

Abstract
Mass spectrometry (MS) has become a crucial technique for the analysis of protein complexes. Native MS has traditionally examined protein subunit arrangements, while proteomics MS has focused on sequence identification. These two techniques are usually performed separately without harvesting the synergies between them. Here we describe the development of an integrated native MS and top-down proteomics method using Fourier transform ion cyclotron resonance (FTICR) to analyze macromolecular protein complexes in a single experiment. We address previous concerns of employing FTICR MS to measure large macromolecular complexes by demonstrating the detection of complexes up to 1.8 MDa, and we demonstrate the efficacy of this technique for direct acquirement of sequence to higher order structural information with several large complexes. We then summarize the unique functionalities of different activation/dissociation techniques. The platform expands the ability of MS to integrate proteomics and structural biology to provide insights into protein structure, function and regulation.
Graphical abstract
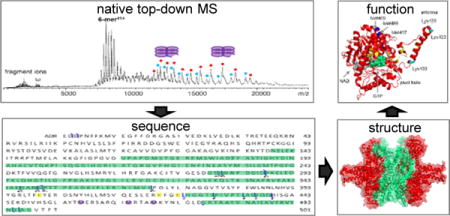
The vast majority of proteins are assembled into large noncovalent complexes to function. How proteins assemble into functional macromolecular complexes remains an intriguing process in molecular life sciences. Identifying and characterizing structure of the components involved in these machines and understanding how they accomplish their biological functions is crucial for understanding their biological processes at the molecular level. Obtaining high resolution three-dimensional structures of complexes by methods such as nuclear magnetic resonance spectroscopy, X-ray crystallography, or microscopy remains a major challenge in structural biology due to the sensitivity of the methods, problems associated with sample heterogeneity, the flexibility of protein structures, and the large size of the complexes1. As a complementary approach, mass spectrometry (MS) has emerged as an important technique to characterize protein complexes in a rapid, sensitive, and selective manner2–4. The MS study of a protein complex usually involves two sets of experiments, proteomics MS and native MS, which are often performed separately due to sample complexity and technical limitations. Proteomics MS, either at the peptide level (bottom-up) or at the intact protein level (top-down), allows for the identification of individual protein components in the protein complex; native MS studies the upper levels of protein organization, intact protein complexes, and provide structural information such as stoichiometry and spatial information of subunit arrangements which is complementary to information obtained from conventional biophysical techniques for structural biology5.
Although native MS can be considered a bridge between proteomics and structural biology, further developments are needed to fulfill its potential. Conventionally, native MS studies of protein complexes have been mostly performed using quadrupole time-of-flight (Q-TOF) MS instruments due to the high transmission efficiency of TOF for high m/z ions6, 7. With the recent development of Orbitrap MS with extended mass range (EMR), the analysis of macromolecular complexes has been extended significantly. The improved m/z range8, 9 and resolving power of Orbitrap EMR have allowed the measurement of the binding of ligands to macromolecular protein complexes10, 11 and the differentiation of proteoforms12, 13. However, both Q-TOF and Orbitrap EMR instruments currently have limited capacity for direct sequence determination, which limits their applications to known protein complex systems. In addition, although subunit connection information can be established, the direct binding or interfacial regions within complexes cannot be obtained easily, separate experiments such as crosslinking have to be performed for establishing the architecture of macromolecular assemblies14, 15. Most importantly, the spatial information of post-translational modifications (PTMs) is lost during most proteomics experiments through denaturation or digestion, while native MS yields no direct PTM information, thus challenging the integration of PTM information with the structural information from native MS. Experimental strategies that can directly target protein complexes to obtain structural information beyond a protein’s sequence would allow connections to be made between proteomics and structural biology and potentially yield insights into the dynamics and functional regulations of protein complexes on disease mechanisms16. Proteoforms and the linkages among different PTMs are often lost in bottom-up experiments, while top-down proteomics approaches allow for the interrogation of different proteoforms, providing detailed information connecting to disease mechanisms17. Therefore, MS instruments having the potential of transmitting high m/z ions for both native MS analysis and top-down sequencing could bridge the gap between proteomics and structural biology.
Due to its high resolving power, mass accuracy, and ability to be coupled with versatile fragmentation techniques, FTICR MS has long been recognized for its top-down capabilities for direct protein sequencing, characterization of PTMs, and probing protein-ligand interactions.18–21 Although the detection of molecules of high mass-to-charge (m/z up to 16,241) using FTICR was demonstrated three decades ago22, its capability to transmit and analyze macromolecular protein complexes has yet to be demonstrated. The previous FTICR record of identifying a 669 kDa thyroglobulin dimer was achieved by recording its fragment ions rather than its intact dimer or denatured form, leaving the transmission and detection ability of FTICR for large complexes still in question23. So far, the largest noncovalent complexes have been detected and characterized are all being smaller than 160 kDa24–28. Major concerns about the high mass performance of FTICR, especially for native MS, are that large ions of high m/z might not be efficiently transmitted to and/or trapped in the ICR cell, and the degradation effect of the low cyclotron frequency of high m/z ions on resolution. In addition, its potential for top-down sequencing of large macromolecular complexes in their native states is largely unexplored.
Here, we report an integrated native top-down MS approach that allows for the acquisition of sequence and higher order structural information of native protein complexes from a single experiment using FTICR. We demonstrate the detection of complexes up to 1.8 MDa. We then fully explored the top-down MS efficiency of different fragmentation techniques for native protein complexes. The versatile activation and dissociation methods available with FTICR MS include in-source dissociation (ISD) in the funnel skimmer region, collisionally activated dissociation (CAD) in the hexapole collision cell, and infrared multiphoton dissociation (IRMPD) or electron capture dissociation (ECD) in the ICR cell, along with the different activated ion (AI) ECD (e.g. ISD-ECD, CAD-ECD, IRMPD-ECD, etc). We first demonstrate the power of this native top-down MS approach with several large macromolecular protein complexes of 195 to 464 kDa. We then summarize the dissociation patterns of 11 protein complexes with different molecular weights and stoichiometry to outline the functionalities of different activation/dissociation techniques. Overall, 31%~87% sequence coverage was directly achieved at native protein-complex level, along with the detection of PTMs, site mutations, and proteoforms that have not been reported previously. More importantly, interfacial/surface residues can be revealed, which significantly improves the structural resolution of protein complexes. PTMs and mutation sites can then be directly mapped onto the structure of protein complexes to probe their biological significance. Altogether, we demonstrate the unique capability of FTICR MS for acquiring from primary to quaternary structural information in a single native top-down experiment.
RESULTS
Measuring macromolecular protein complexes using FTICR MS
FTICR MS analyses of noncovalent protein complexes of ~150 kDa have been previously demonstrated25–27, 41, here we tuned the instrument with CsI for larger complexes analyses (see Experimental Section for details). As shown in Fig. 1a, we observed a charge distribution of 43+ to 35+ at m/z 7,000~10,000 for the 334 kDa glutamate dehydrogenase hexamer. This is similar to the results reported using a Q-TOF MS instrument60, indicating that native-like structure is similarly retained during the FTICR MS measurement. Likewise, the spectrum of chaperone protein GroEL (802 kDa, 14-mer) observed is similar to the ones using Q-TOF or Orbitrap (Fig. 1b). GroEL has been frequently chosen to test the capability of a given type of MS instrument for analyzing macromolecular complexes in their native states8, 52, 61, 62. We further explored the ion transmission and ICR trapping limits of FTICR with an aggregated β-galactosidase (The spectrum of a fresh β-galactosidase sample was provided in Supplementary Fig. 1 for comparison). In addition to its tetrameric form (464 kDa), we observed higher-order oligomers up to 16-mers at m/z ~ 18,000, corresponding to a molecular weight of 1.86 MDa (Fig. 1c). Our results essentially resolved the concern that FTICR might not be suitable for macromolecular protein complex studies.
