

Abstract
Concomitant administration of bevacizumab and pemetrexed‐cisplatin is a common treatment for advanced nonsquamous non‐small cell lung cancer (NSCLC). Vascular normalization following bevacizumab administration may transiently enhance drug delivery, suggesting improved efficacy with sequential administration. To investigate optimal scheduling, we conducted a study in NSCLC‐bearing mice. First, experiments demonstrated improved efficacy when using sequential vs. concomitant scheduling of bevacizumab and chemotherapy. Combining this data with a mathematical model of tumor growth under therapy accounting for the normalization effect, we predicted an optimal delay of 2.8 days between bevacizumab and chemotherapy. This prediction was confirmed experimentally, with reduced tumor growth of 38% as compared to concomitant scheduling, and prolonged survival (74 vs. 70 days). Alternate sequencing of 8 days failed in achieving a similar increase in efficacy, thus emphasizing the utility of modeling support to identify optimal scheduling. The model could also be a useful tool in the clinic to personally tailor regimen sequences.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Bevacizumab is usually administered concomitantly with cytotoxics. However, studies have demonstrated that bevacizumab before cytotoxics yields better efficacy by improving drug uptake, due to a transient phase of vascular normalization following bevacizumab uptake. Determining the best sequence is challenging and could be patient‐dependent.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ What is the optimal schedule for the administration of bevacizumab and pemetrexed‐cisplatin doublet in NSCLC?
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This study shows, using experimental settings and mathematical modeling, that administration of bevacizumab 3 days before cytotoxics yields better efficacy and overall survival compared to concomitant, cytotoxics before bevacizumab, and cytotoxics alone treatments. It also provides a validated model that could be used to individualize combination regimen in the clinic.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study demonstrates that simplified modeling could help to address the issue of finding and personalizing optimal sequences when combining anticancer treatments.
When combined with chemotherapy, antivascular endothelial growth factor‐A bevacizumab achieved improved overall survival (OS) and/or progression‐free survival in multiple phase III trials for several types of solid tumors.1, 2, 3, 4, 5, 6, 7
Although the main effect of bevacizumab is the disruption of the vascularization, increased efficacy with combined administration of cytotoxic agents could also result from transitory vascular normalization, a paradoxical effect of anti‐angiogenics.9, 10, 11, 12 Indeed, preclinical and clinical studies have shown that bevacizumab induces transient changes in the vascular architecture.9, 13, 14 Although the unaltered tumor vasculature is tortuous, chaotic, and poorly functional,15 bevacizumab prunes and remodels tumor vessels to make them resemble normal tissues in terms of structure and function.10, 16 It also proved to induce a more homogeneous distribution of cytotoxics in actively proliferating areas of tumor vessels.17 Therefore, determining alternate scheduling with bevacizumab could improve efficacy.9
In line with these hypotheses, Dickson et al.18 showed that administrating topotecan after bevacizumab to neuroblastoma tumor‐bearing mice resulted in reduced tumor growth as compared to concomitant administration. Another recent study showed that bevacizumab followed 24 hours later by paclitaxel potentiated its antitumor activity in an ovarian cancer model.17 At bedside, Avallone et al.19 showed that administering bevacizumab 4 days prior to chemotherapy in patients with locally advanced rectal cancer led to better OS and progression‐free survival compared to concomitant administration. Although promising, most attempts to revisit bevacizumab scheduling have been made on an empirical basis and a trial‐and‐error mode. Mathematical modeling could help assisting experimental and clinical studies to better understand the combined effects of anti‐angiogenics and cytotoxics on tumor growth.20, 21 Building on the Hahnfeldt et al.22 model for the effect of anti‐angiogenic monotherapy, Wilson et al.23 developed a model to quantify the dynamics of interaction between tumor growth, vasculature generation, and anti‐angiogenic treatment. They demonstrated a possible synergistic interaction between sunitinib and irinotecan.23 Recently, Hutchinson et al.24 developed a model of vascular tumor growth and normalization from breast cancer mice treated with bevacizumab alone. Still in breast cancer, our team tested multiple regimens for combining bevacizumab and paclitaxel. Experimental results combined with mathematical simulations suggested that scheduling bevacizumab 2 days before paclitaxel could improve antitumor efficacy and reduce metastatic spreading.25
The aim of the present modeling and experimental study was to assess predictions of a semimechanistic mathematical model in terms of the optimal sequence of administration for the combination of bevacizumab and pemetrexed‐cisplatin in non‐small cell lung cancer (NSCLC).
MATERIALS AND METHODS
Mice experiments
Cell lines
Human NSCLC cells H460 stably transfected with luciferase (H460 Luc+) were purchased from Perkin Elmer, France. This BioWare light producing cell line was derived from the H460 human adenocarcinoma by stable transfection of the North American firefly gene expressed from the SV40 promoter. Cells were cultured per the manufacturer's recommendation at 37°C in a humidified atmosphere with 5% CO2. H460 Luc+ cells were regularly authenticated based on viability, growth, morphology, and in vitro bioluminescence measuring.
