

Abstract
Aggregated Tau protein is associated with over 20 neurological disorders including Alzheimer’s disease. Previous work has shown that Tau’s sequence segments VQIINK and VQIVYK drive its aggregation, but inhibitors based on the structure of the VQIVYK segment only partially inhibit full-length Tau aggregation and are ineffective at inhibiting seeding by full-length fibrils. Here we show that the VQIINK segment is the more powerful driver of Tau aggregation. Two structures of this segment determined by the cryo EM method MicroED explain its dominant influence on Tau aggregation. Of practical significance, the structures lead to the design of inhibitors that not only inhibit Tau aggregation but also inhibit the ability of exogenous full-length Tau fibrils to seed intracellular Tau in HEK293 biosensor cells into amyloid. We also raise the possibility that the two VQIINK structures represent amyloid polymorphs of Tau that may account for a subset of prion-like strains of Tau.
Graphical Abstract
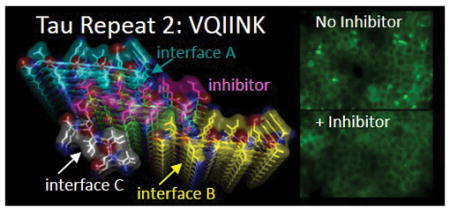
Introduction
The 441-residue protein Tau is abundant in neurons where in its native state it is bound to microtubules. In solution Tau is largely unfolded1,2, and in the numerous neuropathologies known as tauopathies, Tau is aggregated into amyloid fibrils.3 The most prevalent tauopathy is Alzheimer’s disease (AD), where aggregated Tau is found as intracellular “tangles” first reported by Alzheimer. Until recently, tangles were observed only in stained brain sections upon autopsy. The development of PET probes is helping to illuminate the course of AD progression, and it is becoming evident that cognitive decline in AD is tightly coupled to the appearance of Tau aggregates in the brain.4 Furthermore evidence suggests that amyloid β plaques can form early on and may set the stage for Tau aggregation and disease progression.5–7 Once formed, Tau pathology is thought to spread along connected neuronal networks in the brain by trans-cellular propagation of aggregated Tau.8
In normal neurons, Tau promotes microtubule stability by binding tubulin through its microtubule binding domain (K18) comprised of four imperfect repeats. Six Tau isoforms encode variable architectures, the longest of which contains all four repeats (4R) whereas a shorter isoform with 3 repeats (3R) lacks repeat 2. Driving the formation of amyloid aggregates of Tau are two six-residue segments, VQIINK at the start of Repeat 2 and VQIVYK at the start of Repeat 3 (Fig. 1a).2,9
Figure 1.

Atomic structures of amyloid fibrils formed by segments of Tau, viewed down the fibril axes. (a) Schematic of full-length Tau showing the positions of VQIINK and VQIVYK (colored red) in the microtubule binding domain which contains four repeats (R1–4) together termed K18. Shown below is a sequence alignment of Repeats 2 and 3 (R2 and R3) from human Tau with the VQIINK and VQIVYK segments underlined. (b) Comparison of buried surface area Ab and shape complementarity Sc for the VQIINK (interface A; this paper) and VQIVYK (Sawaya et al. 2007; PBD 2ON9) steric zippers. (c) The two steric zipper interfaces, A and B, in the ten residue KVQIINKKLD crystal, shown as stick models with superimposed van der Waals atomic radii. The two interfaces have similar buried areas and shape complementarities. Numbering in C corresponds to the N- and C- termini for the β-sheet colored in cyan. (d) Arrangement of interfaces A and B in the ten residue wild-type KVQIINKKLD structure (left) and the predicted arrangement in the ΔK280 mutant (center and right). Trapezoids in the center diagram represent steric zipper forming residues that are predicted to line the interface between the mated β-strands. The colored arrows show the directions of β-strands forming the steric zippers. Oxygen atoms are red; nitrogen atoms are blue, and mainchain atoms are green for Chains A and C, and cyan for Chain B. In the wild-type structure, interfaces A (red) and B (blue) are formed on opposite faces of the VQIINK β-sheet. Deletion of residue K280 is predicted to reverse the orientation of C-terminal residues by 180º about the β-strand axes (center) merging steric zipper interfaces A and B into a single extended steric zipper interface with greater Ab and Sc as calculated from the ΔK280 model (right and Inset 1).
Highly specific inhibitors of Tau aggregation are needed to definitively determine if, and how Tau aggregates lead to cognitive decline in AD and other tauopathies. Furthermore inhibitors that block seeding by capping the ends of Tau fibrils could halt the progression of Tau pathology in the brain. The atomic structure of an amyloid fibril formed by VQIVYK from Repeat 3 was determined in 200710 and the structure was used to design inhibitors of VQIVYK aggregation.11,12 These studies demonstrated that VQIVYK inhibitors are highly effective at blocking aggregation of 3R Tau isoforms in vitro, but as we show here are ineffective at inhibiting full-length Tau (Tau40) which additionally contains the VQIINK segment of Repeat 2. This observation motivated us to determine the structure of the VQIINK steric zipper.
