

Abstract
Background
Ritodrine is a commonly used tocolytic to prevent preterm labour. However, it can cause unexpected serious adverse reactions, such as pulmonary oedema, pulmonary congestion, and tachycardia. It is unknown whether such adverse reactions are associated with pharmacogenomic variants in patients.
Methods
Whole-exome sequencing of 13 subjects with serious ritodrine-induced cardiac and pulmonary side-effects was performed to identify causal genes and variants. The deleterious impact of nonsynonymous substitutions for all genes was computed and compared between cases (n = 13) and controls (n = 30). The significant genes were annotated with Gene Ontology (GO), and the associated disease terms were categorised into four functional classes for functional enrichment tests. To assess the impact of distributed rare variants in cases with side effects, we carried out rare variant association tests with a minor allele frequency ≤ 1% using the burden test, the sequence Kernel association test (SKAT), and optimised SKAT.
Results
We identified 28 genes that showed significantly lower gene-wise deleteriousness scores in cases than in controls. Three of the identified genes—CYP1A1, CYP8B1, and SERPINA7—are pharmacokinetic genes. The significantly identified genes were categorized into four functional classes: ion binding, ATP binding, Ca2+-related, and ciliopathies-related. These four classes were significantly enriched with ciliary genes according to SYSCILIA Gold Standard genes (P < 0.01), thus representing ciliary genes. Furthermore, SKAT showed a marginal trend toward significance after Bonferroni correction with Joubert Syndrome ciliopathy genes (P = 0.05). With respect to the pharmacokinetic genes, rs1048943 (CYP1A1) and rs1804495 (SERPINA7) showed a significantly higher frequency in cases than controls, as determined by Fisher’s exact test (P < 0.05 and P < 0.01, respectively).
Conclusions
Ritodrine-induced cardiac and pulmonary side effects may be associated with deleterious genetic variants in ciliary and pharmacokinetic genes.
Keywords: Ritodrine, Pulmonary oedema, Whole-exome sequencing, Ciliopathy, Joubert syndrome
Background
Preterm birth (PTB: before 37 weeks gestation) is a major cause of neonatal mortality and morbidity, and can cause long-term health problems [1, 2]. Babies born prematurely are at an increased risk of cerebral palsy, respiratory illnesses, and intellectual disabilities [3]. The incidence of PTB has been reported to range from approximately 4% in Eastern Asia to 17% in the United States [4, 5]. Despite extensive research, advances in obstetrics care, and the development of pharmacological agents designed to reduce PTB, few effective PTB therapies are available.
Tocolytic agents are medications used to delay PTB and suppress uterine contractions. Beta2-adrenergic receptor agonists, such as ritodrine, are widely used tocolytic drugs that are effective for uterine relaxation [6]. However, ritodrine can cause serious adverse effects, such as pulmonary congestion, pulmonary oedema, dyspnoea, and tachycardia for the mother and the foetus [7–9]. In Korea, 13.1% of patients receiving ritodrine experienced side effects [2]. Ritodrine was withdrawn from use in the United States, and is used on a limited basis in Europe and Asia [7, 9]. To our knowledge, few studies have aimed to identify genetic polymorphisms associated with side effects related to ritodrine treatment as a tocolytic therapy to prevent PTB. The relationship between a mutation in calcium voltage-gated channel subunit alpha 1 C (CACNA1C) and ritodrine-induced side effects was recently reported [10].
Whole-exome sequencing (WES) is currently used to identify novel genetic variants that affect protein function [11]. WES is being applied to identify candidate genes in Mendelian disorders, common diseases, and cancer [12]. In addition, rare variants associated with complex diseases have been found by WES [13]. For example, rare variants of identified genes have been found to affect low-density lipoprotein cholesterol levels [14]. However, genetic polymorphisms that lead to ritodrine-induced cardiac and pulmonary side effects have not yet been identified, and the molecular mechanisms underlying the adverse effects of ritodrine remain unclear.
Thus, we investigated genes associated with the side effects of ritodrine using WES from 13 ritodrine-treated subjects with serious side effects in PTB.
Methods
Ethics statement
This research involving human subjects and their genomic data was approved by the Institutional Review Boards of Ewha Womans University Medical Center and Konkuk University Medical Center (IRB Nos. ECT 06–127-7 and KUH1040034). Written Informed consent was obtained from each subject prior to their participation.
Patient and control samples for WES
Thirteen Korean pregnant women were analyzed using next-generation sequencing technology. Four subjects had pulmonary embolism, and nine subjects had a combination of tachycardia, palpitation, and/or dyspnoea. Two subjects were treated at Konkuk University Medical Center and the others were treated at Ewha Womans University Medical Center (Case, Table 1). For the control group, we selected exomes of 30 healthy Korean subjects (Control, 11 females and 19 males) from in-house data, which were provided by volunteers at the Division of Biomedical Informatics at Seoul National University (http://www.snubi.org/). Although the subjects had never been exposed to ritodrine, the exomes were sequenced using the same platform as the cases to minimize platform-specific biases. A total of 43 Koreans were recruited for WES analysis.
Table 1.
