

Abstract
Rheumatoid arthritis (RA) is a chronic inflammatory autoimmune disease that primarily affects the joints. Self-reactive B and T lymphocytes cooperate to promote antibody responses against self proteins and are major drivers of disease. T lymphocytes also promote RA independently of B lymphocytes mainly through the production of key inflammatory cytokines, such as IL-17, that promote pathology. While the innate signals that initiate self-reactive adaptive immune responses are poorly understood, the disease is predominantly caused by inflammatory cellular infiltration and accumulation in articular tissues, and by bone erosions driven by bone-resorbing osteoclasts. Osteoclasts are giant multinucleated cells formed by the fusion of multiple myeloid cells that require short-range signals, such as the cytokines MCSF and RANKL, for undergoing differentiation. The recruitment and positioning of osteoclast precursors to sites of osteoclast differentiation by chemoattractants is an important point of control for osteoclastogenesis and bone resorption. Recently, the GPCR EBI2 and its oxysterol ligand 7a, 25 dihydroxycholesterol were identified as important regulators of osteoclast precursor positioning in proximity to bone surfaces, and of osteoclast differentiation under homeostasis. In chronic inflammatory diseases like RA, osteoclast differentiation is also driven by inflammatory cytokines such as TNFa and IL-1, and can occur independently of RANKL. Finally, there is growing evidence that the chemotactic signals guiding osteoclast precursors to inflamed articular sites contribute to disease and are of great interest. Furthering our understanding of the complex osteoimmune cell interactions should provide new avenues of therapeutic intervention for RA.
Index entries: Rheumatoid arthritis, osteoclasts, osteoclast precursors, cell migration
Introduction
Risk for inflammatory autoimmune diseases, like rheumatoid arthritis (RA), is influenced by genetic variation and environment. Genetic risk loci for autoimmune diseases have been identified by GWAS [1] and recently genetic fine mapping studies coupled with epigenomic analyzes have begun to identify putative causal variants [2,3]. Multiple disease-associated single nucleotide polymorphisms (SNPs) were mapped to genes and regulatory regions that are active in a cell-specific manner. These analyses revealed RA-associated SNP enrichment in genes and regulatory regions active in B cells, T cells, and to a lesser extent, monocytic cells [2,3]. This work provides a contextual framework for understanding the genesis of human autoimmune disease that can be applied to present and future studies in animal models. It also places the adaptive immune system front and center in the pathogenesis of RA.
Pathogenesis of RA
Rheumatoid arthritis is a chronic inflammatory disease characterized by an accumulation of inflammatory leukocytes in the synovium and articular tissues, and autoantibodies (rheumatoid factor and anti-citrullinated protein antibodies (ACPA)). The disease is also comprised of bone and cartilage tissue destruction and by other systemic features presumably caused by flares of inflammation. Even though RA is a common autoimmune disease that has been studied for several decades now, it is still unclear why articular tissues are predominantly attacked. The cellular components of the articular space include cells of the innate and adaptive immune system, chondrocytes, fibroblast-like synoviocytes, and bone remodeling cells, i.e. osteoclasts and osteoblasts. The accumulation of inflammatory leukocytes in articular tissues is a hallmark feature of RA, but this accumulation is thought to occur primarily because of cellular recruitment into inflamed tissues rather than as a consequence of local leukocyte proliferation [4].
The adaptive immune response in RA has been a focus of intense research over the last several decades. The strongest genetic link to RA involves SNPs in the Human Leukocyte Antigen (HLA)-DRB1 gene that likely alter T cell repertoire selection and antigen presentation, amongst other possibilities. The B cell repertoire is also constrained in RA patients, in which defects in early B cell tolerance checkpoints have been described [5], but for which there is still little mechanistic insight. Numerous antigens have been described in RA patients and in RA mouse models, and they include antibodies against the immunoglobulin Fc region (rheumatoid factor) and against citrullinated proteins (ACPA). Citrullination is a post-translational modification in which arginines are deiminated by peptidylarginine deiminases. ACPA are not only prevalent biomarkers for RA diagnosis, they also predict the development of the more debilitating erosive aspects of the disease. To date, it still remains unclear if specific autoantigen epitopes are key for the pathogenesis of RA, but autoantigens are clearly essential for the adaptive immune response in the pathogenesis of RA.
Numerous citrullinated polypeptides have been detected in the synovium of RA patients, populating a “citrullinome” that is comprised of both intracellular and extracellular antigens ranging in size from ~14-100 KDa [6]. ACPAs can be detected years before disease onset and are likely important for shaping the development of disease [7–9]. In support of this possibility it was recently shown that immune complexes with citrullinated histones are potent activators of myeloid cells (macrophages and neutrophils) and induce TNFα secretion [10].
Evidence from several mouse models of inflammation in the joint and of inflammatory arthritis provides valuable insight into the spatio-temporal dynamics of the antibody response [11]. The K/BxN transgenic mouse is a model that allows studying the mechanisms whereby autoantibodies drive inflammation in the joint. In K/BxN transgenic mice there is spontaneous inflammation in the articular space that presents synovial pannus formation and bone and cartilage erosions [12]. K/BxN mice spontaneously produce autoantibodies against glucose 6-phosphate isomerase (GPI) that selectively deposit in the joints as immune complexes and trigger complement fixation [13]. Transfer of serum containing arthritogenic anti-GPI antibodies from K/BxN transgenic mice into naïve wild-type mice leads to the development of inflamed joints clinically and pathologically similar to what is observed in K/BxN transgenic mice [14]. Given that the generation of autoantibodies in the K/BxN model is crucial for disease development, effort has been made to identify the anatomical location(s) and kinetics of anti-GPI antibody production. The anti-GPI antibody response initially localizes to draining LNs and persists until later stages of the disease [15]. This indicates that draining LNs are involved in the initiation and perpetuation of the inflammatory arthritis antibody response in this model. Evidence in the clinical setting for LN involvement in RA patients has long been appreciated, including data showing that immune responses develop in draining LNs prior to the appearance of arthritis symptoms [16]. Importantly, B cell depletion from the draining LNs (popliteal) ameliorates inflammatory arthritis [17,18], and B cell depletion therapies significantly reduce RA symptoms in humans [19].
The activation of the B cell antibody response in the LN requires two antigen-driven events that are segregated in distinct temporal and anatomical locations: 1.) antigen recognition by B cells in follicles 2.) antigen presentation by T-zone dendritic cells and subsequent T cell activation in the T cell zone. Relatively small soluble antigens <70 kDa can gain access to B and T cell areas of the LN by free diffusion via the follicular conduit network [20–22]. Larger antigens are transported into follicles through their initial capture by CD169+ subcapsular sinus macrophages from the incoming lymph, and relay to B cells in follicles in a complement receptor-dependent manner. These large immune complexes become subsequently deposited on follicular dendritic cells (FDCs) where they persist for long periods of time [23–26]. The molecular sizes of autoantigens uncovered in arthritis patients suggests both mechanisms of antigen delivery to T and B cell niches in secondary lymphoid organs are likely to operate in RA. But, T cells can also be activated by DCs that may sample antigens in inflamed articular spaces and migrate to the draining LN for T cell priming. In RA patients, both mature and immature DCs have long been identified in synovium and synovial tissue by histology [27–32]. Plasmacytoid DCs have also been identified in rheumatoid synovium and synovial fluid [33–35]. Whether various DC subsets are recruited to the joints during the earliest stages of synovial inflammation remains unknown.
Shortly after antigen-engagement, activated B cells increase the expression of the T-zone homing chemokine receptor CCR7, and migrate to the T-B border where they interact with a subset of activated (cognate) T cells [36]. At this stage, activated B cells receive further differentiation signals from cognate T cells mostly through CD40/CD40L interactions. Importantly, SNPs in CD40 have been linked to RA [4]. Within 1–2 days after antigen engagement and CD40 signaling B cells increase EBI2 (GPR183) expression [37–40] and together with activated T cells, migrate to outer and inter-follicular areas in an EBI2-dependent manner. The oxysterol ligands of EBI2 (mostly 7α, 25-hydroxycholesterol, (7α, 25-OHC)) are presumably abundant in the follicle perimeter, including interfollicular regions [37–40]. Dendritic cells also express EBI2 and migrate to inter-follicular niches, and signals or cellular interactions occurring at these sites contribute significantly to the development of primary antibody responses by promoting plasmablast differentiation [41,42]. Later in the immune response a subset of activated T and B cells initiates germinal center responses where antibody affinity maturation, isotype switching, and long-lived plasma cell differentiation take place.
The GC reaction is thought to play an important role in RA. Follicular dendritic cells (FDCs) are critical for the formation of autoreactive GC reactions and development of joint inflammation in the K/BxN model [43]. In RA patients, T and B cells form aggregates in the articular space and can form ectopic GCs in about 40% of patients [44]. Given that anti-B cell therapy (Rituximab) is inefficient at depleting antibody-secreting plasma cells, and variably affects antibody titers [19], its efficacy may be associated with other roles B cells play in the organization of ectopic lymphoid follicles and GCs, and in T cell activation through cytokine secretion and autoantigen presentation. These tertiary lymphoid structures do not form in most experimental mouse models of inflammatory arthritis, the exception being in the collagen-induced arthritis (CIA) model [45]. The fact that CIA displays many similar histophathological features with human RA has made it one of the most widely studied models of inflammatory arthritis [46]. In this model, type II collagen (an abundant protein in cartilage), is used as an antigen for immunizing mice, which leads to the development of inflammatory arthritis. CIA is often used to evaluate therapeutic interventions.
Inflammation in the synovial compartment
Despite many decades of intense research very little is still known about the kinetics of cell recruitment into synovial tissues in RA patients and in experimental inflammatory arthritis. In a permutation of the K/BxN inflammatory arthritis model, LaBranche et al. transferred autoreactive KRN TCR transgenic T cells into T cell-deficient mice (B6.TCR.cα−/−H-2b/g7) and were able to induce arthritic lesions that were synchronized and chronic in nature [47]. Interestingly, T cells were not detected in the joint but were present in the draining popliteal LN. Instead, monocyte/macrophage and neutrophil infiltration occurred early in disease progression, and correlated with bone and articular erosions. The influx of monocytes and macrophages, whether by recruitment or local differentiation and activation, highly correlates with inflammation and tissue damage in RA patients [48–50]. Given that monocytes and macrophages secrete numerous chemokines and cytokines, they also could be involved in the recruitment of other inflammatory cells into the joint, such as neutrophils. Neutrophils are abundant in synovial tissues and fluid, and mouse models provide clear evidence for neutrophil involvement in disease. Neutrophil depleted mice are completely resistant to the K/BxN serum transfer model of inflammatory arthritis [51]. A study in an anti-type II collagen antibody induced inflammatory arthritis model reported that the initial cellular infiltrate to the synovium is mainly composed of neutrophils and macrophages [52], and depletion of neutrophils attenuates disease progression [52,53]. However, the mechanisms controlled by neutrophils that exacerbate disease remain poorly understood. Furthermore, whether macrophages also contribute to the onset of disease remains unclear.
Recruitment of innate immune cells
In the synovium and synovial fluid of RA patients numerous homeostatic and inflammatory chemokines with the potential to influence the trafficking of leukocytes into the tissue are upregulated. CCR7+ mature DC (CD83+ and DC-LAMP+) present within synovial infiltrates of RA patients were found in close association with CCR7-ligand (CCL19 and CCL21) expressing cells, suggesting these chemokine pairs may play a role in the recruitment of DCs to the synovium [32]. In this study immature DC (CD1a+) expressed CCR6 and were in close association with CCL20-producing synovial cells [32]. In humans it was also recently reported that pDCs expressed CXCR3 and CXCR4 and accumulated in the inflamed joints of RA patients, [34]. Furthermore, the levels of CXCR3 ligands CXCL10, CXCL11, and of the CXCR4 ligand CXCL12 in the synovial fluid of RA patients were significantly increased when compared to osteoarthritis (OA) patients [34], suggesting the involvement of CXCR3 and CXCR4 in pDC homing or retention in inflamed joints. Besides pDCs, other DC subsets express functional CXCR4 [54], and CXCL12 was found to be highly expressed in synovial tissues [55,56] and synovial fibroblasts from RA patients [57].
In the K/BxN serum transfer model, neutrophils are absolutely required for disease progression [51]. Neutrophils express CXCR2 and its ligand CXCL2 is abundant in arthritic tissues including CXCL2 [58]. Furthermore, CXCR2 was crucial for disease progression in this model [58]. Importantly, blockade of CXCR2 in other animal models of arthritis quantitatively reduces disease severity and the recruitment of neutrophils [59–61]. Given that CXCR2 promotes neutrophil egress from bone marrow cavities [62], it is unclear if CXCR2 deficiency reduced inflammatory arthritis by impairing neutrophil recruitment to the synovium or by reducing their egress from bone marrow, or both. Using intravital 2-photon imaging of LysM-GFP mice and the K/BxN serum transfer model of arthritis, Wang and colleagues were able to visualize neutrophil recruitment to the inflamed synovial space [63]. Intriguingly, they observed that neutrophils were often associated with monocytes at sites of extravasation, and global depletion of monocytes arrested neutrophil extravasation [63].