Efforts in our lab extending over a decade to grow crystals of amyloid fibrils of VQIINK produced only nano-crystals, far too small for single crystal X-ray diffraction. Fortunately with the recent advent of MicroED13–15 we have been able to determine structures of fibrils containing VQIINK by electron diffraction, which we report here. These structures provide the first ever glimpse of the VQIINK aggregation interface. Extrapolating from these structures allowed us to design VQIINK inhibitors that block full-length Tau aggregation and seeding. With these and VQIVYK inhibitors in hand, we tested Tau40 fibrils to determine how each of the respective segments contributes to the spread of Tau aggregation by the seeding. Together our seeding, biochemical, and structural studies suggest that VQIINK is a potent driver of aggregation and seeding, and a superior target for inhibitors of Tau40. Additionally our structures offer insights into Tau fibril polymorphism, which we speculate could relate to the phenomenon of Tau strains.
Results
VQIINK forms an extensive steric zipper interface
To understand the forces that drive aggregation of Tau and to enable the design of inhibitors of aggregation of Tau40, we crystallized a ten residue Tau segment with sequence KVQIINKKLD and determined its 1.5 Å resolution structure by MicroED. Like VQIVYK, this VQIINK-containing segment forms a face-to-face Type 1 homo-steric zipper. That is, the protofilament is formed from the tight mating of identical parallel β-sheets, which are anti-parallel to each other16. The VQIINK zipper shows tighter side chain interdigitation than the VQIVYK zipper (Fig. 1b), having a shape complementarity of 0.77 compared to 0.72 for VQIVYK. Additionally VQIINK buries greater surface area, 168 Å2 compared to 75 Å2 for VQIVYK. These measures of the strength of interfaces suggest that the zipper interface of VQIINK is stronger than the VQIVYK interface.
Interface B
The ten residue VQIINK segment revealed an unanticipated propensity to adopt a second steric zipper interface. The second zipper (referred to as Interface B) is formed by the C-terminal half of the segment, residues KKLD (Fig. 1c). A sequence alignment comparing R2 and R3 shows that the equivalent residues in Repeat 3 are KPVD, which is not expected to adopt a comparable zipper since proline residues have a strong tendency to disrupt β-sheets and break amyloid structures17 (Fig. 1a). Furthermore an overlapping segment with the sequence LDLSN in repeat 2, corresponding to amino acids 282–286, bears the features of an amphiphilic amyloidogenic sequence motif, whereas no additional sequences with these features are found in Repeat 3.18 This suggests that the potential to form a second extended steric zipper interface is limited to the Repeat 2 segment, VQIINK.
ΔK280 mutant
In the wild-type segment structure, interfaces A and B are formed on opposite faces of the VQIINK β-sheet. It is known that the ΔK280 mutation accelerates Tau fiber formation and has been linked to tauopathies.1,19 Lys280 lies at the junction between interfaces A and B in our crystal structure (Fig. 1c and d) and deleting K280 is expected to reverse the orientation of the C-terminal residues by 180º about the β-strand axis20. The result is to rotate the zipper-forming residues in interface B to the same face of the β-sheet as interface A, merging the two separate interfaces that lie on opposite faces in wild-type to a single extended zipper interface in the ΔK280 mutant (Fig. 1d, Inset 1). Modelling the steric zipper formed by the ΔK280 mutant shows that the extended interface formed by these predicted structural changes results in an longer zipper with high shape complementarity (Sc = 0.77) and nearly double the buried surface area (Ab = 299 Å2), explaining how the ΔK280 mutant could promote more rapid Tau aggregation and toxicity.
VQIINK promotes rapid fiber formation
Given the observation that VQIINK forms a more extensive steric zipper interface than VQIVYK, we wondered if VQIINK would be a more potent driver of amyloid formation than VQIVYK. To test the possibility we replaced the residues VY in VQIVYK with IN, converting Tau K18 to an engineered form that contains two copies of VQIINK (named 2xIN). Likewise 2xVY was constructed and compared with wild-type K18 to determine how the rates of fibril formation differ for constructs that contain only VQIINK or VQIVYK zipper segments. As shown by ThT fluorescence in Fig. 2a, 2xIN exhibits a greater rate of aggregation by shaking at 37 ºC with heparin, forming fibrils in half the time of wild-type (75 min vs. 150 min to reach half maximum). 2xVY aggregates most slowly reaching half max after 225 min.
Figure 2.

