

Abstract
Background
Secondary pulmonary alveolar proteinosis (sPAP) is an extremely rare disease. The clinical features of sPAP patients remain to be summarizeds.
Methods
Patients pathologically diagnosed with PAP and with negative results for anti-granulocyte macrophage colony stimulating factor (GM-CSF) autoantibodies from Peking Union Medical College Hospital between January 2000 and July 2016 were retrospectively studied. The PubMed database was also searched for literature to collect published cases.
Results
In our center, nine patients were diagnosed as sPAP with a median age of 37 years. Hematological disorders, including myelodysplastic syndrome (MDS), chronic myelogenous leukemia (CML), and pulmonary tuberculosis (TB) infection were the underlying diseases. Cases secondary to MDS had very poor prognosis as all of them survived less than 2 years after their diagnosis, while those secondary to TB had favorable prognosis. Only 33.3% of cases showed interlobular septal thickening in our sPAP group. Through literature review, 164 sPAP cases were collected. The age at diagnosis was 45.0 ± 14.8 years old and the gender radio was 1.20:1 (M:F). 61.9% of cases were diagnosed by bronchoscopy. MDS and CML were common underlying diseases in 34.1% and 15.2% of patients, respectively. Patients with sPAP secondary to hematological diseases had a short survival time and half of them died within 14.95 months after diagnosis.
Conclusions
MDS and TB infection were the most frequent underlying causes of sPAP in this single-center research in China, with cases secondary to MDS having a poor survival rate. sPAP was more likely to be secondary to hematological disorders, especially MDS and CML and had a fairly poor prognosis in published cases. sPAP should be suspected in PAP patients whose CT scan presents only ground-glass opacities without interlobular septal thickening.
Electronic supplementary material
The online version of this article (10.1186/s12890-018-0590-z) contains supplementary material, which is available to authorized users.
Keywords: Secondary pulmonary alveolar proteinosis (sPAP), Anti-granulocyte macrophage colony stimulating factor autoantibodies (GM-CSF-Ab), Myelodysplastic syndrome (MDS), Tuberculosis (TB)
Background
Pulmonary alveolar proteinosis (PAP), a rare disease with an estimated prevalence of 3.7 to 6.2 cases per million persons [1, 2], is characterized by the accumulation of lipoprotein material in alveoli and terminal respiratory airways [3]. Current knowledge about PAP is based on case studies and individual case reports, and as such, the prevalence of PAP may vary due to differences in medical systems and facilities in different countries. According to the pathogenesis, PAP can be classified in three ways: congenital PAP, secondary PAP (sPAP) and autoimmune PAP (or acquired PAP, or idiopathic PAP) [4]. sPAP mainly develops as a result of hematological diseases, the use of immunosuppression drugs, dust inhalation and certain chronic infections with impaired alveolar macrophage function. Autoimmune PAP, which accounts for over 90% of PAP cases, is an autoimmune disease with autoantibodies targeting granulocyte macrophage colony stimulating factor (GM-CSF) [5].
According to previous reports, sPAP is different from autoimmune PAP in aspects besides the pathogenesis. For example, sPAP has different features on high-resolution computed tomography (HRCT) compared to autoimmune PAP [6] and sPAP has a much poorer prognosis compared with autoimmune PAP [7]. Knowledge of the clinical characteristics of sPAP are still limited as the majority of current literature is in the form of case reports, the largest series being a report by Ishii H which included 40 Japanese patients [7]. As we do not know whether the prevalence of sPAP in China has any differences compared to other countries, we retrospectively collected sPAP patients hospitalized from 2000 to 2016 in our hospital, listed their clinical features and symptoms, and we also reviewed all published cases in literature to learn more about the characteristics of sPAP around the world.
