

Abstract
Analysis of small biological samples would benefit from an efficient micro-scale fractionation strategy that minimizes sample handling, transfer steps and accompanying losses. Here we describe a micro-scale basic reverse phase liquid chromatographic (bRPLC) fractionation method that offers high reproducibility and efficiency for peptide mixtures from small (5–20 μg) samples. We applied our platform to detect differentially expressed proteins from lung tumor cell lines that are sensitive (11-18) and resistant (11-18R) to the tyrosine kinase inhibitor erlotinib. Label-free analyses of 5–20 μg samples yielded identifications of approximately 3,200 to 4,000 proteins with coefficients of variation of 1.9-8.9% in replicate analyses. iTRAQ analyses produced similar protein inventories. Label free and iTRAQ analyses displayed high concordance in identifications of proteins differentially expressed in 11-18 and 11-18R cells. Micro-bRPLC fractionation of cell proteomes increased sensitivity by an average of 4.5-fold in targeted quantitation using parallel reaction monitoring for 3 representative receptor tyrosine kinases (EGFR, PDGFRA, and BMX), which are present at low abundance in 11-18 and 11-18R cells. These data illustrate the broad utility of micro-bRPLC fractionation for global and targeted proteomic analyses. Data are available through Proteome eXchange Accession PXD003604.
Keywords: basic reverse-phase liquid chromatography, drug resistance, label-free quantitation, iTRAQ, protein tyrosine kinase, parallel reaction monitoring
INTRODUCTION
Fractionation of complex peptide mixtures is an important element of proteome analysis platforms.1,2 However, fractionation of microgram protein samples presents major challenges, primarily due to sample loss on chromatographic systems designed for larger samples and to loss during mass transfer steps.3,4 This problem is particularly relevant to the analysis of clinical biospecimens, which may be available in limited amounts. Thus, efficient, reliable micro-scale fractionation strategies for peptide mixtures from small samples prior to MS analysis merit continued development.
A robust, inexpensive system for peptide fractionation is the “stop and go extraction” (Stage-Tip), originally described by Rappsilber and colleagues and which combines reverse phase and ion exchange media in modular disk layers in a disposable pipette tip.5 The Stage-Tip system enables processing of microgram sample amounts5 and has been adapted to serve as a single-vessel reactor for preparation, digestion of proteins and fractionation of the resulting peptides.6
Here, we describe the adaptation of the Stage-Tip system for micro-scale basic pH reverse phase liquid chromatography (micro-bRPLC), a separation method that has proven effective for both global7 and targeted8 proteomic analyses due to high resolution and high orthogonality with low pH reverse phase chromatography. Han et al. applied bRPLC on Stage-Tips to analyze differentially expressed proteins in mouse astrocytes9 and mouse microglia.10 However, this platform required several additional steps for making frits and manual operation with a syringe and also used a relatively high amount of starting protein (200 μg) for the analyses. Here we describe a broader application of the micro-bRPLC platform using simplified processing steps. We demonstrate the application of micro-bRPLC fractionation to label free and iTRAQ proteome analyses of differential protein abundance in tumor cell lines that are sensitive and resistant to the tyrosine kinase inhibitor drug erlotinib. We further demonstrate that micro-bRPLC increases the sensitivity of targeted parallel reaction monitoring (PRM) analysis of receptor tyrosine kinases in cells. The results demonstrate that Stage-Tip based micro-bRPLC fractionation offers a versatile approach to proteomic analysis of small samples.
MATERIALS AND METHODS
Reagents
Alpha-crystallin, dithiothreitol, iodoacetamide and urea were purchased from Sigma-Aldrich (St. Louis, MO). Trypsin (Trypsin Gold) was purchased from Promega (Madison, WI). HALT protease inhibitor cocktail and acetonitrile were purchased from ThermoFisher Scientific (Grand Island, NY). 30 Protein tyrosine kinase (PTK) peptides were obtained from New England Peptide (Gardner, MA) and were of > 95% purity (Table S1).
Preparation of tissues and cells for MS analyses
Frozen colon tumor specimens were obtained from the Cooperative Human Tissue Network-Western Division (Vanderbilt University, Nashville, TN). The deidentified samples and experimental protocol were subject to IRB exempt approval (IRB #080856). The tissue was homogenized in lysis buffer containing 8M urea, 100 mM NH4HCO3, pH 8.0 and 1 × proteinase inhibitor cocktail solution (1 mM 4-benzenesulfonylfluoride hydrochloride, 800 nM aprotinin, 50 μM bestatin, 15 μM [1–[N-[(L-3-trans-carboxyoxirane-2-carbonyl)-L-leucyl]amino]-4-guanidinobutane] (protease inhibitor E64), 20 μM leupeptin, 10 μM pepstatin A) (HALT™, ThermoFisher Scientific, Grand Island, NY). The lysate was sonicated on ice for one min with two second pulses every 30 sec. Insoluble debris was pelleted by centrifugation at 13,500 rpm for 30 min at 4ºC and the supernatant was stored in aliquots at 4ºC. Protein concentrations were determined with the BCA protein assay kit (Thermo Fisher Scientific, Grand Islan, NY) using bovine serum albumin as a standard.
11-18 and 11-18R lung tumor cell lines 11 were provided by Dr. William Pao (Vanderbilt University School of Medicine). Cells were grown in RPMI 1640 medium containing 10% fetal bovine serum and penicillin/streptomycin at 37 °C under 5% CO2. Cells were harvested on ice using cold magnesium- and calcium-free phosphate-buffered saline and supplemented with a phosphatase inhibitor cocktail (1.0 mM sodium orthovanadate, 1.0 mM sodium molybdate, 1.0 mM sodium fluoride, and 10 mM of beta-glycerophosphate (Sigma-Aldrich, St. Louis, MO). The cells were pelleted by centrifugation for 20 min at 500 × g at 4°C and pellets were flash frozen in liquid nitrogen. Denaturation and reduction of proteins was performed in 8 M urea containing 10 mM dithiothreitol and 100 mM NH4HCO3 (pH 8.0) at 37 ºC for 30 min. The solution was stored at room temperature in 10 mM iodoacetamide in the dark for 20 min. The solution was diluted to a urea concentration of 1 M with 50 mM NH4HCO3 and then digested with sequencing grade modified trypsin at an enzyme to substrate ratio of 1:50 at 37°C with shaking for 16 hrs.
iTRAQ labeling
Peptide labeling with iTRAQ 4plex (AB Sciex, Framingham, MA) was performed according to the manufacturer’s protocol. For each analysis, 100 μg of protein from each cell line was labeled with one tube of iTRAQ 4plex reagent. The 11-18 and 11-18R cells were labeled with the iTRAQ 4-plex as follows: 114- and 116-channels, 11-18 cells; 115- and 117-channels 11-18R cells. Lyophilized samples were dissolved in 60 μL of 500 mM triethylammonium bicarbonate, pH 8.5, and the iTRAQ reagent was dissolved in 70 μL of isopropanol. The solution containing peptides and iTRAQ reagent was vortex mixed and then incubated at room temperature for 1h and concentrated to 40 μL under vacuum. Samples labeled with the four different isotopomeric iTRAQ reagents were combined and evaporated to dryness. Peptides then were dissolved in 3% aqueous acetonitrile containing 0.1% formic acid solution before micro-bRPLC fractionation.