For experiment two, tumor growth monitoring was carried out by fluorescence. Therefore, H460 Luc+ cells were transfected with tdTomato gene using pPGK‐tdTomato lentiviral plasmid lentiviral plasmid as vector kindly provided by Dr Valérie Le Morvan (Institut Bergonié, Bordeaux, France). Forty‐eight hours after transfection, blasticidine (10 µg/mL) was added and selection was maintained for 2 weeks. Resulting H460 Luc+ dtTomato+ cells were regularly authenticated by microscopy based on viability, growth, morphology, and in vitro fluorescence monitoring.
Animal experiments
All experiments were approved by the local ethical committee of our institution and registered as #2015110616255292 (French Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche) prior to starting the experiments. Guidelines for animal welfare in experimental oncology as recommended by European regulations were followed. Pathogen‐free, immunocompromised 6‐week‐old female Swiss nude mice (Charles River Laboratories, France) were kept in a sterile environment for 2 weeks upon reception. Mice were maintained in sterilized filter‐topped cages and a sterile thermostatic cabinet throughout the study. Signs of distress, decreased physical activity, and any behavioral change were monitored daily. Bodyweights were monitored twice weekly as a surrogate marker for general toxicity. Water was supplemented with paracetamol (eq. 80 mg/kg/day) to prevent any disease‐related pain. Animals showing signs of distress, pain, cachexia (i.e., loss of 10% of body weight), and tumor mass over 2 g (i.e., ∼2 cm3) were euthanized.
Xenografting
H460 Luc+ (and tdTomato+ for the second experiment) cells were trypsinized, counted, centrifuged (5 minutes, 1,000 g) and washed twice with sterile phosphate‐buffered saline. Cells were resuspended in Roswell Park Memorial Institute‐1640 with 60% Matrigel (BD Sciences, France) and maintained in ice‐cooled conditions until engraftment. A volume of 50 µL containing 80,000 cells (experiment one) and 120,000 cells (experiment two) was injected ectopically in the left flank of each mouse while under anesthesia. In total, 132 tumor‐bearing mice were required to perform both experiments. However, overall, 139 mice were initially xenografted, to ensure that eventually at least 48 (experiment one) + 84 mice (experiment two) presenting positive and measurable tumors could be used, considering an estimated 5% of failure rate during the grafting procedure.
Bioluminescence imaging
In experiment one, monitoring of primary tumor growth started 1 week after engraftment. Acquisitions started 12 minutes after firefly D‐Luciferin (Perkin Elmer, 300 mg/kg) i.p. injection to reach a plateau in bioluminescence signaling.26 Acquisition and data processing were performed using the IVIS spectrum imager equipped with the Living Image 4.2 software (Perkin Elmer). Imaging was performed twice per week. All imaging was performed in anesthetized animals (sevoflurane).
Fluorescence imaging
In experiment two, imaging was performed twice in anesthetized animals (sevoflurane). Acquisition (excitation: 554 nm, emission: 581 nm), and data processing were performed using the IVIS spectrum imager equipped with the Living Image 4.2 software.
Experiment one treatments
In experiment one, 47 xenografted mice were randomized into 4 treatment arms (Supplementary Figure S1a): control (saline injection, n = 12), sequential treatment with bevacizumab administered 4 days before pemetrexed‐cisplatin (“beva then chemo 4 days,” n = 12), sequential treatment arm with bevacizumab administered 4 days after pemetrexed‐cisplatin (“chemo then beva 4 days,” n = 11), and a concomitant treatment arm (“beva + chemo,” n = 12). Treatment started 17 days after xenograft. Doses in each arm were 20 mg · kg−1, 100 mg · kg−1 and 3 mg · kg−1 for bevacizumab, pemetrexed, and cisplatin, respectively. All treatments, including saline, were administered by i.p. route for three 14‐day cycle. All animals were euthanized on day 76 post‐xenografts.
Experiment two treatments
The second experiment was performed with model‐based changes both in scheduling and sample sizes. In experiment two, 77 xenografted mice were randomized into 5 treatment arms (Supplementary Figure S1b): control (saline injection, n = 15), sequential treatment with bevacizumab administered 3 days (see next section below) before pemetrexed‐cisplatin (“beva then chemo 3 days,” n = 16), sequential treatment with bevacizumab administered 8 days before pemetrexed‐cisplatin (“beva then chemo 8 days,” n = 15), concomitant (“beva + chemo,” n = 15), and pemetrexed and cisplatin alone (chemo, n = 15). The administered doses in each arm were and for bevacizumab, pemetrexed, and cisplatin, respectively. All treatments were administered by i.p. route. As for experiment one, 3 cycles (14 days cycle) were administered, starting on day 14 after xenograft. All animals were euthanized at the end of the experiment on day 87 post‐xenografts.
PHARMACOKINETIC/PHARMACODYNAMIC MODELING
Structural model
The tumor size at time is denoted . The function combines plasma concentration of pemetrexed and cisplatin ( . The function represents the plasma concentration of bevacizumab. Pharmacokinetic time courses for bevacizumab, cisplatin, and pemetrexed were modeled each by a one‐compartment model with absorption compartment and pharmacokinetic parameters from the literature.27, 28, 29 Supplementary Figure S2a depicts the concentration profiles for each treatment. A scheme describing the pharmacodynamic model is given in Figure 1. It is based on the following hypotheses:
Figure 1.