Clinical characteristics of 13 pregnant women
| Sample | Maternal age (years) | Gestational age at administration (weeks) | Side effect(s) |
|---|---|---|---|
| S1 | 30–39 | 31.4 | Dyspnea |
| S2 | 30–39 | 30.2 | Pulmonary edema, Pulmonary congestion |
| S3 | 30–39 | 34.0 | Tachycardia, Dyspnea |
| S4 | 30–39 | 35.1 | Dyspnea |
| S5 | 20–29 | 35.4 | Pulmonary edema, Dyspnea |
| S6 | 30–39 | 36.5 | Tachycardia, Dyspnea |
| S7 | 30–39 | 15.2 | Tachycardia, Dyspnea |
| S8 | 20–29 | 33.5 | Dyspnea |
| S9 | 40–49 | 27.6 | Tachycardia, Dyspnea, Palpitation |
| S10 | 20–29 | 20.3 | Tachycardia, Dyspnea, Headache, Sweating |
| S11 | 20–29 | 30.6 | Dyspnea, Palpitation |
| S12 | 20–29 | 30.5 | Pulmonary edema |
| S13 | 40–49 | 32.2 | Pulmonary edema |
WES and variant calls
Genomic DNA extracted from peripheral blood cells was amplified to generate 175–250-base pair (bp) DNA fragments spanning the protein-coding regions of human genome DNA using the Ion AmpliSeq Exome Panel (Thermo Fisher Scientific, Waltham, MA, USA). Library construction was performed to load the DNA samples into the semiconductor chip using the Ion AmpliSeq Exome Library Kit Plus, covering 57,742,646 bp (1.85% of human genomic regions) as described in the manufacturer’s instructions (Thermo Fisher Scientific). The exon-enriched DNA libraries were sequenced using the Ion Proton platform following the manufacturer’s instructions (Thermo Fisher Scientific). All subjects were sequenced with the PI chip, which generated a mean depth of 70× (a sufficient depth to interrogate the exons for mutations). Sequence alignment and MAP files were generated with Torrent Suite (v4.4) software (Thermo Fisher Scientific), and variants were identified using the Genome Analysis Toolkit (GATK; v2.8) software using ‘HaplotypeCaller’ against the GRCh37 version of the human reference genome [15].
Validation with genotyping assay
We validated 11 cases with side effects (out of the 13 cases with sufficient DNA) using an array-based high throughput method for 32 variants in 20 genes. In the single nucleotide polymorphism (SNP) type assay, 40 ng of genomic DNA flanking the SNP of interest was amplified by polymerase chain reaction (PCR) with a specific target amplification primer set. PCR was performed as described in the manufacturer’s instructions (Fluidigm, San Francisco, CA, USA). After amplification, the SNP type assay reaction was carried out according to the manufacturer’s instructions. SNP analysis was performed using Fluidigm SNP Genotyping Analysis software (ver. 4.0.1).
Predicting the deleterious impact of variants
The Sorting Intolerant from Tolerant (SIFT) [16, 17] algorithm predicts the effect of a coding variant on protein function, which is displayed as a single measure based on the conservation and scores precomputed and distributed by the J. Craig Venter Institute (http://sift.jcvi.org/). The SIFT Human database that supports GRCh37 Ensembl release 63 (the latest version) was downloaded. In total, 33,248,232 coding DNA sequence variants with a SIFT score, ranging from 0 to 1, and 19,729 protein-coding genes listed in the SIFT Human database were analyzed. Combined Annotation Dependent Depletion v1.3 (CADD; http://cadd.gs.washington.edu/) [18] and Polymorphism Phenotyping v2.2.2 (PolyPhen2; http://genetics.bwh.harvard.edu/pph2/) [19] were also downloaded to predict the deleteriousness of the annotated variants on protein function.
Aggregation of the impact of variants within genes
The gene deleteriousness score (G), defined as the geometric mean of the SIFT scores for the multitude of deleterious variants in a gene [20], was applied to estimate the aggregate impact of all deleterious variants in the genes. G aggregates the impact of deleterious variants by combining the probabilities of estimation of the likelihood that protein function was altered for each gene. Multiple deleterious variants of the same gene may synergistically impact protein function. We included only variants with a SIFT score < 0.7 as an input of the geometric mean and replaced 0 to 10− 8 for the G score calculation. We assigned a G score of 1 when no variants were reported in a given gene. A lower G score indicates a more damaged function of the gene at the protein level.
Identification of significantly altered genes
Among the 13 cases, a total of 558,091 variants were detected from 117,633 loci (Fig. 1). Initially, we included 86,927 loci with allele frequencies (AFs) ≥ 1/5008, as reported in the 1000 Genomes Project (T1GP; n = 2504), phase 3, with the assumption that variants in a highly curated public database would be less likely to contain errors [21, 22]. Next, we calculated a G score vector for all genes in each sample to assess the impact of deleterious variants on protein function. Student’s t-test was applied to identify altered function genes with deleterious variants by comparing the G score distribution between the case and control groups. We selected genes with at least one variant with SIFT < 0.3, which contributed to the lower G score. Lastly, to exclude likely false-positives, we reviewed each variant of the selected genes by manually inspecting all reads at all candidate loci in BAM files.
Fig. 1.