Time dependence of fibrillization and oligomerization for wild-type K18 construct and 2xIN and 2xVY K18 mutant constructs. (a) Averaged ThT fluorescence curves of wild-type K18 construct and engineered 2xIN and 2xVY K18 constructs at 50 μM in the presence of heparin with shaking at 700 rpm 37 ºC. Lag times determined from the half-maximum values of the curves shown are given for the respective constructs on the right. Error bars show the standard deviation of triplicate ThT measurements. (b) Analysis of oligomers measured by S200 size exclusion chromatography. Peaks corresponding to the soluble oligomer species are marked with an arrow.
To evaluate how the VQIINK and VQIVIYK segments affect formation of soluble oligomers, Tau oligomers were fractionated by gel filtration chromatography. This approach has been used for K18 to separate monomer, oligomer, and fibrillar species.21 Wild-type K18 forms a mixture of aggregates overnight, amyloid fibers and insoluble material are removed by ultracentrifugation and the resulting supernatant injected on a S200 size exclusion column separates into two peaks, one containing monomer, the other one containing oligomer (Fig. 2c, black). 2xVY produces a mixture of species similar to wild-type, whereas no remaining monomer or oligomer is detectable for 2xIN (Fig. 2c blue and green, respectively). Together, these data suggest that 2xIN rapidly fibrillizes in the presence of heparin, shifting the equilibrium away from monomeric and oligomeric species instead favoring rapid amyloid fiber formation.
VQIINK inhibitors reduce Tau fibril formation and block seeding
The VQIINK crystal structure was used as a template for structure-based inhibitor design (Fig. 3a). The native ten residue sequence that we crystallized was used as a starting point, and bulky amino acid sidechains were modelled into the zipper to identify residues that would best disrupt the observed interfaces. As summarized in Table 1, an Ile at position 4 and Leu at position 9 (corresponding to residues I277 and L282 in the Tau40 protein sequence) were identified as key residues in interfaces A and B. Modelling indicated that interface A would be best disrupted by replacing the Ile at position 4 with either a Met or Trp, while interface B appeared to be best disrupted by Arg. Since the native peptide sequence targeted by the inhibitor is dipolar, beginning with a Lys and ending with an Asp, the terminal residues of the inhibitor peptide were charge reversed to promote electrostatic attraction with the binding interface.
Figure 3.
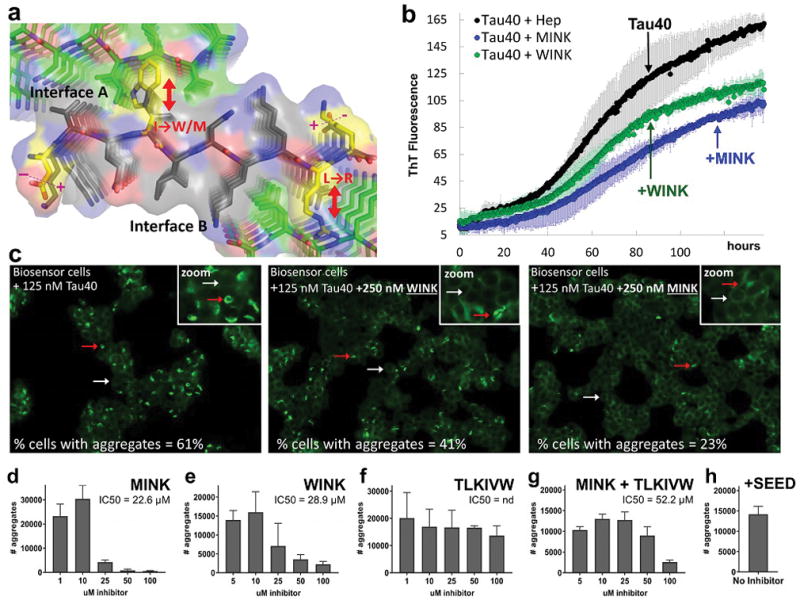
Structure-based design of Phase 1 inhibitors of VQIINK aggregation. (a) Logic of inhibitor design. The inhibitors (Table 1) bind on the tips of VQIINK fibrils and introduce steric clashes that disrupt interfaces A and B. Based on the structures, the inhibitor sequences were designed as follows. Either a tryptophan (inhibitor WINK) or methionine (inhibitor MINK) were incorporated to block interface A. Both inhibitors contain an arginine to disrupt interface B. The ends of the inhibitor were charge reversed to promote electrostatic attraction of the inhibitor peptide to the fibril sequence. (b) The effects of designed inhibitors on the formation of fibrils of 25 μM full-length Tau (Tau40) plus a two-fold molar excess of MINK or WINK inhibitor peptide with shaking at 700 rpm at 37 ºC. Error bars show the standard deviation of triplicate ThT measurements from two independent experiments. (c) The effects of designed inhibitors on the transfer of fibrils of Tau40 into HEK293 biosensor stably expressing a full-length (4R1N P301S) YFP fusion. The cells were seeded with 125 nM Tau40 fiber (final concentration); the seeds were grown for 120 hours in the presence or absence of a two-fold excess of WINK or MINK. Representative cells containing aggregates are marked by red arrows, and cells without by white arrows. Percentages of cells with aggregates were calculated by dividing the number of aggregates in the field of view by the number of cells. Inset box shows a zoomed image from within the presented field of view. (d–h) 4R1N Tau-YFP biosensor cells seeded with pre-capped Tau40 fibers treated with MINK, WINK, TLKIVW, or MINK + TLKIVW inhibitor peptides at indicated concentrations. Bar graphs show the average number of aggregates at the indicated inhibitor concentrations, and error bars show the standard deviation of triplicate measurements. IC50’s were calculated from the dose-response curves shown. Panels B, C and D suggest that MINK is a more effective inhibitor than WINK.