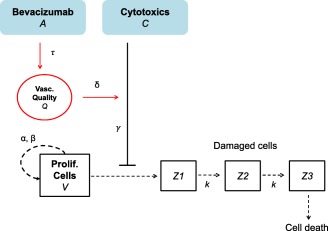
Scheme of the structural mathematical model.
(H1) Without treatment the tumor size kinetics follow a Gompertzian growth governed by parameters (proliferation rate of the tumor cells) and (rate of exponential decrease of the tumor relative growth rate).30
(H2) Cytotoxics act by targeting a fraction of the tumor size (log‐kill effect).31 This effect is driven by the parameter .
(H3) There is a delay between the administration time of cytotoxics and their effects on the tumor. Tumor cells pass through different stages, characterized by different degree of damages, before they get eliminated.32 The transfer rate between these stages is denoted .
(H4) In the absence of data monitoring, the state of the tumor vasculature and the anti‐angiogenic effect of bevacizumab are not explicitly modeled.
- (H5) Bevacizumab increases the drug's delivery by improving the vasculature quality .33 The dynamics of this improvement is assumed to follow the bevacizumab concentration, delayed by a time shift for the normalization to occur. The magnitude of the improvement is controlled by a parameter .The above assumptions are translated into the following system of nonlinear ordinary differential equations:
The initial size was set to 7.04 × 106 photons/second, considering that 80,000 cells were injected (experiment one) and a previously established conversion ratio of 1 cell ≈ 88 photons/second.34
Statistical model and parameters estimation
For a description of the inter‐animal variability, we used the nonlinear mixed‐effects statistical framework.35 It consists in assuming a distribution of the parameters within the animal population, taken here to be log‐normal for each parameter. Importantly, these were the same for all treatment groups. The structural model above depends on six parameters ( After an initial sensitivity analysis showing that not all of these parameters were identifiable from our dataset (Supplementary Methods), we reduced this to the four parameters ( These parameters were then estimated by adapting the result of likelihood maximization performed with the Monolix software (Lixoft, version 2016R1) using visual assessment of the goodness‐of‐fit (visual predictive checks) and consideration of the root mean squared error. See Supplementary Methods and Supplementary Figures S3‐S6 for details. Values of the resulting parameters are reported in Table 1. Model simulations were performed using Matlab software.
Table 1.
Population parameters and interanimal variabilities estimates for experiment one data
| Parameters | Description | Units | Estimates | RSEa (%) | IAVb % | ||||
|---|---|---|---|---|---|---|---|---|---|
|
|
Proliferation rate | Day−1 |
|
8 |
|
||||
|
|
Exponential decay rate of the relative tumor growth rate (Gompertz model) | Day−1 |
|
|
|
||||
|
|
Baseline effect of the chemotherapy | (mg/g)−1.day−1 |
|
|
|
||||
|
|
Cytotoxics efficacy improvement following vascular normalization | (mg/mL)−1 |
|
|
|
||||
|
|
Delay parameter for dynamics of | Day |
|
|
|
||||
|
|
Delay of the tumor cell loss following chemotherapy | Day−1 |
|
|
|
||||
| Σ | Exponential error parameter |
|
|
|
|
IAV, interanimal variabilities; RSE, relative standard error.
Values of the parameters corresponding to the adapted fit. See Supplementary Methods for details on the estimation procedure.
RSE is a measure of the precision of the parameter estimates, expressed as coefficient of variation (CV%).
The IAV is the standard deviation ω estimated using Monolix software.
Statistical analysis
Statistical analyses were performed using R software 3.3.2 (R Core Team, 2016). Intergroup differences in tumor growth were tested by nonparametric Kruskal‐Wallis tests, should the data not meet the assumptions for one‐way analysis of variance. Further between‐group comparisons were performed either by Dunn's multiple comparison tests when treatment groups were compared with the control group, or by Nemenyi post‐hoc tests with Tukey approximation for pairwise multiple comparison between groups. Survival analysis was done using Kaplan‐Meier analysis. Intergroup differences in survival were tested for significance by the log‐rank tests. Owing to sample size, a P value < 0.05 was considered statistically significant.
RESULTS
Administering bevacizumab before pemetrexed + cisplatin improved efficacy and median of survival
Experiment one efficacy
Monitoring of tumor growth for experiment one is shown in Figure 2 a. At the end of the treatment phase (day 53), mean tumor sizes (expressed in × 109 photons/second (p/s)) were 16.9 ± 3.7 (control), 19.2 ± 3.4 (“chemo then beva 4 days”), 15.4 ± 2.5 (“beva + chemo), and 6.9 ± 2.1 (“beva then chemo 4 days”). A statistical difference was found between the groups (P = 0.012). Further Dunn's multiple comparison tests evidenced a significant difference between the sequential administration “beva then chemo 4 days” and control arms (59% tumor growth reduction, P = 0.047). Conversely, concomitant “beva + chemo” and reversed “chemo then beva 4 days” groups had modest effects on tumor growth inhibition (9% tumor reduction (P = 0.98) and 13% higher tumor size (P = 1) as compared with the control group, respectively). The sequential administration of “beva then chemo 4 days” also confirmed its superiority in efficacy when compared with other treatment groups (55% tumor reduction, almost reaching significance compared to “beva + chemo” (P = 0.071) and 64% tumor reduction compared to “chemo then beva 4 days” (P = 0.0073)). All mice in the control group had to be euthanized at day 53.