Peptide fractionation by micro bRPLC
Micro bRPLC columns were prepared by adding slurry of 2 mg (10 mg/1mL acentonitrile) of Jupiter C18 material (5 μm particle diameter, Phenomenex, Torrance, CA) to commercially produced microcolumns (C18 Stage Tip™; SP301, ThermoFisher Scientific, West Palm Beach, FL). All elution steps for column packing, washing, and elution were carried out with benchtop centrifugation (3,000 × g for 3 min) unless otherwise stated. Prior to addition of peptide mixtures, the column was washed with 100% acentonitrile (100 μL), then with 100 μL equilibration buffer (100 mM NH4HCO3, pH 8.0). The sample mixture was fractionated with 100 μL portions of 7 different elution buffers (5%, 10%. 15%, 20%, 25%, 30%, 90% acetonitrile in 100 mM NH4HCO3, pH 8.0).
LC-MS/MS for global identification and targeted quantitation
Nano-high-performance liquid chromatography (nano-LC) analyses were performed using an Easy n-LC 1000 system (Thermo Fisher Scientific, San Jose, CA). The column (30 cm × 75 μm) was packed in-house with Jupiter 3 μm, 100 Å pore size C18 beads (Phenomenex). Mobile phase A for LC separation consisted of 0.1% formic acid in deionized water and the mobile phase B consisted of 0.1% formic acid in acetonitrile. For analysis of fractionated samples, the mobile phase was programmed from 3% B to 5% B over 3 min, 5% B to 32% B over 75 min, 30% B to 60% B over 5 min, and finally to 95% B over 6 min at a flow rate of 450 nL/min. For analysis of unfractionated samples, the mobile phase was programmed from 3% B to 5% B over 3 min, 5% B to 32% B over 140 min, 30% B to 60% B over 10 min, and finally to 95% B over 7 min at a flow rate of 450 nL/min. An LTQ-Orbitrap Elite mass spectrometer (Thermo Fisher) was used for MS analyses and was operated with Xcalibur (version 2.1) to generate peak lists. Full MS scans were acquired on the Orbitrap from m/z 350-1200 at a resolution of 60,000 using an automatic gain control (AGC) value of 5×105. The minimum threshold was set to 50,000 ion counts. Precursor ions were fragmented with the LTQ using an isolation width of 2 m/z units, a maximum injection time of 50 ms and an AGC value of 1×103. For identification of iTRAQ labeled peptides, HCD fragmentation was used with 5 × 104 AGC, 150 ms maximum fill time, and 2 Da isolation widths. Normalized collision energy was set to 40%. A fixed first mass was set to m/z 100.
PRM analyses were performed on a Q-Exactive™ mass spectrometer equipped with an Easy nLC-1000 (Thermo Fisher Scientific, San Jose, CA). For each analysis, 2 μL of each sample was injected onto an in-line solid-phase extraction column (100 μm × 6 cm) packed with ReproSil-Pur C18 AQ 3 μm resin (Dr. Maisch GmbH, Ammerbuch, Germany) and a frit generated with liquid silicate Kasil 1 and washed with 100% solvent A (0.1 % formic acid) at a flow rate of 2 μL/min. After a total wash volume of 7 μL, the precolumn was placed in-line with a 11 cm × 75 μm PicoFrit capillary column (New Objective, Woburn, MA) packed with the same resin. The peptides were separated using a linear gradient of 2% - 35% solvent B (0.1% formic acid in acetonitrile) at a flow rate of 300 nL min-1 over 40 min, followed by an increase to 90% B over 4 min and held at 90% B for 6 min before returning to initial conditions of 2% B. For peptide ionization, 1800 V was applied and a 250°C capillary temperature was used. All samples were analyzed using a multiplexed PRM method based on a scheduled inclusion list containing the target precursor ions and labeled reference peptide (LRP) standard peptides. The full scan event was collected using a m/z 380 - 1500 mass selection, an Orbitrap resolution of 17,500 (at m/z 200), a target automatic gain control (AGC) value of 3 × 106, and a maximum injection time of 30 ms. The PRM scan events used an Orbitrap resolution of 17,500, an AGC value of 1 × 106, and a maximum fill time of 80 ms with an isolation width of 2 m/z. Fragmentation was performed with a normalized collision energy of 27 and MS/MS scans were acquired with a starting mass of m/z 150. Scan windows were set to 4 min for each peptide in the final PRM method to ensure the measurement of 6–10 points per LC peak per transition.
Peptide and protein identification and quantification
Raw data files were analyzed with the Myrimatch algorithm (Version 1.4.133) and MS-GF+ through BumberDash (version 1.4.133) against a decoy protein database consisting of forward and reversed human RefSeq database (Version 20130621, release date June 2013, 69,178 entries).12,13 Database search criteria were as follows: taxonomy, homo sapiens; carboxyamidomethylation (+57) at cysteine residues as a fixed modification; oxidation at methionine (+16) residues as a variable modification; two maximum allowed missed cleavage; 10 ppm precursor MS tolerance; a 0.8 Da CID and 20 ppm HCD MS/MS tolerance. The GraphPad Prism program (version 5.04, LaJolla, CA) was used to perform Spearman’s correlation test. For iTRAQ-labeled peptides, 4-plex iTRAQ modification was added as a fixed modification (+144.1059 Da) at peptide N termini and at lysines. IDPicker software (Version 3.1.592) was used to analyze iTRAQ data. Relative quantitation was performed by calculating the ratio of 116/114 (replicate 1) and 117/115 (replicate 2) generated by IDPicker. IDPicker software was used to filter peptide-spectrum matches and to assemble proteins from peptide identifications. First, IDPicker employed reversed-sequence database-match information to determine thresholds that yield an estimated 1% FDR for the identifications of each charge state by the formula FDR = (2R)/(R + F), where R is the number of passing reversed-peptide identifications and F is the number of passing forward (normal orientation)-peptide identifications. The second round of filtering removed proteins supported by less than two distinct peptide identifications in the analyses. Indistinguishable proteins were recognized and grouped. Parsimony rules were applied to generate a minimal list of proteins that explain all of the peptides that pass the entry criteria.
The Quasitel algorithm was used for statistical analysis of spectral-count based protein quantitation.14 We set a false discovery rate threshold of < 1% for peptide identification. All PRM data analysis was performed using Skyline software. 15 Instrument quality control assessment was done with labeled reference peptide (AAQGDITAPGGA*R) as previous described method.16 The standard mixture (25 fmol of each standard peptide per sample) was added immediately following tryptic digestion. Five transitions for each peptide were extracted from the PRM data. The intensity rank order and chromatographic elution of the transitions were required to match those of a synthetic standard for each peptide measured. Peptide peak area CV was calculated by:
The CV of the three labeled reference peptides was calculated from the three separate injections per sample. Normalized peptide peak areas were calculated by:
Student’s t-test was performed with the pair-wise comparisons to determine statistical significance.
