftor and lumacaftor/ivacaftor marks a major step forward in mutation-specific CFTR modulator-based therapies for CF,3–6,9,10 there is still a significant unmet therapeutic need for patients with the disease. Compared with the efficacy achieved with ivacaftor for patients with the G551D mutation,3,4 there is room for further benefit in lung function for patients with two copies of F508del for whom lumacaftor/ivacaftor is approved.9,10 An even greater need remains for patients with only one copy of F508del, in which their reduced levels of F508del-CFTR protein may prevent them from realizing a clinical benefit with available therapies, owing to the lack of sufficient substrate F508del-CFTR protein for the corrector and potentiator.9 Therapeutics that overcome the substrate limitation of F508del-CFTR, making F508del-CFTR available for other modulators to act upon, have the potential to address the unmet need for the most CF patients with one or two F508del alleles. In addition, if this activity is independent of the underlying CFTR mutation, these types of therapeutics might offer potential for all CF patients.
In this study, we report the results of an HTS campaign aimed at identifying novel pharmacological classes of small molecules that exhibit functional synergy with lumacaftor and ivacaftor. One of the active series identified, represented here by PTI-CH, augments the activity of other CFTR modulators and possesses novel characteristics relative to known modulators. This newly identified class of modulators functions neither as corrector nor as potentiator. We demonstrate that PTI-CH overcomes the inefficiencies of upstream CFTR biosynthesis by selectively increasing the expression of immature CFTR protein across different CFTR mutations, including F508del-CFTR.
No significant increases were detected among cellular stress response genes due to the amplifier-mediated increase in mutant F508del-CFTR expression. Interestingly, ivacaftor and tezacaftor both appear to induce some UPR signaling (see Fig. 5 ). However, the significance of these observations is unclear. PTI-CH appears to selectively increase the amount of CFTR protein available for other modulators to act upon and, thus, enhance the therapeutic benefit afforded by the currently approved corrector and potentiator classes of drugs. We have classified this novel type of small-molecule CFTR modulator as an amplifier.
While the screening strategy used to identify PTI-CH selected against direct transcriptional mechanisms, it did not preclude effects on CFTR biosynthesis and turnover at other points of regulation (e.g., by increasing transcript stability, reducing protein degradation, or improving translational efficiency). The mechanism by which PTI-CH selectively increases CFTR mRNA levels and protein expression is being intensely investigated and may provide further insight into how this novel modulator provides its benefit.
Acknowledgments
The authors thank the Tissue Procurement and Culturing Core at UNC, ChanTest, and Sharp Edge Labs for experimental support. We are grateful to Robert J. Bridges, Amita Thakerar, and Matthew Green from Chicago Medical School; Martin Mense, Hermann Bihler, and Eric Wong from the Cystic Fibrosis Foundation Therapeutics (CFFT); and William Skach from the Cystic Fibrosis Foundation (CFF) for their experimental help, expertise, insight, and helpful discussions. The authors also thank Brigid McEwan for technical support, Ken Longo for statistical analysis support, and Marija Zecevic for helpful comments on the manuscript. We acknowledge Caroline Ritchie for medical writing and F. Ulrich Hartl for editorial support.
Footnotes
Declaration of Conflicting Interests: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are employees of Proteostasis Therapeutics, Inc., and conducted this research as part of the company’s drug discovery efforts. Proteostasis Therapeutics, Inc. has filed several patent applications relating to the amplifier class of CFTR modulators.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded in part by a Therapeutics Development Award from Cystic Fibrosis Foundation Therapeutics, Inc.