Figure 2.
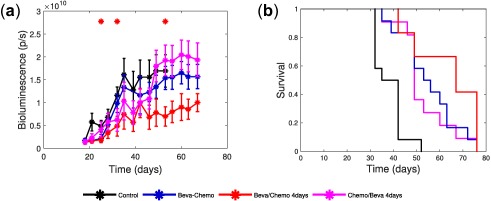
Efficacy and Kaplan‐Meier survival curves of experiment one. (a) Mean tumor growth curves for the four treatment arms of experiment one. Signs above curves indicate statistically significant difference with the control arm (Student's t test, P < 0.05). (b) Kaplan‐Meier plot of the overall survival for the four treatment arms of experiment one.
At study conclusion (day 67; i.e., 18 days after all treatments stopped), mean tumor growth were 19.4 ± 3.7 (“chemo then beva 4 days”), 15.7 ± 2.7 (“beva + chemo”) and 10.0 ± 2.0 (“beva then chemo 4 days”). The sequential administration in the “beva then chemo 4 days” arm had the lowest mean tumor size compared with the other remaining groups (36% lower than concomitant in the “beva + chemo” arm and 49% lower than reversed in the “chemo then beva 4 days” arm), although not statistically significant (P = 0.42 and 0.13, respectively).
Survival curves for experiment one are displayed in Figure 2 b. The median survival times were 39 days (control), 49 days (“chemo then beva 4 days”), 55 days (“beva + chemo”), and 67 days (“beva then chemo 4 days”). A log‐rank test showed a significant difference between all groups (P < 0.0001). Further log‐rank tests showed that each treatment group was significantly different than the control arm (P < 0.001). Moreover, the sequential administration in the “beva then chemo 4 days” arm had greater survival median and was significantly different than concomitant in the “beva + chemo” (P = 0.0485) and reversed in the “chemo then beva 4 days” arms (P = 0.0496). Conversely, no significant difference was observed between the “beva + chemo” and the “chemo then beva 4 days” arms (P = 0.631).
Mathematical modeling predicted an optimal time delay of 3 days between the administration of bevacizumab and pemetrexed + cisplatin
The selected model is a modified version of the Gompertz model with a delay in the treatment effects32 and inclusion of a dynamic variable accounting for the vasculature quality and, thus, the normalization effect. See the Materials and Methods section for a detailed description of the model equation, data fit, and parameters’ estimation method. Population analysis yielded the median parameter and interanimal variability estimates reported in Table 1 with good relative standard errors. Goodness‐of‐fit was assessed by visual predictive check plots (Figure 3 a‐d), which demonstrated a good agreement between the model simulations and the experimental data (see residual analysis in Supplementary Figure S7). Individual simulations also demonstrated the ability of our model to reproduce tumor growth dynamics for each mouse (Supplementary Figure S8).
Figure 3.
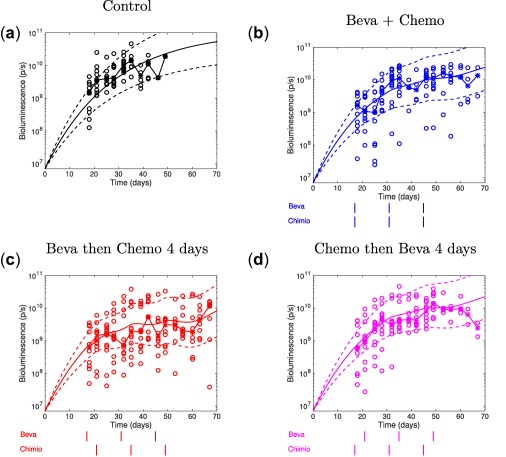
Visual predictive check for experiment one population analysis. (a–d) Visual predictive check plots. Circles: experimental data. Stars with broken lines: median data. Solid lines: tumor growth simulated curves using median parameter values, dashed lines: 95% intervals for interanimal variability, generated from the simulation of 1,000 virtual animals with parameters distributed according to the distribution estimated by the mixed‐effects fit. Beva, bevacizumab; Chemo, chemotherapy.
The model with parameters calibrated on the experimental data allowed us to perform simulations varying the time lag between the administrations of bevacizumab and the pemetrexed‐cisplatin doublet. The criterion for quantification of efficacy was the area under the tumor growth curve. Delays ranging from 1–10 days were tested. Simulation results showed that a 2.8‐days delay between bevacizumab and chemotherapy achieved greater reduction in tumor sizes, with a difference of 76.8% in tumor size as compared with concomitant scheduling (Figure 4 a‐c). Our quantification of the normalization dynamics also predicted that a delay of 8 days would perform substantially worse, with a difference of only 54.3% compared with concomitant administration (Figure 4 c). Quantification of the interanimal variability of the model parameters using our population approach allowed to simulate the resulting interanimal variability of the optimal interdrug administration gap. The optimal gap ranged from 0–10 days with median of 2.8 days and standard deviation of 1.84 days (Figure 4 d).
Figure 4.