RESULTS AND DISCUSSION
Optimization of a micro-bRPLC fractionation system
We employed commercially available C18 Stage Tip™ columns for micro-bRPLC fractionation. The pre-embedded C18 material provides a stationary phase medium that also allows for expanded capacity through packing of additional C18 material. We examined the loading capacity of unmodified C18 stage tip™ columns by loading one each with either 0.5, 1, 3, or 6 μg of colon tissue tryptic digest dissolved in 100 μL of 0.1% formic acid. Each column was washed once with 100 μL 0.1% formic acid. The flow-through fractions collected from loading and washing steps were combined and analyzed by LC-MS/MS. The bound peptide fraction was eluted with 50 μL of 70% aqueous acetonitrile. The experiment then was repeated with the same four columns and the same protein loads. Figure S1A shows mean numbers of peptides identified in the combined flow-through fractions from the 4 different protein digest loads. The mean number of identified flow-through peptides increased over 4-fold between the 0.5 and 1μg sample loads and further increased with loads over 1 μg, thus indicating that these larger loads exceeded the capacity of the Stage Tip™ columns as supplied. Therefore, we added an additional 0.5 mg, 1.0 mg, 1.5 mg, 2.0 mg, or 2.5 mg of Jupiter C18 particles to the Stage Tip™ columns and then repeated the flow-through experiment with a peptide digest load corresponding to 20 μg of colon tumor protein. LC-MS/MS analyses of the flow-through fractions (Figure S1B) identified 1580 and 1905 unique peptides, respectively in the two replicates done on columns with an additional 0.5 mg C18 material. However, columns with additional portions of greater than 1.5 mg C18 yielded peptide inventories that were decreased by 95-98%. Thus, most peptides in the 20 μg colon tumor digests were efficiently retained by Stage Tip™ columns packed with at least 1.5 mg of additional C18 material. Thereafter, we performed all subsequent analyses with Stage Tip™ columns packed with an additional 2.0 mg Jupiter C18.
Efficiency of micro-bRPLC fractionation
Reproducible fractionation with high recovery for peptides from complex mixtures is essential for application of this approach for both shotgun and targeted proteomics analyses. We investigated the fractionation efficiency of micro-bRPLC with regard to reproducibility and recovery of peptides from a purified protein. We subjected tryptic digests corresponding to 2 μg of alpha-crystallin to micro-bRPLC in triplicate. Alpha-crystallin peptides were eluted with a 7 step sequence of elution buffers containg 5-90% acetonitrile in 100 mM NH4HCO3, pH 8.0 and the collected fractions were analyzed by LC-MS/MS. Figure S2 shows extracted ion chromatograms (XICs) for three representative peptides HEERQDEHGFISREFHR, VKVLGDVIEVHGKHEE, and M(ac)DIAIQHPWFK. Each peptide partitioned into a single bRPLC fraction in each of triplicate experiments. This result indicates micro-bRPLC is able to efficiently fractionate peptide mixture with high reproducibility.
Next, we also performed the same fractionation experiment in 4 technical replicates with a complex tryptic digest corresponding to 20 μg colon tumor tissue, which was separated into 7 fractions by micro-bRPLC. The eluted fractions were analyzed by LC-MS/MS as described above. Figure 1A illustrates the consistency and reproducibility of peptide and protein inventories obtained by four replicate bRPLC and LC-MS/MS inventories. Peptide inventories (mean = 16,140 ± 379) and protein inventories (mean=3,652 ± 86) (Table S2) varied by 1.3% and 5.5%, respectively (Supplemental Dataset 1).
Figure 1.
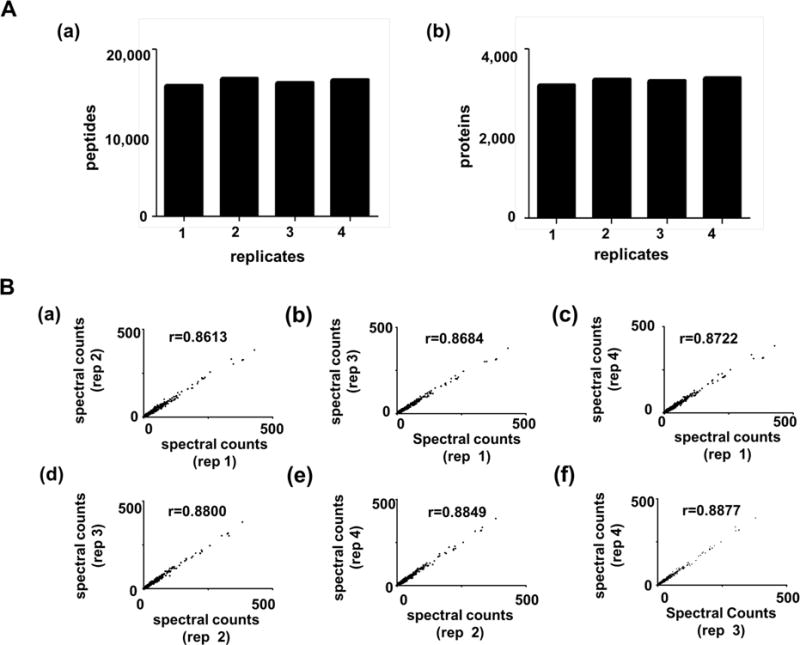
Reproducibility of peptide and protein identifications by LC-MS/MS in four replicate analyses of 20 μg colon tumor protein. A. Number of peptides (a) and proteins (b) identified by LC-MS/MS after micro-bRPLC fractionation of four 20 μg colon tumor protein specimens. B. Pairwise scatter plots and Spearman correlation of spectral counts for proteins identified by LC-MS/MS in four replicate micro-bRPLC fractionations of 20 μg colon tumor tissue protein digest. Colon tumor tissue digests (20 μg) were fractionated by micro-bRPLC (7 fractions). One fourth of each fraction (total 5 μg on-column) was analyzed by LC-MS/MS.
Spectral counts for peptide-spectrum matches have strong correlation with abundance of the corresponding proteins.17,18 Therefore, we compared total spectral counts for proteins identified in the 4 replicate analyses. Figure 1B shows that a strong Spearman correlation was obtained from each comparison (mean 0.876). Thus, simple and complex peptide mixtures were reproducibly fractionated by micro-bRPLC, thus facilitating identification of peptides by LC-MS/MS.
An advantage of fractionation strategies is an increase in the number of identified distinct peptides and spectral counts for proteins. We compared distinct peptides and spectral counts for 15 proteins detected at different abundances with the bRPLC and LC-MS/MS analyses described above compared to a single 160 min gradient LC-MS/MS without fractionation (Figure S3). Fractionation yielded an a mean increase of 39% in the number of distinct peptides per protein and a mean increase of 119% in spectral counts per protein.