Methods
Study design and participants
This is a case study of patients who were diagnosed with PAP in Peking Union Medical College Hospital (PUMCH) between Jan. 1st, 2000 and July 1st, 2016. Medical files were retroactively collected on Feb. 2017. The study was approved by the Ethical Committee of PUMCH (S-K-215), and universal informed consent forms were signed when each subject was admitted to our hospital. The patients or their kin have given written informed consent to publish these case details. Patients had a clinical diagnosis of PAP, which was further confirmed pathologically by testing for amorphous periodic Acid-Schiff (PAS)-positive granules, found either in milky broncho-alveolar lavage fluid (BALF) or in alveolar structures of lung biopsy tissues. Patients under 18 years old were excluded. Serum GM-CSF autoantibodies were evaluated using an enzyme-linked immunosorbent assay, as previously described [8–10]. Patients with positive serum GM-CSF autoantibodies (> 5 μg/mL) were also excluded.
Methods of data analysis
The patients’ quantitative characteristics such as age at diagnosis were summarized by median and range. The qualitative characteristics such as general appearance, imaging features and other organ complications were presented as a frequency distribution or percentage. Authors had no access to information that could identify individual participants during or after data collection.
Results
Demographic characteristics
In total, among 157 hospitalized PAP cases, 9 patients were diagnosed with sPAP. Their clinical characteristics are summarized in Table 1. In these 9 patients, 3 were female and 6 were male, and they ranged in age from 20 to 56 years (median age 37 years). At the time of administration, 5 (5/9) of the cases complained of fever and cough, followed by weakness (4/9), weight loss (3/9), and dyspnea or exertional dyspnea (3/9). Diagnostic procedures included BALF (3/9), CT guided biopsy (2/9), open lung biopsy (2/9), transbronchial lung biopsy (TBLB) plus BALF (1/9), and video-assisted thoracic surgery (VATS) (1/9).
Table 1.
Characteristics of Patients with secondary pulmonary alveolar proteinosis in Peking Union Medical College Hospital
| Gender | Age | Smoking | Clinical Symptoms | Diagnostic Procedure | Primary Disease | PaO2 mmHg |
|
|---|---|---|---|---|---|---|---|
| 1 | Male | 38 | Current | Weight loss, weakness, night sweats | BALF | MDS-MLD | 64.1 |
| 2 | Female | 26 | Never | Fever, productive cough | CT guided percutaneous lung biopsy | TB | 94.5 |
| 3 | Male | 20 | Current | Exertional dyspnea, dry cough | Surgical biopsy | CML | 97.4 |
| 4 | Male | 26 | Never | Weakness, fever | BALF+TBLB | MDS-SLD | NA |
| 5 | Male | 37 | Never | Dyspnea, dry cough | BALF | MDS-U | 48 |
| 6 | Male | 33 | Never | Fever, weight loss, weakness | Surgical biopsy | MDS-EB | 34.6 |
| 7 | Male | 56 | Never | Fever, dry cough | VATS | TB | 84.1 |
| 8 | Female | 54 | Never | Weight loss,weakness, fever | CT guided percutaneous lung biopsy | TB | 39 |
| 9 | Female | 51 | Never | Dyspnea, productive cough | BALF | TB | 35.6 |
| CT Scan Features | Comorbidity/Complication | Treatment | Outcomes | ||||
| 1 | diffused bilateral GGO | None | WLL, Danazol, Retinoic acid, | Die | |||
| 2 | bilateral GGO patchy geographic pattern, interlobular septal thickening,small scattered nodules | Hepatitis | INH, RIF, EMB, Levofloxacin | Recovery | |||
| 3 | bilateral GGO patchy geographic pattern, bilateral cysts | None | Hydroxyurea | NA | |||
| 4 | diffused bilateral GGO, small nodules | Type B viral hepatitis | Prednisone | Die | |||
| 5 | bilateral GGO with a crazy paving pattern,enlarged mediastinal lymph nodes | Mycobacterium cheloneiinfection | Antibiotics, traditional medicine | Die | |||
| 6 | diffused bilateral GGO, interlobular septal thickening,small scattered nodules | Hospital acquired pneumonia | Danazol, Retinoic acid, WLL | Die | |||
| 7 | bilateral GGO with a crazy paving pattern, interlobular septal thickening | None | INH, RIF, EMB,Clarithromycin, Oxifloxacin | Improved | |||