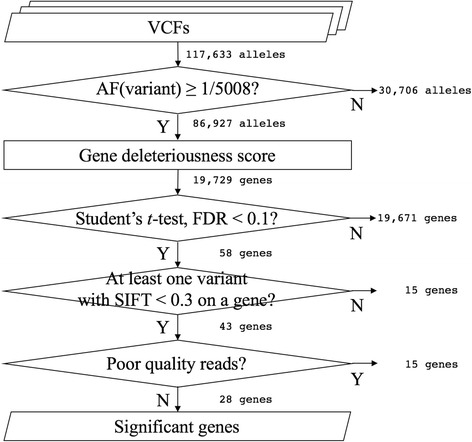
Workflow of data analysis for whole-exome sequencing. Workflow shows the steps used to select the significantly altered genes associated with ritodrine-induced side effects from 13 exomes. AF, allele frequency; FDR, false discovery rate; SIFT, sorting intolerant from tolerant
Gene functional annotation and categorization
To interpret the biological relevance of the significant genes, we used DAVID v6.7 software to annotate Gene Ontology (GO) terms [23, 24]. In addition, we manually reviewed every gene to annotate known gene functions and associated diseases. We categorized the selected genes based on the annotations into four functional categories: ion binding, ATP binding, Ca2+-related, and ciliopathies. We obtained gene sets for the four categories; ion binding (GO: 0043167) and ATP binding (GO: 0005524) had corresponding GO terms that were exactly matched. Thus, we downloaded the gene sets annotated with the GO terms, which were comprised of 4386 and 1495 genes, respectively. However, the GO terms for the remaining two categories did not show an exact match. We searched for ‘calcium’ or ‘cilia’ as keywords to find the GO terms containing either of the keywords. As a result, we collected 1548 and 370 genes annotated to 312 and 42 GO terms for ‘Ca2 + −related’ and ‘ciliopathies’, respectively. We performed hypergeometric distribution tests with the four gene-sets to investigate the statistical significance of the selected genes.
Ciliary genes and Joubert syndrome-related genes
Ciliary genes were extracted from The SYSCILIA Gold Standard Version 1, a high-confidence dataset that provides gene lists associated with ciliopathies (n = 303) [25]. In addition, we downloaded genes known to be involved in ciliopathy (n = 102) and Joubert and Meckel-Gruber syndromes (JBTMKS, n = 30) [26]. As the genes for Joubert syndrome (JBTS, n = 11) and Meckel-Gruber syndrome (MKS, n = 8) were grouped together in the JBTMKS category, we obtained genes for each syndrome from the Genetics Home Reference (GHR) [27, 28]. For each gene list, we carried out three rare-variant association tests: (1) the burden test, (2) sequence Kernel association test (SKAT), and (3) the optimised (SKAT-O) test using the SKAT package in R [13]. Only variants with a minor allele frequency (MAF) < 1% were selected for rare-variant association tests.
Results
Primary WES data analysis and genotyping assays
The Torrent Mapping and Alignment Program aligner in Torrent Suite generated, on average, 20 GB of BAM files per sample. Based on the GATK Best Practices guidelines, we modified a variant-calling pipeline to function with Ion Proton data and obtained an average of 41,819 ± 1976 [mean ± standard deviation (SD)] and 41,661 ± 1848 variants from the case and control groups (P = 0.81), respectively. Additionally, we confirmed that there was no significant difference in the number of variants between males and females in controls (P = 0.89). Next, we filtered out 30,706 variants that were not reported in the T1GP. We merged all variants that were called from individuals and annotated the SIFT score for each variant. Then, we calculated the G score for the genomic profile. The G score distributions were uniform across the different groups, showing a high and sharp peak near the highest score, which indicates a gene with normal function, a small peak near the lowest score, which indicates altered function, and a long plateau between the peaks, which indicates moderate function. Using the G score, we carried out Student’s t-test and found that the distributions of gene scores of 58 genes differed significantly at the significance level (false discovery rate, FDR < 0.1). We selected 43 genes that harboured deleterious and likely deleterious variants predicted by SIFT. For those selected genes, we concluded that the calls in 15 genes were false positive findings due to misalignment and/or off-target regions and that the remaining 28 genes were true positives. In summary, we identified 28 genes SIFT-predicted deleterious variants that were statistically significantly associated with ritodrine-induced cardiac and pulmonary side effects (Table 2). Additionally, all 32 variants that were predicted to be deleterious by either SIFT or CADD, among the 82 variants in the 28 significant genes, were successfully replicated using Fluidigm™ genotyping assays in 11 cases with sufficient DNA.
Table 2.
Genes significantly associated with ritodrine-induced side effects
