

Abstract
Background
Gitelman syndrome (GS) is considered as the most common renal tubular disorder, and we report the first Romanian patient with GS confirmed at molecular level and diagnosed according to genetic testing.
Patient and methods
This paper describes the case of a 27-year-old woman admitted with severe hypokalemia, slight hypomagnesemia, hypocalcemia, hypocalciuria, metabolic alkalosis, hyperreninemia, low blood pressure, limb muscle weakness, marked fatigue and palpitations. Family history revealed a consanguineous family with autosomal-recessive transmission of GS with two cases over five generations.
Results
Next-generation sequencing technology detected two different homozygous mutations c.1805_1806delAT and c.2660+1G>A in the SLC12A3 gene, which encodes the thiazide-sensitive NaCl co-transporter, confirmed by the Sanger method.
Conclusion
Clinicians should be aware of the existence of GS, manage the condition properly and consider the risk of disease recurrence to the next generations.
Keywords: Gitelman syndrome, hypokalemia, hypomagnesemia, SLC12A3 gene, consanguinity, hirsutism
Introduction
Gitelman syndrome (GS) (OMIM 263800), an autosomal-recessive renal tubular salt reabsorption disorder, is characterized by hypokalemic metabolic alkalosis, hypomagnesemia and hypocalciuria;1 elevated levels of plasma renin and aldosterone; and normal blood pressure.2 The electrolyte disturbances are caused by inactivating mutations of the thiazide-sensitive Na+Cl− co-transporter. The SLC12A3 gene located on chromosome 16q13, encodes the transporter that mediates sodium and chloride reabsorption in the distal renal convoluted tubule, target of thiazide diuretics.3 The prevalence of the disease is 1:40,000 in the Caucasian population (1:100 for heterozygotes) (https://www.nih.gov); therefore, it is considered the most common renal tubular disorder.4 Most cases are diagnosed in adulthood, but the first signs of GS corresponding to the GS phenotype are described around the age of 6 years.5
The medical history of patients with GS reveals transient episodes of muscle weakness and tetany, usually in children or young adults,6 but several patients are asymptomatic or have a subclinical disorder.
Genetic diagnosis is often difficult, expensive and not always immediately available in clinics. The aim of the present paper is to describe a case of GS in a young woman, considering that clinicians should be aware of the existence of the GS due to its effects on fluid and electrolyte balance and the risk for future generations.
Case report
Patient history
A 27-year-old woman (Patient V.3, Figure 1) was admitted to the emergency room after a traffic accident, and the ionogram, assessed for the first time, revealed severe hypokalemia and slight hypomagnesemia (Table 1). Her family and past medical history was unremarkable.
Figure 1.
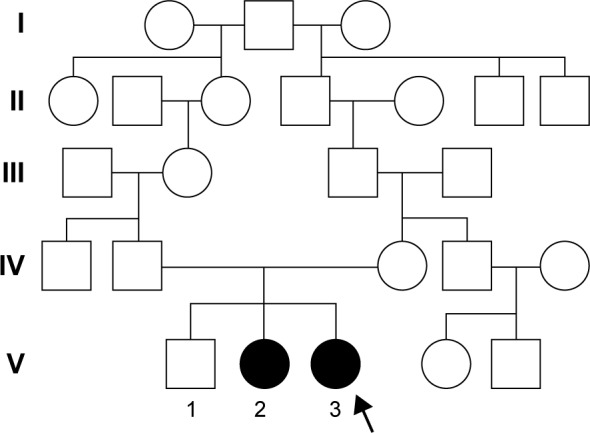
Extended family tree illustrating the transmission of Gitelman syndrome over five generations. Males and females are indicated by squares and circles, respectively. Affected subjects are represented by dark symbols. The index patient is V.3.
Table 1.