Recruitment of lymphocytes
Th1, Th17, and Treg cell types are present in inflamed articular tissues in RA patients, with Th1 cells being the most prevalent T helper cell subset [64,65]. Studies from mouse models in combination with data obtained from RA patients have provided some insight into the trafficking mechanisms for T cells in arthritis. CXCR3 and CCR5 are abundantly expressed by T cells isolated from the synovial fluid and synovial tissues of RA patients, and both receptors are preferentially expressed on Th1 cells [66–68]. In adoptive transfer experiments in a mouse adjuvant arthritis model, T cells deficient in CXCR3 were poorly recruited to inflamed joints, and CXCR3 blockade using antibodies inhibited T cell recruitment and reduced disease severity [69]. Consistent with these findings, the CXCR3 ligands CXCL10 and CXCL9 are abundant in the synovial fluid of RA patients [66] and, interestingly, myeloid DCs isolated from the synovial fluid of RA patients express higher CXCL9 and CXCL10 amounts than myeloid DCs isolated from peripheral blood [70]. CCR5 has also been suggested to contribute to the recruitment of T cells to the synovium. Several studies have reported that a single nucleotide polymorphism (SNP) resulting in a non-functional CCR5 allele was negatively associated with RA [71–74], but this finding was not confirmed in other studies [75,76]. In animal models CCR5 deficiency had no effect on disease outcome [77].
In RA patients, CXCR4 is expressed on memory CD4+ T cells (CD45RO+) infiltrating the synovium [68]. In mice, T cell specific deletion of CXCR4 impaired their infiltration into the joints and decreased the severity of collagen-induced arthritis [78].
CX3CR1 is expressed on T cell subsets, and in RA patients the CX3CR1+ fraction of T cells is increased when compared to healthy controls [79]. The ligand for CX3CR1, fractalkine (CX3CL1), is expressed by synoviocytes and endothelial cells and is elevated in RA patients compared to OA patients [80,79,81]. In the collagen induced mouse model of arthritis, CX3CR1 is also expressed on T cells infiltrating the synovium and CX3CL1 is also upregulated [82]. Given that the depletion of CX3CL1 in synovial fluid in animal models of inflammatory arthritis reduced overall chemotactic potency [83], this chemokine receptor pair likely guides the recruitment of T cells to inflamed synovium.
Th17 cells play a key role in the pathogenesis of RA and these cells express CCR6 [84]. CCR6-expressing cells were identified in synovial tissue from RA patients several years ago [85], and the CCR6 ligand CCL20 is robustly secreted by synovial fibroblasts isolated from arthritic joints (but not secreted by granulocytes and monocytes) [84,85]. CCR6 blockade suppressed the onset and severity of disease and reduced the number of CD4+ T cells that infiltrated the joint [84], although the numbers of Th17 cells were not specifically reported.
Administration of FTY720, a high affinity agonist for sphingosine 1-phosphate (S1P) receptors that induces receptor internalization and renders targeted cells unresponsive to chemotactic gradients of S1P, was protective in mouse models of inflammatory arthritis by reducing lymphocytic infiltration into the joints and bone erosions [86–88]. Lymphocytes (including T cells) [86–88] and monocytes [87] were sequestrated in secondary lymphoid organs and thus, were unavailable to egress into the circulation and migrate to arthritic tissues. Although bone marrow populations were not examined in either study, it highlights the importance of this pathway in lymphocyte circulation and trafficking in inflammatory arthritis. S1P levels are elevated in the synovium and synovial fluid of RA patients (even higher than serum levels) and could contribute to cellular recruitment and retention [89,90].
An essential component of the pathogenesis of RA is B cells, highlighted by the efficacy of B cell depletion therapy [91,92]. In RA, B cell production of ACPA is strongly predictive of erosive bone disease [93]. B cells can be found in abundant numbers organized into follicular structures in the synovium, and in ACPA+ RA patients about 25% of IgG+ B cells isolated from articular space encode antigen receptors that are specific for citrullinated antigens [94]. Whether self-reactive B cells differentiate within ectopic follicles and germinal centers in the synovium, or are recruited from draining LNs is not entirely clear.
The chemokine expression pattern on B cells isolated from the synovium of RA patients includes CCR5, CCR6, CCR7, CXCR3, CXCR4, and CXCR5 [95]. The levels of CCL19, CXCL12, and CXCL13 are all increased in the serum of RA patients [96], CCL19 is increased in the synovium of RA patients [97], and CXCL13 is expressed in ectopic germinal centers in the synovium of RA patients [98]. This combination of homeostatic and inflammation-associated chemoattractant receptors suggests an active involvement in B lymphocyte recruitment and cell organization in synovial spaces.
Inflammation in the subchondral bone marrow
The synovium, and more recently LNs, represent the main focus of the inflammatory response in RA, but nearby compartments also develop inflammation including the subchondral bone marrow. The subchondral bone marrow is located under the subchondral bone that separates the articular cartilage from the bone marrow cavity. The bone marrow cavity is an important site of hematopoiesis but the subchondral bone marrow is populated by adipocytes and lacking abundant hematopoietic activity under homeostatic conditions. However, evidence indicates that subchondral bone marrow participates in the inflammatory response in RA. Magnetic resonance imaging data (MRI) from RA patients identifies edema throughout the bone marrow, even at stages where there is a low level of synovial inflammation [99,100]. Indeed, like synovitis, bone marrow edema is also predictive of subsequent pathologic bone erosion [101,102]. Subchondral bone marrow inflammation, including ectopic germinal center-like structures, has been found in the CIA mouse model [45,103,104]. The subchondral bone marrow undergoes significant changes during the course of RA, including infiltrating leukocytes [105–108] and development of bone resorbing osteoclasts (OC) on the subchondral surface [109,110]. Collectively, these studies indicate that the bone marrow compartment may be important for the pathogenesis of RA, but this compartment has not yet been fully integrated into disease models.
Bone erosion in RA
Periarticular bone erosions are highly prevalent in RA and lead to joint deformities and impaired function [111]. As the only cell capable of bone resorption in the body, osteoclasts have a critical role in inflammatory disorders associated with bone erosions and focal bone loss, including RA [112]. In mouse models of inflammatory arthritis numerous studies have demonstrated that osteoclasts are absolutely required for bone erosions to occur [113–118]. Osteoclasts form early in disease progression in animal models and initially appear at the junction of synovial membrane and periosteal lining and within subchondral bone [119]. Even though osteoclasts can be detected on both the synovial and subchondral bone marrow sides of the joints, osteoblasts (bone synthesizing cells) are only detected on the subchondral side [110,120–122]. Therefore the mechanisms of osteoclast differentiation from these two sides may be distinct.
Osteoclast differentiation under homeostasis
Osteoclasts are hematopoietic cells that differentiate from monocyte progenitors through sequential developmental stages. Although the exact identity of osteoclast precursors still remains unclear, studies have found that cells with osteoclastogenic potential in the bone marrow do not express lymphoid cell surface proteins or the aM integrin (CD11b/MAC1), and instead express the macrophage colony stimulating factor-1 receptor (Cfms, CSF1R, CD115) and the stem cell factor receptor (cKit). These cells overlap with the macrophage/dendritic cell precursor (MDP) subset [123] and can also differentiate into monocytes, macrophages, and dendritic cells [124–127]. More recently, a clonogenic, monocyte- and macrophage-restricted progenitor cell (named cMoP) derived from the MDP was identified [128], but whether the cMoP subset retains osteoclast differentiation potential has not been defined.
The dominant cytokine that governs osteoclast differentiation in vitro and in vivo is RANKL, which signals through its receptor RANK [129–131]. Osteoblasts are key sources of MCSF (a cytokine required for cell survival and osteoclastogenesis) and RANKL [132], and RANKL expressed by osteocytes was recently shown to be critical for osteoclast differentiation in vivo [133]. RANKL is a type II transmembrane receptor and as such requires cell-cell interactions for engaging its receptor RANK expressed on osteoclast precursors. This predicts that positional cues are involved in the recruitment of osteoclast precursors to interact with RANKL+ cells. For many years it has been appreciated that resorbing bone is chemotactic for osteoclast precursors [134]. Recently, osteoblasts were reported to express oxysterol-synthesis enzymes, namely cholesterol 25 hydroxylase (gene name Ch25h) and 25-hydroxycholesterol 7-alpha-hydroxylase (gene name Cyp7b1) [135]. They also secrete the oxysterol 7a, 25 dihydroxycholesterol (7a, 25-OHC), which acts as a potent chemotractant for the pertussis toxin sensitive Gai protein coupled receptor EBI2 [135].
Furthermore, EBI2 is abundantly expressed in murine osteoclast precursors, and its expression increases during osteoclast differentiation [135]. It was reported that EBI2 directs the migration of osteoclast precursors to bone endosteal niches and promotes osteoclastogenesis in vivo [135]. Besides its role in cell positioning at the endosteum, EBI2 signaling also contributes to promote osteoclast precursor motility within bone marrow parenchyma [135], possibly in synergy with CX3CR1 [136]. Importantly, deficiencies in EBI2 or CH25H resulted in similar decreases in osteoclast numbers and increased bone mass in vivo [135], indicating that 7a,25OHC is the main EBI2 ligand controlling osteoclastogenesis and bone mass homeostasis. In summary, this study provided direct in vivo evidence for EBI2 and oxysterols in osteoclast precursor recruitment to endosteal surfaces, and consequently in osteoclast differentiation [135].
The chemokine monocyte chemoattractant protein-1 (MCP-1) is also expressed by osteoblasts in vitro when cultured in inflammatory conditions [137–139] and the MCP-1 receptor, CCR2, is expressed on mouse and human osteoclast precursors [140,141]. These findings led to a model where bone proximal MCP-1, produced by osteoblasts, attracts osteoclast precursors to sites of osteoclast differentiation [142]. In support of this model, CCR2 deficient mice display a mild increase in bone mass and stability, likely due to a reduction in osteoclast differentiation [143]. In humans, a polymorphism in CCR2 (V64I) was associated with increased bone mineral density in middle aged men and postmenopausal women [144], but whether this polymorphism alters CCR2 signaling is unclear [145]. Altogether, the bone mass phenotype reported in CCR2 deficient mice is mild and contrasts with the essential role of CCR2 in promoting monocyte egress from bone marrow, both in homeostasis and during systemic acute inflammation [146–148].
CCR1 and CCR5 have also been reported as abundantly expressed on human, mouse and rat osteoclast precursors, upregulated on osteoclasts, and promoting their chemotaxis in response to several of their ligands, as reviewed elsewhere [149]. Furthermore, CCR1 and its ligands significantly promote osteoclast differentiation in vitro and in vivo (particularly CCL9) and CCR1 deficient mice are mildly osteopenic [150,149]. Several CCR1 ligands are shared by CCR5, but no bone mass phenotypes in CCR5 deficient mice have been reported.
The chemokine receptor pair CX3CR1 and its ligand CX3CL1 are also functionally expressed during osteoclast differentiation, as reviewed elsewhere [149]. Interestingly, CX3CL1 is produced by mouse osteoblasts [151] and CX3CR1 deficient mice have a reduction in osteoclasts that results in a mild increase in bone mass [152]. Furthermore, functional inhibition of CX3CR1 signaling provided protection from bone loss induced by irradiation [153], perhaps by disrupting CX3CR1-mediated migration and retention in the bone marrow [136]. In summary, while these studies suggest that synergy between multiple chemokine receptors controls osteoclastogenesis, only EBI2 was shown to be directly required for osteoclast precursor positioning in bone endosteal niches (Figure 1).
Fig. 1.