Figure 1.
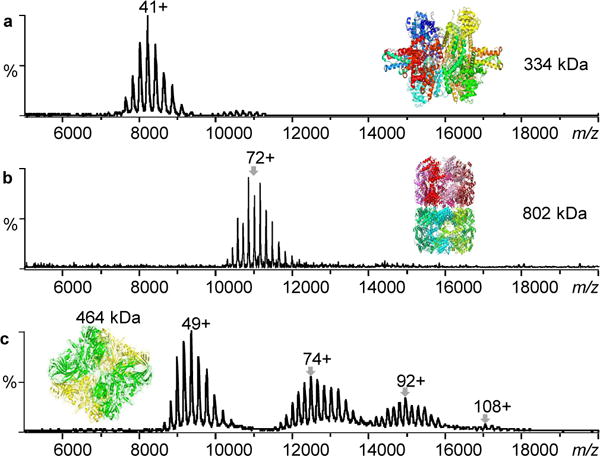
Macromolecular protein complex with molecular weights up to 1.86 MDa can be transmitted and detected by FTICR mass spectrometry under native ESI condition. a) Glutamate dehydrogenase, b) GroEL, and c) aggregated β-galactosidase.
Native top-down analysis of glycogen phosphorylase (GP)
GP is a homodimer that can be found in two different forms, GPa and GPb; both forms of GP can be found in the T (inactive) and R (active) states (Fig. 2a). The activity of the enzyme is regulated by the interconversion between alternative structural states, modulated by either reversible phosphorylation or allosteric interactions. The structure of R state GPa is nearly identical to the structure of sulphate-activated R state GPb, except where there is a covalently bound phosphate group at Ser14 in GPa and a noncovalently bound AMP at Tyr75 in GPb (Fig. 2b)29.
Figure 2.
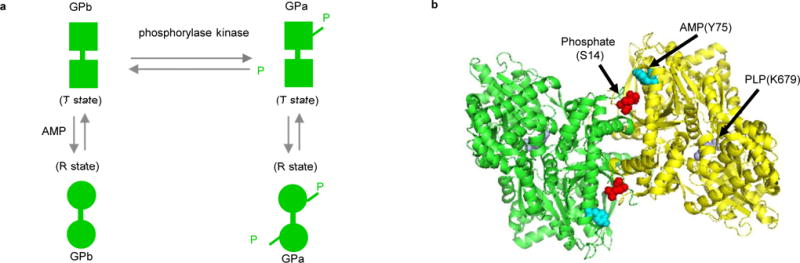
Structure of Glutamate dehydrogenase (GP). a) Structural states of GP controlled by phosphorylation and allosteric interactions. b) Biological assembly of GP structure (PDB 8GPB). The phosphorylation, AMP, and PLP sites are displayed to present their potential locations.
Here, we first performed ISD, CAD, ECD, and AI-ECD experiments. The dissociation of the GP dimer into its monomer components was observed through stepwise increases of energy for ISD experiments. In addition, AI-ECD of GP dimer yielded no fragments, and c/z•-fragment ions were not appeared until the dimer was dissociated into monomer by pre-activation using a combination of ISD and CAD (Fig. 3a). This AI-ECD mode of GP monomer allows the sequencing of the first 89 residues from the N-terminal side and 106 residues from the C-terminus (Fig. 3b). We observed neither phosphorylation at Ser14 nor AMP bound to Y75, suggesting the protein to be GPb in the T state (Figs. 2a). This result is also consistent with the notion that AMP interacts favorably with residues of the allosteric sites of the R state rather than the T state GPb29.
Figure 3.
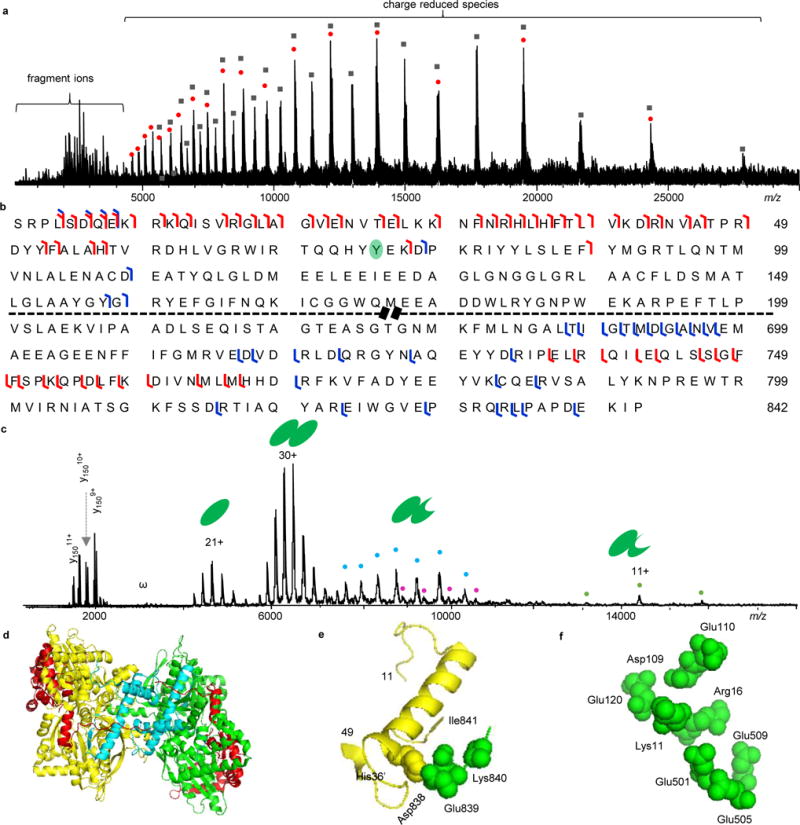
Native top-down MS analysis of GP. a) ISD-CAD-ECD spectrum of GP. The red dots represent charge-reduced monomers and the black-filled squared are charge-reduced dimers. b) Fragmentation map of GP with ISD-CAD-ECD fragmentation sites colored in red and IRMPD sites in blue. c) IRMPD spectrum of GP. “ω” represents harmonic peaks and the cyan, purple, and orange dots represent noncovalently bound fragment peaks. d) GP structure with ISD-CAD-ECD fragmentation sites in red and IRMPD sites in cyan. e) The C-terminal residues 838–841 form salt bridges with His36′ from the other subunit. f) The N-terminal basic residues (K9RKQISVR16) interact intra-molecularly with a pocket of acidic residues Asp109, Glu110, Glu120, Glu501, Glu505, and Glu509. The first 10 amino acid residues were not presented in the PDB (8GPB) structure.
To gain more information on GP, we performed IRMPD experiments (Fig. 3c). Apart from the dissociation of the GP dimer into monomers, we found that backbone cleavages were the major dissociation pathway. IRMPD backbone fragmentation yielded both fragment ions below m/z 3,000 and their complementary pairs (noncovalently-bound complex fragments) above m/z 7,000 (Fig. 3c). A total sequence coverage of 31% was achieved with the assignment of low m/z fragments (Fig. 3b and Supplementary Tab. 1). To compare how well the structural information gained from the native top-down approaches matches the crystal structure of GPb, we overlaid the experimental results onto the biological assembly structure of GPb (PDB 8gpb) with the Al-ECD fragmentation region highlighted in cyan and the IRMPD fragmentation region in red (Fig. 3d). ECD fragment ions were only observed from the GPb monomer rather than the dimer, indicating that the N- and C-termini of GPb are likely involved in noncovalent interactions. Similar results have been previously observed for monomeric proteins, where the separation of c/z• fragments was not achieved until vibrational activation was used to break intramolecular noncovalent interactions30, 31. This result is consistent with the structural features of GPb as the N-terminal residues 10 to 49, 112–118, and the C-terminal residues 838–841 are involved in the subunit interaction interface of the GPb dimer (Fig. 3e)29. In addition, the N-terminus contains a higher percentage of basic residues (K9RKQISVR16) that interact through intramolecular salt bridges with a cluster of acidic residues (Asp109, Glu110, Glu120, Glu501, Glu505, and Glu509) in GPb (Fig. 3f)32. For the IRMPD results, fragments are largely from the C-terminal side and are displayed at the outer region of the GPb structure, whereas the interior regions remain intact (Fig. 3d and Supplementary Tab. 1). This coincides with the fact that IRMPD preferentially cleaves at Asp, Glu, and Pro residues33, 34, and these residues have a much higher tendency to be at the surface-exposed regions of proteins35, while the N-terminal domain of GPb is stabilized by the inter- and intra-subunit interactions (Figs. 3d–3f).