Table 1.
Amino acid sequences of Tau inhibitors, based on two structures of the VQIINK interface. The top row lists the residue number from the native Tau40 sequence. The second row lists the residue position number in the inhibitor peptide. Row 3 is the native sequence from the wild-type human Tau40, and rows 4–10 are inhibitor peptide sequences tested in this paper. Residues at positions 4 and 9 shown in red are critical for forming Interfaces A and B, respectively. Residues 3 and 5 shown in blue comprise Interface C.

|
The effect on fibril formation of Tau40 of the two resulting VQIINK inhibitors, MINK (which contains a Met at position 4 to disrupt interface A) and WINK (which instead contains a Trp) was evaluated in vitro using a ThT assay. At 25 μM Tau40, a two-fold mole excess of either MINK or WINK reduced Tau40 aggregation to about half with MINK inhibiting Tau40 aggregation slightly more effectively than WINK (Fig. 3b). We wondered if the MINK and WINK peptides themselves were capable of aggregating since they derive from the aggregation-prone VQIINK segment. To test the aggregation of these inhibitors, we subjected MINK and WINK to the same conditions used to aggregate Tau40, that is by incubating at 37 ºC with heparin and shaking at 700 rpm. As shown in Supplemental Fig. 2A, WINK showed no detectable aggregation although MINK produced a low ThT signal after 24 hours indicating some degree of aggregation. By comparison, we estimate the level of MINK aggregation to be nearly 20 times less than Tau40.
Recent evidence suggests that aggregated Tau can spread from cell to cell in a prion-like manner, apparently by seeding Tau in the recipient cell into fibrils.8 To test whether the VQIINK inhibitors are capable of blocking the spread of Tau fibers by seeding, Tau40 monomer was incubated at 37 ºC shaking with heparin in the presence or absence of MINK or WINK before seeding HEK293 biosensor cells stably expressing a near full-length isoform, Tau 4R1N P301S-EYFP.22 As shown in Fig. 3c, about 61% of the cells that were seeded with 125 nM unlabeled Tau40 fiber in the absence of inhibitor produced intracellular aggregates. Cells seeded with Tau40 fiber grown with MINK or WINK reduced the percentage of seeded cells to 23 and 41%, respectively. Qualitatively these VQIINK inhibitors improved the appearance of cells resulting in more typical monolayer growth rather than rounded morphology (Fig. 3c).
To test whether VQIINK inhibitors can cap pre-formed fibers to block their spread, Tau40 amyloid fibrils were incubated with varying concentrations of inhibitor before applying the pre-capped fibers to 4R1N Tau biosensor cells and measuring seeding. As shown in Fig. 3d and e, MINK and WINK inhibit seeding by Tau40 fibers in a concentration dependent fashion. From the dose-response curves shown, IC50 values of 22.6 and 28.9 μM were calculated for MINK and WINK. In contrast, the potent VQIVYK capping peptide inhibitor with sequence TLKIVW11 poorly inhibited seeding by Tau40 fibrils (Fig. 3f). Furthermore a combination of MINK and TLKIVW inhibitor failed to synergize, although the pair performed better than TLKIVW alone having an IC50 value of 52.2 μM (Fig. 3g).
Design of Phase 2 inhibitors based on a second polymorph
We sought and discovered a second structural polymorph of amyloid fibrils formed by the VQIINK segment. Our rationale was the report that distinct strains of HEK biosensor cells are apparently caused by seeding with distinct aggregated forms of Tau22, and that K18 which contains VQIINK in addition to VQIVYK exhibits conformational heterogeneity whereas K19, which contains only VQIYVK does not23. We screened for and found nanocrystals of the VQIINK segment and were able to determine their atomic structure by MicroED. This structure reveals a Class 4, face-to-back steric zipper, driven by Gln and Ile sidechains on the opposite face of the VQIINK β-strand. We term this Interface C (Fig. 4B). Interface C buries substantial, albeit marginally lower surface area than Interface A (Ab = 146 Å2), and a Sc = 0.67. Apparently the VQIINK segment of Tau is capable of forming at least three distinct steric zipper interactions, leading to at least three distinct amyloid fibrils and potentially three distinct phenotypic downstream effects.