| 8 | diffused bilateral GGO, small scattered nodules | None | INH, RIF, EMB,Rifapentine | Stable | |||
| 9 | diffused bilateral GGO, septal thickening | Thrombopenia, Sjogren’s syndrome | INH, RIF, EMB,Levofloxacin, WLL | Improved | |||
BALF broncho-alverolar lavage fluid, CML chronic myelogenousleukemia;EMB ethambutol, GGO ground-glass opacities, INH isonicotinic acid hydrazide, MDS myelodysplastic syndrome(SLD:single lineage dysplasia; MLD: multilineage dysplasia; EB: excess blasts; U: unclassifiable), NA not available, RIF rifampicine, TB tuberculosis, TBLB transbronchial lung biopsy, VATS via video-assisted thoracic surgery, WLL whole lung lavage
Primary diseases
The primary diseases of these 9 sPAP cases were variable, as 5 cases arose from hematologic disorders (4 myelodysplastic syndrome (MDS) and 1 chronic myelogenous leukemia (CML)) and 4 cases from tuberculosis (TB). Mycobacterium chelonei infection was detected in one patient with underlying MDS using a subcutaneous nodule biopsy, and another patient was affected by hospital-acquired pneumonia with obscure pathogens, and neither of their sputum nor BALF cultures tested positive.
CT features
As shown in Table 1, all of our cases presented bilateral ground-glass opacities (GGO) on their CT scans. Among them, GGO with a diffuse pattern was the most frequent pattern, presenting in 5 (5/9) patients, while a patchy geographic pattern and crazy paving pattern each presented in 2 (2/9) patients respectively. Small scattered nodules were found in both TB (2/4) and MDS patients (2/4). The patient infected with M. chelonei showed enlarged mediastinal lymph nodes. Interlobular septal thickening, which was thought to be a typical feature of PAP, could only be recognized in three (3/9) of our sPAP patients.
Pulmonary function tests
At the time of diagnosis, 8 patients underwent arterial blood gas tests and the mean arterial partial oxygen pressure (PaO2) was 62.2 mmHg (34.1, 97.4 mmHg) in room air. In 7 patients who performed pulmonary function tests during administration, 6 had normal forced vital capacity (FVC) and forced expiratory volume in the first 1 s (FEV1). All 5 cases evaluated for diffusing function showed a remarkable reduction in diffusing capacity for carbon monoxide (DLCO) with a mean of 43.1% (22.1%, 58.9%) of the normal predicted value.
Treatment and prognosis
The median follow-up duration was 4.7 years in these patients. All 4 patients with underlying MDS died within 2 years after diagnosis, and whole lung lavage (WLL) was performed in 2 of them, but symptoms only improved temporarily. Both the symptoms and the CT scans improved in 3 of the 4 patients with TB after anti-TB therapy, and the remaining patient with TB remains stable throughout 8 years of follow-up. Unfortunately, the patient with CML could not be followed-up on. The patient with M.chelonei complications (patient 5) had his condition worsen rapidly even with antibiotic administration based on drug sensitivity tests and died 3 months after diagnosis.
Literature reviews
Texts were searched for using the terms “pulmonary alveolar lipoproteinosis [Title/abstract]” or “pulmonary alveolar proteinosis [Title/abstract]” in the PubMed database. Cases published only in abstract without full-text were excluded. For cases with a history of dust inhalation, only those that tested negative for GM-CSF autoantibodies were included as dust exposure was not a specified factor of sPAP [11].
Using the above search strategy, 1291 articles on PAP were identified. These articles were carefully reviewed to remove reviews, abstracts, commitments, animal researches, relevant pathophysiological studies or pharmaceutical researches, articles about congenital PAP, and articles about autoimmune PAP. We also removed those diagnosed by autopsy post-mortem and pediatric patients less than 18 years old. In the end, 155 cases of sPAP with underlying diseases were found (Additional file 1: Figure S1). In Table 2, we summarized the available characteristics of the 164 cases in total (including our 9 cases).