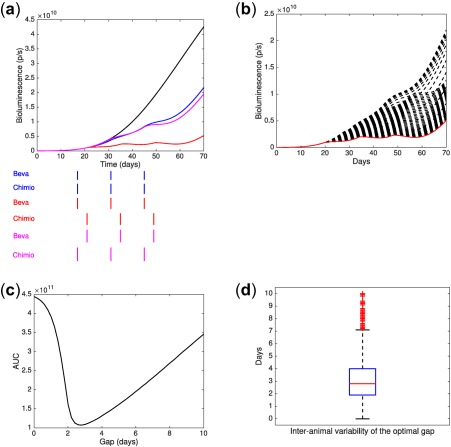
Data‐informed modeling simulations of various gaps between bevacizumab and pemetrexed‐cisplatin administrations. (a) Median tumor growth curves. (b) Simulations of the tumor growth using different time lags between the administration of bevacizumab and pemetrexed‐cisplatin (“beva then chemo”). The red curve corresponds to a time lag of 3 days. (c) Area under the tumor growth curve (AUC) as a function of the time lag. (d) Interanimal variability on the optimal lag time between bevacizumab and chemotherapy (2.8 ± 1.84 days, median ± SD).
Validation of the optimal delay predicted by the model
To test the predictions from the mathematical model, we designed a new experiment that implemented the above‐mentioned schedules (3 and 8 day lag for sequential administrations of “beva then chemo”).
Tumor growth for experiment two is shown in Figure 5 a. At the end of the treatment phase (day 54), mean tumor size (expressed in p/s) were 12,567 ± 2,461 (control), 8,692 ± 543 (“beva then chemo 8 days”), 8,446 ± 1,253 (chemo), 7,486 ± 1,106 (“beva + chemo”), and 4,626 ± 868 (“beva then chemo 3 days”). A statistically significant difference between all arms was obtained (P = 0.0016). Further, Dunn's multiple comparison tests confirmed the superiority of the sequential administration “beva then chemo 3 days,” and showed a statistically significant difference in efficacy between the “beva then chemo 3 days” arm and the control arm (63% of tumor growth reduction (P = 0.002)). Other treatment sequences led to more modest effects on tumor growth as compared with the control arm. Furthermore, mean tumor size for the sequential administration in the “beva then chemo 3 days” arm was markedly lower and almost reached significant difference compared to all other treatment arms (i.e., 38% compared with concomitant “beva + chemo” (P = 0.072), 47% tumor growth reduction as compared with “beva then chemo 8 days” (P < 0.001), and 45% compared with the chemo group (P = 0.016)).
Figure 5.
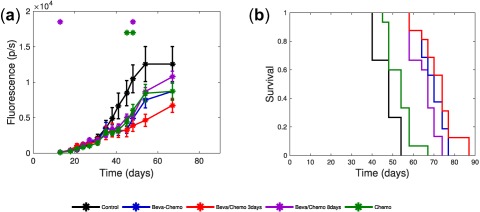
Efficacy and Kaplan‐Meier survival curves of experiment two. (a) Mean tumor growth curve for the five treatment arms of experiment two. Signs above curves indicate statistically significant difference with the control arm (Student's t test, P < 0.05). (b) Kaplan‐Meier plot of the overall survival for the five treatment arms of experiment two.
At the end of the experiment (day 67; i.e. 9 days after all treatment stopped), a statistically significant difference was found between the four remaining arms (P = 0.015). Further, Dunn's multiple comparison tests showed that “beva then chemo 8 days” was significantly different than the optimized “beva then chemo 3 days” group (48% tumor growth reduction, P = 0.007). However, no more statistically significant difference was evidenced among the other treatment arms.
The median survival times were 48 days (control), 54 days (chemo), 67 days (“beva then chemo 8 days”), 70 days (“beva + chemo”), and 74 days (“beva then chemo 3 days”), as presented in Figure 5 b. A log‐rank test showed a statistically significant difference between each treatment arm and the control arm (P < 0.0001). Consistently with our mathematical model predictions, further log‐rank tests showed a statistically significant difference between the “beva then chemo 3 days” and “beva then chemo 8 days” arms (P = 0.0056). Conversely, the difference did not reach statistical significance between the sequential “beva then chemo 3 days” and concomitant “beva + chemo” arms (P = 0.32).
DISCUSSION
Bevacizumab induces a transient phase of vascular normalization. If correctly identified, this could increase drug delivery and improve treatment outcome. In this study, we proposed an integrative strategy that combined experiments on NSCLC tumor‐bearing mice and mathematical modeling to explore and validate improved scheduling of the sequential administration of bevacizumab with cytotoxics. In our first experiment, our data confirmed the benefits, in terms of survival and efficacy, of administering bevacizumab before cytotoxics. To analyze these data, we developed a semimechanistic mathematical model with a critical component quantifying the dynamics of vascular normalization. Model parameters were estimated using a nonlinear mixed effects approach and simulations predicted that giving bevacizumab 3 days before cytotoxics would yield better efficacy. Subsequent experiments confirmed the superiority of this optimized sequence compared with other sequential and concomitant administrations. This proves that beyond a simple shift to sequential administration to achieve better efficacy, the precise timing of the administration of each drug does matter and that mathematical modeling may help to identify optimized alternate scheduling that would require too much resources to be explored empirically.