Native top-down analysis of glutamate dehydrogenase (GDH)
GDH is a homohexameric enzyme that reversibly catalyzes the oxidative deamination using either NADP(H) or NAD(H) to convert L-glutamate into β-ketoglutarate. GDH is composed of a stacked dimer of trimers (Fig. 4a); each subunit has 501 amino acids with a molecular weight of 56 kDa. We performed ISD, CAD, and AI-ECD experiments. However, neither ISD nor CAD was sufficient to decompose the hexameric GDH. ISD-ECD yielded no c/z•-product ions while IR-ECD generated only b/y ions rather than c/z•-product ions. The results suggest the strong involvement of N- and/or C-termini in inter- and/or intra-molecular noncovalent interactions. This hypothesis is supported by the structural features of GDH as shown in Fig. 4b; Asp6 and Asn8 form direct contacts with Lys329 through H-bonds and salt bridges, which therefore prevents the separation of fragments from the N-terminal region36. In addition, limited digestion experiments by Banerjee et al. showed that the C-terminus is located at the interior of the structure and is shielded from digestion37.
Figure 4.
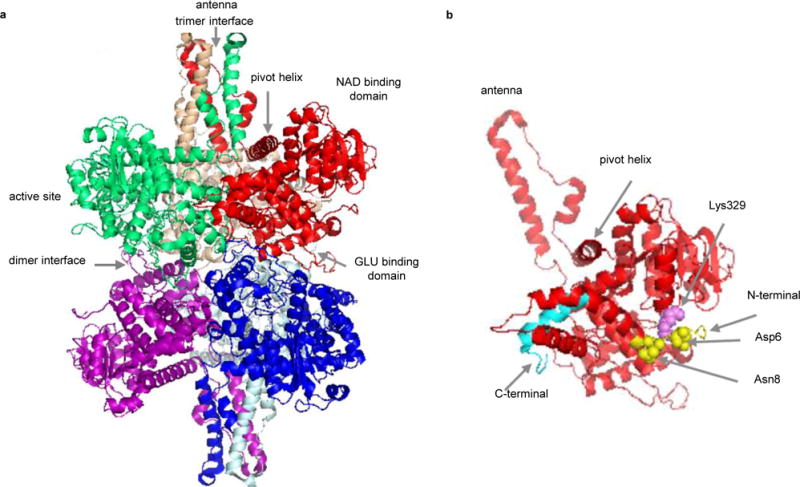
Structure of bovine glutamate dehydrogenase (GDH) (PDB 1HWZ). a) Hexameric GDH with six subunits displayed in different colors. b) Cartoon structure of one GDH subunit. The N-terminus is shown in yellow and the C-terminus in cyan. Asp6 and Asn8 (yellow spheres) form direct contacts with Lys329 (violet sphere).
In energy-resolved IRMPD experiments, low energy activation resulted in the removal of salt and ligand adducts. Further increases in the IR laser power caused backbone cleavages to yield b/y-product ions at the lower m/z region (Fig. 5a & 5b) (A detailed fragmentation map and peak-list table are provided in Supplementary Fig. 2 and Supplementary Tab. 2) and large noncovalently bound complex fragment ions at the higher m/z region (Fig. 5a). We detected two PTMs, methylation and oxidation, which have not been previously reported in bovine GDH. Based on the assignments of internal fragments from double backbone cleavages, the methylation site was narrowed down to residues 369 to 423 (possibly at Arg396, Arg403, Arg419, Lys400, Lys420, or Lys423) and the oxidation site to residues 447 to 476 (potentially at Met457, Met465, or Met469). As shown in Fig. 5c, these two PTMs yield two GDH proteoforms, a methylated form, and a methylated and oxidized form.
Figure 5.
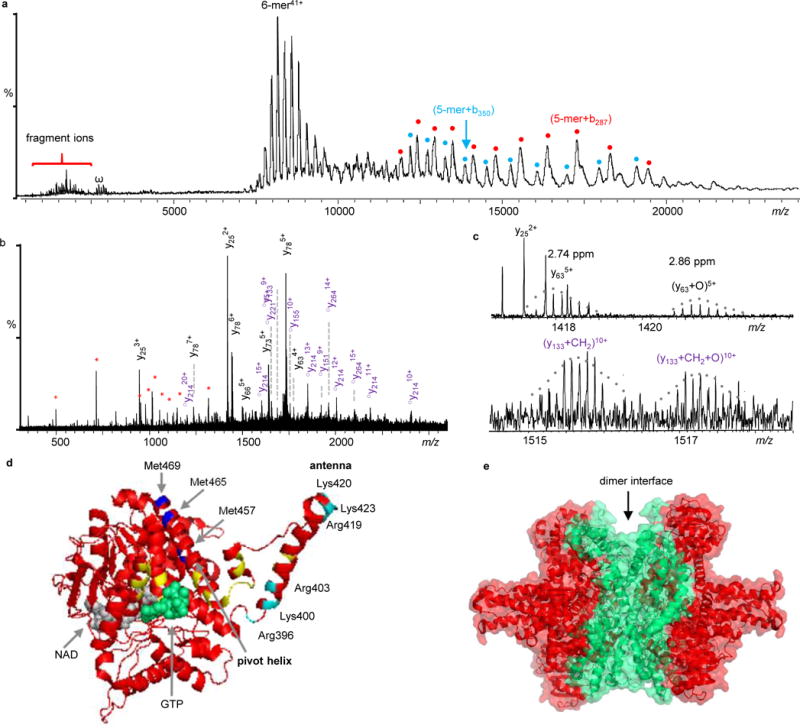
Native top-down MS analysis of GDH. a) IRMPD mass spectrum of GDH. Fragment ions labeled in cyan and red dots are 5-mer+b287 and 5-mer+b350, respectively. They are complementary ions of y214 and y151, respectively. b) Expanded spectrum of the low-m/z region of Fig. 5a. The y-ions in black are unmodified, ○ym represent y-ions with methylation (○ym= ym+CH2), and internal fragments are in red *. Detailed peak lists are available in Supplementary Table 3. c) Expanded spectra to show GDH proteoforms with different PTMs. d) Locations of observed PTMs. The potential oxidation sites (Met457, Met465, or Met469) are colored in blue and methylation site (Lys400, Lys420, Lys423, Arg396, Arg403, or Arg419) are in cyan. GTP is in green and NAD is in grey. Mutation sites reported in human GDH are labeled in yellow. e) Structure of GDH with the sequence coverage regions by IRMPD colored in red.
GDH is regulated by nucleotides and plays a crucial role in metabolic pathways; a few GDH mutations that abrogate GTP inhibition have been reported in hyperinsulinemia/hyperammonemia syndrome patients38. To explore how the PTMs we detected might affect the function of GDH, we aligned the sequences of human and bovine GDH (human GDH shares 97.5% sequence identity with bovine GDH) and labeled the previously reported human mutations onto the bovine GDH structure. These mutations (in yellow) are mostly adjacent to the GTP binding site and in the antenna region (Fig. 5d). It has been previously postulated that the first group of mutations affect GTP inhibition through their impacts on GTP binding, and the second group of mutations that lie in the antenna may affect the open and close of the active site38. In addition, it has been confirmed that the antenna is required for allosteric regulation and the removal of the antenna from human GDH causes the protein to lose its regulatory function38. Interestingly, the methylation site (in cyan) and the oxidation site (in blue) that we detected fall into the same regions (Fig. 5d).