| Gene symbol | Gene name | FDR | P | G (mean ± SD) | Gene functional category | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Case, (n = 13) | Control, (n = 30) | ION | ATP | Ca2+ | CP | PK | ||||
| AASDH | Aminoadipate-Semialdehyde Dehydrogenase | 0.019104 | 0.000041 | 0.16 ± 0.19 | 0.58 ± 0.44 | ✓ | ✓ | |||
| ARMC9 | Armadillo Repeat Containing 9 | 0.025396 | 0.000057 | 0.06 ± 0.03 | 0.44 ± 0.46 | ✓ | ||||
| B9D2 | B9 Protein Domain 2 | 0.012597 | 0.000017 | 0.25 ± 0.33 | 0.82 ± 0.36 | ✓ | ||||
| BLK | B lymphoid tyrosine kinase | 0.007009 | 0.000004 | 0.16 ± 0.00 | 0.58 ± 0.43 | ✓ | ||||
| CARS2 | Cysteinyl-TRNA Synthetase 2, Mitochondrial (Putative) | 0.068711 | 0.000227 | 0.23 ± 0.35 | 0.73 ± 0.43 | ✓ | ✓ | |||
| CD1A | CD1a Molecule | 0.061968 | 0.000192 | 0.35 ± 0.37 | 0.86 ± 0.33 | |||||
| CSPG5 | Chondroitin Sulfate Proteoglycan 5 (Neuroglycan C) | 0.075577 | 0.000336 | 0.24 ± 0.00 | 0.49 ± 0.36 | |||||
| CYP1A1 | Cytochrome P450, Family 1, Subfamily A, Polypeptide 1 | 0.065067 | 0.000209 | 0.03 ± 0.03 | 0.36 ± 0.46 | ✓ | ✓ | ✓ | ||
| CYP8B1 | Cytochrome P450, Family 8, Subfamily B, Polypeptide 1 | 0.012973 | 0.000020 | 0.45 ± 0.09 | 0.71 ± 0.26 | ✓ | ✓ | |||
| FAT4 | FAT Atypical Cadherin 4 | 0.092483 | 0.000539 | 0.23 ± 0.10 | 0.43 ± 0.28 | ✓ | ✓ | |||
| FUT6 | Fucosyltransferase 6 (Alpha (1,3) Fucosyltransferase) | 0.051144 | 0.000147 | 0.14 ± 0.04 | 0.46 ± 0.42 | |||||
| GALNT10 | Polypeptide N-Acetylgalactosaminyltransferase 10 | 0.092483 | 0.000549 | 0.35 ± 0.21 | 0.66 ± 0.35 | ✓ | ||||
| HHATL | Hedgehog Acyltransferase-Like | 0.076828 | 0.000369 | 0.27 ± 0.11 | 0.50 ± 0.30 | |||||
| IFT74 | Intraflagellar Transport 74 | 0.011408 | 0.000014 | 0.25 ± 0.10 | 0.56 ± 0.32 | ✓ | ||||
| ICE1 | Interactor Of Little Elongation Complex ELL Subunit 1 | 0.014088 | 0.000026 | 0.49 ± 0.08 | 0.70 ± 0.23 | |||||
| KNDC1 | Kinase Non-Catalytic C-Lobe Domain (KIND) Containing 1 | 0.099453 | 0.000617 | 0.34 ± 0.03 | 0.53 ± 0.29 | |||||
| NKAIN3 | Na+/K+ Transporting ATPase Interacting 3 | 0.014088 | 0.000026 | 0.21 ± 0.08 | 0.56 ± 0.40 | ✓ | ||||
| OR6B1 | Olfactory Receptor, Family 6, Subfamily B, Member 1 | 0.092483 | 0.000562 | 0.28 ± 0.02 | 0.50 ± 0.33 | |||||
| PSMD9 | Proteasome (Prosome, Macropain) 26S Subunit, Non-ATPase, 9 | 0.092483 | 0.000511 | 0.43 ± 0.40 | 0.91 ± 0.25 | |||||
| PXT1 | Peroxisomal, Testis Specific 1 | 0.075577 | 0.000336 | 0.02 ± 0.00 | 0.35 ± 0.47 | |||||
| RBBP8NL | RBBP8 N-Terminal Like | 0.092483 | 0.000564 | 0.21 ± 0.25 | 0.58 ± 0.43 | |||||
| RSPH3 | Radial Spoke 3 Homolog (Chlamydomonas) | 0.012770 | 0.000018 | 0.27 ± 0.07 | 0.61 ± 0.37 | ✓ | ||||
| SERPINA7 | Serpin Peptidase Inhibitor, Clade A (Alpha-1 Antiproteinase, Antitrypsin), Member 7 | 0.077599 | 0.000400 | 0.31 ± 0.48 | 0.90 ± 0.30 | ✓ | ||||
| SLC12A7 | Solute Carrier Family 12 (Potassium/Chloride Transporter), Member 7 | 0.051144 | 0.000148 | 0.36 ± 0.04 | 0.60 ± 0.31 | ✓ | ||||
| SPTA1 | Spectrin, Alpha, Erythrocytic 1 | 0.008266 | 0.000007 | 0.32 ± 0.08 | 0.49 ± 0.15 | ✓ | ✓ | |||
| TNKS | Tankyrase, TRF1-Interacting Ankyrin-Related ADP-Ribose Polymerase | 0.077599 | 0.000401 | 0.38 ± 0.19 | 0.68 ± 0.33 | ✓ | ||||
| ZDHHC12 | Zinc Finger, DHHC-Type Containing 12 | 0.075577 | 0.000332 | 0.17 ± 0.15 | 0.41 ± 0.27 | ✓ | ||||
| ZNF273 | Zinc Finger Protein 273 | 0.092483 | 0.000527 | 0.10 ± 0.05 | 0.38 ± 0.42 | ✓ | ||||
FDR false discovery rate, G gene deleteriousness score, ION ion binding, ATP adenosine triphosphate binding, Ca2+ calcium-related, CP ciliopathy, PK pharmacokinetics–relate
Genes associated with ritodrine-induced side effects
The hypergeometric test failed to identify a significantly enriched GO term, perhaps due to the small number of significant genes obtained. However, the hypergeometric distribution test revealed that the 28 genes showed significant enrichment with the SYSCILIA Gold Standard genes (P = 0.009). SYSCILIA is a multi-national consortium for a biology systems approach to dissect cilia function and its disruption in human genetic disease, and the SYSCILIA Gold Standard contains highly curated ciliary and ciliopathy genes. Even after lowering the definition stringency of deleterious genes between cases and controls from FDR < 0.01 to P < 0.01 and P < 0.05, the statistical significance of the SYSCILIA Gold Standard gene enrichment remained robust and sustained (P0.01 = 0.047 and P0.05 = 0.001, respectively). As shown in Table 2, the 28 significant genes (FDR < 0.1) were categorized into four functional classes: ion binding, ATP binding, Ca2+-related, and ciliopathies.