Human NSCLC H460 cells are a canonical model when performing experimental therapeutics studies in lung cancer. In this respect, following previously published experimental studies, we have chosen the H460 model as a paradigm for mimicking NSCLC tumors.36, 37, 38
Compared to our previous studies,21, 24 the mathematical model presented here was simplified to focus on a minimal number of equations and parameters. We abandoned a more mechanistic description of the vasculature quality in terms of stable and unstable vessels to the benefit of a more phenomenological but more parsimonious and robust model that implements normalization in terms of a simple delay from the bevacizumab concentration.
Mean tumor growth curves obtained in experiment one highlight the high interanimal variability observed within each treatment group (Figure 2 a). This variability can partially explain the high residual variability (Σ = 0.951). Moreover, comparing standard errors of tumor growth data between experiments one and two emphasizes the higher variability of bioluminescence measurements compared to fluorescence imaging. A likely explanation for this discrepancy is the interanimal variability of pharmacokinetics of luciferin used as a tracer for bioluminescence.39
In experiment two, although differences in survival between the “beva + chemo” and “beva then chemo 3 days” groups did not reach statistical significance, median survival was still larger in the latter than in the former (74 days vs. 70 days). Moreover, final tumor size was 38% smaller in the “beva then chemo 3 days” as compared to the “beva + chemo” group. The P value of a Kruskal‐Wallis test for this difference was not below the arbitrarily level of 0.05, but was close to it (P = 0.072), thus still supporting superiority of the “beva then chemo 3 days” group. We hypothesize that this lack of statistical significance in our results is due to a limited power of the study, itself linked to the restricted number of animals per group for ethical constraints.
Several studies have already explored alternate sequences for administering anti‐angiogenics with other drugs, with sometimes contradictory results. Rocchetti et al.40 found that giving bevacizumab after targeted therapy in tumor‐bearing mice yielded better efficacy. Recently, Hutchinson et al.24 inferred (using a mathematical model and experimental data from breast cancer models) a vessel normalization window beginning 15 days after the start of anti‐angiogenics which is a much larger optimal delay compared to other studies.23, 25 In addition, such lag‐time could hardly meet the requirements of clinical testing, because bevacizumab is usually administered on a Q2W or a Q3W basis at bedside. The optimal lag we identified in our study is in line with other experimental studies that explored the normalization window after bevacizumab or other anti‐angiogenics administration, most of these reporting an optimal delay ranging between 2 and 5 days.18 , 33, 41 In patients, Avallone et al.19 showed that giving bevacizumab 4 days prior to chemotherapy yielded better efficacy compared to concomitant administration.
Intriguingly, two clinical studies in NSCLC and metastatic colorectal cancer showed reduced tumor drug delivery of cytotoxic agents when administered 1 and 4 days after bevacizumab,42 which contrasts with our preclinical findings and direct clinical observation of vessel normalization after bevacizumab administration by means of interstitial fluid pressure measurements and functional computed tomography.43 More precisely, a study conducted on 10 patients with NSCLC treated with a single dose of 15 mg/kg of bevacizumab followed by 11C labeled‐docetaxel found a decreased tumor uptake of the chemotherapy after 4 days.44 These findings were obtained on a small group, with a different bevacizumab dose, no repeated cycle, and a different chemotherapeutic drug than here, which altogether might explain the discrepancy. Interestingly, in this same study a wide interpatient variability was observed in the reduction of perfusion and uptake of docetaxel. To this regard, our mathematical model could be of help by characterizing and quantifying the tumor response in a patient‐specific fashion and integrate relevant biomarkers of vessel normalization into a predictive numerical tool for individually optimized scheduling.
Together, these findings highlight the importance of drug scheduling and advocate further studies to optimize scheduling of anti‐angiogenic drugs. In this respect, our approach in conducting experimental studies assisted by semimechanistic mathematical modeling proved to be efficient and both time‐effective and cost‐effective. This proof‐of‐concept study suggests that simplified modeling could help to address the issue of finding the optimal dosing and scheduling of anticancer treatments to improve efficacy.
Critically, our model could be used in a biomarker‐based strategy for improving anti‐angiogenic therapy. As observed in Figure 4 d, even in a homogenous controlled animal population, variability in the optimal gap was observed. Although no predictive biomarker has yet been clearly validated with anti‐angiogenics,45 our model parameters (possibly included in a broader modeling of metastatic disease46) could be quantitatively linked to imaging data and/or predictive circulating biomarkers acting as covariates.47, 48 Consequently, this would provide personalized simulations of response to treatment allowing to individually adapt the dose and timing in order to maximize the efficacy and reduce the toxicity at the patient's bedside.
Conflict of Interest
J.C. received fees for participating to Boards from Roche Laboratory. D.B., F.B., S.B., A.B., R.E.C., D.‐C.I., C.M., and B.L. have no conflicts of interest to declare.
Source of Funding
This study was supported by grants from Canceropole PACA, Roche Laboratory (BevaLung Project), and AM*IDEX (Mars Project, AMU). D.‐C.I. and R.E.C. post‐doc positions were funded by the AM*IDEX (Mars Project, AMU) grant.
Supporting information
Supplementary Methods Parameters’ estimation detailed procedure.
Supplementary Figures
Model Code
ACKNOWLEDGMENTS
We thank Dr Valerie Le Morvan and Professor Jacques Robert from the Institut Bergonié, France, for their kind assistance.