The noncovalently bound complex fragment ions detected at high m/z ((5-mer + b350), red dots) and ((5-mer + b287), cyan dots) (Fig. 5a) from IRMPD are complementary to y151 and y214, respectively. From the position of the IRMPD fragments on the GDH structure, it can be observed that the IRMPD fragments originate largely from the outer regions of the structure, with the dimer interface remaining intact (Fig. 5e). Similar IRMPD fragmentation patterns were also observed in GPb, indicating that IRMPD could be a novel way to disassemble protein complexes. Overall, we achieved up to 87% of sequence coverage (Supplementary Fig. 2 and Tab. 2) for GDH along with all of the above structural information in a single experiment.
Native top-down analysis of β-galactosidase (β-GTD)
The enzyme β-GTD from E. coli is a 464 kDa homotetramer with each monomer constituting 1023 amino acids. Each monomer folds into five sequential domains plus ~50 residues at the N-terminus that are relatively extended. We observed the intact tetramer at m/z 8,500 ~10,500 and found it is sensitive to dissociation upon ISD or CAD activation. Both dissociation approaches yielded a similar fragmentation pattern. Apart from subunit ejection upon activation, bn-ions derived from backbone cleavages assigned to the first 125 amino acid residues of the N-terminus were observed (Fig. 6a and Supplementary Fig. 3 and Table 3). However, all ions had a mass shift of +18 Da (H2O, see Supplementary Fig. 4 for details). Furthermore, we observed multiple proteoforms with different PTMs or site mutations in the residue 1~54 region, including +15.98930 Da (oxidation), +30.98345 Da (a combination of oxidation and N to E mutation), and +43.99052 Da (a combination of oxidation and formylation, or A to D mutation, or carboxylation) (Fig. 6b). From the peak intensities of the b54, b95, b102, and b125 ions, the relative ratio of the four β-GTD proteoforms were estimated to be 1 : 0.13 : 0.10 : 0.19 (Fig. 6c). As shown in Fig. 6d, the constituent monomers of β-GTD form two different monomer-monomer contacts that are referred to as the “activating” interface and the “long” interface. Residues 1–50 from each subunit contribute to the “activating” interface (Fig. 6e), and the deletion of residues 23–31 or 11–41 has been found to result in inactive dimers39, 40. The PTMs and mutations that we observed fall directly in the residue 1–54 region, which may affect the stability, activity, or even function of β-GTD. Additionally, we found that ECD yielded no fragments, which is consistent with the fact that both the N- and C-termini are involved in the interaction interfaces (Figs. 6e and 6f)39, 40. With IRMPD, we were able to sequence 420 amino acid residues from the C-terminus without observing any PTMs (Supplementary Table 3) and the fragments are consistently found to be from the outer region of the structure rather than from random regions (Fig. 6d). A sequence coverage of 49% was achieved for the 464 kDa noncovalently-bound protein complex.
Figure 6.
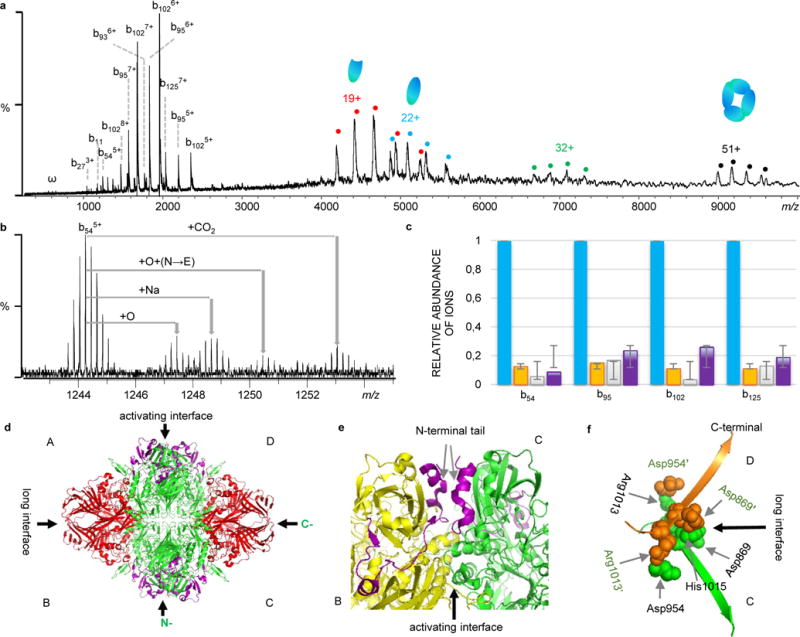
Native top-down MS analysis of β-galactosidase (β-GTD). a) CAD mass spectrum of β-GTD. b) Observed proteoforms with different PTMs and site mutations. c) Relative ratio of proteoforms based on the intensity of fragment ions (Blue: unmodified; Orange: oxidized (+15.98930); Green: +30.98345; and Purple: +43.9905. d) Structure of β-GTD (PDB 1F4A) with the regions sequenced by IRMPD in red, and CAD in purple. e) The activating interface between chain B (yellow) and chain C (green) with the N-terminal tail highlighted in purple (AA 1-50). f) The long interface between chain C (green) and chain D (orange). The C-terminal residues Asp869, Asp954, Arg1013, and His1015 from C chain interact with residues Asp869′, Asp954′, Arg1013′, and His1015′ from D chain through salt bridges.
ECD for obtaining quaternary structural information of protein complexes
Although several groups including ours have demonstrated that direct sequence information can be obtained from protein complexes in their native states using either ECD25–27, 41 or the similar technique, electron transfer dissociation (ETD)42, ECD of protein complexes gave no fragment ions has also been previously reported28 and observed here. To better explore the factors that govern ECD dissociations of native protein complexes, we extended the research to 11 protein complexes of different molecular weights and stoichiometry (Supplementary Table 4). Here, we listed the structures of all 11 protein complexes in Fig. 7, and ECD fragmentation regions are colored in red on their structures whenever fragments are available. In addition, we displayed the corresponding monomeric structure of each complex with the N- and C-termini, the subunit interaction interfaces, and the interfacial residues indicated. Based on their dissociation patterns, we categorized them into two groups, group I which yielded direct sequence information (Fig. 7a and Supplementary Fig. 5) and group II which produced no fragments even though ECD reaction happened as indicated by the charge reduced species (Fig. 7b and Supplementary Fig. 5). After carefully examining the structure of each complex, we found that the results are in line with the unique feature of ECD technique, namely breaking protein backbone bonds but preserving noncovalent interactions. Most importantly, ECD results reflect the structural properties of these complexes. More specifically, for group I (Fig. 7a), ECD fragments were observed from the N-terminal of horse liver alcohol dehydrogenase (hADH, 2-mer, 80 kDa), yeast alcohol dehydrogenase (hADH, 4-mer, 147 kDa)26, and human transferrin receptor (hTfR, 2-mer, 196 kDa), and the C-terminal of rabbit aldolase (ALD, 4-mer, 158 kDa)25, and all these are all from the terminal residues that are solvent-exposed. In contrast, the C-terminal residues of hADH, yADH, hTfR, and the N-terminal residues of ALD are constrained by noncovalent interactions, with the C-terminal residues of hADH involved in ligand (NAD) binding, and the rest involved in subunit interaction interfaces. For group II (Fig. 7b), inter- and intra-subunit interactions are the contributing factors limiting the separation of fragment ions. Both of the N- and C-termini of superoxide dismutase (SOD1, 2-mer, 32 kDa)43, GroES (7-mer, 84 kDa), pyruvate kinase (PK, 4-mer, 232 kDa), GPb, β-GTD, and GroEL are engaged in subunit interaction interfaces, and the N- and C-termini of GDH and the N-terminal of GPb are involved in intra-subunit interactions. Previously, the location of the termini has been examined for 425 monomeric proteins and it was found that the average solvent accessibility of terminal residues is 87%; 80% of the N-terminal residues and 86% of the C-terminal residues are located on the surface and exposed44. Evolution, thermodynamic, and kinetic selections are major contributors for this phenomenon. The fact that terminal residues are predominantly located on the surfaces of monomeric proteins is well reflected in our ECD results. Therefore ECD result can be a unique indicator to infer whether terminal residues are at surfaces or interfaces of protein complexes.