Figure 4.

The inhibition of Tau aggregation by Phase 2 designed inhibitors. (a and b) Comparison of the 10-residue (Polymorph 1) and 6-residue (Polymorph 2) VQIINK MicroED structures, viewed down the fibril axes. 2fo-fc electron density maps are contoured to 1.0 σ, and an alternate conformation modelled at 50% occupancy rendered in line format is shown for Lys281 in polymorph 1. (a) The 10-residue segment forms a face-to-face Class 1 zipper, whereas (b) the 6-residue VQIINK segment forms a face-to-back Class 4 zipper10. (c–i) Seeding by Tau40 fibers pre-capped with Phase 2 VQIINK inhibitors (described in Table 1) designed to block interfaces A, B and C, measured in 4R1N Tau-YFP biosensor cells. Bar graphs show the average number of aggregates at the indicated inhibitor concentrations, and error bars show the standard deviation of triplicate measurements. Fluorescence images in E show seeded cells treated with 0.3 or 2.5 μM W-MINK inhibitor. Representative cells containing aggregates are marked by red arrows, and cells without by white arrows. Panel I shows quantified levels of aggregation in unseeded 4R1N Tau biosensor cells, and cells seeded by untreated Tau40 fibrils.
We hypothesize that the interfaces observed in our two crystal structures represent the actual surfaces used by Tau40 to form different aggregate subpopulations. To test this, we re-engineered MINK to incorporate a third steric clash that would disrupt Interface C, in addition to Interfaces A and B. We reasoned that if multiple subpopulations of aggregates can simultaneously exist as a mixture in solution, or perhaps in a mixture of cells, then one inhibitor capable of targeting Interfaces A, B, and C should be more potent than the MINK and WINK inhibitors alone, which target only interfaces A and B.
This hypothesis led to our Phase 2 inhibitors (Table 1). As shown in Fig. 4b, a Gln and Ile at positions 3 and 5 form the basis of Interface C. Modelling indicated that substituting these positions with a tryptophan would be most destabilizing to the steric zipper structure. As summarized in Table 1, the resulting iterations of MINK incorporate a Trp steric clash at position 3, or positions 3 and 5.
As shown in Fig. 4c the Phase 2 inhibitor W-MINK, which contains a Trp steric clash at position 3, blocked Tau40 seeding more effectively than MINK, having an IC50 of 1.1 μM compared to 22.6 μM for MINK. Substituting the Arg at position 9 of W-MINK with a Trp (inhibitor peptide M4W39) also exhibited improved potency over MINK, having an IC50 of 2.9 μM (Fig. 4f). In spite of their improved IC50s, the Phase 2 inhibitors still did not synergize with TLKIVW (Fig. 4d). Fluorescence images of seeded cells at two selected W-MINK concentrations, 0.3 and 2.5 μM shown in Fig. 4e illustrate the effect W-MINK has on seeding by Tau40 fibers. At 0.3 μM W-MINK, Tau40 fibers are still able to induce seeding in 4R1N Tau biosensor cells whereas at 2.5 μM W-MINK, seeding by Tau40 fibers is no longer observed.
By contrast to W-MINK, the WMW peptide which produces a Trp steric clash at both positions 3 and 5, and WWW which contains Trp at positions 3, 4 and 5 failed to show any inhibition (Fig. 4g–h), possibly owing to a destabilizing effect that multiple tryptophans in series may have on capping onto the amyloid chain. These data suggest that the Ile at position 5 in Interface C is insensitive to substitution, whereas the Gln at position 3 (Gln 276 in the native sequence) plays an important role in stabilizing interface C.
Discussion
Prior to the two atomic structures of the VQIINK segment reported here, the atomic-level understanding of how repeat 2 contributes to Tau aggregation was limited. The crystals of Tau that included the VQIINK segment were invariably some ten thousand times smaller in volume than crystals of the other fibril-driving segment VQIVYK, and thus too tiny for manipulation and X-ray data collection. Electron diffraction came to the rescue, providing atomic resolution structures of fibrils of both the 10-residue amyloid forming segment with sequence KVQIINKKLD and the 6-residue structure of VQIINK itself.
Both structures reveal a propensity of VQIINK to form tightly interdigitated steric-zipper interfaces that appear to be more adhesive than the interface of VQIVYK. Biochemical studies support this interpretation showing that engineered Tau constructs containing only VQIINK aggregate more rapidly than wild-type, and constructs containing only VQIVYK instead accrue a mixture of monomer, oligomer and fiber. These data indicate that VQIINK is a potent driver of Tau amyloid aggregation, and that it is likely an excellent target for inhibitors of full-length Tau fibril formation.