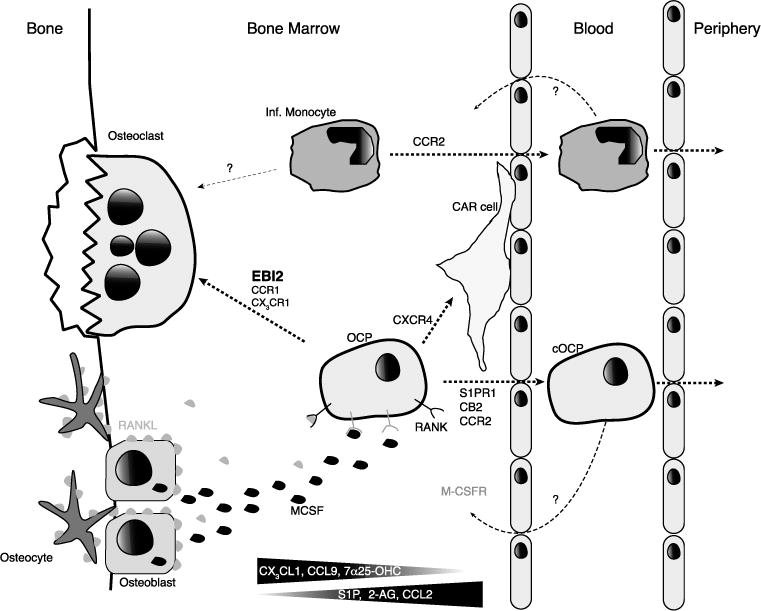
Trafficking of osteoclast precursors in the bone marrow (BM), blood, and peripheral tissues. Monocytic osteoclast precursor cells (OCP) and inflammatory monocytes (Inf Mono) migrate into osteoclastogenic niches where osteoblasts (OB) and osteocytes provide important signals for osteoclast differentiation (MCSF and RANKL). EBI2 directs the migration of OCP to the endosteum, where the EBI2 ligand 7α, 25-OHC is presumably abundant. CX3CR1 promotes the retention of OCP in BM parenchyma, and possibly directs cells to the endosteum in response to CX3CL1 produced by OB. OB also express CCR1 ligands (e.g. CCL9) and CCR1 may also direct OCP to sites of osteoclast differentiation. S1PR1 is essential for the egress of monocytic cells in the bone marrow as they follow S1P gradients into the circulation and inflammatory monocytes egress via CCR2. CXCR4 is likely to guide OCP away from sites of osteoclastogenesis given that its ligand CXCL12 is abundant parenchymal and perivacular mesenchymal stromal cells, while it is reduced in OBs. The CB2 ligand 2-AG is likely abundant in the BM sinusoids, and may guide OCP away from osteoclastogenic niches. Both circulatory OCP and inflammatory monocytes re-enter into BM by unknown mechanisms.
In contrast to chemoattractants derived from bone-proximal niches, chemoattractants emanating from bone-distal niches can also influence the positioning of osteoclast precursors in the bone marrow. Furthermore, the balance between bone marrow retention and egress cues is likely an important point of control for osteoclast precursor positioning and differentiation. In favor of such a model is the fact that osteoclasts are predominantly in close proximity to blood vessels in bone marrow thus making it likely that osteoclast precursors encounter bone and blood vessel chemoattractants (Figure 2) [149].
Fig. 2.
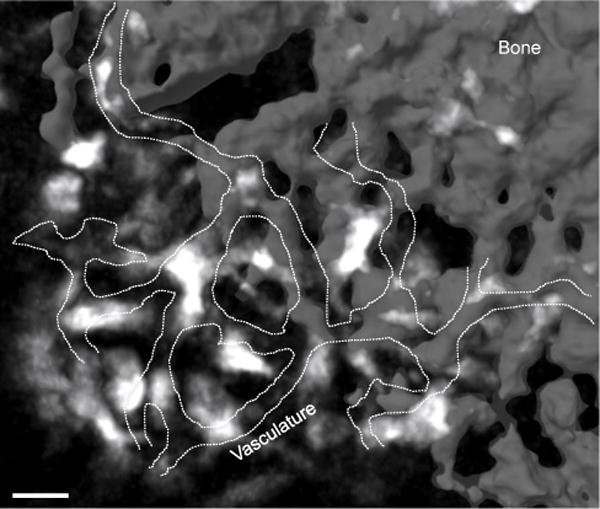
Osteoclasts are located in close proximity to blood vessels in the bone marrow. Image acquired using two-photon microscopy of calvaria bone marrow of TRAPRed reporter mouse. Osteoclasts (white); blood vessels (dashed line) were visualized with injection of dextran-FITC; and bone (gray). Scale bar= 30μm
Emerging evidence has provided insight into leukocyte migration and dynamic behavior in steady-state conditions, including the mechanisms controlling their egress from bone marrow. Leukocytes are thought to egress from bone marrow through a vast network of blood vessels often referred to as bone marrow sinusoids. The majority of bone marrow sinusoids are characterized by a single layer of endothelial cells often separated by small gaps and without a basement membrane. Several hematopoietic cells exit bone marrow parenchyma by moving towards bone marrow sinusoids in a chemoattractant-dependent manner. For example, natural killer cells, eosinophils and B lymphocytes express S1P receptors and migrate towards high concentrations of S1P within sinusoids [154–157]. Other hematopoietic cells, such as monocytes and neutrophils depend on chemokine receptors, namely CCR2 and CXCR2, for exiting bone marrow parenchyma, and CCR2 ligands are abundant around sinusoids [147,62,146]. Osteoclast precursors have also been shown to use S1P receptors for exiting bone marrow, and defects in S1PR signaling alter osteoclast precursor differentiation [158,159]. CXCR4 is also expressed in osteoclast precursors and directs their migration towards CXCL12 in vitro [160,161]. CXCR4, and its ligand CXCL12, have a well-described role in the retention of multiple hematopoietic cell subsets in bone marrow, including hematopoietic stem and progenitor cells [162]. Even though CXCR4 is one of the most studied chemokine receptors, its role in steady-state osteoclastogenesis in vivo remains controversial. Several studies reported that osteoblasts express CXCL12 in vivo [163,164] and it was proposed that CXCR4 promotes osteoclast precursor migration towards sites of osteoclast differentiation in vivo [158,165]. However, studies using CXCR4 deficient and CXCL12 reporter mouse strains provided results that are not compatible with this model. For example, CXCL12 expression is highest in rare mesenchymal stromal cells capable of multilineage differentiation (osteoblasts, chondrocyte, and adipocytes) distributed in parenchyma and some adjacent to blood vessels [166–168]. Furthermore, CXCL12 expression is reduced by nearly 100-fold in osteoblasts in vivo [169], making it unlikely that CXCR4 would direct osteoclast precursors towards osteoblasts. In agreement with this possibility, osteoclast differentiation and bone resorption were increased when hematopoietic cells lack CXCR4 expression, [170], which favors a model where the balance between responsiveness to bone chemoattractants and to other chemoattractants influences osteoclast development in vivo (Figure 1).
Cannabinoid receptor-2 (CB2) is abundantly expressed in various hematopoietic cells, including monocytes/osteoclast precursors and it promotes cell migration to its ligand 2-arachidonoylglycerol (2-AG) [171]. In vivo, 2-AG activity is likely high in (or near) bone marrow sinusoids given that CB2 promotes immature B lymphocyte positioning in these locations, a process that is also dependent on α4β1 adhesion to VCAM-1 [172]. It is, thus, conceivable that monocytes and osteoclast precursors are similarly attracted and/or retained within bone marrow sinusoids via CB2 chemotactic activity. In support of this hypothesis, CB2 deficient mice exhibit low trabecular bone mass, and CB2 antagonists inhibit osteoclast differentiation in vitro [173,174]. Furthermore, in humans a single nucleotide polymorphism in CNR2 (encoding CB2) was significantly associated with osteoporosis [175–177].
Leukocytes can exit bone marrow through mechanisms that are independent of pertussis toxin-sensitive Gαi protein coupled receptors, and presumably independent of chemoattractant gradient sensing and cell intrinsic motility [178]. B-lineage lymphocytes enforced to express pertussis toxin or deficient in CXCR4 expression were found to be largely non-motile within bone marrow cavities of live mice, and were rapidly mobilized from bone marrow parenchyma into blood [178]. It was also noted that the bone marrow parenchyma is under shear stress induced by plasma perfusion and interstitial fluid flow [178]. It is plausible that the highly fenestrated nature of the sinusoidal network in combination with plasma and interstitial fluid flow back to collecting sinusoids allows non-motile cells (e.g. red blood cells) to exit the bone marrow in a passive manner, and that such unconventional exit routes are used by essentially all leukocytes, including osteoclast precursors.
Osteoclast differentiation within inflamed synovial space
The identity of osteoclast precursors in arthritis may be distinct from steady-state osteoclast precursors, but presumably belongs to the myeloid cell compartment. Myeloid cells are accumulated in synovial tissue and synovial fluid in RA [48,49]. Some studies have interrogated the phenotype of osteoclast precursors in inflammatory arthritis models. The hTNFα transgenic strain develops synovial hyperplasia and lymphocytic infiltrate, pannus formation, articular cartilage destruction, and osteoclast driven bone erosion [179]. In this model, a cell population expressing the aM integrin CD11b but not Gr-1 displayed osteoclastogenic potential, and this population was increased in the bone marrow and blood of hTNFα transgenic mice [180]. Another study utilizing the SKG model of spontaneous inflammatory arthritis identified a population of cells with osteoclastogenic potential that had low to negative expression for CD11b and expressed high levels of Ly6C [125], and these cells may overlap with cMoPs (Nevius and Pereira unpublished observations). Dendritic cells have also been reported to contain osteoclast differentiation potential. Specifically, immature DCs were able to form osteoclasts in response to MCSF and RANKL, and unidentified soluble factors in human synovial fluid increased the DC differentiation into osteoclasts. These findings indicate that DCs may contribute to arthritis not only by acting as antigen-presenting cells and promoting T cell activation, but also by their potential to differentiate into bone-resorbing osteoclasts [181,182]. Collectively these studies suggest that multiple myeloid cell populations contain osteoclast differentiation potential (Figure 3).
Fig. 3.
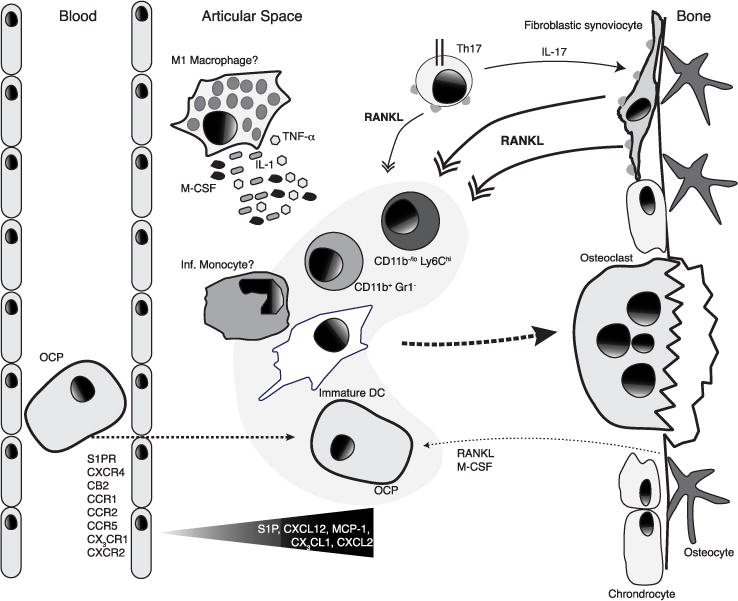
Trafficking of monocytic osteoclast precursors (OCP) into inflamed joints. Cells with osteoclastogenic potential include CD11b−/loLy6Chi, CD11b+GR-1−, and DCs. In RA, sinusoidal fibroblastic cells provide RANKL, which can be induced by IL-17 provided by Th17 cells. The cytokines TNF-α, IL-1, and IL-6, which may be locally secreted by macrophages also promote osteoclast differentiation under inflammatory conditions. S1P receptor expression on OCPs possibly directs cells into the synovial tissue where S1P is upregulated during inflammation. CXCR4 also likely directs cells into the synovial tissue with fibroblasts, and possibly other cells, express high levels of CXCL12. Selective antagonism of CB2 inhibits the migration of monocytes into the synovium, indicating that 2-Ag levels may be present in synovial fluid. CXCR2, CX3CR1, CCR1, CCR2, and CCR5 are also implicated in inflammatory cell recruitment into the inflamed articular space.
RANKL is expressed on activated T cells, B cells, DCs, and synovial fibroblasts, besides bone-producing cells, RANKL expressed on T cells [133,183] and B cells [184] is dispensable for osteoclast differentiation and skeletal development and maintenance under homeostatic conditions. However, in mouse models of inflammatory arthritis, and in RA patients, the expression of RANKL on T cells and synovial fibroblasts is robust [113,185–187]. In murine inflammatory arthritis it has been established that synovial fibroblasts support the conversion of FOXP3+ Tregs into pathogenic Th17 cells, which express higher RANKL amounts than other CD4+ T cell subsets [188]. Furthermore, Th17-derived RANKL is able to support osteoclastogenesis in vitro [188,189]. That the cytokine IL-17A is an important driver of osteoclastogenesis in arthritis has been appreciated for some time now [190], but the mechanisms of its action are still being elucidated. Interestingly, IL-17A has been shown to induce RANK on human and murine osteoclast precursors and promotes osteoclastogenesis [191–193]. In addition, some evidence suggests IL-17A may increase RANKL expression on fibroblastic synoviocytes and/or mesenchymal-derived cells [189]. A recent study has dissected the relative contributions of RANKL derived from T cells and synovial fibroblasts and found that synovial fibroblasts provide the relevant sources of RANKL in models of inflammatory arthritis [194]. In humans, RANKL blockade with denosumab reduces bone erosion possibly through inhibition of osteoclast differentiation and function [195]. In mice, RANKL-expressing Th17 cells have been reported to interact directly with osteoclasts in vivo. By fluorescently labeling osteoclasts and Th17 cells it was observed that Th17 cells preferentially located in contact with mature osteoclasts and this interaction was partially dependent on RANKL [196]. Finally, by using a pH sensitive fluorescent probe and an indicator of local pH decreases associated with osteoclast H+ secretion and bone resorption it was observed that Th17 and osteoclast interactions occurred in sites of active bone resorption [196]. Collectively through these studies a picture emerges in which Th17 derived IL-17A regulates the expression of RANKL on synovial fibroblasts, enhances the differentiation of osteoclasts in the inflamed joint, and exacerbates bone resorption in vivo (Figure 3).