Clinical and biological data of the Gitelman syndrome patient and her family members
| Variable | Standards | Proband | Affected sister | Unaffected brother | Unaffected father | Unaffected mother |
|---|---|---|---|---|---|---|
| Age, years | 28 | 44 | 46 | 75 | 69 | |
| Age at which hypokalemia was detected, years | 28 | 44 | ||||
| Treatment (dose/day) | – | – | – | – | ||
| Potassium, mg/d | 3,000 | |||||
| Magnesium, mg/d | 100 | |||||
| Spironolactone, mg/d | 25 | |||||
| Standing blood pressure, mmHg (cardiac frequency, beats/min) | 95/65 mmHg 78/min |
|||||
| Serum parameters | ||||||
| Sodium Na+, mmol/L | 136–145 | 140 mmol/L | 138 | 142 | 141 | 139 |
| Potassium K+, mmol/L | 3.4–4.5 | 2.3–2.5 mmol/L | 3 | 4.1 | 4.1 | 4.1 |
| Magnesium Mg++, mmol/L | 1.8–2.4 | 0.72 mmol/L | 1.1 | 2 | 2 | 1.9 |
| Total Ca++, mmol/L | 8.8–10.8 | 8.5 mmol/L | 8.6 | |||
| Chloride Cl−, mmol/L | 95–106 | 105 mmol/L | ||||
| Bicarbonate HCO3++, mmol/L | 21–32 | 31 mmol/L | ||||
| pH | 7.35–7.45 | 7.36 | ||||
| Standing renin, 1 μUI/mL | 4.4–46.1 | 220–910 μUI/mL | ||||
| Standing aldosterone, ng/dL | 2.27–35 | 28.4 ng/dL | ||||
| Cortisol μg/dL | 11–35l | 16.6 μg/dL | ||||
| Urine parameters | ||||||
| Potassium K+, mmol/24 h | K+: 25–125 | 53 | ||||
| Sodium Na+, mmol/24 h | Na+: 40–220 | 111 | ||||
| Magnesium Mg++, mmol/24 h | Mg++: 30–50 | 25 | ||||
| Calcium Ca++, mmol/24 h | Ca++: 1.1–8.8 | 0.65 | ||||
| Chloride Cl−, mmol/24 h | Cl−: 110–250 | 365.8 | ||||
| pH | 4.8–7.4 | 8 |
She was transferred to the Department of Internal Medicine for clinical diagnosis and therapy. Laboratory investigations revealed, besides severe hypokalemia (2.3–2.5 mmol/L) and slight hypomagnesemia (1.48 mg/dL), hypocalcemia, hypocalciuria, metabolic alkalosis and hyperreninemia (220–910 μIU/mL). Blood pressure was low (95/65 mmHg), heart rate was 78 beats/min and body mass index was 17.55 kg/m2. The patient also reported limb muscle weakness, marked fatigue and palpitations. Her ECG revealed a normal sinus rhythm (78 beats/min) with normal QT interval. Abdominal ultrasound did not reveal renal stones or nephrocalcinosis. A renal tubular disorder was suspected. The prescribed therapy included potassium chloride daily, spironolactone and magnesium. Endocrinology consultation showed hyperandrogenism (increased total testosterone, DHEAS, 17-OH progesterone) and reduced cortisol level. Biochemical data are summarized in Table 1.
Previously, an attempt to administer thiazide was made, but the patient was not compliant regarding oral fluid intake. So, she suffered from severe symptoms of hypovolemia, requiring diuretic withdrawal. Subsequently, the patient was admitted to our center and underwent genetic testing. Therapy was changed in order to normalize biological values. We measured the 24-h urinary electrolyte excretion levels since her renal function was normal. The patient was treated with oral KCl 3,000 mg/day, spironolactone 25 mg/day and magnesium oxide 100 mg/day. Oral treatment with spironolactone and KCl was titrated up to the maximum tolerated doses (as already mentioned), since the patient experienced dizziness when spironolactone was increased and gastrointestinal symptoms (nausea, abdominal pain) when KCl was increased. Although the serum K was not in the normal range, the levels of 2.3–2.5 mmol/L obtained with oral treatment kept the patient symptom free and with a good quality of life. We consider the highly activated renin–aldosterone system to be secondary to hypovolemia; however, we were not able to confirm by remeasuring the relevant parameters since the national medical insurance policy would not cover the additional tests.
Family history revealed two sisters with hypokalemia, severe in one of them, with a potassium level of 2.3 mmol/L, and a brother with normal serum potassium level (Figure 1).
Analysis of the family tree revealed that the father’s grandmother and the mother’s grandfather were siblings (Figure 2). The phenotypes of the two sisters V.2 and V.3 were similar and strongly suggested GS.
Figure 2.