Large-scale label-free comparison of cell proteomes by micro-bRPLC and LC-MS/MS
We further evaluated application of this platform to analyze differentially expressed proteins in a lung adenocarcinoma cell model of resistance to the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor erlotinib.11 Although we have recently documented protein tyrosine kinase expression changes associated with resistance19, we hypothesized that alteration of protein expression also might be associated with erlotinib resistance. Therefore, we compared proteome inventories of erlotinib-sensitive cells (11-18) and erlotinib-resistant cells (11-18R). To compare the performance of micro-bRPLC for different sample sizes, we analyzed both 5 μg and 20 μg samples of cell protein. To minimize differences due to on-column LC-MS/MS loads, we analyzed 100% of each fraction from the 5 μg sample and 25% of each fraction from the 20 μg sample by LC-MS/MS. Three separate cultures each of the 11-18 and 11-18R cells each were analyzed. Identified proteins are listed in Table S3. As shown in Figure 2A, a seven-fraction micro-bRPLC and LC-MS/MS analysis of the 20 μg sample identified 4,154 ± 98 proteins (CV 2.3%) from three 11-18 biological replicates, respectively whereas 3,943 ± 263 proteins (CV 5.4%) were identified from identical analyses of three 11-18R cultures. Three replicate analyses of 5 μg samples of 11-18 cells yielded 3,515 ± 129 identifications (CV 3.0%), whereas 3,229 ± 348 proteins (CV 8.8%) were identified from the 11-18R cells (Supplemental Dataset 2). These inventories were 81-86% of those from 20 μg fractionations. The results demonstrate that micro-bRPLC and LC-MS/MS yielded highly reproducible proteome inventories and that micro-bRPLC fractionation was nearly as efficient with the low 5 μg sample load as with the higher 20 μg load.
Figure 2.
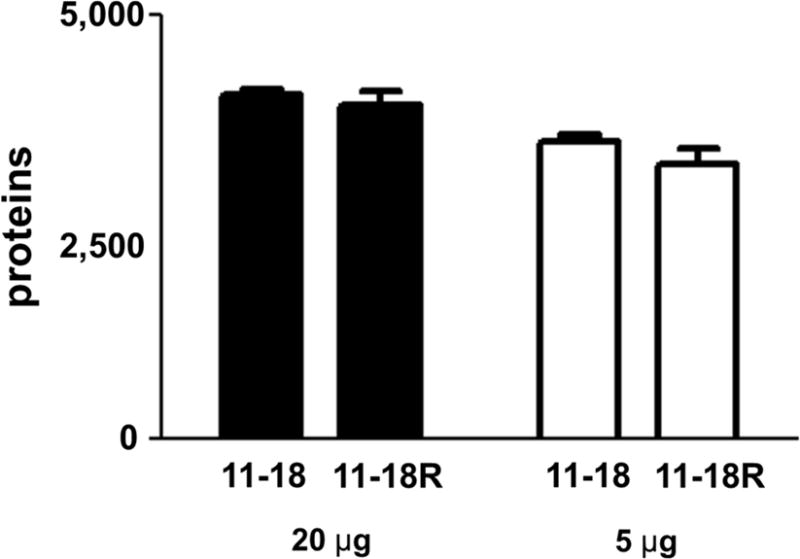
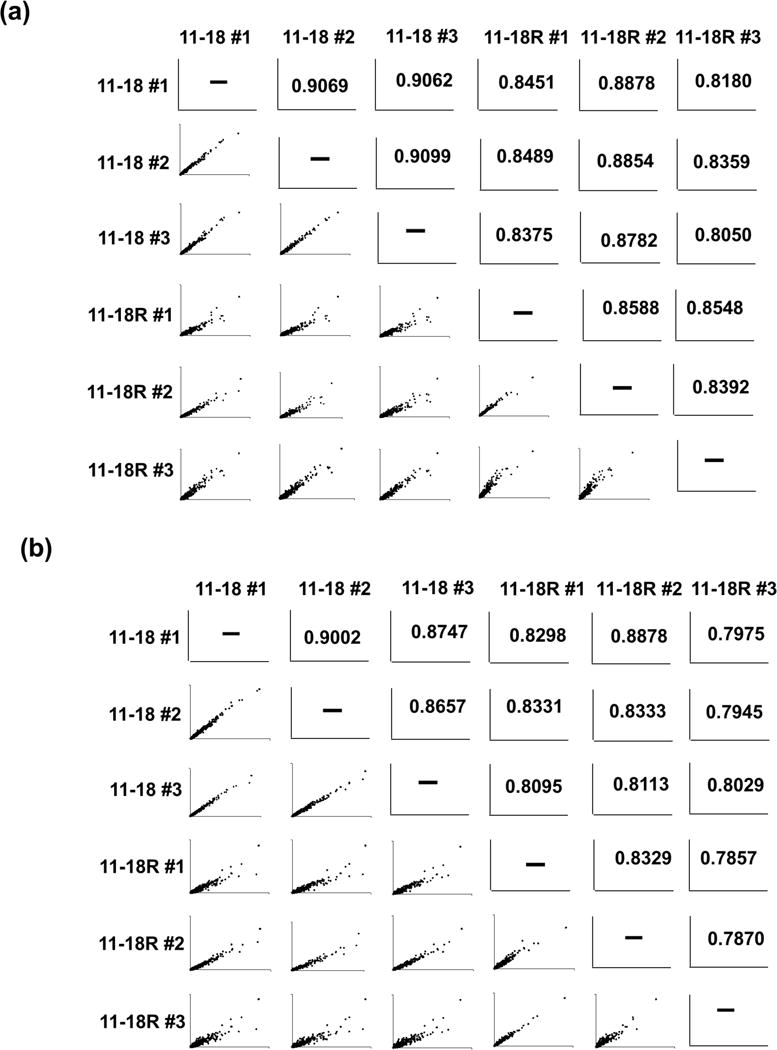
Reproducibility of proteomic inventories of 11-18 and 11-18R cells. (A) Comparison of number of proteins obtained from analysis of three replicate cultures of 11-18 and 11-18R cells for with 20 μg or 5 μg sample inputs. (B) Pairwise scatter plots and Spearman’s correlation coefficients based on spectra counts calculated from replicate analyses of 11-18 and 11-18R cultures with 20 μg (a) and 5 μg (b) inputs.
Next, we assessed the reproducibility of relative protein abundance in replicate analyses of 3 separate cultures each of 11-18 and 11-18R cells. Figure 2B shows pairwise comparisons and their Spearman’s correlation coefficient (r) for spectral counts for identified proteins. For the 20 μg fractionations of 11-18 cells, the average Spearman’s correlation coefficient between replicates was 0.9065, whereas the average Spearman’s correlation for 11-18R cell fractionations was 0.8505. Fractionation of 5 μg samples yielded similar results, with 11-18 cell fractionations having average Spearman’s correlation coefficients of 0.8802 and 11-18R replicates having coefficients of 0.8222.