Analysis of ciliopathy genes and their subsets
The SKAT rare variant association test between the case (n = 13) and control (n = 30) groups showed marginal significance for JBTMKS after Bonferroni correction (P = 0.1054, Table 3) from the Invitae. Since JBTMKS harbour both JBTS- and MKS-related genes, we further evaluated the association signals for JBTS and MKS separately using GHR. Only JBTS genes showed a marginal trend toward significance, whereas MKS genes did not. This result suggests that genetic variations identified in patients with serious ritodrine-induced cardiac and pulmonary side effects may be associated with JBTS. Table 4 presents the rare (MAF < 0.01) and deleterious (predicted to be deleterious by at least one Bioinformatics tools: SIFT, CADD, or PolyPhen2) variants in JBTS genes (n = 11) in patients with serious ritodrine-induced side effects. Of these, four genes (AHI1, ARL13B, KIF7, and RPGRIP1L) exhibited five heterozygous mutations, but no homozygous mutations. Pulmonary embolism, considered to be the most serious side effect induced by ritodrine, was observed in four patients (SN1923, SN3230, SN8592 and SN9899). Rs193219215 on ADP ribosylation factor-like GTPase 13B (ARL13B), which interacts with Intraflagellar transport 74 (IFT74) [29], was found in two cases with pulmonary embolism (SN3230 and SN8592). This nonsynonymous variant, predicted to be deleterious by CADD, is a rare variant (MAF < 0.001) that has been found only in East Asian (EAS) populations according to T1GP. Rs146925098 on RPGRIP1 like (RPGRIP1L) was found in one case with pulmonary embolism (SN3230). SN1923 and SN9899 cases with pulmonary embolism exhibited none of the above variants.
Table 3.
Rare-variant association tests for genes associated with ciliopathies and subsets
| Database | Gene list (No. of genes) | Bonferroni corrected P | Reference | ||
|---|---|---|---|---|---|
| Burden | SKAT | SKAT-O | |||
| Invitae | Ciliopathies (102) | 1.00000 | 1.00000 | 1.00000 | [26] |
| Joubert and Meckel-Gruber Syndromes (JBTMKS; 30) | 0.39528 | 0.10536 | 0.19548 | [26] | |
| GHR | Joubert Syndrome (JBTS; 11) | 0.09908 | 0.05076 | 0.08020 | [27] |
| Meckel Syndrome (MKS; 8) | 1.00000 | 1.00000 | 1.00000 | [28] | |
GHR Genetics home references, SKAT sequence kernel association test
Table 4.
Distribution of rare and deleterious variants on four Joubert Syndrome genes from GHR in 13 cases with ritodrine-induced serious cardiac and pulmonary side effects
| Gene Symbol | rsID | Variant deleteriousness score | MAF | SN1275 | SN1923 | SN2931 | SN2956 | SN3230 | SN3828 | SN4181 | SN4211 | SN4407 | SN5021 | SN5209 | SN8592 | SN9899 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | CADD | PolyPhen2 | ||||||||||||||||
| AHI1 | rs148000791 | 0.25 | 23.6 | NA | 0.0077 | HET | WT | HET | HET | WT | WT | WT | WT | WT | WT | WT | HET | WT |
| ARL13B | rs193219215 | 0.12 | 20.9 | 0.41 | 0.0008 | WT | WT | WT | WT | HET | WT | WT | WT | WT | WT | WT | HET | WT |
| KIF7 | rs546772749 | 0.01 | 9.2 | 0.01 | 0.0002 | HET | WT | HET | HET | HET | WT | WT | WT | HET | HET | WT | WT | WT |
| rs536773143 | 0.16 | 15.4 | 0.89 | 0.0002 | WT | WT | WT | WT | WT | WT | WT | WT | WT | WT | WT | HET | WT | |
| RPGRIP1L | rs146925098 | 0 | 19.7 | 0.99 | 0.0002 | WT | WT | WT | WT | HET | WT | WT | WT | WT | WT | WT | WT | WT |
SIFT Sorting Intolerant from Tolerant, CADD Combined Annotation Dependent Depletion, PolyPhen2 Polymorphism Phenotyping v2.2.2, AF allele frequency, HET heterozygous, WT wild type, AHI1 Abelson Helper Integration Site 1, ARL13B ADP Ribosylation Factor Like GTPase 13B, KIF7 Kinesin Family Member 7, RPGRIP1L RPGRIP1 Like
Drug metabolism-related genes
Two cytochrome P450 genes [family 1, subfamily A, polypeptide 1 (CYP1A1) and family 8, subfamily B, polypeptide 1 (CYP8B1)] and serpin family A member 7 (SERPINA7), which are involved in drug metabolism phases I and II, respectively, exhibited significantly lower G scores in the case group compared to those in the control group. Rs1048943 on CYP1A1 was found in nine patients including two patients with pulmonary embolism (SN1932 and SN9899), and the carrier frequency in cases was significantly higher than in controls using Fisher’s exact test (P = 0.046, Table 5). The carrier frequency of rs143070677 on CYP1A1 was significantly higher in cases than in the EAS population (P = 0.050), whereas it was not significant compared to the control group (P = 0.518). This allele was found in one patient with pulmonary embolism (SN3230) and is a singleton in T1GP (MAF < 0.0002). Rs4646422 on CYP1A1 was found in four patients including one patient with pulmonary embolism (SN8592). In summary, either one of the two functional variants of CYP1A1 was found in all patients with pulmonary embolism. These alleles are East Asian-specific, in that their frequencies are relatively higher in EAS than in other populations. Although a patient with rs202192572 on CYP8B1 did not have pulmonary embolism, this is also a rare East Asian-specific variant that was found in only five EAS subjects in T1GP. The carrier frequency of rs1804495 on SERPINA7 was significantly higher in cases than in control groups (PControl < 0.001, PEAS = 0.018, and PT1GP = 0.001), and nine patients carried this allele including two with pulmonary embolism. Rs1804495 was also over-represented in the EAS population.