Sequencing chromatograms of the two detected mutations in the SLC12A3 gene are shown and confirmed by classical sequencing Sanger method. (A) NM_000339.2 (SLC12A3): c.1805_1806delAT (p.Tyr602Cysfs*31); EX14; Hom et (B) NM_000339.2 (SLC12A3): c.2660+1G>A; IVS22; Hom zygosity: Hom represents a homozygous mutation, Het represents a heterozygous mutation.
The clinical and biological signs of the patient suggested a genetic mutation. The phenotypes of patients V.2 and V.3 were similar and strongly suggested GS. The five generational pedigree highlighted the parenting relationship, demonstrating the autosomal-recessive inheritance and reflecting consanguinity in the family.
Genetic analysis
Before genetic testing, written informed consent was obtained from the patient to have the case details published.
Next-generation sequencing technology (NGS) was used, and whole exome sequencing (WES) focused on 2,752 genetic disorders and exons of 20,370 genes. Sequencing also covered the intron–exon junction. Microarrays allowed high-throughput analysis of focused regions in the genome, with a sensitivity of 99%. Point mutations, microinsertions and 20 base pairs small deletions can be detected simultaneously. Bioinformatical analysis of the results using international data bases and own database enabled association of mutations with clinical signs.
Two homozygous mutations in the SLC12A3 gene, located at chromosome 16q13 (NCBI number is NT-024766.4, GI: 14778846), which encodes the thiazide-sensitive NaCl co-transporter, were detected, both considered pathogenic. The two detected mutations by NGS were confirmed by the Sanger method (Figure 2). In silico softs identify certain pathogenic mutations.
The first mutation (Figure 2A), homozygous, is a frameshift c.1805_1806delAT (with reference sequence NM_000339.2), producing a deletion of two nitrogenous bases: A and T, in positions 1,805 and 1,806, respectively, located in DNA exon 14 from the SLC12A3 gene, causing a shift in mRNA reading frame and a premature stop codon. As a consequence, tyrosine is replaced by cysteine in position 602 (pY602Cfs*31/p.Tyr602Cysfs*31) resulting in a truncated protein. The mutation is located on chromosome 16:56918096. In silico prediction is that the frameshift mutations are pathogenic.
The second mutation (Figure 2B), c.2660+1G>A causes substitution of guanine and adenine at a 5′ splice site of intron 22 (IVS22+1G>A) deletion of exon 22, and frameshift at exon 23, causing aberrantly spliced transcripts. The mutation is located on chromosome 16:56928555. Four different bioinformatic methods integrated in the Alamut V.2 software predict a loss of donor splice site (Interactive Biosoftware, Rouen, France).7
Genetic analysis of the family demonstrated that the two mutations were present in the two homozygous sisters exhibiting symptoms. Other family members were not genetically tested. The five generational pedigree highlighted the parenting relationship, demonstrating the autosomal-recessive inheritance.
Allele frequency in the dbSNP database, 1,000 genomes project, ESP 6500 database and local database of BGI is unreported, and so we can conclude that its frequencies is very low.
These mutations are absent in normal control databases used in the analysis pipeline.
Additional findings obtained with the clinical WES method
Our patient also has hirsutism. Due to the clinical WES analysis, the etiology for hirsutism has been identified. Another frame-shift mutation (p.N273Kfs*16|p.Asn273Lysfs*16), heterozygous, was identified in exone 9 of AKR1C2 gene (c.819delT). OMIM identified in 1995 AKR1C2 gene (10p15.1), encoding enzyme Aldo-keto reductase family 1, also called 3-alpha- hydroxysteroid dehydrogenase, type III (3-alpha-HSD). The enzyme 3-alpha-HSD is involved in glucocorticoid, progesterone, prostaglandin and biliary acid metabolism. The normal enzyme plays a role in transforming dihydrotestosterone. The role of 3-alpha-HSD was tested in women with hirsutism, considering their pathogenic role.
In hirsute women, reduced AKR1C2 gene expression in skin results in reduced 3-alpha-HSD activity, decreased DHT metabolism and elevated tissue levels of DHT, and diminished DHT metabolism may play an important role in the pathogenesis of hirsutism.8
Differential diagnosis
Measurement of urinary K+ excretion and an assessment of the acid–base status may be helpful in the differential diagnosis.9 A very low rate of K excretion and the absence of a metabolic acid–base disorder suggests hypokalemic periodic paralysis (HPP), whereas a high rate of excretion of K accompanied by either metabolic alkalosis or metabolic acidosis favors non-HPP. Persistent high urine Na+ and Cl− concentrations, extremely low urine calcium excretion rate, and hypomagnesemia in the absence of diuretics suggested a diagnosis of GS.6 Hypokalemia in GS is usually mild to moderate in degree (>2 mmol/L).5,10 In this patient, molecular analysis identified two mutations causing GS.