As mentioned previously, MCSF and RANKL are critical cytokines for osteoclastogenesis, and while MCSF is a soluble cytokine, RANKL is a type II membrane protein. Therefore osteoclast precursors in the joint presumably receive RANK signaling directly from fibroblastic synoviocytes, and possibly from T cells, osteoblasts, or from interaction with osteocyte processes at the bone surface (Figure 3). Given that osteoclasts can form bi-directionally in the joint (synovium and bone marrow), the specific cell type presenting RANKL (and other cytokines) on the synovial or bone marrow side may differ, and may be under the influence of distinct cell recruitment mechanisms.
RANKL independent osteoclast differentiation in RA
It is now appreciated that in inflammatory settings osteoclast differentiation can occur independently of RANKL signaling [197]. Inflammatory cytokines such as TNFα [198], TNFα with IL-6 [199], and IL-1α [200] can induce osteoclast differentiation when in the presence of MCSF, but non-inflammatory cytokines like TGFβ have also been shown to trigger osteoclast differentiation [201]. Collectively these data identify cytokine-driven alternative pathways of osteoclast differentiation not dependent on the RANK/RANKL signaling pathway. Many of these pro-osteoclastogenic cytokines can be produced by macrophages, as reviewed elsewhere [197], and synovial macrophage numbers correlate with severity of inflammatory activity in RA [202]. Although pro-osteoclastogenic cytokines are not exclusively produced by synovial macrophages, these cytokines are present in the synovial milieu and may contribute to alternative pathways of osteoclast differentiation. Recent work from Harre and colleagues found that ACPAs enhanced the differentiation of osteoclasts in vitro [203]. They further showed that administration of ACPAs to lymphocyte deficient Rag1−/− mice was able to increase osteoclast numbers and reduce bone mass in vivo [203]. The mechanism by which ACPAs promote osteoclast differentiation is in part dependent on the Fc sialylation state of IgG in immune complexes [204]. Alternative pathways of osteoclast differentiation, driven by the unique inflammatory cytokine milieu (e.g. TNFα) and pathogenic autoantibody responses likely synergize with the dominant RANK/RANKL signaling pathway to induce bone erosions associated with RA. Indeed, TNFα induces RANKL expression on stromal cells via IL-1α signaling and enhance RANKL-dependent pathways of osteoclast differentiation [205].
Recruitment of osteoclast precursors in RA
It is clear in murine inflammatory arthritis and human RA that osteoclast precursors populate the synovium and synovial fluid. Conceptually, osteoclast differentiation in RA is likely controlled by multiple guidance cues balancing osteoclast precursor retention in bone marrow, migration from the bone marrow to egress sites, and migration into tissues, i.e. the synovium (Figure 3). The chemotactic signals guiding cells to these sites are incompletely defined and are of great interest. Some studies indicate that the cytokine TNFα, which is abundant in synovial tissue, can expand osteoclast precursors in inflammatory arthritis models and can also induce the mobilization of osteoclast precursors from the bone marrow into the periphery [206,207,180]. Given that in RA patients S1P levels are elevated in the synovium and synovial fluid (even higher than serum levels) [89,90], S1P receptor signalling could contribute to monocyte recruitment and retention in the joints. Consistent with this hypothesis, blockade of S1P signaling by administration of FTY720 reduces lymphocyte [86–88] and monocyte [87] migration to the joints.
Human and mouse monocytes/osteoclast precursors and osteoclasts also express functional CCR1, CCR5, and CCR2. The CCR2 and MCP-1 chemokine receptor pair not only promotes monocyte egress from bone marrow under homeostatic and inflammatory conditions [146–148], but MCP-1 expression is increased in inflamed synovial fluids [208,209]. Therefore, these collective reports suggest that CCR2 attracts monocytes/osteoclast precursors from blood circulation to inflamed joints. It remains to be completely dissected the relative contribution of CCR2 in the egress of monocytic cells and in the recruitment of those cells into tissues like the inflamed synovium. Potential sources for MCP-1 include osteoblasts and mesenchymal stromal cells.
Early studies in mouse models showed that the severity of inflammatory arthritis is mitigated by administration of receptor antagonists to CCR2 and CCR5 [210,211]. It was subsequently elucidated that the kinetics of CCR2 blockade are crucial for a beneficial outcome [212]. However, genetic deletion of these receptors had an opposite effect. Mice genetically deficient in CCR2 and CCR5 displayed either enhanced severity of inflammatory arthritis (including enhanced monocyte recruitment) or had no effect on disease outcome, respectively [77]. More recently, inflammatory arthritis induced in CCR2 deficient mice were described to have increased Th17 cells, which may partially explain the disease exacerbation [213]. The blockade of another GPCR, CCR1, in inflammatory arthritis mouse models showed both inhibition of chemotaxis and reduction of inflammation [214,215,211,216].
The promising data from mouse models prompted the development and testing of CCR1, CCR2, and CCR5 antagonists as a therapeutic approach for the treatment of RA [217–221]. However, the results of the clinical trials were variable and did not show an overt efficacy in the treatment of RA patients. This could be in part due to the redundancy of the chemokine receptor and chemokine system. For example, CCR1 has 13 distinct chemokine ligands, some of which are shared ligands for CCR2, CCR3, CCR5, and CCR10. There are additional chemokine receptors that have been suggested to regulate the recruitment of monocytes/osteoclast precursors in RA.
Other chemokine receptor pairs that may regulate the recruitment of osteoclast precursors to the bone surfaces in homeostasis may also play a role in inflammatory conditions, like RA. For instance, CXCL12 is highly expressed in synovial tissues [55,56] and synovial fibroblasts from RA patients [57] and the migration of monocytes to supernatants from synovial fibroblasts was significantly decreased after neutralization of CXCL12 [222]. In the context of RA, CB2 is more abundantly expressed in the synovial tissues of RA patients, and administration of a selective CB2 antagonist in a mouse model ameliorated inflammatory arthritis and bone destruction [223]. Although the distribution of 2-AG within bone marrow has not been exactly defined, the fact that CB2 signaling occurs in the vicinity of (or within) bone marrow sinusoids under normal homeostatic conditions, and that it can be detected in synovial tissues, suggests that 2-AG may act as a chemoattractant or a retention signal. Finally, the neutralization of the CX3CR1 ligand, CX3CL1 (fractalkine), in synovial fluid in murine inflammatory arthritis reduced overall chemotactic potency [83]. Blockade of CX3CR1 in a murine model decreased the recruitment of monocytic cells to synovial tissues and decreased disease incidence and severity [82]. CX3CL1 is elevated in RA patients compared to OA patients [81,80] and CX3CL1 has been shown to be upregulated in mouse bone marrow endothelial cells in inflammatory conditions, such as full body irradiation [153].
CXCR2 and CXCL2 have also been implicated in inflammatory bone remodeling [224]. In osteoclast precursors, CXCL2 is induced by RANKL and promotes their chemotaxis to CXCL2 [224]. The expression of CXCL2 was increased in the synovial fluid of RA patients and CXCL2 significantly enhanced bone resorption in vivo [224]. Therapeutic neutralization of this chemokine receptor pair had positive results in animal models of arthritis [225,60,226], possibly owing to the inhibition of CXCL2-mediated recruitment of osteoclast precursors to inflamed joints. In the K/BxN serum transfer model, the relevant chemokine receptor for arthritis progression was shown to be CXCR2, specifically for the recruitment of neutrophils [58]. But this study does not preclude an important role for CXCR2 in the recruitment of osteoclast precursors [58].
Under homeostasis, EBI2 and CH25H are required for osteoclast precursor recruitment and osteoclast differentiation. But, EBI2 and CH25H are also required for the generation of antibody responses, and have also been implicated in a variety of inflammatory diseases. SNPs in Ebi2 or in regulatory elements of Ebi2 expression are associated with cardiac inflammation, type I diabetes and inflammatory bowel disease [227–229]. Furthermore, Ch25h is one of the most induced interferon-stimulated genes [230]. Future studies should determine if CH25H activity is increased in the inflamed articular space, and if EBI2 ligands play roles in inflammatory cell recruitment and osteoclast differentiation in inflamed joints.
While many studies interrogated functional roles for chemoattractant receptors in osteoclast differentiation and bone erosions in RA patients, and mouse models of RA, little remains understood about the manner in which they contribute to disease. It is likely that osteoclast precursor differentiation within inflamed synovium requires sequential chemotactic signals: at the first stage cells must be captured from blood circulation and extravasate into the synovium. Subsequently, osteoclast precursors likely follow chemotactic gradients that not only promote retention within the articular space but may also direct their migration towards sites of osteoclast differentiation. It is possible that redundancy between chemokine and other chemoattractants control such multi-step process of osteoclast differentiation.
Inflammation induced changes in bone homeostasis
Hematopoiesis has been proposed to be regulated locally by cytokines [231]. Cytokines are capable of instructing hematopoietic lineage choice by acting on 1) HSC, 2) Hematopoietic progenitor cells (HPC), and 3) mesenchymal stromal cells that support HSC. Furthermore, inflammatory cytokines, such as type I interferons may also function as rheostats of HSC proliferation [232,233]. During stress response, HSC enter in cycle and self-renew but when homeostasis is reestablished, HSC return to a quiescent state [234].
The inflammatory cytokine signature of RA includes several cytokines that have profound effects in hematopoiesis by biasing cell lineage choices. Two of the most notorious are TNFα and IL-1, and these cytokines can induce hematopoietic shifts that favor the production of inflammatory, innate-type cells, such as neutrophils and monocyte-lineage cells, at the expense of lymphopoiesis [235,236]. Of note, systemic TNFα increases the number of CD11bhigh osteoclast precursors in circulation [237], and this effect may in part be due to an increase in osteoclast precursor proliferation and differentiation in bone marrow [180]. Such inflammation-induced shifts in hematopoiesis are most likely dependent on local G-CSF production, although it is possible that other inflammatory signals contribute to this effect [238]. In experimental mouse models of RA, genetic and pharmacologically induced G-CSF deficiency prevented acute and chronic arthritis [239], possibly because it reduced myeloid cell differentiation in bone marrow.
Concluding remarks
It is becoming increasingly understood that a variety of processes controlled by the adaptive immune system are the major determinants of RA. But, adaptive immunity requires instructive signals provided by innate immune effector mechanisms, thus leaving open the question of which is/are the triggers that inadvertently fire self-reactive lymphocytes. Furthermore, even though RA does not develop without lymphocytes, disease only manifests when innate immune effector cells infiltrate articular spaces and cause pathology. Of note, the resorptive activity of synovial osteoclasts is a hallmark feature of chronic, advanced stage RA and is a major contributor to articular damage.
Considering the multiple pathways EBI2 signaling is involved in (namely osteoclast development and the generation of adaptive immune responses), which are central to the development of inflammation in articular spaces, antagonists of EBI2 signaling may provide significant protection against RA. Understanding the multitude of osteoclast differentiation pathways and mechanisms of osteoclast precursor recruitment in homeostasis and during inflammation is thus of utmost importance.
Acknowledgments
We thank L.K. Bockenstedt for critical review of the manuscript. This work was funded by the NIH (RO1AI113040) and by the Careers in Immunology Fellowship program of the American Association of Immunologists.
A. C. Gomes was supported by a PhD fellowship (SFRH/BD/73782/2010) from
Footnotes
Fundação para a Ciência e Tecnologia (FCT, Portugal).
Conflict of Interest: E. Nevius, A.C. Gomes, and J.P. Pereira declare that they have no conflict of interest.