Table 2.
Summary of characteristics of 159 secondary pulmonary alveolar proteinosis cases
| sPAP | 164 | Underlying disease | |
|---|---|---|---|
| Age in years | 45.0 ± 14.8 | Myelodysplastic syndrome | 56 (34.1) |
| Sex ratio (Male: Female) | 1.20:1 | Chronic myeloid leukemia | 25 (15.2) |
| Clinical symptoms | 99 | Acute myeloid leukemia | 9 (5.5) |
| Asymptomatic | 11 (11.1) | Acute lymphoid leukemia | 5 (3.0) |
| Dyspnea with effort | 53 (53.5) | Lymphoma | 6 (3.7) |
| Cough | 32 (32.3) | Myelofibrosis | 3 (1.8) |
| fever | 38 (38.3) | Other hematological disorders | 9 (5.5) |
| Diagnostic procedure | Infection diseases | 13 (7.9) | |
| Bronchoscopy | 101 (61.6) | Autoimmune diseases | 11 (6.7) |
| CT guided biopsy | 3 (1.8) | Post transplantation | 17 (10.4) |
| VATS | 29 (17.7) | non-hematological tumors | 5 (3.0) |
| Surgery lung biopsy | 31 (18.9) | Dust inhalation | 5 (3.0) |
Data is present by number (percentage, %)
VATS via video-assisted thoracic surgery
The patients were diagnosed at an age of 45.0 ± 14.8. The male to female ratio was 1.20:1. Bronchoscopy, including BALF and/or TBLB, was the most frequent method used to diagnose sPAP, with 61.9% of cases diagnosed in this manner. Other procedures used included VATS and open lung biopsy, with 17.7% and 18.9% of cases being diagnosed in those manners respectively. Approximately 70% of cases were secondary to hematological diseases, such as MDS (34.1%), CML (15.2%) and AML (acute myelogenous leukemia) (5.5%).
Underlying infectious diseases, mainly due to TB, non-TB mycobacteria, human immunodeficiency virus, and Aspergillosis, consisted of 7.9% of all cases. Autoimmune diseases including Behcet’s disease, vasculitis, Sjogren’s syndrome, and dermatomyositis accounted for 6.7% of the underlying causes. 10.4% of sPAP patients were secondary to lung or kidney transplantation. The other rare underlying diseases were non-hematological tumors and dust inhalation. As shown in Fig. 1, the median survival in 92 cases secondary to hematological diseases was only 14.95 months, which was significantly shorter than sPAP secondary to other causes (p < 0.001).
Fig. 1.
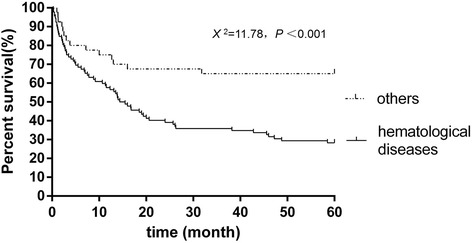
Survival probability of secondary pulmonary alveolar proteinosis cases. In our 9 cases and 155 cases from literatures, 132 cases had the survival information. In 92 cases secondary to hematological diseases, the median survival was 14.95 months as 66 (71.3%) died within 5 years. In 40 cases secondary to other causes, the median survival was to months and 14 (35%) died within 5 years. Log-rank (Mantel-Cox) test X 2 = 11.78, P < 0.001
Discussion
Since sPAP is a rare disease, less than 200 cases have been reported throughout the world, with most of them in form of case reports. H. Ishii [7] summarized the characteristics of 40 Japanese sPAP patients, which was the largest retrospective observational study about sPAP that we found. To the best of our knowledge, our study is the largest series of sPAP from a single center in China. We present some interesting findings. Firstly, TB is also a frequent underlying disease of sPAP in China, and fortunately, after effective anti-TB therapy, sPAP can be improved concurrently. Furthermore, hematologic disorders, especially MDS, are the predominant primary disease of sPAP, and these patients have a terrible prognosis with the majority dying within 2 years after diagnosis. Complications from other infections may accelerate the advancement of the disease.