Our VQIINK structures explain observations that have been made previously. The structure of the ten residue segment shows that Ile277 projects into Interface A, forming the centerpiece for the aggregation of this motif. Previous studies highlighted the importance of Ile277 by showing that I277P disrupts Tau amyloid fiber structure.19 Additionally as described in Figure 1D, our structure provides insight into how deletion of Lys280 can enhance Tau fibril formation by extending interface A, which nearly doubles the contiguous buried surface area of the steric zipper interface for the ΔK280 mutant.
These two VQIINK-containing structures enabled the design of VQIINK inhibitors that act on full-length Tau by capping the ends of Tau fibrils to prevent template-assisted filament growth11,24. The VQIINK inhibitors tested here slow full-length Tau aggregation in vitro and furthermore, for the first time, block seeding induced by Tau40 fibers in 4R1N Tau HEK293 biosensor cells. This is significant because as we show, VQIVYK inhibitors were unable to inhibit seeding by Tau40. The failure of VQIVYK inhibitors to block seeding might be viewed as conflicting with the observation that VQIVYK is central to the core of Tau fibrils extracted from AD tissue25. As we discuss in the following paragraph however, our findings may be consistent with this observation.
Tau fibrils extracted from the AD brain adopt a fibril core comprising Repeats 3 and 4, in addition to ten residues extending beyond the end of Repeat 4. VQIINK, along with the rest of Repeat 2 apparently reside in the fuzzy coat of this AD fibril. This would seem to suggest that VQIVYK capping inhibitors should be highly effective at blocking seeding by Tau40 fibers. To the contrary, our results show that VQIINK is the more prominent effector of seeding by Tau40, at least for recombinant fibrils. How can these two observations be reconciled? One possibility is the core of the Tau fibril formed in vitro differs from the one that is found in AD. Alternatively it is possible that the fibril core itself is not the primary driver of seeding, but rather serves as a solvent-excluded scaffold that clusters VQIINK segments together in the fuzzy coat, thereby poising the solvent-exposed VQIINK steric zippers for seeding. From this perspective, VQIINK localization to the fuzzy coat would make it more accessible to protein monomers in the cellular milieu, in turn explaining its prominent role in seeding and its exquisite sensitivity to inhibition. Interpreted this way, our VQIINK structures are consistent with the AD-derived Tau fibril model reported by Fitzpatrick et al (2017).
Our studies emphasize that the effectiveness of inhibitor treatment depends on the stage of administration of the inhibitor. That is, the concentration of inhibitor required to block seeding by pre-formed fibers is about 100 times greater than the amount required when inhibitor is added to monomeric Tau prior to initiating aggregation by shaking and the addition of heparin, when 250 nM inhibitor effectively inhibited seeding. The greater amount of inhibitor required to block pre-formed fibers likely stems from the propensity of mature fibers to extend unabated, producing longer fibers with more surface area for seeding by secondary nucleation and/or fragmentation into smaller species that produce new sites for primary nucleation. This observation may highlight the importance of early disease intervention with amyloid inhibitors, underscoring the need for sensitive disease biomarkers.
Our structures of VQIINK may suggest a structural basis for the mysterious phenomenon of Tau strains.22,26 Whereas strains in microorganisms are different phenotypes encoded by different nucleic acid sequences, strains in amyloid diseases are different phenotypes encoded by different fibril structures. Different fibril structures, in turn, are likely encoded by formation of different steric zippers.27 Three distinctly different steric zippers emerged from our two VQIINK structures: interfaces A, B and C. The existence of these distinct interfaces shows how conformationally distinct fibrils of Tau can form, and we speculate that these different structures may begin to reveal how a subset of different Tau strains could originate at the molecular level, although future studies will be needed to address this possibility directly.
Methods
Crystallization, data collection, and structure solution
Nanocrystals of KVQIINKKLD synthetic peptide (purchased from Genscript) were prepared from 30 mg/ml stocks dissolved in water by the addition of 45 mM arachidonic acid to concentrated peptide. Subsequently crystals were grown in batch at 37 ºC by the addition of 10 μl of precipitant (32% PEG10K in 100mM phosphate citrate, pH 4.2) to 5 μl of protein. After 18 hours nanocrystals were resuspended by pipetting, spun down in an epitube and the resulting crystalline pellet was washed three times with water. Nanocrystals of VQIINK peptide (purchased from Genscript) were grown by vapor diffusion from peptide at 15 mg/ml as a 1:2 drop ratio (protein:reservoir) from 0.29 M LiNO3, 24% (w/v) PEG 3350 at 18 ºC and similarly harvested for data collection.