Figure 7.

Native top-down ECD reveals the structural features of protein complexes. a) Group I: ECD yielded direct sequence information for Horse liver alcohol dehydrogenase (hADH, PDB 2OHX), yeast alcohol dehydrogenase (yADH, PDB 4W6Z), aldolase (PDB 1ZAH), and human transferrin receptor (hTfR, PDB 1SUV). b) Group II: No c/z• fragments were observed for GPb (PDB 8GPB), pyruvate kinase (PK, PDB 1F3W), GDH (PDB 1HWZ), β-GTD (PDB 1F4A), SOD1 (PDB 2C9V), GroES (PDB 1PCQ), and GroEL (PDB 4HEL). The ECD fragmented residues are displayed in red on the structures; interfacial residues were calculated using PISA (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) and colored in salmon. The corresponding subunit structure is shown at the right-hand of the each complex structure to demonstrate the relative positions of N- and C-termini to the subunit interaction interfaces (yellow dash-lines).
Although the structural information we obtained is unique, information about the number, types, or strengths of inter- or intra-molecular interactions that prevent fragment ion separation cannot be directly obtained from a single ECD experiment31. AI-ECD is therefore a great complementary tool to provide more extensive structural information. We performed pre-activation ECD using ISD, CAD, or IRMPD to gradually remove noncovalent interactions and concomitant- or post-ECD activation using IRMPD to separate c/z• fragment ions, respectively. Neither concomitant- or post-ECD activation appeared to be as efficient as for monomic proteins31, especially for complexes in group II, which can be rationalized by the extra constraints of inter-subunit interactions. Here we focused our discussion on pre-activation ECD, and we found that the structural information obtained appears to be more versatile and highly structural dependent. Based on how protein complex ions respond to pre-activation, we categorized them into three types. (1) Improvement of sequencing prior to subunit ejection or backbone cleavage. This is observed for the solvent-exposed terminal residues of all four complexes in group I (Fig. 7a), including hADH, yADH, ALD, and hTfR. This result indicates that inter-subunit interactions of each complex in group I are held tighter than the intra-subunit interactions between the exposed terminal residues and the rest of chains. (2) Direct subunit ejection prior to yielding c/z• fragments. The ejection of a subunit was found for SOD1, GroES, and GroEL at different energy levels, respectively, with SOD1 requiring the least activation (ISD 10V). In contrast, GPb required much higher level of activation (ISD 120V or a combination of ISD and CAD) to separate the dimer into monomers. These results reflect the relative strength of inter-subunit interactions among different protein complexes. As noticed from the structures of SOD1 and GPb (Fig. 7b), in which the two subunits of GPb wrap arms around one another, while the two protein chains of SOD1 only form weak contacts. Consecutive ECD can then yield sequence information as demonstrated in the GPb experiment (Fig. 3a). (3) Direct backbone cleavage but preserving noncovalent interactions. Interestingly, rather than breaking noncovalent bonds, we observed that IRMPD leads to direct backbone cleavages and yields backbone fragments and their complementary ion pairs, the noncovalently bound complex fragment ions shown at higher m/z region (Fig. 3c and Fig. 5a). This phenomenon can be attributed to the following reasons. First, evidence from ion mobility spectrometry (IMS) MS and molecular dynamics simulations suggests that proteins or protein complexes experience structural contraction when transferred from solution to the gas-phase during electrospray ionization process45, 46. Hydrogen bonds and salt bridges in the gas phase are therefore enhanced and hold protein complexes much tighter by forming large noncovalent bond networks. Second, such backbone cleavage sites are mostly found at the N-terminal of Pro and the C-terminal of Asp and Glu residues, namely the preferential cleavage sites of IRMPD and CAD33, 34, 47, and these residues have a much higher tendency to be at the surface-exposed regions of protein complexes. Third, since IRMPD is not a collision-based activation process, the separation of the interior of large noncovalent bond network can consequently become difficult, and the preferential cleavage sites on the surface become more facile. Subunit ejection and backbone cleavage were sometimes observed simultaneously, but nevertheless, these two types of patterns were only found for complexes in group II (Fig. 7b)
ISD, CAD, and IRMPD for sequencing and PTMs
Apart from ECD, slow heating techniques such as ISD, CAD, and IRMPD are also valuable complementary approaches for direct sequencing and PTMs identification. In our FTICR MS instrument, IRMPD was found most effective for native top-down sequencing and delivered sequence information in all complexes; while ISD and CAD appeared to offer less sufficient energy and were not as efficient. The main advantage of IRMPD over ISD and CAD is the inherent non-resonant nature of the photoactivation process. All ions that reside on the optical path of the laser are excited and absorb energy continuously, IRMPD thus promotes more extensive dissociation and leads to a richer array of secondary fragment ions (internal fragments)48. It therefore helped us with improved sequencing and PTMs characterization. Additionally, in contrast to CAD or IRMPD of tryptic peptides in bottom-up experiments that often ejects PTMs, labile PTMs are retained in top-down MS experiments as demonstrated in β-GTD, GDH, and other examples23, 26, 49–51. This is likely due to the higher-order structure of gaseous complex ions preferentially driving fragmentation at amide backbone sites over PTM ejections19.
DISCUSSION
Our experimental platform and strategy addressed major challenges in bridging the gap between the proteomics and structural biology for protein complexes. With a single native top-down MS experiment, we were able to directly achieve sequence coverage of 31% – 87% at the protein-complex level, along with the identification of PTMs, site mutations, and proteoforms. Additionally, ECD allows for the probing of the N- and C-terminal positions of subunits in relation to the complex structure (interface vs surface), which could be used as spatial restraints to improve the speed and accuracy of integrated structural modeling in the near future. Our native top-down MS strategy allowed us to directly link fragmentation mass spectra to the higher order structure of the protein complexes25–27, 41. This is different from the existing method using a modified Orbitrap instrument for native top-down MS52, in which MS2 via CAD is used to eject an intact subunit from a complex, and an additional stage of MS (i.e., MS3) of the intact subunit further generates sequence information. In this case, the subunit generated through collision has lost its native structure, therefore the sequence information obtained in MS3 therefore cannot be linked to the complex’s quaternary structure.
There are several important aspects to our approach. The capability to obtain sequence information at the protein-complex level opens up new possibilities not only to directly link PTMs to their specific functions but also to directly study endogenous protein assemblies. This approach yields information at the residue level for interfacial and surface regions of protein complexes. Interactions of proteins are at the heart of almost every biological process; the delivery of residues that accounts for the binding of proteins and stabilizing the complexes can greatly improve our understanding of how protein complexes assemble and function. With continuing efforts to develop additional activation/dissociation techniques43, 53, and software for the assignment of internal fragments54, higher-resolution structural information along with improved sequence coverage and the precision of PTM/mutation locations should result in the future. With the simultaneous delivery of PTMs and higher-order structural information, the spatial location information of PTMs can be retained and directly mapped onto the structures of protein complexes to provide insights into the dynamics and functional regulations of PTMs.