We then examined the ability of the micro-bRPLC and LC-MS/MS analyses to detect protein abundance differences between 11-18 and 11-18R cells. For 11-18 versus 11-18R, we observed Spearman’s correlations of 0.8982 and 0.8955 for average spectral counts for proteins in the 20 μg or 5 μg analyses, respectively (Figure S4A). Most proteins thus exhibited no significant abundance difference between the erlotinib-sensitive and resistant cells. Similarly, average protein spectral counts in 11-18 versus 11-18R were highly correlated, even when the 20 μg and 5 μg fractionations were compared (r = 0.8806 and 0.8852 respectively; Figure S4B). These results indicated that micro-bRPLC fractionation with 5 μg samples and 20 μg samples produced highly reproducible comparisons of the proteomes of 11-18 and 11-18R cells.
We then asked whether the relatively small number of differentially abundant proteins between 11-18 and 11-18R cells were consistently detected in analyses of the 5 and 20 μg samples. Significant differences in protein spectral counts (at least 2-fold and p<0.05) between 11-18 and 11-18R were calculated with Quasitel14 for 20 μg and 5 μg fractionations, respectively. These comparisons only included proteins for which at least 20 spectral counts were acquired, because low abundance proteins acquired with few spectral counts may yield inaccurate quantitation values.20 In the 20 μg fractionation, 22 proteins were more abundant and 43 were less abundant in 11-18R compared to 11-18 cells (p < 0.05) (Table S5A). Similarly, 28 proteins were more abundant and 71 proteins were less abundant in 11-18R in the 5μg sample comparison (p < 0.05) (Table S5B). In these comparisons, 16 proteins displayed consistent differential abundance in both 20 μg and 5 μg fractionation analyses (Figure S5). To further verify the spectral count-based expression difference for these 16 proteins, relative MS1 intensity for each peptide was measured in unfractionated samples with a Q-Exactive™ instrument. Supplemental Dataset 3A shows base-peak chromatograms obtained from 3 biological replicates and 2 technical replicates showing high reproducibility. These MS1 analyses successfully quantified 15 of the 16 proteins identified as differential abundant in the fractionated samples. Supplemental Dataset 3(B-M) shows XICs for two representative peptides from each protein, as well as for the internal standard actin peptide. As shown in Supplemental Dataset 3B, the MS1 intensity of the actin peptide SYELPDGQVITIGNER (m/z 895.8472) did not show significant differences between 11-18 and 11-18R cells or replicates. However, we found that MS1intensities for each of the two peptides measured from the 15 differential proteins were consistently different between 11-18 and 11-18R. Moreover, the intensity differences were consistent with fold-changes calculated by spectral counts obtained from micro-bRPLC platform. These results collectively demonstrate that micro-bRPLC affords high reproducibility in proteome inventories and detection of differential protein abundance for label-free quantitation from both 20 μg and 5 μg fractionations.
iTRAQ-based profiling of 11-18 and 11-18R cells by micro-bRPLC and LC-MS/MS
Isobaric labeling (e.g., iTRAQ and TMT) is one of the most popular approaches for MS2-based relative quantification.21 Efficient fractionation prior to LC-MS/MS analysis can benefit these analyses by simplifying peptide mixtures, thereby reducing quantitation inaccuracy due to contamination of reporter ion signals by co-eluting isobaric interferences.21 Therefore, we asked whether our micro-bRPLC platform could improve iTRAQ analyses of small samples. We analyzed differentially expressed proteins in 11-18 and 11-18R cells. Two biological replicate 5 μg samples of each cell line were labeled with 4-plex iTRAQ (114, 11–18; 115, 11–18’; 116, 11–18R; 117, 11–18R’) respectively and mixed in equal proportions. A similar sample set was prepared from 20 μg samples. Each sample set was fractionated by micro-bRPLC and the fractions were analyzed by LC-MS/MS on the Orbitrap Elite system. The abundance of proteins was measured by summing the normalized reporter ion intensities for all peptides that were assigned to that protein. Only proteins that were identified and quantified both in both biological replicate samples were included in the analysis (Supplemental Dataset 4).
The 20 μg sample yielded 2,850 quantifiable proteins (Table S5A), whereas the 5 μg sample yielded 2,571 quantifiable proteins (Table S5B). To compare quantitative reproducibility for the two biological replicates, we plotted iTRAQ intensities measured between either 114 and 115 or 116 and 117. As shown in Figure S6A, the Spearman’s correlation coefficient between 2 biological replicates in the 20 μg or 5 μg fractionations averaged 0.9988. The global distribution of average iTRAQ ratios calculated from the two biological replicates centered on unity (Figure S6B), which indicates that most of proteins were not significantly different in their abundance. These results are consistent with our label-free analyses.
In the 20 μg fractionation, we identified 9 proteins that were up-regulated (≥ 1.5-fold) and 27 that were down-regulated (≥ 1.5-fold) in 11-18R. Similarly, in the 5 μg fractionation, 12 proteins were up-regulated (≥ 1.5 fold) and 24 proteins were down-regulated (≥ 1.5-fold) in 11-18R. Moreover, 3 proteins (CRABP2, MT2A, and SLC2A1) and 6 proteins (MVP, HADH, S100P, CTSA, ALDH3A1, and GSTP1) were consistently up and down-regulated (≥ 1.5-fold) in both fractionation experiments. Three differential proteins (MVP, S100P, and ALDH3A1) found in the iTRAQ experiment were also differential in the label-free LC-MS/MS analysis (both 20 μg and 5 μg). Figure S7 shows relative iTRAQ ratios of representative peptides from MVP, S100P, ALDH3A1, and ACTIN acquired from both 20 μg and 5 μg fractionations. The ACTIN peptide SYELPDGQVITIGNER showed no significant difference between 11-18 and 11-18R cells in both 20 μg and 5 μg fractionations, whereas iTRAQ reporter ions for representative peptides for the other 3 proteins were significantly decreased in 11-18R, which is very consistent results obtained by label-free LC-MS/MS analyses. We note that, although 3 differential proteins were identified in both label-free and iTRAQ comparisons, these were the proteins with the highest level of differential expression. The lack of concordance in identifications of other differential proteins may reflect the relatively small abundance differences (approximately 2-fold) and possibly differences in the ionization, intensity and detection of unlabeled peptides compared to iTRAQ-labeled peptides. Our results thus demonstrate the utility of micro-bRPLC for fractionating small amounts of iTRAQ labeled peptide mixtures.