The Bartter syndrome is, similar to GS, another autosomal-recessive inherited salt-losing tubulopathy, characterized by increased urinary loss of sodium and potassium, low blood pressure and metabolic alkalosis, hyperreninemia and secondary hyperaldosteronism.11 A phenotypic overlap with GS is frequently noticed, especially in clinical signs and laboratory findings, especially hypokalemia, hypomagnesemia and metabolic alkalosis.11 The differences are related to the impaired co-transporter, which is the NaCl co-transporter in the renal distal convoluted tube and the sodium-potassium-chloride co-transporter in the GS and Bartter syndrome, respectively. GS is much more common than Bartter syndrome and mimics the effects of thiazides. Urinary calcium excretion is normal or increased in Bartter syndrome, renal prostaglandin E2 production is increased, the usual age at presentation is early childhood and neuromuscular symptoms are uncommon or mild. Hypomagnesemia may or may not be present in Bartter syndrome, but is always present in GS.
Pseudo-Bartter syndrome including certain conditions that cause the same symptoms and signs of Bartter syndrome and GS without renal tubular dysfunction was excluded considering genetic tests.
Primary and secondary hyperaldosteronism were eliminated considering hypotension and hyperreninemia.
Vomiting, diuretics and laxative abuse were excluded by history, urine chloride, hypocalcemia and urine assays for diuretics.12
Pathophysiology of GS
The distal convoluted renal tubule plays an important role in renal excretion of NaCl and divalent cations such as calcium and magnesium.13 Under normal conditions, NaCl is reabsorbed by the apical thiazide-sensitive NaCl co-transporter in the distal convoluted tube. Na reabsorption is driven by low intracellular sodium and chloride concentrations generated by the Na/K-ATPase and an basolateral chloride channel.14 The loss of function mutations of the thiazide-sensitive NaCl symporter impairs Na reabsorption in the distal convoluted tubes, increasing solute delivery to the collecting duct and causes mild volume depletion, activating the renin–angiotensin–aldosterone system. Secondary hyperaldosteronism enables augmentation of salt reabsorption at the expense of increasing secretion of potassium and protons, accounting for the hypokalemia and metabolic alkalosis noticed in patients with GS.14,15 The coexistence of hypokalemia, hypocalciuria and hypomagnesemia represents the hallmark of GS.16 Angiotensin II and aldosterone are chronically elevated, but activation of the renin–angiotensin–aldosterone system is insufficient to sustain hypertension in the presence of renal salt wasting and patients with GS show reduced responsiveness to several vasoactive substances, including angiotensin II.15 Different mechanisms were proposed to explain hypomagnesemia and hypocalciuria in GS subjects. Loss of function of the NaCl co-transporter leads not only to decreased reabsorption of NaCl, but also to an increased reabsorption of calcium.14,15 Decreased intracellular Na facilitates calcium efflux via the basolateral sodium–calcium exchanger, resulting in hyperpolarization of the cell, enabling calcium reabsorption due to apical, voltage-activated calcium channels.14 Another explanation is based on the increased proximal paracellular calcium reabsorption occurring as a result of compensatory increased Na transport in the proximal tubule.15 It has been speculated that hypomagnesemia in GS may reflect loss of distal convoluted tube mass15 and is due to the mutation of genes encoding magnesium channels in the thiazide-sensitive segments of the distal convoluted tubule.17
Discussion
The present paper describes the case of a patient with hypokalemia, hypomagnesemia and hypocalcemia, diagnosed as GS, confirmed at molecular level after genetic testing.
Patients with GS are usually diagnosed in adulthood during routine investigation, because the patients generally display mild symptoms including muscle cramps and fatigue at presentation.5 Hypokalemia was also accidentally discovered in our patient.