The performance of our method is highly sensitive to the structural features of protein complexes. This, by itself, is a unique feature of native top-down MS. Gas phase complexes stabilized by hydrogen bonds and salt bridges require more energetic activation methods to disrupt these forces to yield sequence-informative fragment ions55. Fortunately, there are many types of fragmentation techniques (e.g., collisional-based, electron-based, and photon-based) available with FTICR MS. Each provides unique structural information that is complementary to the others and extends the potential structural information content compared to more conventional native MS experiments using only collision-based dissociation techniques. Additionally, the current limit on quadrupole isolation (m/z 6000) can be easily ratified using well-documented high m/z quadrupole technology, which will then allow us to directly pinpoint how PTMs or sequence mutations affect quaternary structures of protein complexes as demonstrated previously for smaller proteins56.
The task of assembling and characterizing macromolecular protein complexes is becoming more and more dependent on integrated techniques. Our method is broadly applicable to be integrated with different types of biophysical techniques, such as cryo-electron microscopy, modeling, etc. Such integration will allow the study of large and complex molecular machines in greater detail, providing insight into the functional dynamics of the system. Native top-down MS will be a promising approach to advance structural biology and hasten drug discovery and development.
METHODS
Materials
Protein complex samples, including glycogen phosphorylase b from rabbit muscle (195 kDa), glutamate dehydrogenase from bovine liver (334 kDa), beta-galactosidase from Escherichia coli (464 kDa), horse liver alcohol dehydrogenase (80 kDa), yeast alcohol dehydrogenase (147 kDa), rabbit aldolase (158 kDa), GroES (84 kDa), and pyruvate kinase (232 kDa) were purchased from Sigma-Aldrich (St. Louis, MO). GroEL from Escherichia coli (802 kDa) was prepared57 and provided by Dr. I.D.G.C.; human superoxide dismutase (32 kDa) was provided by Dr. Y. Sheng and human transferrin receptor (157 kDa) was provided by Dr. M. Penichet.
Sample Preparation
Protein complexes were dissolved in MilliQ water to a concentration of 100 μM and then buffer exchanged three times with 200 mM ammonium acetate solution (300 μL each time) using Amicon centrifugal filters (Millipore Inc., Billerica, MA) with a molecular weight cut-off (MWCO) of 50~100 K. The buffer exchanged protein samples were diluted with 200 mM ammonium acetate solution to a concentration of 3~20 μM for native nano-ESI MS analysis.
FT-ICR MS Analysis of Native Protein Complexes
Protein solutions were loaded into metal-coated borosilicate capillaries (Au/Pd-coated, 1 μm I.D.; Thermo Fisher Scientific, West Palm Beach, FL) and sprayed at a flow rate of 10 – 40 nL/min through a nanospray ion source. The experiments were performed using a 15 Tesla Bruker SolariX FTICR MS with an infinity cell. The ESI capillary voltage was set to 0.9~1.2 kV. The temperature of dry gas was 80 °C and the flow rate was 2.5 L/min. The RF amplitude of the ion-funnels was 300 Vpp, and the applied voltages were 210 V and 6 V for funnels 1 and 2, respectively. The voltage of skimmer 1 was 30 V and the voltage of skimmer 2 was kept at 20 V. The lowest values of RF frequencies were used in all ion-transmission regions: multipole 1 (2 MHz), quadrupole (1.4 MHz), and transfer hexpole (1MHz). Ions were accumulated for 500 ms in the hexapole collision cell before being transmitted to the infinity ICR cell. The time-of-flight was varied to allow the best transmission of complex ions appeared at different m/z ranges, with 1.5 ms used for the smallest complex SOD1 and 3.5 ms used for the largest complex beta-galactosidase aggregates. The vacuum pressures for different regions were ~2 mbar for the source region, ~2×10−6 mbar for the quadrupole region, and ~2×10−9 mbar for the UHV-chamber. In-source-dissociation (ISD) was performed by varying the voltage of skimmer 1 up to 200 V and the skimmer 2 voltage was kept at 20 V. IRMPD was performed with a Synrad 30-W CO2 laser (Mukilteo, WA) that was interfaced to the back of the mass spectrometer. The laser power was varied up to 95% (30 W) for all IRMPD experiments, and the irradiation time was kept at 0.5 s. ECD experiments were performed with an ECD pulse length of 0.02 s, ECD bias of 1.5 V, ECD lens of 15 V, and hollow-cathode current of 1.6 A. No charge state isolation was performed as most of the complex ions appear at m/z above the quadrupole isolation limit of 6,000. Data were collected with 512k data points for full spectra and 4M data points for tandem spectra. Spectrum at each experimental condition was recorded with 300-scan averaged. All spectra were externally calibrated with cesium iodide.
Transmission and trapping limits of FTICR MS
In order for macromolecular complex ions to be detected, they must be efficiently transmitted and trapped in the ICR cell. To address these concerns, we first calculated the trapping limit of our Bruker solariX 15-Tesla FTICR MS instrument and then experimentally tested its transmission and trapping limit. The calculation of the trapping limit of our system with an infinity cell was based on the equation: (a in m, refers to the length of the infinity cell; B in T; m in u; Vtrap in volts; z in multiples of elementary charge)58. Given that B=15 T, a=0.06, and α=2.8404, the trapping limit of our FTICR instrument should be approximately 5.73 MDa for a singly charged ion using a trapping potential Vtrap of 0.6 V, and ~3.43 MDa with a Vtrap of 1 V. Thus, the calculation indicates that the 15 T FTICR MS instrument with an infinity cell should be able to trap large molecules over one MDa.
Based on the Rayleigh equation ,59 the maximum number of charge (zR) a 3.43 MDa complex can carry is 144; the complex ions should therefore be detected at roughly m/z 24,000. To address the issue whether such large m/z macromolecular ions can be efficiently transmitted into our ICR cell, we first experimentally tested the transmission limit using cesium iodide (CsI). High molecular weight CsI cluster ions up to m/z 17,000 were detected (Supplementary Fig. 6). Detection was achieved by adjusting the excitation power, time-of-flight, RF frequencies and RF amplitudes for all ion-transmission regions (Supplementary Fig. 7).
FTICR MS workflow for native top-down analysis of protein complexes
After recording the full mass spectra of each protein complex (MS1), we then perform native top-down analysis (MS2) using various fragmentation techniques such as ISD, CAD, IRMPD, ECD, and their combinations to probe protein structure. A distinct feature of ECD over ISD, CAD, or IRMPD is that the cleavage of backbones happens prior to the vibrational energy redistribution, ECD therefore retains the integrity of noncovalent interactions and labile modifications such as PTMs63, which therefore makes it a unique approach for obtaining structural information of native proteins and protein complexes21, 25–27, 31, 41. Moreover, more extensive and complementary structural information can be obtained using activated ion (AI) ECD30. Pre-activation is carried out using either ISD, CAD, or IRMPD to unfold protein complexes prior to ECD, while concomitant- or post-activation can only be performed using IRMPD, and this allows for the separation of ECD fragment ions held by noncovalent interactions. Unless otherwise stated, we refer to all proteins in their functional oligomeric states. Individual charge states are not isolated for tandem MS experiments because most of the complexes appear at m/z close to or above the isolation limit (m/z 6000) of the quadrupole.
Data Analysis
Data were processed in DataAnalysis (Bruker Daltonics Inc.). Peaks were detected using the SNAP centroid peak detection algorithm (version 2.0) with quality factor threshold of 0.5, signal-to-noise threshold of 3, and maximum charge state of 20. Spectra were only smoothed using Savitzky Golay algorithm in DataAnalysis for better display of large complex peaks; high resolution spectra were used for top-down data analysis. The top-down data were interpreted manually with the help of a home-build program to facilitate the assignments of internal fragments. Manual isotopic peak determination was performed for verification; the assignments of fragment ions were accepted with a mass accuracy cut-off of ± 10 ppm. The “sequence coverage” describes the percentage of the protein’s sequence represented by the residues identified in the MS/MS. The calculation of backbone cleavages were based on the total number of observed non-redundant backbone cleavages in the protein.