References
- 1. Egan M. E. Genetics of Cystic Fibrosis: Clinical Implications. Clin. Chest Med. 2016, 37, 9–16. [DOI] [PubMed] [Google Scholar]
- 2. Ong T., Ramsey B. W. New Therapeutic Approaches to Modulate and Correct Cystic Fibrosis Transmembrane Conductance Regulator. Pediatr. Clin. North Am. 2016, 63, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramsey B. W., Davies J., McElvaney N. G., et al. A CFTR Potentiator in Patients with Cystic Fibrosis and the G551D Mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Davies J. C., Wainwright C. E., Canny G. J., et al. ; VX08-770-103 (ENVISION) Study Group. Efficacy and Safety of Ivacaftor in Patients Aged 6 to 11 Years with Cystic Fibrosis with a G551D Mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Boeck K., Munck A., Walker S., et al. Efficacy and Safety of Ivacaftor in Patients with Cystic Fibrosis and a Non-G551D Gating Mutation. J. Cyst. Fibros. 2014, 13, 674–680. [DOI] [PubMed] [Google Scholar]
- 6. Moss R. B., Flume P. A., Elborn J. S., et al. Efficacy and Safety of Ivacaftor Treatment: Randomized Trial in Subjects with Cystic Fibrosis Who Have an R117H-CFTR Mutation. Lancet Resp. Med. 2015, 32, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cystic Fibrosis Foundation. Patient Registry Annual Data Report 2015; Cystic Fibrosis Foundation: Bethesda, Maryland, 2016. [Google Scholar]
- 8. US Cystic Fibrosis Foundation, Johns Hopkins Medicine, The Hospital for Sick Children. Clinical and Functional Translation of CFTR. https://www.cftr2.org (accessed September 1, 2017).
- 9. Boyle M. P., Bell S. C., Konstan M. W., et al. ; VX09-809-102 Study Group. A CFTR Corrector (Lumacaftor) and a CFTR Potentiator (Ivacaftor) for Treatment of Patients with Cystic Fibrosis Who Have a Phe508del CFTR Mutation: A Phase 2 Randomised Controlled Trial. Lancet Respir. Med. 2014, 2, 527–538. [DOI] [PubMed] [Google Scholar]
- 10. Wainwright C. E., Elborn J. S., Ramsey B. W., et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lukacs G. L., Mohamed A., Kartner N., et al. Conformational Maturation of CFTR but Not Its Mutant Counterpart (ΔF508) Occurs in the Endoplasmic Reticulum and Requires ATP. EMBO J. 1994, 13, 6076–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meng Q. H., Springall D. R., Bishop A. E., et al. Lack of Inducible Nitric Oxide Synthase in Bronchial Epithelium: A Possible Mechanism of Susceptibility to Infection in Cystic Fibrosis. J Pathol. 1998, 184, 323–331. [DOI] [PubMed] [Google Scholar]
- 13. Bebok Z., Collawn J. F., Wakefield J., et al. Failure of cAMP Agonists to Activate Rescued ΔF508 CFTR in CFBE41o¯ Airway Epithelial Monolayers. J. Physiol. 2005, 569, 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wachter R. M., Remington S. J. Sensitivity of the Yellow Variant of Green Fluorescent Protein to Halides and Nitrate. Curr. Biol. 1999, 9, R628–R629. [DOI] [PubMed] [Google Scholar]
- 15. Galietta L. J., Haggie P. M., Verkman A. S. Green Fluorescent Protein–Based Halide Indicators with Improved Chloride and Iodide Affinities. FEBS Lett. 2001, 499, 220–224. [DOI] [PubMed] [Google Scholar]
- 16. Sondo E., Tomati V., Caci E., et al. Rescue of the Mutant CFTR Chloride Channel by Pharmacological Correctors and Low Temperature Analyzed by Gene Expression Profiling. Am. J. Physiol. Cell Physiol. 2011, 301, C872–C885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vijftigschild L. A. W., van der Ent C. K., Beekman J. M. A Novel Fluorescent Sensor for Measurement of CFTR Function by Flow Cytometry. Cytometry Part A.2013, 83A, 576–584. [DOI] [PubMed] [Google Scholar]
- 18. Lin S., Sui J., Cotard S., et al. Identification of Synergistic Combinations of F508del Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators. Assay Drug Dev. Technol. 2010, 8, 669–684. [DOI] [PubMed] [Google Scholar]
- 19. Sui J., Cotard S., Andersen J., et al. Optimization of a Yellow Fluorescent Protein–Based Iodide Influx High-Throughput Screening Assay for Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Modulators. Assay Drug Dev. Technol. 2010, 8, 656–668. [DOI] [PubMed] [Google Scholar]