AUTHOR CONTRIBUTIONS
D.‐C. I., R.E.C., A.B., J.C., and S.B. wrote the article. J.C., C.M., B.L., F.B., D.B., and S.B. designed the research. A.B. and J.C. performed the research. D.‐C.I., R.E.C., D.B., and S.B. analyzed the data.
References
- 1. Hurwitz, H. et al Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 3, 2335–2342 (2004). [DOI] [PubMed] [Google Scholar]
- 2. Sandler, A. et al Paclitaxel‐carboplatin alone or with bevacizumab for non‐small‐cell lung cancer. N. Engl. J. Med. 355, 2542–2550 (2006). [DOI] [PubMed] [Google Scholar]
- 3. Miller, K. et al Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 357, 2666–2676 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Tewari, K.S. et al Improved survival with bevacizumab in advanced cervical cancer. N. Engl. J. Med. 370, 734–743 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zalcman, G. et al Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): a randomised, controlled, open‐label, phase 3 trial. Lancet 387, 1405–1414 (2016). [DOI] [PubMed] [Google Scholar]
- 6. Fu, P. et al Bevacizumab treatment for newly diagnosed glioblastoma: systematic review and meta‐analysis of clinical trials. Mol. Clin. Oncol. 4, 833–838 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perren, T.J. et al A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 365, 2484–2496 (2011). [DOI] [PubMed] [Google Scholar]
- 8. Li, Q. , Yan, H. , Zhao, P. , Yang, Y. & Cao, B. Efficacy and safety of bevacizumab combined with chemotherapy for managing metastatic breast cancer: a meta‐analysis of randomized controlled trials. Sci. Rep. 5, 15746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jain, R.K. Normalizing tumor vasculature with anti‐angiogenic therapy: a new paradigm for combination therapy. Nat. Med. 7, 987–989 (2001). [DOI] [PubMed] [Google Scholar]
- 10. Jain, R.K. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J. Clin. Oncol. 31, 2205–2218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jain, R.K. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fuso Nerini, I. , Cesca, M. , Bizzaro, F. & Giavazzi, R. Combination therapy in cancer: effects of angiogenesis inhibitors on drug pharmacokinetics and pharmacodynamics. Chin. J. Cancer 35, 61 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kerbel, R.S. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science 312, 1171–1175 (2006). [DOI] [PubMed] [Google Scholar]
- 14. Cesca, M. et al Bevacizumab‐induced inhibition of angiogenesis promotes a more homogeneous intratumoral distribution of paclitaxel, improving the antitumor response. Mol. Cancer Ther. 15, 125–135 (2016). [DOI] [PubMed] [Google Scholar]
- 15. Carmeliet, P. & Jain, R.K. Angiogenesis in cancer and other diseases. Nature 407, 249–257 (2000). [DOI] [PubMed] [Google Scholar]
- 16. Carmeliet, P. & Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 10, 417–427 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Cesca, M. et al Abstract 3377: Bevacizumab‐improved distribution of paclitaxel in ovarian cancer xenografts potentiates antitumor efficacy. Cancer Res. 76(14 Suppl), abstract 3377 (2016). [Google Scholar]
- 18. Dickson, P.V. et al Bevacizumab‐induced transient remodeling of the vasculature in neuroblastoma xenografts results in improved delivery and efficacy of systemically administered chemotherapy. Clin. Cancer Res. 13, 3942–3950 (2007). [DOI] [PubMed] [Google Scholar]
- 19. Avallone, A. et al Critical role of bevacizumab scheduling in combination with pre‐surgical chemo‐radiotherapy in MRI‐defined high‐risk locally advanced rectal cancer: results of the BRANCH trial. Oncotarget 6, 30394–30407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ciccolini, J. , Benzekry, S. , Lacarelle, B. , Barbolosi, D. & Barlési, F. Improving efficacy of the combination between antiangiogenic and chemotherapy: time for mathematical modeling support. Proc. Natl. Acad. Sci. USA 112, E3453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benzekry, S. , Chapuisat, G. , Ciccolini, J. , Erlinger, A. , & Hubert, F. A new mathematical model for optimizing the combination between antiangiogenic and cytotoxic drugs in oncology. C R Acad Sci Paris, Ser I 350, 23–28 (2012). [Google Scholar]
- 22. Hahnfeldt, P. , Panigrahy, D. , Folkman, J. & Hlatky, L. Tumor development under angiogenic signaling: a dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 59, 4770–4775 (1999). [PubMed] [Google Scholar]
- 23. Wilson, S. et al Modeling and predicting optimal treatment scheduling between the antiangiogenic drug sunitinib and irinotecan in preclinical settings. CPT Pharmacometrics Syst. Pharmacol. 4, 720–727 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hutchinson, L. et al Modeling longitudinal preclinical tumor size data to identify transient dynamics in tumor response to antiangiogenic drugs. CPT Pharmacometrics Syst. Pharmacol. 5, 636–645 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mollard, S. , Ciccolini, J. , Imbs, D.C. , El Cheikh, R. , Barbolosi, D. & Benzekry, S. Model driven optimization of antiangiogenics + cytotoxics combination: application to breast cancer mice treated with bevacizumab + paclitaxel doublet leads to reduced tumor growth and fewer metastasis. Oncotarget 8, 23087–23098 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mollard, S. , Fanciullino, R. , Giacometti, S. , Serdjebi, C. , Benzekry, S. & Ciccolini, J. In vivo bioluminescence tomography for monitoring breast tumor growth and metastatic spreading: comparative study and mathematical modeling. Sci. Rep. 6, 36173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnsson, A. , Olsson, C. , Nygren, O. , Nilsson, M. , Seiving, B. & Cavallin‐Stahl, E. Pharmacokinetics and tissue distribution of cisplatin in nude mice: platinum levels and cisplatin‐DNA adducts. Cancer Chemother. Pharmacol. 37, 23–31 (1995). [DOI] [PubMed] [Google Scholar]