In our center, 5.73% of PAP cases were determined to be sPAP, which is slightly lower than the percentage of sPAP (8.3% to 10%) in the Japanese case series [6, 7]. Both our study and existing literature illustrates that hematologic disorders are the most frequent underlying disease of sPAP. The study including 31 cases of sPAP with MDS found that sPAP is an important risk factor predicting adverse outcome of MDS with a 2-year survival of 46.2% [12]. In our cases, all of the 4 patients with MDS as the underlying disease died within 2 years. The pathomechanism of sPAP secondary to hematological diseases remains unknown. Mutations in GATA2 may be associated with sPAP [13, 14] since an autosomal dominant familial MDS and AML carried a heritable GATA2 mutation [15] and approximately one third of GATA2 mutation carriers also complicated sPAP [16]. It is speculated that a GATA2 deficiency leads to the alteration of alveolar cellular immunity because these patients had neither GM-CSF receptor mutations nor anti-GM-CSF autoantibodies. In the mice model, T-bet–over expressing T cells act to initiate the pathogenesis of sPAP [17]. Treatment of sPAP should focus on curing the underlying diseases. Bone marrow transplantation or cord blood transplantation can cure sPAP in these patients, but at the cost of an increased risk of infection and mortality [12, 18].
Some infectious diseases such as TB can cause sPAP, possibly due to microorganisms impairing the function of macrophages. The heavy burden of TB in China makes it a significant underlying disease in our cases. In this circumstance, treating the underlying infection(s) can ameliorate sPAP, as shown in our cases, 3 cases improved and 1 case remained stable after anti-TB therapy. However, autoimmune PAP might cause complications with infections including TB, Nocardia, and Aspergillus. An anti-GM-CSF autoantibody test is effective to distinguish these two situations since it is negative in the former while positive in the later. Besides antibiotics, GM-CSF replacement is ineffective to the former but of benefit to the latter.
In our study, only 33.3% of cases show interlobular septal thickening, which is regarded as a typical feature in the CT scan of autoimmune PAP patients [19]. Additionally, only 22.2% of them present GGO with patchy geographic pattern, which is more common in autoimmune PAP [6]. As a result, sPAP should be suspected in PAP patients whose CT scans only present GGO without interlobular septal thickening or in the absence of a geographic pattern. More detailed evaluation about infectious and hematologic diseases should be performed. Moreover, qualification of anti-GM-CSF-Ab is valuable for the differentiation of autoimmune or secondary PAP.
Several limitations must be stressed with regards to this study. First, since PAP is a rare disease, the value of this study is limited by its small sample size. Our study was also limited in scope in that the underlying disease in our sPAP cases only included hematological diseases and infections, without covering dust inhalation, autoimmune disease or tumors. Secondly, as a retrospective study, some patient characteristics and information are not comprehensive, such as arterial blood gas and lung function. Furthermore, because our hospital is the largest rare disease treatment center in China, patients admitted are usually more complex and serious cases, and as such, we cannot avoid selection bias. Meanwhile, occupational diseases were restrictively diagnosed and treated in designated hospitals in China, so PAP secondary to occupational exposure is difficult to diagnose in our hospital. It is also difficult to compare the result of a single Chinese center with those all over the world. Thus, we conducted a comprehensive review of all the available cases published in available literature to minimize selection bias. Finally, in the review, we did not account for other factors that may affect the variation of sPAP, such as the geographical features of the patients.