References
- 1.Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nature reviews Genetics. 2013;14(9):661–673. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- 2.Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, Hatan M, Carrasco-Alfonso MJ, Mayer D, Luckey CJ, Patsopoulos NA, De Jager PL, Kuchroo VK, Epstein CB, Daly MJ, Hafler DA, Bernstein BE. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518(7539):337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. The New England journal of medicine. 2011;365(23):2205–2219. doi: 10.1056/NEJMra1004965. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 5.Samuels J, Ng YS, Coupillaud C, Paget D, Meffre E. Impaired early B cell tolerance in patients with rheumatoid arthritis. The Journal of experimental medicine. 2005;201(10):1659–1667. doi: 10.1084/jem.20042321. doi: jem.20042321 [pii] 10.1084/jem.20042321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Beers JJ, Schwarte CM, Stammen-Vogelzangs J, Oosterink E, Bozic B, Pruijn GJ. The rheumatoid arthritis synovial fluid citrullinome reveals novel citrullinated epitopes in apolipoprotein E, myeloid nuclear differentiation antigen, and beta-actin. Arthritis and rheumatism. 2013;65(1):69–80. doi: 10.1002/art.37720. [DOI] [PubMed] [Google Scholar]
- 7.Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis and rheumatism. 2003;48(10):2741–2749. doi: 10.1002/art.11223. [DOI] [PubMed] [Google Scholar]
- 8.Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis and rheumatism. 2004;50(2):380–386. doi: 10.1002/art.20018. [DOI] [PubMed] [Google Scholar]
- 9.Schellekens GA, Visser H, de Jong BA, van den Hoogen FH, Hazes JM, Breedveld FC, van Venrooij WJ. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis and rheumatism. 2000;43(1):155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 10.Hyun Sohn D, Rhodes C, Onuma K, Zhao X, Sharpe O, Gazitt T, Shiao R, Fert-Bober J, Cheng D, Lahey LJ, Wong HH, Van Eyk J, Robinson WH, Sokolove J. Local joint inflammation and histone citrullination provides a murine model for the transition from preclinical autoimmunity to inflammatory arthritis. Arthritis & rheumatology. 2015 doi: 10.1002/art.39283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevaart L, Vervoordeldonk MJ, Tak PP. Evaluation of therapeutic targets in animal models of arthritis: how does it relate to rheumatoid arthritis? Arthritis Rheum. 2010;62(8):2192–2205. doi: 10.1002/art.27503. [DOI] [PubMed] [Google Scholar]
- 12.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3(4):360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 14.Maccioni M, Zeder-Lutz G, Huang H, Ebel C, Gerber P, Hergueux J, Marchal P, Duchatelle V, Degott C, van Regenmortel M, Benoist C, Mathis D. Arthritogenic monoclonal antibodies from K/BxN mice. J Exp Med. 2002;195(8):1071–1077. doi: 10.1084/jem.20011941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandik-Nayak L, Wipke BT, Shih FF, Unanue ER, Allen PM. Despite ubiquitous autoantigen expression, arthritogenic autoantibody response initiates in the local lymph node. Proc Natl Acad Sci U S A. 2002;99(22):14368–14373. doi: 10.1073/pnas.182549099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly CA, Malcolm AJ, Griffiths I. Lymphadenopathy in rheumatic patients. Annals of the rheumatic diseases. 1987;46(3):224–227. doi: 10.1136/ard.46.3.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Kuzin I, Moshkani S, Proulx ST, Xing L, Skrombolas D, Dunn R, Sanz I, Schwarz EM, Bottaro A. Expanded CD23(+)/CD21(hi) B cells in inflamed lymph nodes are associated with the onset of inflammatory-erosive arthritis in TNF-transgenic mice and are targets of anti-CD20 therapy. J Immunol. 2010;184(11):6142–6150. doi: 10.4049/jimmunol.0903489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Ju Y, Bouta EM, Xing L, Wood RW, Kuzin I, Bottaro A, Ritchlin CT, Schwarz EM. Efficacy of B cell depletion therapy for murine joint arthritis flare is associated with increased lymphatic flow. Arthritis and rheumatism. 2013;65(1):130–138. doi: 10.1002/art.37709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350(25):2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 20.Pape KA, Catron DM, Itano AA, Jenkins MK. The humoral immune response is initiated in lymph nodes by B cells that acquire soluble antigen directly in the follicles. Immunity. 2007;26(4):491–502. doi: 10.1016/j.immuni.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 21.Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, von Andrian UH, Carroll MC. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity. 2009;30(2):264–276. doi: 10.1016/j.immuni.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sixt M, Kanazawa N, Selg M, Samson T, Roos G, Reinhardt DP, Pabst R, Lutz MB, Sorokin L. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity. 2005;22(1):19–29. doi: 10.1016/j.immuni.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity. 2007;27(1):160–171. doi: 10.1016/j.immuni.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, van Rooijen N, Mempel TR, Whelan SP, von Andrian UH. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450(7166):110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 25.Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8(9):992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- 26.Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol. 2009;10(7):786–793. doi: 10.1038/ni.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoh J, Kinjoh K, Ohyama A, Nose M, Kyogoku M. Application of two-color immunofluorescence staining to demonstration of T-cells and HLA-DR-bearing cells in rheumatoid synovitis. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1992;40(11):1675–1683. doi: 10.1177/40.11.1431056. [DOI] [PubMed] [Google Scholar]
- 28.van Dinther-Janssen AC, Pals ST, Scheper R, Breedveld F, Meijer CJ. Dendritic cells and high endothelial venules in the rheumatoid synovial membrane. The Journal of rheumatology. 1990;17(1):11–17. [PubMed] [Google Scholar]
- 29.de Vere Tyndall A, Knight SC, Edwards AJ, Clarke JB. Veiled (dendritic) cells in synovial fluid. Lancet. 1983;1(8322):472–473. doi: 10.1016/s0140-6736(83)91468-x. [DOI] [PubMed] [Google Scholar]
- 30.Zvaifler NJ, Steinman RM, Kaplan G, Lau LL, Rivelis M. Identification of immunostimulatory dendritic cells in the synovial effusions of patients with rheumatoid arthritis. The Journal of clinical investigation. 1985;76(2):789–800. doi: 10.1172/JCI112036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas R, Davis LS, Lipsky PE. Rheumatoid synovium is enriched in mature antigen-presenting dendritic cells. J Immunol. 1994;152(5):2613–2623. [PubMed] [Google Scholar]
- 32.Page G, Lebecque S, Miossec P. Anatomic localization of immature and mature dendritic cells in an ectopic lymphoid organ: correlation with selective chemokine expression in rheumatoid synovium. J Immunol. 2002;168(10):5333–5341. doi: 10.4049/jimmunol.168.10.5333. [DOI] [PubMed] [Google Scholar]
- 33.Cavanagh LL, Boyce A, Smith L, Padmanabha J, Filgueira L, Pietschmann P, Thomas R. Rheumatoid arthritis synovium contains plasmacytoid dendritic cells. Arthritis research & therapy. 2005;7(2):R230–240. doi: 10.1186/ar1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lande R, Giacomini E, Serafini B, Rosicarelli B, Sebastiani GD, Minisola G, Tarantino U, Riccieri V, Valesini G, Coccia EM. Characterization and recruitment of plasmacytoid dendritic cells in synovial fluid and tissue of patients with chronic inflammatory arthritis. J Immunol. 2004;173(4):2815–2824. doi: 10.4049/jimmunol.173.4.2815. [DOI] [PubMed] [Google Scholar]
- 35.Van Krinks CH, Matyszak MK, Gaston JS. Characterization of plasmacytoid dendritic cells in inflammatory arthritis synovial fluid. Rheumatology. 2004;43(4):453–460. doi: 10.1093/rheumatology/keh115. [DOI] [PubMed] [Google Scholar]
- 36.Pereira JP, Kelly LM, Cyster JG. Finding the right niche: B-cell migration in the early phases of T-dependent antibody responses. International immunology. 2010;22(6):413–419. doi: 10.1093/intimm/dxq047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity. 2009;31(2):259–269. doi: 10.1016/j.immuni.2009.06.016. doi: S1074-7613(09)00313-6 [pii] 10.1016/j.immuni.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 38.Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP, Guerini D, Baumgarten BU, Roggo S, Wen B, Knochenmuss R, Noel S, Gessier F, Kelly LM, Vanek M, Laurent S, Preuss I, Miault C, Christen I, Karuna R, Li W, Koo DI, Suply T, Schmedt C, Peters EC, Falchetto R, Katopodis A, Spanka C, Roy MO, Detheux M, Chen YA, Schultz PG, Cho CY, Seuwen K, Cyster JG, Sailer AW. Oxysterols direct immune cell migration via EBI2. Nature. 2011;475(7357):524–527. doi: 10.1038/nature10280. doi:nature10280 [pii] 10.1038/nature10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu C, Yang XV, Wu J, Kuei C, Mani NS, Zhang L, Yu J, Sutton SW, Qin N, Banie H, Karlsson L, Sun S, Lovenberg TW. Oxysterols direct B-cell migration through EBI2. Nature. 2011;475(7357):519–523. doi: 10.1038/nature10226. doi: nature10226 [pii] 10.1038/nature10226. [DOI] [PubMed] [Google Scholar]
- 40.Pereira JP, Kelly LM, Xu Y, Cyster JG. EBI2 mediates B cell segregation between the outer and centre follicle. Nature. 2009;460(7259):1122–1126. doi: 10.1038/nature08226. doi:nature08226 [pii] 10.1038/nature08226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gatto D, Wood K, Caminschi I, Murphy-Durland D, Schofield P, Christ D, Karupiah G, Brink R. The chemotactic receptor EBI2 regulates the homeostasis, localization and immunological function of splenic dendritic cells. Nature immunology. 2013;14(5):446–453. doi: 10.1038/ni.2555. doi: ni.2555 [pii] 10.1038/ni.2555. [DOI] [PubMed] [Google Scholar]
- 42.Yi T, Cyster JG. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. Elife. 2013;2:e00757. doi: 10.7554/eLife.00757. 00757 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Victoratos P, Kollias G. Induction of autoantibody-mediated spontaneous arthritis critically depends on follicular dendritic cells. Immunity. 2009;30(1):130–142. doi: 10.1016/j.immuni.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 44.Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O’Fallon WM, Goronzy JJ, Weyand CM. Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001;167(2):1072–1080. doi: 10.4049/jimmunol.167.2.1072. [DOI] [PubMed] [Google Scholar]
- 45.Han S, Cao S, Bheekha-Escura R, Zheng B. Germinal center reaction in the joints of mice with collagen-induced arthritis: an animal model of lymphocyte activation and differentiation in arthritis joints. Arthritis and rheumatism. 2001;44(6):1438–1443. doi: 10.1002/1529-0131(200106)44:6<1438::AID-ART239>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 46.Courtenay JS, Dallman MJ, Dayan AD, Martin A, Mosedale B. Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980;283(5748):666–668. doi: 10.1038/283666a0. [DOI] [PubMed] [Google Scholar]
- 47.LaBranche TP, Hickman-Brecks CL, Meyer DM, Storer CE, Jesson MI, Shevlin KM, Happa FA, Barve RA, Weiss DJ, Minnerly JC, Racz JL, Allen PM. Characterization of the KRN cell transfer model of rheumatoid arthritis (KRN-CTM), a chronic yet synchronized version of the K/BxN mouse. The American journal of pathology. 2010;177(3):1388–1396. doi: 10.2353/ajpath.2010.100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burmester GR, Stuhlmuller B, Keyszer G, Kinne RW. Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis and rheumatism. 1997;40(1):5–18. doi: 10.1002/art.1780400104. [DOI] [PubMed] [Google Scholar]
- 49.Tak PP, Smeets TJ, Daha MR, Kluin PM, Meijers KA, Brand R, Meinders AE, Breedveld FC. Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis and rheumatism. 1997;40(2):217–225. doi: 10.1002/art.1780400206. [DOI] [PubMed] [Google Scholar]
- 50.Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis and rheumatism. 1996;39(1):115–124. doi: 10.1002/art.1780390116. [DOI] [PubMed] [Google Scholar]