Enhanced sensitivity for targeted peptide quantification with micro-bRPLC
Selective measurement of proteotypic peptides in complex mixtures is an essential technique for protein quantitation. Although this method is frequently done without prior fractionation, enhanced sensitivity can be achieved by antibody-based targeted peptide enrichment22 or by fractionation of complex digest mixtures.23 Shi and colleagues described a highly sensitive bRPLC fractionation approach guided by in-line MS detection called PRISM.8 Therefore, we explored the application of our micro-bRPLC method to enhance measurement of low abundance proteins by parallel reaction monitoring (PRM) analysis24,25 of their proteotypic peptides. We recently described a multiplexed PRM assay panel to measure 83 protein tyrosine kinases (PTKs).19 In our previous report, many PTKs were not detected because of low abundance in cell models, even though corresponding mRNAs were detected.19
We evaluated the impact of micro-bRPLC on detection of PTK peptides in a targeted acquisition LC-MS/MS experiment, in which measurement estimates were based on spectral counts for the targeted peptides. We measured 30 peptides corresponding to 23 PTK proteins (Table S5). Using unlabeled synthetic peptide standards to establish LC retention times, we constructed a targeted acquisition MS method to analyze unfractionated tryptic digests from three separate cultures of 11-18 cells (2 μg protein) on the Orbitrap Elite system. We then fractionated each 11-18 culture (20 μg protein) by micro-bRPLC and analyzed 25% of each fraction (5 μg on-column) with the targeted acquisition method. The resulting datafiles were searched for peptide identification with Myrimatch. Table S6 lists detected PTK peptides and the corresponding spectral counts from the unfractionated and micro-bRPLC fractionated analyses. Only three PTK peptides were identified in the unfractionated samples and were detected with low spectral counts, whereas 28 of the 30 target peptides were successfully identified in the micro-bRPLC fractionated samples and with high spectral counts. Moreover, each detected peptide was reproducibly eluted in the same micro-bRPLC fraction in the three replicate analyses. As shown in Fig. S7, representative two PTK peptides IPLENIQIIR and ELDIFGLNPADESTR partitioned into a single bRPLC fraction in each of triplicate experiments.
Next, we performed PRM analyses with the Q-Exactive™ instrument of unfractionated and micro-bRPLC fractionated 11-18 and 11-18R cells. We targeted three proteotypic peptides representing the PTK proteins EGFR (IPLENLQIIR), PDGFR (ELDIFGLNPADESTR) and BMX (VPDSVSLGNGIWELK). Quantitative normalization was done with the labeled reference peptide method26 with the heavy labeled peptide AAQGDITAPGGA*R as the reference standard. Three separate cultures of 11-18 and 11-18R cells were analyzed without fractionation (1 μg protein digest on column) or with micro-bRPLC fractionation (20 μg protein digest, 7 fractions with half of each fraction injected on-column). Figure 3A shows comparison of normalized signal for the three target peptides acquired in PRM analyses of unfractionated and micro-bRPLC fractionated 11-18 and 11-18R cells. Micro-bRPLC fractionation enhanced the normalized signals an average of 4.5 fold compared to the unfractionated samples. Figure 3B compares XICs for the three PTK peptides generated from unfractionated and micro-bRPLC fractionated mixtures. Whereas the product ion traces generated from unfractionated samples displayed low intensities and poorly resolved transition order, signals from micro-bRPLC fractionated samples displayed clear co-elution and order matching those of the synthetic peptide standards. The measured differences for EGFR, PDGFR and BMX peptides were consistent with the values we reported previously19.
Figure 3.
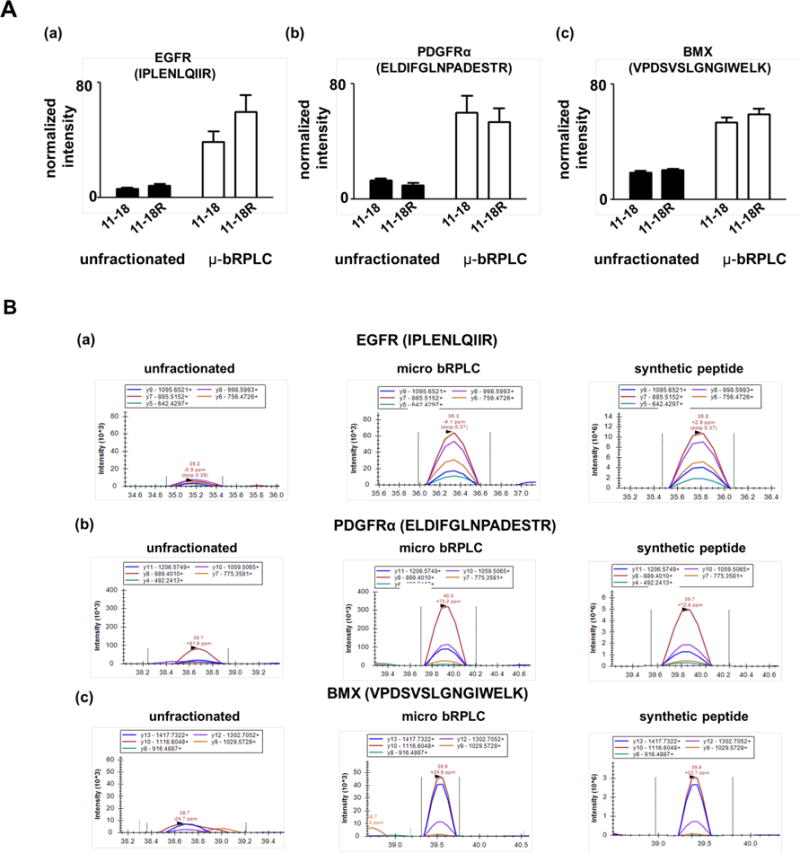
Improvement in sensitivity of targeted peptide analysis by PRM with micro-bRPLC fractionation. (A) Comparison of normalized abundance ratio obtained from unfractionated (filled bars) and micro-bRPLC (open bars) for 3 target peptides (IPLENLQIIR from EGFR; b, ELDIFGLNPADESTR from PDGFRA; c, VPDSVSLGNGIWELK from BMX) from three separate cultures of 11-18 and 11-18R cells. Target peptide signals were normalized to the same labeled reference peptide standard (AAQGDITAPGGA*R), which was spiked into each sample. (B) Comparison of extracted PRM transitions from the three target PTK peptides (a, IPLENLQIIR from EGFR; b, ELDIFGLNPADESTR from PDGFRA; c, VPDSVSLGNGIWELK from BMX). PRM analyses were done with unfractionated digests (left), micro-bRPLC fractionated digests (middle), and with synthetic standards for the three peptides (right).