Genetic diagnosis is often difficult, considering the large number of known mutations and costs. Gene diagnosis is not always immediately available in clinics, and clinicians should be aware of the existence of GS and manage the condition properly. Electrolyte homeostasis and water balance are vital for the organism. The phenotypic symptoms are stronger in male patients.
Hypokalemia in GS is usually mild to moderate (>2 mmol/L).10 Severe hypokalemia (1.8 mmol/L in the absence of extrarenal K+ loss) with muscle paralysis,6 tetany and rhabdomyolysis is uncommon in GS.5
Before the post-genomic era, histologic findings were useful in confirming the diagnosis of Bartter syndrome and GS. Renal biopsy is not usually required, especially if GS is confirmed by the pathogenic mutation in the SLC12A3 gene.
The SLC12A3 gene located at 16q13 encodes the thiazide-sensitive NaCl co-transporter in the distal convoluted tubules, and is considered to be a functional defect in GS. The SLC12A3 gene consists of 26 exons encoding a protein of 1021 amino acids.18 Its transcript is expressed mainly in the kidney.19 The identified mutations were located in exon 14, more precisely in codon 602 and intron 22. Understanding the mechanisms involved in mis-splicing may lead to advancements in diagnosis of the disease.
We report a Romanian consanguineous family with autosomal-recessive transmission of GS with two cases over five generations. The disease occurred due to the union of two healthy heterozygous individuals, carrying two mutations in one allele, both descendants of a common ancestor, currently deceased, clinically healthy but carrier of the two mutations. From the time of marriage to testing, the two did not know they were second-degree cousins. The proband (patient V.3) was found homozygous for two different mutations in the same gene, a situation that has not been reported so far, reflecting consanguinity in the family.
The mutations identified as c.1805_1806delAT, p.Tyr602Cysfs*31 (Hom) and c.2660+1G>A (Hom) i n the SLC12A3 gene were previously reported in several patients with GS.
The two mutations found together on the same allele were previously described in a French non-consanguineous family. There were 5 cases showing the two mutations in the same allele (cis), all in heterozygosity, present in one healthy carrier and four composite heterozygous patients.7,20
The first mutation (Figure 2A), c.1805_1806delAT, was first identified in Japan21 and the second mutation (Figure 2B), c.2660+1G>A (intron 22), detected in the intron was described relatively recently in a French family.7 The two mutations detected in the described family, one frameshift and the other splice-site mutation, are pathogenic and rare and resulted in truncated proteins and/or unstable/abnormal messenger RNAs. Each of these homozygous mutations could have determined the GS phenotype providing the molecular diagnosis of GS.
There is a great genetic heterogeneity, considering that 172 distinct mutations were identified up to 2011, and among them 100 newly reported. Considering the type of mutation, 64% were missense type, 14% frameshift, 2% in-frame small deletions/insertions, 14% splice and 6% nonsense.22
Patients with GS generally show two mutations (70%), out of which 25% are homozygous and 75% heterozygous. The gene mutation of the majority patients with GS is compound heterozygous (two different mutations in the same allele).23 Extensive mutations were not associated with a more severe phenotype.
No genotype–phenotype correlations were established, considering that a certain mutation may cause clinical types of different severity,22 and therefore we consider useful reporting any new documented case. A familial aggregation of mutations was noticed and therefore efforts to establish genotype–phenotype correlations in GS have been limited.21 This is the first GS in Romania confirmed at molecular level trough NGS. The five-generation family tree suggests a autosomal-recessive transmission, and due to consanguinity (the father’s grandmother was the sister of the mother’s grandfather), the homozygous state appeared in our patient.
There are several reasons to choose clinical WES, including the ability to test each gene associated with hypokalaemia, in a cost-effective manner. Clinical WES, considering associated symptoms, enables a personalized search including clinical symptoms and signs. In the present case, the associated symptom was excessive hair growth since adolescence and it enabled discovery of the gene responsible for its symptoms.
Whole exome sequencing takes into account three types of information regarding the target genes: considering the main symptoms, considering the guidelines of the American College of Medical Genetics and any incidental findings. It accelerates the possible diagnosis, enables a personalized therapy, management and risk assessment for several disorders and reduces costs of multiple test use.
Currently, it is well established that GS is transmitted as an autosomal-recessive trait, and patients presenting with typical GS phenotype are homozygous or compound heterozygous (ie, bearing two pathogenic mutations in trans). The estimated prevalence of SLC12A3 heterozygous carriers is about 1%,24 and the prevalence of disease is 1–9/100,000 (http://www.orpha.net/consor/).