Conclusions
In summary, this study found that MDS and TB infection were the most frequent underlying causes of sPAP in this single center research in China and cases that secondary to MDS had a poor survival rate. However, after taking all the published cases into consideration, hematological disorders, especially MDS and CML, are the major primary disease in 68.9% of cases. sPAP was often middle age onset, but it had a fairly poor prognosis when the underlying diseases were hematologic disorders. The diagnosis of sPAP should be considered in PAP patients whose CT scan only presents GGO without interlobular septal thickening.
Acknowledgements
The authors would like to thank Tao Liu, Yongjian Liu, Juhong Shi, Chi Shao, and Xiaoming Huang for their efforts in performing and aiding the clinical diagnosis and following of sPAP patients.
Funding
This work was supported by “State Key Project, Clinical Cohort Study of Rare Diseases. Rare Lung Disease Research Group in China” (2016YFC0901502) and The Capital of the Public Health Program: Study on the Evaluation of Diagnosis and Treatment on Seven Treatable Rare Diseases (Z151100003915126)
Availability of data and materials
The data of our patients is available in the department of medical records in PUMCH. This data can be released with the agreement of patients. Identifying patient data cannot be shared.
Abbreviations
- AML
Acute myelogenous leukemia
- BALF
Bronchoalveolar lavage fluid
- CML
Chronic myelogenous leukemia
- CT
Computed tomography
- DLCO
Diffusing capacity for carbon monoxide
- FEV1
Forced expiratory volume in the first 1 second
- FVC
Forced vital capacity
- GGO
Ground-glass opacities
- GM-CSF-Ab
Granulocyte macrophage colony stimulating factor autoantibodies
- HRCT
High-resolution computed tomography
- MDS
Myelodysplastic syndrome
- PAP
Pulmonary alveolar proteinosis
- PAS
Periodic Acid-Schiff
- PUMCH
Peking Union Medical College Hospital
- sPAP
Secondary pulmonary alveolar proteinosis
- TB
Tuberculosis
- TBLB
Transbronchial lung biopsy
- VATS
Video-assisted thoracic surgery
- WLL
Whole lung lavage.
Additional file
Trial inclusion flow chart. (TIFF 200 kb)
Authors’ contributions
DZ and XT conception, design, obtaining consent, and drafting manuscript; RF reviewed all the pathologicalslides of all the patients, PW detected pathogenic microbes of all the patients. XG collection of data; YS design and drafting manuscript; YX and KFX data analysis and interpretation. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study has been approved by the Ethical Committee of PUMCH with reference number S-K-215, and informed consent forms and contents for publication forms were signed by all participants.
Consent for publication
The patients or their kin have given written informed consent to publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12890-018-0590-z) contains supplementary material, which is available to authorized users.
Contributor Information
Dongmei Zhang, Email: zhdm89@126.com.
Xinlun Tian, Email: xinlun_t@sina.com.
Ruie Feng, Email: fengruie1@163.com.
Xiaobei Guo, Email: guoxiaobeibeibei@163.com.
Peng Wang, Email: pumch2005@126.com.
Yusen Situ, Email: situ@ualberta.ca.
Yi Xiao, Email: xiaoyipumch@sina.com.
Kai-Feng Xu, Email: kaifeng.xu@gmail.com.