- 28. Lin, Y.S. et al Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of a humanized monoclonal antibody against vascular endothelial growth factor. J. Pharmacol. Exp. Ther. 288, 371–378 (1999). [PubMed] [Google Scholar]
- 29. Woodland, J.M. et al Metabolism and disposition of the antifolate LY231514 in mice and dogs. Drug Metab. Dispos. 25, 693–700 (1997). [PubMed] [Google Scholar]
- 30. Benzekry, S. et al Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput Biol. 10, e1003800 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skipper, H.E. The effects of chemotherapy on the kinetics of leukemic cell behavior. Cancer Res. 25, 1544–1550 (1965). [PubMed] [Google Scholar]
- 32. Simeoni, M. et al Predictive pharmacokinetic‐pharmacodynamic modeling of tumor growth kinetics in xenograft models after administration of anticancer agents. Cancer Res. 64, 1094–1101 (2004). [DOI] [PubMed] [Google Scholar]
- 33. Tong, R.T. , Boucher, Y. , Kozin, S.V. , Winkler, F. , Hicklin, D.J. & Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 64, 3731–3736 (2004). [DOI] [PubMed] [Google Scholar]
- 34. Hartung, N. et al Mathematical modeling of tumor growth and metastatic spreading: validation in tumor‐bearing mice. Cancer Res. 74, 6397–6407 (2014). [DOI] [PubMed] [Google Scholar]
- 35. Lavielle, M. Mixed Effects Models for the Population Approach: Models, Tasks, Methods and Tools (Chapman and Hall/CRC Press, Boca Raton, FL, 2014).
- 36. Li, H. et al Addition of bevacizumab enhances antitumor activity of erlotinib against non‐small cell lung cancer xenografts depending on VEGF expression. Cancer Chemother. Pharmacol. 74, 1297–1305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nukatsuka, M. et al Combination therapy using oral S‐1 and targeted agents against human tumor xenografts in nude mice. Exp. Ther. Med. 3, 755–762 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu, Y. , Chang, E. , Liu, H. , Jiang, H. , Gambhir, S.S. & Cheng, Z. Proof‐of‐concept study of monitoring cancer drug therapy with cerenkov luminescence imaging. J. Nucl. Med. 53, 312–317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Winnard, P.T. Jr , Kluth, J.B. & Raman, V. Noninvasive optical tracking of red fluorescent protein‐expressing cancer cells in a model of metastatic breast cancer. Neoplasia 8, 796–806 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rocchetti, M. et al Predictive pharmacokinetic–pharmacodynamic modeling of tumor growth after administration of an anti‐angiogenic agent, bevacizumab, as single‐agent and combination therapy in tumor xenografts. Cancer Chemother. Pharmacol. 71, 1147–1157 (2013). [DOI] [PubMed] [Google Scholar]
- 41. Winkler, F. et al Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoietin‐1, and matrix metalloproteinases. Cancer Cell. 6, 553–563 (2004). [DOI] [PubMed] [Google Scholar]
- 42. Arjaans, M. , Schröder, C.P. , Oosting, S.F. , Dafni, U. , Kleibeuker, J.E. & de Vries, E.G. VEGF pathway targeting agents, vessel normalization and tumor drug uptake: from bench to bedside. Oncotarget 7, 21247–21258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Willett, C.G. et al Direct evidence that the VEGF‐specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat. Med. 10, 145–147 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Van der Veldt, A.A. et al Rapid decrease in delivery of chemotherapy to tumors after anti‐VEGF therapy: implications for scheduling of anti‐angiogenic drugs. Cancer Cell. 21, 82–91 (2012). [DOI] [PubMed] [Google Scholar]
- 45. Collinson, F. et al Biomarkers and response to bevacizumab—response. Clin. Cancer Res. 20, 1058 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Benzekry, S. , Tracz, A. , Mastri, M. , Corbelli, R. , Barbolosi, D. & Ebos, J.M. Modeling spontaneous metastasis following surgery: an in vivo‐in silico approach. Cancer Res. 76, 535–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Heist, R.S. et al Improved tumor vascularization after anti‐VEGF therapy with carboplatin and nab‐paclitaxel associates with survival in lung cancer. Proc. Natl. Acad. Sci. USA 112, 1547–1552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lassau, N. et al Selection of an early biomarker for vascular normalization using dynamic contrast‐enhanced ultrasonography to predict outcomes of metastatic patients treated with bevacizumab. Ann. Oncol. 27, 1922–1928 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods Parameters’ estimation detailed procedure.
Supplementary Figures
Model Code