- 51.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167(3):1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka D, Kagari T, Doi H, Shimozato T. Essential role of neutrophils in anti-type II collagen antibody and lipopolysaccharide-induced arthritis. Immunology. 2006;119(2):195–202. doi: 10.1111/j.1365-2567.2006.02424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eyles JL, Hickey MJ, Norman MU, Croker BA, Roberts AW, Drake SF, James WG, Metcalf D, Campbell IK, Wicks IP. A key role for G-CSF-induced neutrophil production and trafficking during inflammatory arthritis. Blood. 2008;112(13):5193–5201. doi: 10.1182/blood-2008-02-139535. [DOI] [PubMed] [Google Scholar]
- 54.Penna G, Sozzani S, Adorini L. Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J Immunol. 2001;167(4):1862–1866. doi: 10.4049/jimmunol.167.4.1862. [DOI] [PubMed] [Google Scholar]
- 55.Burman A, Haworth O, Hardie DL, Amft EN, Siewert C, Jackson DG, Salmon M, Buckley CD. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J Immunol. 2005;174(3):1693–1700. doi: 10.4049/jimmunol.174.3.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blades MC, Ingegnoli F, Wheller SK, Manzo A, Wahid S, Panayi GS, Perretti M, Pitzalis C. Stromal cell-derived factor 1 (CXCL12) induces monocyte migration into human synovium transplanted onto SCID Mice. Arthritis and rheumatism. 2002;46(3):824–836. doi: 10.1002/art.10102. [DOI] [PubMed] [Google Scholar]
- 57.Bradfield PF, Amft N, Vernon-Wilson E, Exley AE, Parsonage G, Rainger GE, Nash GB, Thomas AM, Simmons DL, Salmon M, Buckley CD. Rheumatoid fibroblast-like synoviocytes overexpress the chemokine stromal cell-derived factor 1 (CXCL12), which supports distinct patterns and rates of CD4+ and CD8+ T cell migration within synovial tissue. Arthritis and rheumatism. 2003;48(9):2472–2482. doi: 10.1002/art.11219. [DOI] [PubMed] [Google Scholar]
- 58.Jacobs JP, Ortiz-Lopez A, Campbell JJ, Gerard CJ, Mathis D, Benoist C. Deficiency of CXCR2, but not other chemokine receptors, attenuates autoantibody-mediated arthritis in a murine model. Arthritis and rheumatism. 2010;62(7):1921–1932. doi: 10.1002/art.27470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barsante MM, Cunha TM, Allegretti M, Cattani F, Policani F, Bizzarri C, Tafuri WL, Poole S, Cunha FQ, Bertini R, Teixeira MM. Blockade of the chemokine receptor CXCR2 ameliorates adjuvant-induced arthritis in rats. Br J Pharmacol. 2008;153(5):992–1002. doi: 10.1038/sj.bjp.0707462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Podolin PL, Bolognese BJ, Foley JJ, Schmidt DB, Buckley PT, Widdowson KL, Jin Q, White JR, Lee JM, Goodman RB, Hagen TR, Kajikawa O, Marshall LA, Hay DW, Sarau HM. A potent and selective nonpeptide antagonist of CXCR2 inhibits acute and chronic models of arthritis in the rabbit. J Immunol. 2002;169(11):6435–6444. doi: 10.4049/jimmunol.169.11.6435. [DOI] [PubMed] [Google Scholar]
- 61.Grespan R, Fukada SY, Lemos HP, Vieira SM, Napimoga MH, Teixeira MM, Fraser AR, Liew FY, McInnes IB, Cunha FQ. CXCR2-specific chemokines mediate leukotriene B4-dependent recruitment of neutrophils to inflamed joints in mice with antigen-induced arthritis. Arthritis and rheumatism. 2008;58(7):2030–2040. doi: 10.1002/art.23597. [DOI] [PubMed] [Google Scholar]
- 62.Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. The Journal of clinical investigation. 2010;120(7):2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang B, Zinselmeyer BH, Runnels HA, LaBranche TP, Morton PA, Kreisel D, Mack M, Nickerson-Nutter C, Allen PM, Miller MJ. In vivo imaging implicates CCR2(+) monocytes as regulators of neutrophil recruitment during arthritis. Cell Immunol. 2012;278(1–2):103–112. doi: 10.1016/j.cellimm.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Snir O, Backlund J, Bostrom J, Andersson I, Kihlberg J, Buckner JH, Klareskog L, Holmdahl R, Malmstrom V. Multifunctional T cell reactivity with native and glycosylated type II collagen in rheumatoid arthritis. Arthritis and rheumatism. 2012;64(8):2482–2488. doi: 10.1002/art.34459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Janson PC, Linton LB, Bergman EA, Marits P, Eberhardson M, Piehl F, Malmstrom V, Winqvist O. Profiling of CD4+ T cells with epigenetic immune lineage analysis. J Immunol. 2011;186(1):92–102. doi: 10.4049/jimmunol.1000960. [DOI] [PubMed] [Google Scholar]
- 66.Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, Koch AE, Moser B, Mackay CR. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101(4):746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ruth JH, Rottman JB, Katschke KJ, Jr, Qin S, Wu L, LaRosa G, Ponath P, Pope RM, Koch AE. Selective lymphocyte chemokine receptor expression in the rheumatoid joint. Arthritis and rheumatism. 2001;44(12):2750–2760. doi: 10.1002/1529-0131(200112)44:12<2750::aid-art462>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 68.Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, Yavuz S, Lipsky PE. Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol. 2000;165(11):6590–6598. doi: 10.4049/jimmunol.165.11.6590. [DOI] [PubMed] [Google Scholar]
- 69.Mohan K, Issekutz TB. Blockade of chemokine receptor CXCR3 inhibits T cell recruitment to inflamed joints and decreases the severity of adjuvant arthritis. J Immunol. 2007;179(12):8463–8469. doi: 10.4049/jimmunol.179.12.8463. [DOI] [PubMed] [Google Scholar]
- 70.Moret FM, Hack CE, van der Wurff-Jacobs KM, de Jager W, Radstake TR, Lafeber FP, van Roon JA. Intra-articular CD1c-expressing myeloid dendritic cells from rheumatoid arthritis patients express a unique set of T cell-attracting chemokines and spontaneously induce Th1, Th17 and Th2 cell activity. Arthritis research & therapy. 2013;15(5):R155. doi: 10.1186/ar4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rossol M, Pierer M, Arnold S, Keysser G, Burkhardt H, Baerwald C, Wagner U. Negative association of the chemokine receptor CCR5 d32 polymorphism with systemic inflammatory response, extra-articular symptoms and joint erosion in rheumatoid arthritis. Arthritis research & therapy. 2009;11(3):R91. doi: 10.1186/ar2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pokorny V, McQueen F, Yeoman S, Merriman M, Merriman A, Harrison A, Highton J, McLean L. Evidence for negative association of the chemokine receptor CCR5 d32 polymorphism with rheumatoid arthritis. Annals of the rheumatic diseases. 2005;64(3):487–490. doi: 10.1136/ard.2004.023333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prahalad S. Negative association between the chemokine receptor CCR5-Delta32 polymorphism and rheumatoid arthritis: a meta-analysis. Genes and immunity. 2006;7(3):264–268. doi: 10.1038/sj.gene.6364298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zapico I, Coto E, Rodriguez A, Alvarez C, Torre JC, Alvarez V. CCR5 (chemokine receptor-5) DNA-polymorphism influences the severity of rheumatoid arthritis. Genes and immunity. 2000;1(4):288–289. doi: 10.1038/sj.gene.6363673. [DOI] [PubMed] [Google Scholar]
- 75.Lindner E, Nordang GB, Melum E, Flato B, Selvaag AM, Thorsby E, Kvien TK, Forre OT, Lie BA. Lack of association between the chemokine receptor 5 polymorphism CCR5delta32 in rheumatoid arthritis and juvenile idiopathic arthritis. BMC medical genetics. 2007;8:33. doi: 10.1186/1471-2350-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kohem CL, Brenol JC, Xavier RM, Bredemeier M, Brenol CV, Dedavid e Silva TL, de Castilhos Mello A, Canedo AD, Neves AG, Chies JA. The chemokine receptor CCR5 genetic polymorphism and expression in rheumatoid arthritis patients. Scandinavian journal of rheumatology. 2007;36(5):359–364. doi: 10.1080/03009740701393999. [DOI] [PubMed] [Google Scholar]
- 77.Quinones MP, Ahuja SK, Jimenez F, Schaefer J, Garavito E, Rao A, Chenaux G, Reddick RL, Kuziel WA, Ahuja SS. Experimental arthritis in CC chemokine receptor 2-null mice closely mimics severe human rheumatoid arthritis. The Journal of clinical investigation. 2004;113(6):856–866. doi: 10.1172/JCI20126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung SH, Seki K, Choi BI, Kimura KB, Ito A, Fujikado N, Saijo S, Iwakura Y. CXC chemokine receptor 4 expressed in T cells plays an important role in the development of collagen-induced arthritis. Arthritis research & therapy. 2010;12(5):R188. doi: 10.1186/ar3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nanki T, Imai T, Nagasaka K, Urasaki Y, Nonomura Y, Taniguchi K, Hayashida K, Hasegawa J, Yoshie O, Miyasaka N. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis and rheumatism. 2002;46(11):2878–2883. doi: 10.1002/art.10622. [DOI] [PubMed] [Google Scholar]
- 80.Blaschke S, Koziolek M, Schwarz A, Benohr P, Middel P, Schwarz G, Hummel KM, Muller GA. Proinflammatory role of fractalkine (CX3CL1) in rheumatoid arthritis. The Journal of rheumatology. 2003;30(9):1918–1927. [PubMed] [Google Scholar]
- 81.Yano R, Yamamura M, Sunahori K, Takasugi K, Yamana J, Kawashima M, Makino H. Recruitment of CD16+ monocytes into synovial tissues is mediated by fractalkine and CX3CR1 in rheumatoid arthritis patients. Acta medica Okayama. 2007;61(2):89–98. doi: 10.18926/AMO/32882. [DOI] [PubMed] [Google Scholar]
- 82.Nanki T, Urasaki Y, Imai T, Nishimura M, Muramoto K, Kubota T, Miyasaka N. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J Immunol. 2004;173(11):7010–7016. doi: 10.4049/jimmunol.173.11.7010. [DOI] [PubMed] [Google Scholar]
- 83.Ruth JH, Volin MV, Haines GK, 3rd, Woodruff DC, Katschke KJ, Jr, Woods JM, Park CC, Morel JC, Koch AE. Fractalkine, a novel chemokine in rheumatoid arthritis and in rat adjuvant-induced arthritis. Arthritis and rheumatism. 2001;44(7):1568–1581. doi: 10.1002/1529-0131(200107)44:7<1568::AID-ART280>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 84.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204(12):2803–2812. doi: 10.1084/jem.20071397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsui T, Akahoshi T, Namai R, Hashimoto A, Kurihara Y, Rana M, Nishimura A, Endo H, Kitasato H, Kawai S, Takagishi K, Kondo H. Selective recruitment of CCR6-expressing cells by increased production of MIP-3 alpha in rheumatoid arthritis. Clinical and experimental immunology. 2001;125(1):155–161. doi: 10.1046/j.1365-2249.2001.01542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsunemi S, Iwasaki T, Kitano S, Imado T, Miyazawa K, Sano H. Effects of the novel immunosuppressant FTY720 in a murine rheumatoid arthritis model. Clinical immunology. 2010;136(2):197–204. doi: 10.1016/j.clim.2010.03.428. [DOI] [PubMed] [Google Scholar]
- 87.Wang F, Tan W, Guo D, He S. Reduction of CD4 positive T cells and improvement of pathological changes of collagen-induced arthritis by FTY720. European journal of pharmacology. 2007;573(1–3):230–240. doi: 10.1016/j.ejphar.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 88.Matsuura M, Imayoshi T, Okumoto T. Effect of FTY720, a novel immunosuppressant, on adjuvant- and collagen-induced arthritis in rats. International journal of immunopharmacology. 2000;22(4):323–331. doi: 10.1016/s0192-0561(99)00088-0. [DOI] [PubMed] [Google Scholar]
- 89.Kitano M, Hla T, Sekiguchi M, Kawahito Y, Yoshimura R, Miyazawa K, Iwasaki T, Sano H, Saba JD, Tam YY. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis and rheumatism. 2006;54(3):742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- 90.Lai WQ, Irwan AW, Goh HH, Howe HS, Yu DT, Valle-Onate R, McInnes IB, Melendez AJ, Leung BP. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J Immunol. 2008;181(11):8010–8017. doi: 10.4049/jimmunol.181.11.8010. [DOI] [PubMed] [Google Scholar]
- 91.Edwards JC, Cambridge G. Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology. 2001;40(2):205–211. doi: 10.1093/rheumatology/40.2.205. [DOI] [PubMed] [Google Scholar]
- 92.Cohen SB, Emery P, Greenwald MW, Dougados M, Furie RA, Genovese MC, Keystone EC, Loveless JE, Burmester GR, Cravets MW, Hessey EW, Shaw T, Totoritis MC, Group RT Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: Results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis and rheumatism. 2006;54(9):2793–2806. doi: 10.1002/art.22025. [DOI] [PubMed] [Google Scholar]