MicroED data were collected by spotting 2–3 μl of crystals onto TEM grids with carbon film support and plunge frozen in liquid ethane using a Vitrobot Mark IV (FEI). Diffraction images were collected on an FEI Tecnai F20 TEM at 200 kV as a continuous rotation tilt series at a rate of 0.2° per second with 2 second exposures per image. Diffraction data from several crystals were indexed, merged and scaled with XDS and XSCALE.28 Molecular replacement was performed with Phaser29 using a polyalanine search model composed of an ideal β-strand. The structure of VQIINK was solved using a truncated version of KVQIINKKLD comprising residues 2–7 (VQIINK) as a search model in Phaser. Crystallographic refinements were performed using the programs Refmac30, Buster31, and Coot32.
Inhibitor peptide design
VQIINK inhibitor peptides were designed using the native structure as a starting point. Bulky sidechains were modelled at sites in the VQIINK structure that were in close contact with residues in the mated sheet of the steric zipper interface as shown in Fig. 3a. Capping residues were chosen by modelling all possible rotamers to find sidechains without any compatible conformer with the steric zipper interface (that is, sidechains that clashed with the mated beta sheet at every rotamer conformer were selected). All of the inhibitor peptides shown in Table 1 were synthesized by Genscript with minimum purities of 90% and dissolved in deionized water to a final stock concentration of 6 mM.
Tau fibril incubation with inhibitors for biosensor cell seeding assays
Tau40 fibers were prepared by shaking 25 μM Tau40 in PBS buffer (pH 7.4) with 0.5 mg/ml heparin (Sigma cat. H3393), 1 mM DTT for 3–6 days. Presence of fibers was confirmed with an endpoint ThT reading, and then fibrils were diluted 20-fold to 1.25 μM in OptiMEM (Life Technologies, cat. 31985070). Inhibitors dissolved in water were added to 20 μl of diluted Tau40 fibers at a concentration 20-fold greater than the final desired concentration. For combination VQIINK inhibitor treatments with TLKIVW, the indicated concentration is the total amount of inhibitor, which is the sum of each individual inhibitor present at an equimolar ratio. The d-TLKIVW peptide was first dissolved to a high concentration in DMSO (>25 mM), and then diluted in water before addition to fibers. Fibers were incubated 16 hours with inhibitor, and subsequently were sonicated in a cuphorn water bath for 3 minutes before seeding cells. The resulting “pre-capped fibrils” were mixed with 1 volume of Lipofectamine 2000 (Life Technologies, cat. 11668027) prepared by diluting 1 μl of Lipofectamine in 19 μl of OptiMEM. After twenty minutes, 10 μl of fibrils were added to 90 μl of 4R1N Tau biosensor cells to achieve the final indiated ligand concentration.
ThT plate reader assay
Concentrated Tau protein was diluted into PBS buffer (pH 7.4) to a final concentration as indicated in the text. Proteins were shaken as solutions containing 75 μM ThT, 0.5 mg/ml heparin (Sigma cat. H3393), 1 mM DTT, and a two-fold molar excess of inhibitor (when indicated) in 96-well plates with a plastic bead to enhance agitation. ThT fluorescence was measured with excitation and emission wavelengths of 440 and 480 nm, and averaged curves were generated from triplicate measurements as indicated in the figure legend. Error bars show the standard deviation of replicates measurements.
Supplementary Material
Acknowledgments
We thank Marc Diamond for discussion and for gifting the monoclonal biosensor HEK293 cell line expressing Tau 4R1N P301S-EYFP for our inhibitor assay, and Hilda Mirbaha for assistance and advice conducting biosensor seeding experiments and purifying K18 Tau oligomers by gel filtration chromatography. We also thank awards 1R01 AG029430 and RF1 AG054022 from the National Institute on Aging, 1F32 NS095661 from the National Institute of Neurological Disorders and Stroke, A2016588F from the BrightFocus Foundation, and HHMI and the Janelia Visiting Scientist Program for support.
Abbreviations
- Ab
solvent-accessible surface area buried
- MicroED
micro-electron diffraction
- Sc
shape complementarity
- Tau40
full-length Tau protein
- nd
not determined
Footnotes
Data Availability
Atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 5V5B (KVQIINKKLD) and 5K7N (VQIINK).
Author Contributions
PS conceived and designed the experiments; PS, DRB, JAR, and KM performed the experiments; PS, DRB, MRS, and DC analyzed data; and PS, DSE and TG co-wrote the paper.