Data availability
Data supporting the findings of this study are available within the Article and its Supplementary Information and from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
The authors thank Dr. C. Wan, and Dr. R. Malmirchegini for helpful discussions, Dr. M. Penichet for the hTfR sample. Support from the US National Institutes of Health (R01 GM103479 and S10 RR028893 to JAL), the US Department of Energy (UCLA/DOE Institute for Genomics and Proteomics; DE-FC03-02ER63421), and the American Society for Mass Spectrometry (ASMS) Postdoctoral Research Award (to HL) are acknowledged.
Footnotes
AUTHOR CONTRIBUTIONS
H.L. and J.A.L. conceived and designed the experiments; H.L. performed experiments and analyzed data; H.H.N. contributed to analysis tools; I.D.G.C. prepared and contributed GroEL sample; H.L., R.R.O.L, and J.A.L co-wrote the paper. All authors discussed the results and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Supplementary information is available in the online version of the paper. Reprints and permissions information is available online at www.nature.com/reprints. Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sharon M. How Far Can We Go with Structural Mass Spectrometry of Protein Complexes? J Am Soc Mass Spectrom. 2010;21:487–500. doi: 10.1016/j.jasms.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 2.Heck AJR. Native mass spectrometry: a bridge between interactomics and structural biology. Nat Meth. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 3.Lorenzen K, Duijn EV. Current Protocols in Protein Science. John Wiley & Sons, Inc; 2001. [Google Scholar]
- 4.van Duijn E. Current Limitations in Native Mass Spectrometry Based Structural Biology. J Am Soc Mass Spectrom. 2010;21:971–978. doi: 10.1016/j.jasms.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 5.Benesch JLP, Ruotolo BT, Simmons DA, Robinson CV. Protein Complexes in the Gas Phase: Technology for Structural Genomics and Proteomics. Chem Rev. 2007;107:3544–3567. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- 6.Snijder J, Rose RJ, Veesler D, Johnson JE, Heck AJR. Studying 18 MDa Virus Assemblies with Native Mass Spectrometry. Angew Chem Int Ed. 2013;52:4020–4023. doi: 10.1002/anie.201210197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Berkel WJH, Van Den Heuvel RHH, Versluis C, Heck AJR. Detection of intact megaDalton protein assemblies of vanillyl-alcohol oxidase by mass spectrometry. Protein Sci. 2000;9:435–439. doi: 10.1110/ps.9.3.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rose RJ, Damoc E, Denisov E, Makarov A, Heck AJR. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat Meth. 2012;9:1084–1086. doi: 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- 9.van de Waterbeemd M, et al. High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat Meth. 2017;14:283–286. doi: 10.1038/nmeth.4147. [DOI] [PubMed] [Google Scholar]
- 10.Snijder J, et al. Defining the Stoichiometry and Cargo Load of Viral and Bacterial Nanoparticles by Orbitrap Mass Spectrometry. J Am Chem Soc. 2014;136:7295–7299. doi: 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gault J, et al. High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat Meth. 2016;13:333–336. doi: 10.1038/nmeth.3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dyachenko A, et al. Tandem Native Mass-Spectrometry on Antibody–Drug Conjugates and Submillion Da Antibody–Antigen Protein Assemblies on an Orbitrap EMR Equipped with a High-Mass Quadrupole Mass Selector. Anal Chem. 2015;87:6095–6102. doi: 10.1021/acs.analchem.5b00788. [DOI] [PubMed] [Google Scholar]
- 13.Rosati S, Yang Y, Barendregt A, Heck AJR. Detailed mass analysis of structural heterogeneity in monoclonal antibodies using native mass spectrometry. Nat Protocols. 2014;9:967–976. doi: 10.1038/nprot.2014.057. [DOI] [PubMed] [Google Scholar]
- 14.Walzthoeni T, Leitner A, Stengel F, Aebersold R. Mass spectrometry supported determination of protein complex structure. Curr Opin Struct Biol. 2013;23:252–260. doi: 10.1016/j.sbi.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Shi Y, et al. Structural Characterization by Cross-linking Reveals the Detailed Architecture of a Coatomer-related Heptameric Module from the Nuclear Pore Complex. Molecular & Cellular Proteomics: MCP. 2014;13:2927–2943. doi: 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savaryn J, Catherman A, Thomas P, Abecassis M, Kelleher N. The emergence of top-down proteomics in clinical research. Genome Med. 2013;5:53. doi: 10.1186/gm457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith LM, Kelleher NL. Proteoform: a single term describing protein complexity. Nat Meth. 2013;10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li H, et al. Use of Top-Down and Bottom-Up Fourier Transform Ion Cyclotron Resonance Mass Spectrometry for Mapping Calmodulin Sites Modified by Platinum Anticancer Drugs. Anal Chem. 2011;83:9507–9515. doi: 10.1021/ac202267g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat Meth. 2007;4:817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian Z, et al. Enhanced top-down characterization of histone post-translational modifications. Genome Biol. 2012;13:R86. doi: 10.1186/gb-2012-13-10-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie Y, Zhang J, Yin S, Loo JA. Top-Down ESI-ECD-FT-ICR Mass Spectrometry Localizes Noncovalent Protein-Ligand Binding Sites. J Am Chem Soc. 2006;128:14432–14433. doi: 10.1021/ja063197p. [DOI] [PubMed] [Google Scholar]
- 22.Castro ME, Russell DH, Amster IJ, McLafferty FW. Detection of mass 16241 ions by Fourier-transform mass spectrometry. Anal Chem. 1986;58:483–485. doi: 10.1021/ac00293a049. [DOI] [PubMed] [Google Scholar]
- 23.Karabacak NM, et al. Sensitive and Specific Identification of Wild Type and Variant Proteins from 8 to 669 kDa Using Top-down Mass Spectrometry. Molecular & Cellular Proteomics: MCP. 2009;8:846–856. doi: 10.1074/mcp.M800099-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Cui W, Gross ML, Blankenship RE. Native mass spectrometry of photosynthetic pigment–protein complexes. FEBS Lett. 2013;587:1012–1020. doi: 10.1016/j.febslet.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H, Wolff JJ, Van Orden SL, Loo JA. Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 kDa Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal Chem. 2014;86:317–320. doi: 10.1021/ac4033214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Wongkongkathep P, Van Orden S, Ogorzalek Loo R, Loo J. Revealing Ligand Binding Sites and Quantifying Subunit Variants of Noncovalent Protein Complexes in a Single Native Top-Down FTICR MS Experiment. J Am Soc Mass Spectrom. 2014;25:2060–2068. doi: 10.1007/s13361-014-0928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Native Electrospray and Electron-Capture Dissociation FTICR Mass Spectrometry for Top-down Studies of Protein Assemblies. Anal Chem. 2011;83:5598–5606. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geels RBJ, van der Vies SM, Heck AJR, Heeren RMA. Electron Capture Dissociation as Structural Probe for Noncovalent Gas-Phase Protein Assemblies. Anal Chem. 2006;78:7191–7196. doi: 10.1021/ac060960p. [DOI] [PubMed] [Google Scholar]