CONCLUSION
We established an efficient micro-scale bRPLC platform that enables multi-sample processing with simple steps. Our platform provides high reproducibility in global proteome profiling for both spectral-count based label-free quantitation and iTRAQ quantitation in samples as small as 5 μg protein. Moreover, the sensitivity of targeted PRM analysis is significantly enhanced by micro-bRPLC fractionation. These results further suggest that micro-bRPLC can be combined with other peptide fractionation or enrichment methods to create multidimensional workflows. This platform offers performance enhancements similar to previously described bRPLC systems5,8,10, but can easily be deployed at modest cost. An advantage of the current version micro-bRPLC platform is speed of the fractionation steps, which are performed in a spin column format with eution steps done by brief centrifugation. A six fraction sample prep by micro-bRPLC can be done in less than 30 min, whereas a conventional bRPLC separation typically requires 1–2 hrs. Moreover, the micro-bRPLC fractionation could be implemented in a multi-well plate format, thus enabling automated, high-throughput sample preparation. In our present study, LC-MS/MS analyses were done with an Orbitrap-Elite MS instrument, which produced low resolution CID spectra at moderate acquisition rates. Combination of bRPLC fractionation with state of the art MS instruments (e.g., Q-Exactive™ or TripleTOF™) would allow considerably greater depth of proteome coverage.
Perhaps the most important application of the micro-bRPLC platform may be to analyze clinical specimens of limited protein amounts, such as circulating tumor cells, embryonic stem cells, laser capture microdissected cells, and needle biopsies. Indeed, the simplicity and reproducibility of peptide fractionation with the micro-bRPLC system should allow application in clinical biospecimen analysis workflows, which can bring the power of MS-based protein analysis technology to clinical diagnostics. Moreover, an automated μ-bRPLC platform.
Supplementary Material
Supplemental Dataset 1. IDPicker report containing summary of peptides and protein identification data by LC-MS/MS in four replicate analysis of 20 μg colon tumor protein (file: Supplemental dataset 1_Colon tumor proteome inventories.xls)
Supplemental Dataset 2. IDPicker report containing summary of peptides and protein identification data by LC-MS/MS for three replicate cultures of 11-18 and 11-18R cells for with 20 μg or 5 μg sample input. (file: Supplemental Dataset 2_ 11-18 and 11-18R cell proteome inventories_revised_032216.xls)
Supplemental Dataset 3. (A) Base-peak chromatograms obtained from 3 biological replicates and 2 technical replicates showing high reproducibility. (B-M) XICs for two representative peptides from each protein, as well as an internal standard actin peptide. (file: Supplemental Dataset 3_Base peak and XIC for 11-18 and 11-18R cell comparisons.pdf)
Supplemental Dataset 4. IDPicker report containing summary of peptides and protein identification data by LC-MS/MS for 4-Plex iTRAQ labeled 11-18 and 11-18R cells for with 20 μg or 5 μg sample input. (file: Supplemental Dataset 4_iTRAQ protein ID and quantitation in 11-18 and 11-18R cells.xls)
Figure S1. Evaluation of binding capacity of unmodified (A) and C18-supplemented (B) Stage-Tips™.
Figure S2. Efficiency and reproducibility of micro-bRPLC fractionation of alpha-crystallin peptides.
Figure S3. Pairwise scatter plots and Spearman correlation coefficient (r) for average spectral counts per protein acquired from analyses of three replicate cultures of 11-18 and 11-18R cells with either 20 μg and 5 μg inputs.
Figure S4. Comparison of the abundance ratio for 16 proteins in 11-18 vs. 11-18R cells from analyses of either 20 μg or 5 μg cell protein.
Figure S5. Reproducibility of 4-plex iTRAQ quantitation for analysis of two replicate cultures of 11-18 and 11-18R cells.
Figure S6. Relative iTRAQ reporter ion ratios for representative peptides corresponding to ACTIN (A), which is equally expressed in 11-18 vs 11-18R cells and ALDH3A1 (B), MVP (C) and S100P (D), which are significantly decreased in 11-18R.
Figure S7. Efficient and reproducible fractionation of PTK peptides in 20 μg 11-18 cell by micro-bRPLC.
Table S1. Proteotypic peptides from 30 protein tyrosine kinase (PTK) proteins
Table S2. Proteins identified by LC-MS/MS after micro bRPLC fractionation for 20 μg colon tumor tissue with 4 technical replicates
Table S3. Proteins identified by LC-MS/MS after micro bRPLC for 11-18 and 11-18R with 3 biological replicates between 20 μg and 5 μg samples.
Table S4A. Differentially expressed proteins from micro bRPLC LC-MS/MS of 11-18 and 11-18R with 3 biological replicates of 20 μg samples.
Table S4B. Differentially expressed proteins from micro bRPLC LC-MS/MS of 11-18 and 11-18R with 3 biological replicates of 5 μg samples.
Table S5A. iTRAQ quantified proteins from 11-18 and 11-18R cells with 2 biological replicates for 20 μg samples.
Table S5B. iTRAQ quantified proteins from 11-18 and 11-18R cells with 2 biological replicates for 5 μg samples.
Table S6. Detected PTK peptides and their spectral counts obtained from unfractionated and micro bRPLC fractionated 11-18 cells.
Acknowledgments
This work was supported by National Institutes of Health Grant U24CA159988.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website at DOI: (to be provided).
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
References
- 1.Camerini S, Mauri P. The role of protein and peptide separation before mass spectrometry analysis in clinical proteomics. J Chromatogr A. 2015;1381:1–12. doi: 10.1016/j.chroma.2014.12.035. [DOI] [PubMed] [Google Scholar]
- 2.Mesmin C, van Oostrum J, Domon B. Complexity reduction of clinical samples for routine mass spectrometric analysis. Proteomics Clin Appl. 2015 doi: 10.1002/prca.201500135. [DOI] [PubMed] [Google Scholar]
- 3.Feist P, Hummon AB. Proteomic challenges: sample preparation techniques for microgram-quantity protein analysis from biological samples. Int J Mol Sci. 2015;16:3537–3563. doi: 10.3390/ijms16023537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin JG, Rejtar T, Martin SA. Integrated microscale analysis system for targeted liquid chromatography mass spectrometry proteomics on limited amounts of enriched cell populations. Anal Chem. 2013;85:10680–10685. doi: 10.1021/ac401937c. [DOI] [PubMed] [Google Scholar]
- 5.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2:1896–1906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 6.Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 7.Stein DR, Hu X, McCorrister SJ, Westmacott GR, Plummer FA, Ball TB, Carpenter MS. High pH reversed-phase chromatography as a superior fractionation scheme compared to off-gel isoelectric focusing for complex proteome analysis. Proteomics. 2013;13:2956–2966. doi: 10.1002/pmic.201300079. [DOI] [PubMed] [Google Scholar]