The risk of recurrence of the disease in the next generation depends on the genotype of the patient’s future partner. If the patient, homozygous for both mutations, marries a healthy heterozygous person, there is a 50% chance of disease manifestation in offspring. If the patient marries an individual with no mutations in the SLC12A3 gene, the risk is 0%. We recommend sequencing testing for the SLC12A3 gene for the future partner, and, depending on the outcome, a prenatal diagnosis may be required.
The present paper has several limitations, including some missing tests, such as thiazide challenge tests due to hypotension and hypovolemia, no results of DNA direct sequencing in the parents of the patient and no functional assays to test if the mutations impair the activity of thiazide-sensitive NaCl co-transporter. We believe that functional assays to test mutations impairing the activity of thiazide-sensitive NaCl co-transporter are not necessary if GS is confirmed by SLC12A3 gene mutation.
DNA direct sequencing in the parents of the patient was not possible considering that, at the time of writing the paper, one of the parents died (the father) and the other (the mother) was in a poor state of health and therefore hardly transportable. The cost of genetic analysis was paid by the patient, which is why it was not possible to extend genetic testing to other family members considering that expenses are not covered by the Romanian medical insurance policy.
Conclusion
GS is a rare condition, but, due to a good prognosis and few symptoms, it is underdiagnosed. Detecting hypokalemia, associated or not with kaliuresis, and hyperchloruria in a patient requires genetic analysis for accurate diagnosis of the GS.
Genetic diagnosis is essential in order to confirm or rule out the disorder in doubtful cases and provide genetic counseling for the patient.
Whole exome sequencing enables improvement of the diagnosis speed and achievement of accuracy in diagnosis of hereditary diseases, including GS. Besides confirming the diagnosis, it can provide additionally genomic information.
Diagnosis of GS requires a multidisciplinary medical team including an internist, a nephrologist and a geneticist.
Further reading
OMIM, Online Mendelian Inheritance in Man (OMIM), https://www.omim.org/.
NIH Gov, National Institutes of Health (NIH), https://www.nih.gov/.
NCBI, The National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/.
Interactive Biosoftware, Rouen, France; http://www.interactive-biosoftware.com/.
Orphanet, http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=358.
dbSNP database, https://www.ncbi.nlm.nih.gov/projects/SNP/.
1000 genomes project, http://www.1000genomes.org/.
ESP 6500 database, http://evs.gs.washington.edu/EVS/.
Acknowledgments
We would like to thank all the members of this family for their participation. We thank Raul Patrascu for the helping in the translation and editing of text.
Footnotes
Author contributions
All authors contributed equally toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235. [PubMed] [Google Scholar]
- 2.Bettinelli A, Bianchetti MG, Girardin E, et al. Use of calcium excretion value to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr. 1992;120(1):38–43. doi: 10.1016/s0022-3476(05)80594-3. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann GF, Zschocke J, Nyhan WL. Inherited Metabolic Diseases A Clinical Approach. 2nd ed. New York: Springer; 2017. [Google Scholar]
- 4.Melander O, Orho-Melander M, Bengtsson K, et al. Genetic variants of thiazide-sensitive NaCl-cotransporter in Gitelman’s syndrome and primary hypertension. Hypertension. 2000;36(3):389–394. doi: 10.1161/01.hyp.36.3.389. [DOI] [PubMed] [Google Scholar]
- 5.Cruz DN, Shaer AJ, Bia MJ, Lifton RP, Simon DB, Yale Gitelman’s and Bartter’s Syndrome Collaborative Study Group Gitelman’s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int. 2001;59(2):710–717. doi: 10.1046/j.1523-1755.2001.059002710.x. [DOI] [PubMed] [Google Scholar]