- 29.Barford D, Hu SH, Johnson LN. Structural mechanism for glycogen phosphorylase control by phosphorylation and AMP. J Mol Biol. 1991;218:233–260. doi: 10.1016/0022-2836(91)90887-c. [DOI] [PubMed] [Google Scholar]
- 30.Horn DM, Ge Y, McLafferty FW. Activated Ion Electron Capture Dissociation for Mass Spectral Sequencing of Larger (42 kDa) Proteins. Anal Chem. 2000;72:4778–4784. doi: 10.1021/ac000494i. [DOI] [PubMed] [Google Scholar]
- 31.Schennach M, Breuker K. Probing Protein Structure and Folding in the Gas Phase by Electron Capture Dissociation. J Am Soc Mass Spectrom. 2015;26:1059–1067. doi: 10.1007/s13361-015-1088-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson LN. Glycogen phosphorylase: control by phosphorylation and allosteric effectors. The FASEB Journal. 1992;6:2274–2282. doi: 10.1096/fasebj.6.6.1544539. [DOI] [PubMed] [Google Scholar]
- 33.Tsaprailis G, Somogyi Á, Nikolaev EN, Wysocki VH. Refining the model for selective cleavage at acidic residues in arginine-containing protonated peptides2. Int J Mass spectrom. 2000;195–196:467–479. [Google Scholar]
- 34.Breci LA, Tabb DL, Yates JR, Wysocki VH. Cleavage N-Terminal to Proline: Analysis of a Database of Peptide Tandem Mass Spectra. Anal Chem. 2003;75:1963–1971. doi: 10.1021/ac026359i. [DOI] [PubMed] [Google Scholar]
- 35.Rose G, Geselowitz A, Lesser G, Lee R, Zehfus M. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 36.Carrigan JB, Engel PC. The structural basis of proteolytic activation of bovine glutamate dehydrogenase. Protein Sci. 2008;17:1346–1353. doi: 10.1110/ps.034785.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerjee S, Schmidt T, Fang J, Stanley CA, Smith TJ. Structural Studies on ADP Activation of Mammalian Glutamate Dehydrogenase and the Evolution of Regulation. Biochemistry (Mosc) 2003;42:3446–3456. doi: 10.1021/bi0206917. [DOI] [PubMed] [Google Scholar]
- 38.Smith TJ, Stanley CA. Untangling the glutamate dehydrogenase allosteric nightmare. Trends Biochem Sci. 2008;33:557–564. doi: 10.1016/j.tibs.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Jacobson RH, Zhang XJ, DuBose RF, Matthews BW. Three-dimensional structure of [beta]-galactosidase from E. coli. Nature. 1994;369:761–766. doi: 10.1038/369761a0. [DOI] [PubMed] [Google Scholar]
- 40.Matthews BW. The structure of E. coli β-galactosidase. C R Biol. 2005;328:549–556. doi: 10.1016/j.crvi.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 41.Cui W, Zhang H, Blankenship RE, Gross ML. Electron-capture dissociation and ion mobility mass spectrometry for characterization of the hemoglobin protein assembly. Protein Sci. 2015;24:1325–1332. doi: 10.1002/pro.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lermyte F, et al. ETD Allows for Native Surface Mapping of a 150 kDa Noncovalent Complex on a Commercial Q-TWIMS-TOF Instrument. J Am Soc Mass Spectrom. 2014;25:343–350. doi: 10.1007/s13361-013-0798-3. [DOI] [PubMed] [Google Scholar]
- 43.Li H, et al. Structural Characterization of Native Proteins and Protein Complexes by Electron Ionization Dissociation-Mass Spectrometry. Anal Chem. 2017;89:2731–2738. doi: 10.1021/acs.analchem.6b02377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacob E, Unger R. A tale of two tails: why are terminal residues of proteins exposed? Bioinformatics. 2007;23:e225–e230. doi: 10.1093/bioinformatics/btl318. [DOI] [PubMed] [Google Scholar]
- 45.van der Spoel D, Marklund EG, Larsson DSD, Caleman C. Proteins, Lipids, and Water in the Gas Phase. Macromolecular Bioscience. 2011;11:50–59. doi: 10.1002/mabi.201000291. [DOI] [PubMed] [Google Scholar]
- 46.Faull PA, et al. Gas-phase metalloprotein complexes interrogated by ion mobility-mass spectrometry. Int J Mass spectrom. 2009;283:140–148. [Google Scholar]
- 47.Haverland NA, et al. Defining Gas-Phase Fragmentation Propensities of Intact Proteins During Native Top-Down Mass Spectrometry. J A Soc Mass Spectrom. 2017;28:1203–1215. doi: 10.1007/s13361-017-1635-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brodbelt JS, Wilson JJ. Infrared multiphoton dissociation in quadrupole ion traps. Mass Spectrom Rev. 2009;28:390–424. doi: 10.1002/mas.20216. [DOI] [PubMed] [Google Scholar]
- 49.Bourgoin-Voillard S, Leymarie N, Costello CE. Top-Down Tandem Mass Spectrometry on RNase A and B Using a Qh/FT-ICR Hybrid Mass Spectrometer. PROTEOMICS. 2014;14:1174–1184. doi: 10.1002/pmic.201300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahlf D, et al. Evaluation of the compact high-field orbitrap for top-down proteomics of human cells. J Proteome Res. 2012;11:4308–4314. doi: 10.1021/pr3004216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holzmann J, Hausberger A, Rupprechter A, Toll H. Top-down MS for rapid methionine oxidation site assignment in filgrastim. Analytical and Bioanalytical Chemistry. 2013;405:6667–6674. doi: 10.1007/s00216-013-7138-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belov ME, et al. From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal Chem. 2013;85:11163–11173. doi: 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- 53.Brodbelt JS. Ion Activation Methods for Peptides and Proteins. Anal Chem. 2016;88:30–51. doi: 10.1021/acs.analchem.5b04563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Durbin KR, Skinner OS, Fellers RT, Kelleher NL. Analyzing Internal Fragmentation of Electrosprayed Ubiquitin Ions During Beam-Type Collisional Dissociation. J Am So Mass Spectrom. 2015;26:782–787. doi: 10.1007/s13361-015-1078-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogorzalek Loo RR, Loo JA. Protein Complexes: Breaking Up is Hard to Do Well. Structure (London, England: 1993) 2013;21:1265–1266. doi: 10.1016/j.str.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schennach M, Breuker K. Proteins with Highly Similar Native Folds Can Show Vastly Dissimilar Folding Behavior When Desolvated. Angew Chem Int Ed. 2014;53:164–168. doi: 10.1002/anie.201306838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campuzano I, Giles K. In: Nanoproteomics: Methods and Protocols. Toms SA, Weil RJ, editors. Humana Press; Totowa, NJ: 2011. pp. 57–70. [Google Scholar]
- 58.Marshall AG, Hendrickson CL, Jackson GS. Fourier transform ion cyclotron resonance mass spectrometry: A primer. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 59.Rayleigh LXX. On the equilibrium of liquid conducting masses charged with electricity. Philosophical Magazine Series 5. 1882;14:184–186. [Google Scholar]
- 60.Ma X, Zhou M, Wysocki V. Surface Induced Dissociation Yields Quaternary Substructure of Refractory Noncovalent Phosphorylase B and Glutamate Dehydrogenase Complexes. J Am Soc Mass Spectrom. 2014;25:368–379. doi: 10.1007/s13361-013-0790-y. [DOI] [PubMed] [Google Scholar]
- 61.Rostom AA, Robinson CV. Detection of the Intact GroEL Chaperonin Assembly by Mass Spectrometry. J Am Chem Soc. 1999;121:4718–4719. [Google Scholar]
- 62.Sobott F, Robinson CV. Characterising electrosprayed biomolecules using tandem-MS—the noncovalent GroEL chaperonin assembly. Int J Mass Spectrom. 2004;236:25–32. [Google Scholar]
- 63.Zubarev RA. Electron-capture dissociation tandem mass spectrometry. Curr Opin Biotechnol. 2004;15:12–16. doi: 10.1016/j.copbio.2003.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the findings of this study are available within the Article and its Supplementary Information and from the corresponding author upon reasonable request.