- 8.Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, Xie F, Wu S, Kim JS, Jones N, Moore RJ, Pasa-Tolic L, Kagan J, Rodland KD, Liu T, Tang K, Camp DG, 2nd, Smith RD, Qian WJ. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc Natl Acad Sci U S A. 2012;109:15395–15400. doi: 10.1073/pnas.1204366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han D, Jin J, Woo J, Min H, Kim Y. Proteomic analysis of mouse astrocytes and their secretome by a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics. 2014;14:1604–1609. doi: 10.1002/pmic.201300495. [DOI] [PubMed] [Google Scholar]
- 10.Han D, Moon S, Kim Y, Kim J, Jin J, Kim Y. In-depth proteomic analysis of mouse microglia using a combination of FASP and StageTip-based, high pH, reversed-phase fractionation. Proteomics. 2013;13:2984–2988. doi: 10.1002/pmic.201300091. [DOI] [PubMed] [Google Scholar]
- 11.Ohashi K, Sequist LV, Arcila ME, Moran T, Chmielecki J, Lin YL, Pan Y, Wang L, de Stanchina E, Shien K, Aoe K, Toyooka S, Kiura K, Fernandez-Cuesta L, Fidias P, Yang JC, Miller VA, Riely GJ, Kris MG, Engelman JA, Vnencak-Jones CL, Dias-Santagata D, Ladanyi M, Pao W. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109:E2127–2133. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holman JD, Ma ZQ, Tabb DL. Identifying proteomic LC-MS/MS data sets with Bumbershoot and IDPicker. Curr Protoc Bioinformatics. 2012 doi: 10.1002/0471250953.bi1317s37. Chapter 13, Unit13 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim S, Pevzner PA. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun. 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M, Gray W, Zhang H, Chung CH, Billheimer D, Yarbrough WG, Liebler DC, Shyr Y, Slebos RJ. Comparative shotgun proteomics using spectral count data and quasi-likelihood modeling. J Proteome Res. 2010;9:4295–4305. doi: 10.1021/pr100527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 2013;52:3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou W, Liotta LA, Petricoin EF. The spectra count label-free quantitation in cancer proteomics. Cancer Genomics Proteomics. 2012;9:135–142. [PMC free article] [PubMed] [Google Scholar]
- 18.Lundgren DH, Hwang SI, Wu L, Han DK. Role of spectral counting in quantitative proteomics. Expert Rev Proteomics. 2010;7:39–53. doi: 10.1586/epr.09.69. [DOI] [PubMed] [Google Scholar]
- 19.Kim HJ, Lin, Lee HJ, Li M, Liebler DC. Quantitative profiling of protein tyrosine kinases in human cancer cell lines by multiplexed parallel reaction monitoring assays. Mol Cell Proteomics. 2015 doi: 10.1074/mcp.O115.056713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulze WX, Usadel B. Quantitation in mass-spectrometry-based proteomics. Annu Rev Plant Biol. 2010;61:491–516. doi: 10.1146/annurev-arplant-042809-112132. [DOI] [PubMed] [Google Scholar]
- 21.Rauniyar N, Yates JR., 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J Proteome Res. 2014;13:5293–309. doi: 10.1021/pr500880b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3:235–244. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 23.Keshishian H, Addona T, Burgess M, Mani DR, Shi X, Kuhn E, Sabatine MS, Gerszten RE, Carr SA. Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2009;8:2339–2349. doi: 10.1074/mcp.M900140-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallien S, Bourmaud A, Kim SY, Domon B. Technical considerations for large-scale parallel reaction monitoring analysis. J Proteomics. 2014;100:147–159. doi: 10.1016/j.jprot.2013.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Lesur A, Domon B. Advances in high-resolution accurate mass spectrometry application to targeted proteomics. Proteomics. 2015;15:880–890. doi: 10.1002/pmic.201400450. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Liu Q, Zimmerman LJ, Ham AJ, Slebos RJ, Rahman J, Kikuchi T, Massion PP, Carbone DP, Billheimer D, Liebler DC. Methods for peptide and protein quantitation by liquid chromatography-multiple reaction monitoring mass spectrometry. Mol Cell Proteomics. 2011;10:M110 006593. doi: 10.1074/mcp.M110.006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Dataset 1. IDPicker report containing summary of peptides and protein identification data by LC-MS/MS in four replicate analysis of 20 μg colon tumor protein (file: Supplemental dataset 1_Colon tumor proteome inventories.xls)
Supplemental Dataset 2. IDPicker report containing summary of peptides and protein identification data by LC-MS/MS for three replicate cultures of 11-18 and 11-18R cells for with 20 μg or 5 μg sample input. (file: Supplemental Dataset 2_ 11-18 and 11-18R cell proteome inventories_revised_032216.xls)
Supplemental Dataset 3. (A) Base-peak chromatograms obtained from 3 biological replicates and 2 technical replicates showing high reproducibility. (B-M) XICs for two representative peptides from each protein, as well as an internal standard actin peptide. (file: Supplemental Dataset 3_Base peak and XIC for 11-18 and 11-18R cell comparisons.pdf)
Supplemental Dataset 4. IDPicker report containing summary of peptides and protein identification data by LC-MS/MS for 4-Plex iTRAQ labeled 11-18 and 11-18R cells for with 20 μg or 5 μg sample input. (file: Supplemental Dataset 4_iTRAQ protein ID and quantitation in 11-18 and 11-18R cells.xls)
Figure S1. Evaluation of binding capacity of unmodified (A) and C18-supplemented (B) Stage-Tips™.
Figure S2. Efficiency and reproducibility of micro-bRPLC fractionation of alpha-crystallin peptides.
Figure S3. Pairwise scatter plots and Spearman correlation coefficient (r) for average spectral counts per protein acquired from analyses of three replicate cultures of 11-18 and 11-18R cells with either 20 μg and 5 μg inputs.
Figure S4. Comparison of the abundance ratio for 16 proteins in 11-18 vs. 11-18R cells from analyses of either 20 μg or 5 μg cell protein.
Figure S5. Reproducibility of 4-plex iTRAQ quantitation for analysis of two replicate cultures of 11-18 and 11-18R cells.
Figure S6. Relative iTRAQ reporter ion ratios for representative peptides corresponding to ACTIN (A), which is equally expressed in 11-18 vs 11-18R cells and ALDH3A1 (B), MVP (C) and S100P (D), which are significantly decreased in 11-18R.
Figure S7. Efficient and reproducible fractionation of PTK peptides in 20 μg 11-18 cell by micro-bRPLC.
Table S1. Proteotypic peptides from 30 protein tyrosine kinase (PTK) proteins
Table S2. Proteins identified by LC-MS/MS after micro bRPLC fractionation for 20 μg colon tumor tissue with 4 technical replicates
Table S3. Proteins identified by LC-MS/MS after micro bRPLC for 11-18 and 11-18R with 3 biological replicates between 20 μg and 5 μg samples.
Table S4A. Differentially expressed proteins from micro bRPLC LC-MS/MS of 11-18 and 11-18R with 3 biological replicates of 20 μg samples.
Table S4B. Differentially expressed proteins from micro bRPLC LC-MS/MS of 11-18 and 11-18R with 3 biological replicates of 5 μg samples.
Table S5A. iTRAQ quantified proteins from 11-18 and 11-18R cells with 2 biological replicates for 20 μg samples.
Table S5B. iTRAQ quantified proteins from 11-18 and 11-18R cells with 2 biological replicates for 5 μg samples.
Table S6. Detected PTK peptides and their spectral counts obtained from unfractionated and micro bRPLC fractionated 11-18 cells.