References
- 1.Margittai M, Langen R. Side Chain-dependent Stacking Modulates Tau Filament Structure. Journal of Biological Chemistry. 2006;281:37820–37827. doi: 10.1074/jbc.M605336200. [DOI] [PubMed] [Google Scholar]
- 2.von Bergen M, et al. Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proceedings of the National Academy of Sciences. 2000;97:5129–5134. doi: 10.1073/pnas.97.10.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, Eisenberg DS, Crowther RA. Annual Review of Neuroscience. 2017;40 doi: 10.1146/annurev-neuro-072116-031153. [DOI] [PubMed] [Google Scholar]
- 4.Schwarz AJ, et al. Regional profiles of the candidate tau PET ligand recapitulate key features of Braak histopathological stages. Brain. 2016;139:1539–1550. doi: 10.1093/brain/aww023. [DOI] [PubMed] [Google Scholar]
- 5.Manczak M, Reddy PH. Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. Journal of Alzheimer’s disease: JAD. 2013;36:285–295. doi: 10.3233/JAD-130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seward ME, et al. Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. Journal of Cell Science. 2013;126:1278–1286. doi: 10.1242/jcs.1125880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brier MR, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Science Translational Medicine. 2016;8:338ra366–338ra366. doi: 10.1126/scitranslmed.aaf2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular Propagation of Tau Aggregation by Fibrillar Species. The Journal of Biological Chemistry. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Bergen M, et al. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta-structure. Journal of Biological Chemistry. 2001 doi: 10.1074/jbc.M105196200. [DOI] [PubMed] [Google Scholar]
- 10.Sawaya MR, et al. Atomic structures of amyloid cross-[bgr] spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. http://www.nature.com/nature/journal/v447/n7143/suppinfo/nature05695_S1.html. [DOI] [PubMed] [Google Scholar]
- 11.Sievers SA, et al. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. http://www.nature.com/nature/journal/v475/n7354/abs/nature10154-f1.2.html#supplementary-information. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng J, et al. Macrocyclic β-Sheet Peptides That Inhibit the Aggregation of a Tau-Protein-Derived Hexapeptide. Journal of the American Chemical Society. 2011;133:3144–3157. doi: 10.1021/ja110545h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi D, Nannenga BL, Iadanza MG, Gonen T. Three-dimensional electron crystallography of protein microcrystals. eLife. 2013;2:e01345. doi: 10.7554/eLife.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez JA, et al. Structure of the toxic core of [agr]-synuclein from invisible crystals. Nature. 2015;525:486–490. doi: 10.1038/nature15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez JA, Gonen T. In: Methods in Enzymology. Crowther RA, editor. Vol. 579. Academic Press; 2016. pp. 369–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenberg DS, Sawaya MR. Annu Rev Biochem. 2017:3.1–3.27. doi: 10.1146/annurev-biochem-061516-045104. [DOI] [PubMed] [Google Scholar]
- 17.Wood SJ, Wetzel R, Martin JD, Hurle MR. Prolines and Aamyloidogenicity in Fragments of the Alzheimer’s Peptide.beta./A4. Biochemistry. 1995;34:724–730. doi: 10.1021/bi00003a003. [DOI] [PubMed] [Google Scholar]
- 18.Moore CL, et al. Secondary nucleating sequences affect kinetics and thermodynamics of tau aggregation. Biochemistry. 2011;50:10876–10886. doi: 10.1021/bi2014745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Bergen M, et al. Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local β-Structure. Journal of Biological Chemistry. 2001;276:48165–48174. doi: 10.1074/jbc.M105196200. [DOI] [PubMed] [Google Scholar]
- 20.Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Quarterly Reviews of Biophysics. 2008;41:265–297. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 21.Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. Journal of Biological Chemistry. 2015 doi: 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders David W, et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. http://dx.doi.org/10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siddiqua A, et al. Conformational Basis for Asymmetric Seeding Barrier in Filaments of Three- and Four-Repeat Tau. Journal of the American Chemical Society. 2012;134:10271–10278. doi: 10.1021/ja303498q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margittai M, Langen R. Template-assisted filament growth by parallel stacking of tau. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10278–10283. doi: 10.1073/pnas.0401911101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzpatrick AWP, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–190. doi: 10.1038/nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaufman Sarah K, et al. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron. 2016;92:796–812. doi: 10.1016/j.neuron.2016.09.055. http://dx.doi.org/10.1016/j.neuron.2016.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wiltzius JJW, et al. Molecular mechanisms for protein-encoded inheritance. Nat Struct Mol Biol. 2009;16:973–978. doi: 10.1038/nsmb.1643. http://www.nature.com/nsmb/journal/v16/n9/suppinfo/nsmb.1643_S1.html. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W, XDS Acta Crystallographica Section D: Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCoy AJ, et al. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica Section D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Bricogne G, BE, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER 2.10.0. Cambridge, United Kingdom: Global Phasing Ltd; 2016. [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biernat J, et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. The EMBO Journal. 1992;11:1593–1597. doi: 10.1002/j.1460-2075.1992.tb05204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Meth. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.