Table 5.
Carrier frequencies and Fisher’s exact test results of variants on the drug metabolism-related genes
| Gene Symbol | rsID | Variant deleteriousness score | Case, n = 13 | Control, n = 30 | EAS, n = 504 | T1GP, n = 2504 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | CADD | PolyPhen2 | Freq | P | Freq | P | Freq | P | |||
| CYP1A1 | rs1048943 | 0.01 | 21.9 | 0.49 | 9 (69%) | 10 (33%) | 0.0457 | 221 (44%) | 0.0902 | 568 (23%) | 0.0005 |
| rs143070677 | 0.03 | 24.4 | 0.51 | 1 (8%) | 1 (3%) | 0.5183 | 1 (0.2%) | 0.0497 | 1 (0.04%) | 0.0103 | |
| rs4646422 | 0.08 | 24.0 | 0.19 | 4 (31%) | 12 (40%) | 0.7349 | 111 (22%) | 0.4986 | 116 (5%) | 0.0025 | |
| CYP8B1 | rs202192572 | 0.04 | 23.9 | 0.05 | 1 (8%) | 0 (0%) | 0.3023 | 5 (1%) | 0.1423 | 5 (0.2%) | 0.0306 |
| SERPINA7 | rs1804495 | 0.01 | 23.8 | 0.46 | 9 (69%) | 3 (10%) | 0.0002 | 179 (36%) | 0.0180 | 623 (25%) | 0.0010 |
Freq carrier frequency, SIFT Sorting Intolerant from Tolerant, CADD Combined Annotation Dependent Depletion, PolyPhen2 Polymorphism Phenotyping v2.2.2, EAS East Asian in the 1000 Genomes Project, T1GP The 1000 Genomes Project, CYP1A1 cytochrome P450, family 1, subfamily A, polypeptide 1, CYP8B1 cytochrome P450, family 8, subfamily B, polypeptide 1, SERPINA7 serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 7
Discussion
Little is known about the physiological mechanism of ritodrine; ADRB2 is the only known target. Here, we analyzed the whole-exomes of 13 cases with serious side effects and identified 28 genes that increase the risk of ritodrine-induced cardiac and pulmonary side effects. To the best of our knowledge, this study was the first analysis using next-generation sequencing data to identify associations between ritodrine side effects and genomic variants. Using the G scoring method, ion binding, ATP binding, Ca2+-related, and ciliopathy genes were found to be significantly altered in protein function. In particular, ion binding, ATP binding, and Ca2+-related genes are important and well-known for their roles in tocolysis. In addition, rare variants in cases were associated with JBTS genes using RVATs. Two out of four pulmonary embolism cases carried at least one rare and deleterious variant on JBTS genes. Consequently, we speculate that rare variants on JBTS genes may contribute to the ritodrine-induced side effects related to muscle flexibility.
In addition to identifying ciliopathy genes, we detected ion- and ATP-binding, and calcium-related, genes that were significantly associated with ritodrine-induced side effects. These genes have not been reported as being disease-related genes and are not members of a pathway, implying that they may not play a key role in pathogenicity. The results suggest that, the significant genes may be important in drug responses; however, further investigations are required. The molecular functions of these genes have been well-studied. Calcium, ATP and inorganic phosphate play key roles in the physiological functions of proteins, including muscle contraction [30]. In particular, myometrium contraction is affected by increased calcium levels and activation of the myosin light chain kinase via phosphorylation [31]. Ritodrine causes relaxation of smooth muscles in the uterus while stimulating cardiac muscles [31, 32]. In addition, mutations in sulfotransferase 1A3 were determined to affect the metabolic activity of ritodrine [33]. Collectively, we speculate that genetic predisposition related to ion- and ATP-binding and calcium plays an important role in the side effects of ritodrine treatment of PTB. An association between rs10774053 in CACNA1C and ritodrine side effects was reported recently [10]; however, this genetic association was not replicated in this study as no significant difference (P = 0.1962) was detected in CACNA1C; G score (mean ± SD) between the case and the control groups (0.28 ± 0.2 and 0.35 ± 0.3, respectively). However, they selected five SNPs in CACNA1C, including three and two variants in intron and exon regions, respectively. The two exonic variants are predicted to be benign according to SIFT, and a recessive model was applied for statistical tests.
The distribution of deleterious variants of pharmacokinetic genes differs markedly across ethnic groups. Two rare Asian-specific functional alleles (rs143070677 and rs202192572) were reported only in an EAS population with MAF < 0.001. Rs1048943, rs4646422, and rs1804495 are common alleles (MAF > 0.05), and MAFs in EAS are relatively higher than those in other ethnic groups. CYP1A1 and CYP8B1, which are xenobiotic enzymes, are involved in the metabolism of drug and toxicants [34]. Elevated CYP1A1 activity causes the formation of DNA adducts in pulmonary tissue and cellular damage [35, 36]. A particularly interesting finding was that at least one of the functional variants of CYP1A1 was identified in pulmonary embolism cases in this study. An association of CYP1A1 polymorphisms with an increase in CYP1A1 activity has been confirmed by functional studies [37, 38]. In particular, the catalytic activity in oestrogen metabolism was significantly higher in those with rs1048943 than wild-type [39]. The tocolytic effect of ritodrine was enhanced by adding natural progesterone in pregnant women [40]. A more recent study revealed the loss of a transcription factor binding site at Sp7 due to rs1048943 at exon 7 of CYP1A1; this variant was predicted to be deleterious by SIFT and CADD [41]. We also identified rare variants in drug metabolism-related genes with a relatively higher frequency in Asian subjects, despite the small sample size. Therefore, overrepresented and/or significantly higher deleterious variants in drug metabolism genes may also increase the risk of ritodrine-induced side effects, such as pulmonary embolism, in the Korean population.