- 6.Cheng NL, Kao MC, Hsu YD, Lin SH. Novel thiazide-sensitive Na–Cl cotransporter mutation in a Chinese patient with Gitelman’s syndrome presenting as hypokalaemic paralysis. Nephrol Dial Transplant. 2003;18(5):1005–1008. doi: 10.1093/ndt/gfg073. [DOI] [PubMed] [Google Scholar]
- 7.de La Faille R, Vallet M, Venisse A, et al. A pseudo-dominant form of Gitelman’s syndrome. NDT Plus. 2011;4(6):386–389. doi: 10.1093/ndtplus/sfr094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steiner AZ, Chang L, Ji Q, Ookhtens M, Stolz A, Paulson RJ, Stanczyk FZ. 3alpha-hydroxysteroid dehydrogenase type III deficiency: a novel mechanism for hirsutism. J Clin Endocrinol Metab. 2008;93(4):1298–1303. doi: 10.1210/jc.2007-2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin SH, Lin YF, Halperin ML. Hypokalemia and paralysis. QJM. 2001;94(3):133–139. doi: 10.1093/qjmed/94.3.133. [DOI] [PubMed] [Google Scholar]
- 10.Peters M, Jeck N, Reinalter SS, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med. 2002;112(3):183–190. doi: 10.1016/s0002-9343(01)01086-5. [DOI] [PubMed] [Google Scholar]
- 11.Lee JW, Lee J, Heo NJ, Cheong HI, Han JS. Mutations in SLC12A3 and CLCNKB and their correlation with clinical phenotype in patients with Gitelman and Gitelman-like syndrome. J Korean Med Sci. 2016;31(1):47–54. doi: 10.3346/jkms.2016.31.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coleman WB, Tsongalis GJ. Essential Concepts In Molecular Pathology. Burlington: Elsevier; 2010. [Google Scholar]
- 13.Seyberth HW, Schlingmann KP. Bartter- and Gitelman-like syndromes: salt-losing tubulopathies with loop or DCT defects. Pediatr Nephrol. 2011;26(10):1789–1802. doi: 10.1007/s00467-011-1871-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar P, Clark M. Clinical Medicine. 7th ed. Philadelphia: Saunders Elsevier; 2009. [Google Scholar]
- 15.Lifton RP, Somlo S, Giebisch GH, et al. Genetic Diseases of the Kidney. Burlington: Elsevier; 2009. [Google Scholar]
- 16.Ashton Acton Q. Gitelman syndrome: New insights for the healthcare professional. 2012. Dec 1–17, (Edition: Scholarly Paper). [Google Scholar]
- 17.Graziani G, Fedeli C, Moroni L, Cosmai L, Badalamenti S, Ponticelli C. Gitelman syndrome: pathophysiological and clinical aspects. QJM. 2010;103(10):741–748. doi: 10.1093/qjmed/hcq123. [DOI] [PubMed] [Google Scholar]
- 18.Simon DB, Nelson-Williams C, Bia MJ, et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na–Cl cotransporter. Nat Genet. 1996;12(1):24–30. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 19.Chang H, Tashiro K, Hirai M, Ikeda K, Kurokawa K, Fujita T. Identification of a cDNA encoding a thiazide-sensitive sodium–chloride cotransporter from the human and its mRNA expression in various tissues. Biochem Biophys Res Commun. 1996;223(2):324–328. doi: 10.1006/bbrc.1996.0893. [DOI] [PubMed] [Google Scholar]
- 20.Tseng MH, Yang SS, Hsu YJ, et al. Genotype, phenotype, and follow-up in Taiwanese patients with salt-losing tubulopathy associated with SLC12A3 mutation. J Clin Endocrinol Metab. 2012;97(8):E1478–E1482. doi: 10.1210/jc.2012-1707. [DOI] [PubMed] [Google Scholar]
- 21.Maki N, Komatsuda A, Wakui H, et al. Four novel mutations in the thiazide-sensitive Na–Cl co-transporter gene in Japanese patients with Gitelman’s syndrome. Nephrol Dial Transplant. 2004;19(7):1761–1766. doi: 10.1093/ndt/gfh239. [DOI] [PubMed] [Google Scholar]
- 22.Vargas-Poussou R, Dahan K, Kahila D, et al. Spectrum of mutations in Gitelman syndrome. J Am Soc Nephrol. 2011;22(4):693–703. doi: 10.1681/ASN.2010090907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo J, Yang X, Liang J, Li W. A pedigree analysis of two homozygous mutant Gitelman syndrome cases. Endocr J. 2015;62(1):29–36. doi: 10.1507/endocrj.EJ14-0289. [DOI] [PubMed] [Google Scholar]
- 24.Knoers NV, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008;30:3–22. doi: 10.1186/1750-1172-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]