- 20. Tait B. D., Cullen M. D. (Proteostasis Therapeutics, Inc.). Methods of Modulating CFTR Activity. International Patent Appl. WO 2014/210159 A1. December 31, 2014. [Google Scholar]
- 21. Pettit R. S., Fellner C. CFTR Modulators for the Treatment of Cystic Fibrosis. P. T. 2014, 39, 500–511. [PMC free article] [PubMed] [Google Scholar]
- 22. Fulcher M. L., Gabriel S., Burns K. A., et al. Well-Differentiated Human Airway Epithelial Cell Cultures. Methods Mol. Med. 2005, 107, 183–206. [DOI] [PubMed] [Google Scholar]
- 23. Neuberger T., Burton B., Clark H., et al. Use of Primary Cultures of Human Bronchial Epithelial Cells Isolated from Cystic Fibrosis Patients for the Pre-clinical Testing of CFTR Modulators. Methods Mol. Biol. 2011, 741, 39–54. [DOI] [PubMed] [Google Scholar]
- 24. Kleyman T. R., Cragoe E. J., Jr. Amiloride and Its Analogs as Tools in the Study of Ion Transport. J. Membr. Biol. 1988, 105, 1–21. [DOI] [PubMed] [Google Scholar]
- 25. Wang X. F., Reddy M. M., Quinton P. M. Effects of a New Cystic Fibrosis Transmembrane Conductance Regulator Inhibitor on Cl¯ Conductance in Human Sweat Ducts. Exp. Physiol. 2004, 89, 417–425. [DOI] [PubMed] [Google Scholar]
- 26. Holleran J., Brown D., Fuhrman M. H., et al. Fluorogen-Activating Proteins as Biosensors of Cell-Surface Proteins in Living Cells. Cytometry A 2010, 77, 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Van Goor F., Hadida S., Grootenhuis P. D. J., et al. Correction of the F508del-CFTR Protein Processing Defect In Vitro by the Investigational Drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18843–18848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Goor F., Hadida S., Grootenhuis P. D., et al. Rescue of CF Airway Epithelial Cell Function In Vitro by a CFTR Potentiator, VX-770. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18825–18830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang X-B., Mengos A., Hou Y-X., et al. Role of N-Linked Oligosaccharides in the Biosynthetic Processing of the Cystic Fibrosis Membrane Conductance Regulator. J. Cell Sci. 2008, 121, 2814–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng S. H., Gregory R. J., Marshall J., et al. Defective Intracellular Transport and Processing of CFTR Is the Molecular Basis of Most Cystic Fibrosis. Cell 1990, 63, 827–834. [DOI] [PubMed] [Google Scholar]
- 31. Molinski S. V., Ahmadi S., Ip W., et al. Orkambi® and Amplifier Co-Therapy Improves Function from a Rare CFTR Mutation in Gene-Edited Cells and Patient Tissue. EMBO Mol. Med. 2017. June 30. pii: e201607137. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang P., Leu J. I-J., Murphy M. E., et al. Crystal Structure of the Stress-Inducible Human Heat Shock Protein 70 Substrate-Binding Domain in Complex with Peptide Substrate. PLoS ONE 2014, 9, e103518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stricher F., Macri C., Ruff M., et al. HSPA8/HSC70 Chaperone Protein: Structure, Function, and Chemical Targeting. Autophagy 2013, 9, 1937–1954. [DOI] [PubMed] [Google Scholar]
- 34. Zuehlke A. D., Beebe K., Neckers L., et al. Regulation and Function of the Human HSP90AA1 Gene. Gene 2015, 570, 8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gewirth D. T. Paralog Specific Hsp90 Inhibitors—A Brief History and a Bright Future. Curr. Top Med. Chem. 2016, 16, 2779–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Takayanagi S., Fukuda R., Takeuchi Y., et al. Gene Regulatory Network of Unfolded Protein Response Genes in Endoplasmic Reticulum Stress. Cell Stress Chaperones 2013, 18, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Matsumura K., Sakai C., Kawakami S., et al. Inhibition of Cancer Cell Growth by GRP78 siRNA Lipoplex via Activation of Unfolded Protein Response. Biol. Pharm. Bull. 2014, 37, 648–653. [DOI] [PubMed] [Google Scholar]
- 38. Rutkowski D. T., Kang S-W., Goodman A. G., et al. The Role of p58IPK in Protecting the Stressed Endoplasmic Reticulum. Mol. Biol. Cell 2007, 18, 3681–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]