Conclusions
Using WES, this study identified rare deleterious variants associated with ritodrine-induced serious cardiac and pulmonary side effects in Korean preterm labour subjects. Most importantly, rare variants on ciliopathy genes were demonstrated to be significantly associated with JBTS. Asian-specific rare and common variants related to the pharmacokinetics of ritodrine may elicit serious cardiac and pulmonary side effects. Further studies are needed to validate the rare variants in a larger cohort for replication, and to elucidate the role of these variants in the molecular mechanisms of the side effects.
Acknowledgements
JHK would like to gratefully acknowledge the Education and Research Encouragement Fund of Seoul National University Hospital.
Funding
This study was supported by the Ministry of Health & Welfare of the Republic of Korea (HI14C0306 and HI15C2059) through the Korea Health Industry Development Institute, and grants from the Korean Health Technology R&D Project, Ministry of Health and Welfare (HI16C11280000).
Availability of data and materials
The SRA (Sequence Read Archive) accession number for the exome sequences reported in this paper is SRP127651.
Authors’ contributions
HS, EJK, YAY, YP, and KY performed the research, wrote the manuscript and contributed to revision of the paper. BJM performed the sequencing experiments, assisted in data analysis, and wrote the manuscript. HSH and YJK conducted the clinical design and sample acquisition. JHK conceived of the research idea and designed the study. HSH, JHK and YJK managed and supervised the study. All authors reviewed, edited and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Institutional Review Boards of Ewha Womans University Medical Center and Konkuk University Medical Center (IRB Nos. ECT 06–127-7 and KUH1040034). All participants provided written informed consent prior to participation in the study.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Heewon Seo, Email: lootpiz@snu.ac.kr.
Eun Jin Kwon, Email: friendkej1004@hanmail.net.
Young-Ah You, Email: yerang02@naver.com.
Yoomi Park, Email: yoomip@snu.ac.kr.
Byung Joo Min, Email: muz1978@snu.ac.kr.
Kyunghun Yoo, Email: yookyunghun@snu.ac.kr.
Han-Sung Hwang, Email: 20080251@kuh.ac.kr.
Ju Han Kim, Phone: +82-2-740-8320, Email: juhan@snu.ac.kr.
Young Ju Kim, Phone: +82-2-2650-5029, Email: kkyj@ewha.ac.kr.
References
- 1.Beck S, Wojdyla D, Say L, Betran AP, Merialdi M, Requejo JH, et al. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity. Bull World Health Organ. 2010;88:31–38. doi: 10.2471/BLT.08.062554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hwang HS, Na SH, Hur SE, Lee SA, Lee KA, Cho GJ, et al. Practice patterns in the management of threatened preterm labor in Korea: a multicenter retrospective study. Obstet Gynecol Sci. 2015;58:203–209. doi: 10.5468/ogs.2015.58.3.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371:75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merritt TA, Deming DD, Boynton BR. The ‘new’ bronchopulmonary dysplasia: challenges and commentary. Semin Fetal Neonatal Med. 2009;14:345–357. doi: 10.1016/j.siny.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 5.Horbar JD, Carpenter JH, Badger GJ, Kenny MJ, Soll RF, Morrow KA, et al. Mortality and neonatal morbidity among infants 501 to 1500 grams from 2000 to 2009. Pediatrics. 2012;129:1019–1026. doi: 10.1542/peds.2011-3028. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Zhang Y, Shi Z. Ritodrine in the treatment of preterm labour: a meta-analysis. Indian J Med Res. 2005;121:120–127. [PubMed] [Google Scholar]
- 7.Canadian Preterm Labor Investigators Group Treatment of preterm labor with the beta-adrenergic agonist ritodrine. N Engl J Med. 1992;327:308–312. doi: 10.1056/NEJM199207303270503. [DOI] [PubMed] [Google Scholar]
- 8.Kagabu M, Murai S, Ogasawara T. Acute coronary syndrome associated with oral administration of ritodrine hydrochloride during pregnancy: a case report. J Obstet Gynaecol Res. 2001;27:337–340. doi: 10.1111/j.1447-0756.2001.tb01282.x. [DOI] [PubMed] [Google Scholar]
- 9.Park JY, Lee NR, Lee KE, Park S, Kim YJ, Gwak HS. Effects of β2-adrenergic receptor gene polymorphisms on ritodrine therapy in pregnant women with preterm labor: prospective follow-up study. IJMS. 2014;15:12885–12894. doi: 10.3390/ijms150712885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baek MY, Hwang HS, Park JY, Chung JE, Lee KE, Lee GY, et al. Association between CACNA1C gene polymorphisms and ritodrine-induced adverse events in preterm labor patients. Eur J Clin Pharmacol. 2017;73:837–842. doi: 10.1007/s00228-017-2222-6. [DOI] [PubMed] [Google Scholar]
- 11.Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. The next-generation sequencing revolution and its impact on genomics. Cell. 2013;155:27–38. doi: 10.1016/j.cell.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaname T, Yanagi K, Naritomi K. A commentary on the promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59:117–118. doi: 10.1038/jhg.2014.7. [DOI] [PubMed] [Google Scholar]
- 13.Lee S, Abecasis GR, Boehnke M, Lin X. Rare-variant association analysis: study designs and statistical tests. Am J Hum Genet. 2014;95:5–23. doi: 10.1016/j.ajhg.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lange LA, Hu Y, Zhang H, Xue C, Schmidt EM, Tang ZZ, et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genet. 2014;94:233–245. doi: 10.1016/j.ajhg.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 18.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee KH, Baik SY, Lee SY, Park CH, Park PJ, Kim JH. Genome sequence variability predicts drug precautions and withdrawals from the market. PLoS One. 2016;11:e0162135. doi: 10.1371/journal.pone.0162135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.1000 Genomes Project Consortium. Abecasis GR, Auton A, Brooks LD, MA DP, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.1000 Genomes Project Consortium. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 25.van Dam TJ, Wheway G, Slaats GG, SYSCILIA Study Group. Huynen MA, Giles RH. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia. 2013;2:7. doi: 10.1186/2046-2530-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Invitae, “Ciliopathies”, Invitae Corporation, https://www.invitae.com/en/physician/ny-category/CAT000016/ (Accessed 20 June 2017).