- 93.van Gaalen FA, Linn-Rasker SP, van Venrooij WJ, de Jong BA, Breedveld FC, Verweij CL, Toes RE, Huizinga TW. Autoantibodies to cyclic citrullinated peptides predict progression to rheumatoid arthritis in patients with undifferentiated arthritis: a prospective cohort study. Arthritis and rheumatism. 2004;50(3):709–715. doi: 10.1002/art.20044. [DOI] [PubMed] [Google Scholar]
- 94.Amara K, Steen J, Murray F, Morbach H, Fernandez-Rodriguez BM, Joshua V, Engstrom M, Snir O, Israelsson L, Catrina AI, Wardemann H, Corti D, Meffre E, Klareskog L, Malmstrom V. Monoclonal IgG antibodies generated from joint-derived B cells of RA patients have a strong bias toward citrullinated autoantigen recognition. The Journal of experimental medicine. 2013;210(3):445–455. doi: 10.1084/jem.20121486. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 95.Nanki T, Takada K, Komano Y, Morio T, Kanegane H, Nakajima A, Lipsky PE, Miyasaka N. Chemokine receptor expression and functional effects of chemokines on B cells: implication in the pathogenesis of rheumatoid arthritis. Arthritis research & therapy. 2009;11(5):R149. doi: 10.1186/ar2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sellam J, Rouanet S, Hendel-Chavez H, Miceli-Richard C, Combe B, Sibilia J, Le Loet X, Tebib J, Jourdan R, Dougados M, Taoufik Y, Mariette X. CCL19, a B cell chemokine, is related to the decrease of blood memory B cells and predicts the clinical response to rituximab in patients with rheumatoid arthritis. Arthritis and rheumatism. 2013;65(9):2253–2261. doi: 10.1002/art.38023. [DOI] [PubMed] [Google Scholar]
- 97.Pickens SR, Chamberlain ND, Volin MV, Pope RM, Talarico NE, Mandelin AM, 2nd, Shahrara S. Role of the CCL21 and CCR7 pathways in rheumatoid arthritis angiogenesis. Arthritis and rheumatism. 2012;64(8):2471–2481. doi: 10.1002/art.34452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi K, Hayashida K, Kaneko M, Hashimoto J, Tomita T, Lipsky PE, Yoshikawa H, Ochi T. Lymphoid chemokine B cell-attracting chemokine-1 (CXCL13) is expressed in germinal center of ectopic lymphoid follicles within the synovium of chronic arthritis patients. J Immunol. 2001;166(1):650–655. doi: 10.4049/jimmunol.166.1.650. [DOI] [PubMed] [Google Scholar]
- 99.Bugatti S, Caporali R, Manzo A, Sakellariou G, Rossi S, Montecucco C. Ultrasonographic and MRI characterisation of the palindromic phase of rheumatoid arthritis. Annals of the rheumatic diseases. 2012;71(4):625–626. doi: 10.1136/annrheumdis-2011-200077. [DOI] [PubMed] [Google Scholar]
- 100.Conaghan PG, O’Connor P, McGonagle D, Astin P, Wakefield RJ, Gibbon WW, Quinn M, Karim Z, Green MJ, Proudman S, Isaacs J, Emery P. Elucidation of the relationship between synovitis and bone damage: a randomized magnetic resonance imaging study of individual joints in patients with early rheumatoid arthritis. Arthritis and rheumatism. 2003;48(1):64–71. doi: 10.1002/art.10747. [DOI] [PubMed] [Google Scholar]
- 101.Boyesen P, Haavardsholm EA, Ostergaard M, van der Heijde D, Sesseng S, Kvien TK. MRI in early rheumatoid arthritis: synovitis and bone marrow oedema are independent predictors of subsequent radiographic progression. Annals of the rheumatic diseases. 2011;70(3):428–433. doi: 10.1136/ard.2009.123950. [DOI] [PubMed] [Google Scholar]
- 102.McQueen FM, Benton N, Perry D, Crabbe J, Robinson E, Yeoman S, McLean L, Stewart N. Bone edema scored on magnetic resonance imaging scans of the dominant carpus at presentation predicts radiographic joint damage of the hands and feet six years later in patients with rheumatoid arthritis. Arthritis and rheumatism. 2003;48(7):1814–1827. doi: 10.1002/art.11162. [DOI] [PubMed] [Google Scholar]
- 103.Hayashida K, Ochi T, Fujimoto M, Owaki H, Shimaoka Y, Ono K, Matsumoto K. Bone marrow changes in adjuvant-induced and collagen-induced arthritis. Interleukin-1 and interleukin-6 activity and abnormal myelopoiesis. Arthritis and rheumatism. 1992;35(2):241–245. doi: 10.1002/art.1780350219. [DOI] [PubMed] [Google Scholar]
- 104.Proulx ST, Kwok E, You Z, Papuga MO, Beck CA, Shealy DJ, Calvi LM, Ritchlin CT, Awad HA, Boyce BF, Xing L, Schwarz EM. Elucidating bone marrow edema and myelopoiesis in murine arthritis using contrast-enhanced magnetic resonance imaging. Arthritis and rheumatism. 2008;58(7):2019–2029. doi: 10.1002/art.23546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wyllie JC. Histopathology of the subchondral bone lesion in rheumatoid arthritis. The Journal of rheumatology Supplement. 1983;11:26–28. [PubMed] [Google Scholar]
- 106.Watson WC, Tooms RE, Carnesale PG, Dutkowsky JP. A case of germinal center formation by CD45RO T and CD20 B lymphocytes in rheumatoid arthritic subchondral bone: proposal for a two-compartment model of immune-mediated disease with implications for immunotherapeutic strategies. Clinical immunology and immunopathology. 1994;73(1):27–37. doi: 10.1006/clin.1994.1166. [DOI] [PubMed] [Google Scholar]
- 107.O’Connell JX, Nielsen GP, Rosenberg AE. Subchondral acute inflammation in severe arthritis: a sterile osteomyelitis? The American journal of surgical pathology. 1999;23(2):192–197. doi: 10.1097/00000478-199902000-00008. [DOI] [PubMed] [Google Scholar]
- 108.Kaneko M, Tomita T, Nakase T, Ohsawa Y, Seki H, Takeuchi E, Takano H, Shi K, Takahi K, Kominami E, Uchiyama Y, Yoshikawa H, Ochi T. Expression of proteinases and inflammatory cytokines in subchondral bone regions in the destructive joint of rheumatoid arthritis. Rheumatology. 2001;40(3):247–255. doi: 10.1093/rheumatology/40.3.247. [DOI] [PubMed] [Google Scholar]
- 109.Bromley M, Woolley DE. Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis and rheumatism. 1984;27(9):968–975. doi: 10.1002/art.1780270902. [DOI] [PubMed] [Google Scholar]
- 110.Bugatti S, Caporali R, Manzo A, Vitolo B, Pitzalis C, Montecucco C. Involvement of subchondral bone marrow in rheumatoid arthritis: lymphoid neogenesis and in situ relationship to subchondral bone marrow osteoclast recruitment. Arthritis and rheumatism. 2005;52(11):3448–3459. doi: 10.1002/art.21377. [DOI] [PubMed] [Google Scholar]
- 111.McQueen FM, Stewart N, Crabbe J, Robinson E, Yeoman S, Tan PL, McLean L. Magnetic resonance imaging of the wrist in early rheumatoid arthritis reveals a high prevalence of erosions at four months after symptom onset. Annals of the rheumatic diseases. 1998;57(6):350–356. doi: 10.1136/ard.57.6.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. The American journal of pathology. 1998;152(4):943–951. [PMC free article] [PubMed] [Google Scholar]
- 113.Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, Wong T, Campagnuolo G, Moran E, Bogoch ER, Van G, Nguyen LT, Ohashi PS, Lacey DL, Fish E, Boyle WJ, Penninger JM. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402(6759):304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 114.Romas E, Sims NA, Hards DK, Lindsay M, Quinn JW, Ryan PF, Dunstan CR, Martin TJ, Gillespie MT. Osteoprotegerin reduces osteoclast numbers and prevents bone erosion in collagen-induced arthritis. The American journal of pathology. 2002;161(4):1419–1427. doi: 10.1016/S0002-9440(10)64417-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lubberts E, Oppers-Walgreen B, Pettit AR, Van Den Bersselaar L, Joosten LA, Goldring SR, Gravallese EM, Van Den Berg WB. Increase in expression of receptor activator of nuclear factor kappaB at sites of bone erosion correlates with progression of inflammation in evolving collagen-induced arthritis. Arthritis and rheumatism. 2002;46(11):3055–3064. doi: 10.1002/art.10607. [DOI] [PubMed] [Google Scholar]
- 116.Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. The American journal of pathology. 2001;159(5):1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Redlich K, Hayer S, Ricci R, David JP, Tohidast-Akrad M, Kollias G, Steiner G, Smolen JS, Wagner EF, Schett G. Osteoclasts are essential for TNF-alpha-mediated joint destruction. The Journal of clinical investigation. 2002;110(10):1419–1427. doi: 10.1172/JCI15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, Turk B, Pietschmann P, Woloszczuk W, Haralambous S, Kollias G, Steiner G, Smolen JS, Schett G. Tumor necrosis factor alpha-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis and rheumatism. 2002;46(3):785–792. doi: 10.1002/art.10097. [DOI] [PubMed] [Google Scholar]
- 119.Schett G, Stolina M, Bolon B, Middleton S, Adlam M, Brown H, Zhu L, Feige U, Zack DJ. Analysis of the kinetics of osteoclastogenesis in arthritic rats. Arthritis and rheumatism. 2005;52(10):3192–3201. doi: 10.1002/art.21343. [DOI] [PubMed] [Google Scholar]
- 120.Walsh NC, Reinwald S, Manning CA, Condon KW, Iwata K, Burr DB, Gravallese EM. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J Bone Miner Res. 2009;24(9):1572–1585. doi: 10.1359/jbmr.090320. [DOI] [PubMed] [Google Scholar]
- 121.Jimenez-Boj E, Redlich K, Turk B, Hanslik-Schnabel B, Wanivenhaus A, Chott A, Smolen JS, Schett G. Interaction between synovial inflammatory tissue and bone marrow in rheumatoid arthritis. J Immunol. 2005;175(4):2579–2588. doi: 10.4049/jimmunol.175.4.2579. [DOI] [PubMed] [Google Scholar]
- 122.Gortz B, Hayer S, Redlich K, Zwerina J, Tohidast-Akrad M, Tuerk B, Hartmann C, Kollias G, Steiner G, Smolen JS, Schett G. Arthritis induces lymphocytic bone marrow inflammation and endosteal bone formation. J Bone Miner Res. 2004;19(6):990–998. doi: 10.1359/JBMR.040205. [DOI] [PubMed] [Google Scholar]
- 123.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661. doi: 10.1126/science.1178331. doi: 327/5966/656 [pii] 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Arai F, Miyamoto T, Ohneda O, Inada T, Sudo T, Brasel K, Miyata T, Anderson DM, Suda T. Commitment and differentiation of osteoclast precursor cells by the sequential expression of c-Fms and receptor activator of nuclear factor kappaB (RANK) receptors. J Exp Med. 1999;190(12):1741–1754. doi: 10.1084/jem.190.12.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Charles JF, Hsu LY, Niemi EC, Weiss A, Aliprantis AO, Nakamura MC. Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J Clin Invest. 2012;122(12):4592–4605. doi: 10.1172/JCI60920. doi: 60920 [pii] 10.1172/JCI60920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jacome-Galarza CE, Lee SK, Lorenzo JA, Aguila HL. Identification, characterization, and isolation of a common progenitor for osteoclasts, macrophages, and dendritic cells from murine bone marrow and periphery. J Bone Miner Res. 2013;28(5):1203–1213. doi: 10.1002/jbmr.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Jacquin C, Gran DE, Lee SK, Lorenzo JA, Aguila HL. Identification of multiple osteoclast precursor populations in murine bone marrow. J Bone Miner Res. 2006;21(1):67–77. doi: 10.1359/JBMR.051007. [DOI] [PubMed] [Google Scholar]
- 128.Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, Feuerer M. Origin of monocytes and macrophages in a committed progenitor. Nat Immunol. 2013;14(8):821–830. doi: 10.1038/ni.2638. doi:ni.2638 [pii] 10.1038/ni.2638. [DOI] [PubMed] [Google Scholar]
- 129.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sarosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle WJ. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93(2):165–176. doi: 10.1016/s0092-8674(00)81569-x. doi:S0092-8674(00)81569-X [pii] [DOI] [PubMed] [Google Scholar]
- 130.Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397(6717):315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 131.Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, Tan HL, Elliott G, Kelley MJ, Sarosi I, Wang L, Xia XZ, Elliott R, Chiu L, Black T, Scully S, Capparelli C, Morony S, Shimamoto G, Bass MB, Boyle WJ. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U S A. 1999;96(7):3540–3545. doi: 10.1073/pnas.96.7.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, Koga T, Martin TJ, Suda T. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A. 1990;87(18):7260–7264. doi: 10.1073/pnas.87.18.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–1234. doi: 10.1038/nm.2452. doi:nm.2452 [pii] 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 134.Mundy GR, Varani J, Orr W, Gondek MD, Ward PA. Resorbing bone is chemotactic for monocytes. Nature. 1978;275(5676):132–135. doi: 10.1038/275132a0. [DOI] [PubMed] [Google Scholar]