References
- 1.Ben-Dov I, Kishinevski Y, Roznman J, Soliman A, Bishara H, Zelligson E, et al. Pulmonary alveolar proteinosis in Israel: ethnic clustering. Isr Med Assoc J. 1999;1:75–78. [PubMed] [Google Scholar]
- 2.Inoue Y, Trapnell BC, Tazawa R, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177(7):752–762. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258:1123–1142. doi: 10.1056/NEJM195806052582301. [DOI] [PubMed] [Google Scholar]
- 4.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 5.Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis. Eur Respir Rev. 2011;20(120):98–107. doi: 10.1183/09059180.00001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ishii H, Trapnell BC, Tazawa R, Inoue Y, Akira M, Kogure Y, et al. Comparative study of high-resolution CT findings between autoimmune and secondary pulmonary alveolar proteinosis. Chest. 2009;136:1348–1355. doi: 10.1378/chest.09-0097. [DOI] [PubMed] [Google Scholar]
- 7.Ishii H, Tazawa R, Kaneko C, Saraya T, Inoue Y, Hamano E, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J. 2011;37:465–468. doi: 10.1183/09031936.00092910. [DOI] [PubMed] [Google Scholar]
- 8.Xu KF, Chen Y, Guo ZJ, Zhu YJ. Autoantibody against granulocyte-macrophage colony-stimulating factor and other serum markers in pulmonary alveolar proteinosis. Zhonghua Jie He He Hu Xi Za Zhi. 2004;27:824–828. [PubMed] [Google Scholar]
- 9.Uchida K, Nakata K, Trapnell BC, Terakawa T, Hamano E, Mikami A, et al. High-affinity autoantibodies specifically eliminate granulocyte-macrophage colony-stimulating factor activity in the lungs of patients with idiopathic pulmonary alveolar proteinosis. Blood. 2004;103:1089–1098. doi: 10.1182/blood-2003-05-1565. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Tian X, Gui Y, Ma A, Li X, Zeng N, Zhang P, Li G, Xu K. Serum markers in patients with idiopathic pulmonary alveolar proteinosis. Zhonghua Jie He He Hu Xi Za Zhi. 2014;37(7):497–501. [PubMed] [Google Scholar]
- 11.Costabel U, Nakata K. Pulmonary alveolar proteinosis associated with dust inhalation: not secondary but autoimmune. Am J Respir Crit Care Med. 2010;181:427–428. doi: 10.1164/rccm.200912-1800ED. [DOI] [PubMed] [Google Scholar]
- 12.Ishii H, Seymour JF, Tazawa R, Inoue Y, Uchida N, Nishida A, et al. Secondary pulmonary alveolar proteinosis complicating myelodysplastic syndrome results in worsening of prognosis: a retrospective cohort study in Japan. BMC Pulm Med. 2014;14:37. doi: 10.1186/1471-2466-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ballerie A, Nimubona S, Meunier C, et al. Association of pulmonary alveolar proteinosis and fibrosis: patient with GATA2 deficiency. Eur Respir J. 2016;48(5):1510–1514. doi: 10.1183/13993003.00252-2016. [DOI] [PubMed] [Google Scholar]
- 14.Griese M, Zarbock R, Costabel U, Hildebrandt J, Theegarten D, Albert M, et al. GATA2 deficiency in children and adults with severe pulmonary alveolar proteinosis and hematologic disorders. BMC Pulm Med. 2015;15:87. doi: 10.1186/s12890-015-0083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaulagain CP, Pilichowska M, Brinckerhoff L, Tabba M, Erban JK. Secondary pulmonary alveolar proteinosis in hematologic malignancies. Hematol Oncol Stem Cell Ther. 2014;7:127–135. doi: 10.1016/j.hemonc.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Iriguchi S, Kikuchi N, Kaneko S, et al. T-cell-restricted T-bet overexpression induces aberrant hematopoiesis of myeloid cells and impairs function of macrophages in the lung. Blood. 2015;125(2):370–382. doi: 10.1182/blood-2014-05-575225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tabata S, Shimoji S, Murase K, Takiuchi Y, Inoue D, Kimura T, et al. Successful allogeneic bone marrow transplantation for myelodysplastic syndrome complicated by severe pulmonary alveolar proteinosis. Int J Hematol. 2009;90:407–412. doi: 10.1007/s12185-009-0404-4. [DOI] [PubMed] [Google Scholar]
- 19.Mehrian P, Homayounfar N, Karimi MA, Jafarzadeh H. Features of idiopathic pulmonary alveolar proteinosis in high resolution computed tomography. Pol J Radiol. 2014;79:65–69. doi: 10.12659/PJR.890218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data of our patients is available in the department of medical records in PUMCH. This data can be released with the agreement of patients. Identifying patient data cannot be shared.