- 27.Genetics Home Reference, “Joubert syndrome”, Lister Hill National Center for Biomedical Communications, U.S. National Library of Medicine, National Institutes of Health, Department of Health & Human Services, https://ghr.nlm.nih.gov/condition/joubert-syndrome#genes (Accessed 28 June 2017).
- 28.Genetics Home Reference, “Meckel syndrome”, Lister Hill National Center for Biomedical Communications, U.S. National Library of Medicine, National Institutes of Health, Department of Health & Human Services, https://ghr.nlm.nih.gov/condition/meckel-syndrome#genes (Accessed 28 June 2017).
- 29.Cevik S, Sanders AA, Van Wijk E, Boldt K, Clarke L, van Reeuwijk J, et al. Active transport and diffusion barriers restrict Joubert syndrome-associated ARL13B/ARL-13 to an inv-like ciliary membrane subdomain. PLoS Genet. 2013;9:e1003977. doi: 10.1371/journal.pgen.1003977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wray S. Uterine contraction and physiological mechanisms of modulation. Am J Phys. 1993;264:C1–18. doi: 10.1152/ajpcell.1993.264.1.C1. [DOI] [PubMed] [Google Scholar]
- 31.Word RA, Stull JT, Casey ML, Kamm KE. Contractile elements and myosin light chain phosphorylation in myometrial tissue from nonpregnant and pregnant women. J Clin Invest. 1993;92:29–37. doi: 10.1172/JCI116564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wray S, Jones K, Kupittayanant S, Li Y, Matthew A, Monir-Bishty E, et al. Calcium signaling and uterine contractility. J Soc Gynecol Investig. 2003;10:252–264. doi: 10.1016/S1071-5576(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 33.Hui Y, Liu MC. Sulfation of ritodrine by the human cytosolic sulfotransferases (SULTs): effects of SULT1A3 genetic polymorphism. Eur J Pharmacol. 2015;761:125–129. doi: 10.1016/j.ejphar.2015.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 35.Nebert DW. Role of genetics and drug metabolism in human cancer risk. Mutat Res. 1991;247:267–281. doi: 10.1016/0027-5107(91)90022-G. [DOI] [PubMed] [Google Scholar]
- 36.Bartsch H, Petruzzelli S, De Flora S, Hietanen E, Camus AM, Castegnaro M, et al. Carcinogen metabolism in human lung tissues and the effect of tobacco smoking: results from a case--control multicenter study on lung cancer patients. Environ Health Perspect. 1992;98:119–124. doi: 10.1289/ehp.9298119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crofts F, Taioli E, Trachman J, Cosma GN, Currie D, Toniolo P, et al. Functional significance of different human CYP1A1 genotypes. Carcinogenesis. 1994;15:2961–2963. doi: 10.1093/carcin/15.12.2961. [DOI] [PubMed] [Google Scholar]
- 38.Nerurkar PV, Okinaka L, Aoki C, Seifried A, Lum-Jones A, Wilkens LR, et al. CYP1A1, GSTM1, and GSTP1 genetic polymorphisms and urinary 1-hydroxypyrene excretion in non-occupationally exposed individuals. Cancer Epidemiol. 2000;9:1119–1122. [PubMed] [Google Scholar]
- 39.Kisselev P, Schunck W-H, Roots I, Schwarz D. Association of CYP1A1 polymorphisms with differential metabolic activation of 17beta-estradiol and estrone. Cancer Res. 2005;65:2972–2978. doi: 10.1158/0008-5472.CAN-04-3543. [DOI] [PubMed] [Google Scholar]
- 40.Chanrachakul B, Broughton Pipkin F, Warren AY, Arulkumaran S, Khan RN. Progesterone enhances the tocolytic effect of ritodrine in isolated pregnant human myometrium. Am J Obstet Gynecol. 2005;192:458–463. doi: 10.1016/j.ajog.2004.07.077. [DOI] [PubMed] [Google Scholar]
- 41.Zou J-G, Ma Y-T, Xie X, Yang Y-N, Pan S, Adi D, et al. The association between CYP1A1 genetic polymorphisms and coronary artery disease in the Uygur and Han of China. Lipids Health Dis. 2014;13:145. doi: 10.1186/1476-511X-13-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The SRA (Sequence Read Archive) accession number for the exome sequences reported in this paper is SRP127651.