- 135.Nevius E, Pinho F, Dhodapkar M, Jin H, Nadrah K, Horowitz MC, Kikuta J, Ishii M, Pereira JP. Oxysterols and EBI2 promote osteoclast precursor migration to bone surfaces and regulate bone mass homeostasis. J Exp Med. 2015 doi: 10.1084/jem.20150088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jacquelin S, Licata F, Dorgham K, Hermand P, Poupel L, Guyon E, Deterre P, Hume DA, Combadiere C, Boissonnas A. CX3CR1 reduces Ly6Chigh-monocyte motility within and release from the bone marrow after chemotherapy in mice. Blood. 2013;122(5):674–683. doi: 10.1182/blood-2013-01-480749. doi:blood-2013-01-480749 [pii] 10.1182/blood-2013-01-480749. [DOI] [PubMed] [Google Scholar]
- 137.Rahimi P, Wang CY, Stashenko P, Lee SK, Lorenzo JA, Graves DT. Monocyte chemoattractant protein-1 expression and monocyte recruitment in osseous inflammation in the mouse. Endocrinology. 1995;136(6):2752–2759. doi: 10.1210/endo.136.6.7750500. [DOI] [PubMed] [Google Scholar]
- 138.Williams SR, Jiang Y, Cochran D, Dorsam G, Graves DT. Regulated expression of monocyte chemoattractant protein-1 in normal human osteoblastic cells. Am J Physiol. 1992;263(1 Pt 1):C194–199. doi: 10.1152/ajpcell.1992.263.1.C194. [DOI] [PubMed] [Google Scholar]
- 139.Zhu JF, Valente AJ, Lorenzo JA, Carnes D, Graves DT. Expression of monocyte chemoattractant protein 1 in human osteoblastic cells stimulated by proinflammatory mediators. J Bone Miner Res. 1994;9(7):1123–1130. doi: 10.1002/jbmr.5650090721. [DOI] [PubMed] [Google Scholar]
- 140.Kim MS, Day CJ, Selinger CI, Magno CL, Stephens SR, Morrison NA. MCP-1-induced human osteoclast-like cells are tartrate-resistant acid phosphatase, NFATc1, and calcitonin receptor-positive but require receptor activator of NFkappaB ligand for bone resorption. The Journal of biological chemistry. 2006;281(2):1274–1285. doi: 10.1074/jbc.M510156200. doi:M510156200 [pii] 10.1074/jbc.M510156200. [DOI] [PubMed] [Google Scholar]
- 141.Kim MS, Magno CL, Day CJ, Morrison NA. Induction of chemokines and chemokine receptors CCR2b and CCR4 in authentic human osteoclasts differentiated with RANKL and osteoclast like cells differentiated by MCP-1 and RANTES. J Cell Biochem. 2006;97(3):512–518. doi: 10.1002/jcb.20649. [DOI] [PubMed] [Google Scholar]
- 142.Li X, Qin L, Bergenstock M, Bevelock LM, Novack DV, Partridge NC. Parathyroid hormone stimulates osteoblastic expression of MCP-1 to recruit and increase the fusion of pre/osteoclasts. The Journal of biological chemistry. 2007;282(45):33098–33106. doi: 10.1074/jbc.M611781200. doi: M611781200 [pii] 10.1074/jbc.M611781200. [DOI] [PubMed] [Google Scholar]
- 143.Binder NB, Niederreiter B, Hoffmann O, Stange R, Pap T, Stulnig TM, Mack M, Erben RG, Smolen JS, Redlich K. Estrogen-dependent and C-C chemokine receptor-2-dependent pathways determine osteoclast behavior in osteoporosis. Nat Med. 2009;15(4):417–424. doi: 10.1038/nm.1945. doi: nm.1945 [pii] 10.1038/nm.1945. [DOI] [PubMed] [Google Scholar]
- 144.Yamada Y, Ando F, Niino N, Shimokata H. Association of a polymorphism of the CC chemokine receptor-2 gene with bone mineral density. Genomics. 2002;80(1):8–12. doi: 10.1006/geno.2002.6793. doi: S0888754302967935 [pii] [DOI] [PubMed] [Google Scholar]
- 145.Lee B, Doranz BJ, Rana S, Yi Y, Mellado M, Frade JM, Martinez AC, O’Brien SJ, Dean M, Collman RG, Doms RW. Influence of the CCR2-V64I polymorphism on human immunodeficiency virus type 1 coreceptor activity and on chemokine receptor function of CCR2b, CCR3, CCR5, and CXCR4. J Virol. 1998;72(9):7450–7458. doi: 10.1128/jvi.72.9.7450-7458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7(3):311–317. doi: 10.1038/ni1309. doi: ni1309 [pii] 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 147.Shi C, Jia T, Mendez-Ferrer S, Hohl TM, Serbina NV, Lipuma L, Leiner I, Li MO, Frenette PS, Pamer EG. Bone Marrow Mesenchymal Stem and Progenitor Cells Induce Monocyte Emigration in Response to Circulating Toll-like Receptor Ligands. Immunity. 2011;34(4):590–601. doi: 10.1016/j.immuni.2011.02.016. doi: S1074-7613(11)00074-4 [pii] 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. The Journal of clinical investigation. 2007;117(4):902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kikuta J, Nevius E, Ishii M, Pereira JP. Trafficking of Osteoclast Precursors. In: Lorenzo J, editor. Osteoimmunology: Interactions of the Immune and Skeletal Systems. 2. Elsevier; 2015. pp. 25–40. [Google Scholar]
- 150.Hoshino A, Iimura T, Ueha S, Hanada S, Maruoka Y, Mayahara M, Suzuki K, Imai T, Ito M, Manome Y, Yasuhara M, Kirino T, Yamaguchi A, Matsushima K, Yamamoto K. Deficiency of chemokine receptor CCR1 causes osteopenia due to impaired functions of osteoclasts and osteoblasts. The Journal of biological chemistry. 2010;285(37):28826–28837. doi: 10.1074/jbc.M109.099424. doi: M109.099424 [pii] 10.1074/jbc.M109.099424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Koizumi K, Saitoh Y, Minami T, Takeno N, Tsuneyama K, Miyahara T, Nakayama T, Sakurai H, Takano Y, Nishimura M, Imai T, Yoshie O, Saiki I. Role of CX3CL1/fractalkine in osteoclast differentiation and bone resorption. J Immunol. 2009;183(12):7825–7831. doi: 10.4049/jimmunol.0803627. doi: jimmunol.0803627 [pii] 10.4049/jimmunol.0803627. [DOI] [PubMed] [Google Scholar]
- 152.Hoshino A, Ueha S, Hanada S, Imai T, Ito M, Yamamoto K, Matsushima K, Yamaguchi A, Iimura T. Roles of chemokine receptor CX3CR1 in maintaining murine bone homeostasis through the regulation of both osteoblasts and osteoclasts. J Cell Sci. 2013;126(Pt 4):1032–1045. doi: 10.1242/jcs.113910. doi: jcs.113910 [pii] 10.1242/jcs.113910. [DOI] [PubMed] [Google Scholar]
- 153.Han KH, Ryu JW, Lim KE, Lee SH, Kim Y, Hwang CS, Choi JY, Han KO. Vascular expression of the chemokine CX3CL1 promotes osteoclast recruitment and exacerbates bone resorption in an irradiated murine model. Bone. 2014;61:91–101. doi: 10.1016/j.bone.2013.12.032. doi: S8756-3282(13)00554-1 [pii] 10.1016/j.bone.2013.12.032. [DOI] [PubMed] [Google Scholar]
- 154.Sugita K, Kabashima K, Sakabe J, Yoshiki R, Tanizaki H, Tokura Y. FTY720 regulates bone marrow egress of eosinophils and modulates late-phase skin reaction in mice. Am J Pathol. 2010;177(4):1881–1887. doi: 10.2353/ajpath.2010.100119. doi: S0002-9440(10)60239-8 [pii] 10.2353/ajpath.2010.100119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Allende ML, Tuymetova G, Lee BG, Bonifacino E, Wu YP, Proia RL. S1P1 receptor directs the release of immature B cells from bone marrow into blood. The Journal of experimental medicine. 2010;207(5):1113–1124. doi: 10.1084/jem.20092210. doi:jem.20092210 [pii] 10.1084/jem.20092210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jenne CN, Enders A, Rivera R, Watson SR, Bankovich AJ, Pereira JP, Xu Y, Roots CM, Beilke JN, Banerjee A, Reiner SL, Miller SA, Weinmann AS, Goodnow CC, Lanier LL, Cyster JG, Chun J. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206(11):2469–2481. doi: 10.1084/jem.20090525. doi: jem.20090525 [pii] 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Walzer T, Chiossone L, Chaix J, Calver A, Carozzo C, Garrigue-Antar L, Jacques Y, Baratin M, Tomasello E, Vivier E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol. 2007;8(12):1337–1344. doi: 10.1038/ni1523. doi: ni1523 [pii] 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- 158.Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J, Proia RL, Germain RN. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature. 2009;458(7237):524–528. doi: 10.1038/nature07713. doi: nature07713 [pii] 10.1038/nature07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ishii M, Kikuta J, Shimazu Y, Meier-Schellersheim M, Germain RN. Chemorepulsion by blood S1P regulates osteoclast precursor mobilization and bone remodeling in vivo. J Exp Med. 2010;207(13):2793–2798. doi: 10.1084/jem.20101474. doi: jem.20101474 [pii] 10.1084/jem.20101474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Grassi F, Piacentini A, Cristino S, Toneguzzi S, Cavallo C, Facchini A, Lisignoli G. Human osteoclasts express different CXC chemokines depending on cell culture substrate: molecular and immunocytochemical evidence of high levels of CXCL10 and CXCL12. Histochem Cell Biol. 2003;120(5):391–400. doi: 10.1007/s00418-003-0587-3. [DOI] [PubMed] [Google Scholar]
- 161.Wright LM, Maloney W, Yu X, Kindle L, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 binding to its chemokine receptor CXCR4 on precursor cells promotes the chemotactic recruitment, development and survival of human osteoclasts. Bone. 2005;36(5):840–853. doi: 10.1016/j.bone.2005.01.021. doi: S8756-3282(05)00017-7 [pii] 10.1016/j.bone.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 162.Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nat Rev Immunol. 2006;6(2):107–116. doi: 10.1038/nri1780. doi: nri1780 [pii] 10.1038/nri1780. [DOI] [PubMed] [Google Scholar]
- 163.Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, Levesque JP, Chappel J, Ross FP, Link DC. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106(9):3020–3027. doi: 10.1182/blood-2004-01-0272. doi: 2004-01-0272 [pii] 10.1182/blood-2004-01-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016. doi: S1074-7613(06)00515-2 [pii] 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 165.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J Bone Miner Res. 2003;18(8):1404–1418. doi: 10.1359/jbmr.2003.18.8.1404. [DOI] [PubMed] [Google Scholar]
- 166.Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-Receptor-Expressing Mesenchymal Stromal Cells Represent the Main Source of Bone Formed by Adult Bone Marrow. Cell Stem Cell. 2014 doi: 10.1016/j.stem.2014.06.008. doi:S1934-5909(14)00256-2 [pii] 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, Nagasawa T. The Essential Functions of Adipo-osteogenic Progenitors as the Hematopoietic Stem and Progenitor Cell Niche. Immunity. 2010;33(3):387–399. doi: 10.1016/j.immuni.2010.08.017. doi: S1074-7613(10)00322-5 [pii] 10.1016/j.immuni.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 168.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. doi: 10.1038/nature09262. doi: nature09262 [pii] 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013 doi: 10.1038/nature11885. doi:nature11885 [pii] 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hirbe AC, Rubin J, Uluckan O, Morgan EA, Eagleton MC, Prior JL, Piwnica-Worms D, Weilbaecher KN. Disruption of CXCR4 enhances osteoclastogenesis and tumor growth in bone. Proc Natl Acad Sci U S A. 2007;104(35):14062–14067. doi: 10.1073/pnas.0705203104. doi: 0705203104 [pii] 10.1073/pnas.0705203104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pandey R, Mousawy K, Nagarkatti M, Nagarkatti P. Endocannabinoids and immune regulation. Pharmacological research : the official journal of the Italian Pharmacological Society. 2009;60(2):85–92. doi: 10.1016/j.phrs.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pereira JP, An J, Xu Y, Huang Y, Cyster JG. Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat Immunol. 2009;10(4):403–411. doi: 10.1038/ni.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Idris AI, Sophocleous A, Landao-Bassonga E, van’t Hof RJ, Ralston SH. Regulation of bone mass, osteoclast function, and ovariectomy-induced bone loss by the type 2 cannabinoid receptor. Endocrinology. 2008;149(11):5619–5626. doi: 10.1210/en.2008-0150. doi: en.2008-0150 [pii] 10.1210/en.2008-0150. [DOI] [PubMed] [Google Scholar]
- 174.Ofek O, Karsak M, Leclerc N, Fogel M, Frenkel B, Wright K, Tam J, Attar-Namdar M, Kram V, Shohami E, Mechoulam R, Zimmer A, Bab I. Peripheral cannabinoid receptor, CB2, regulates bone mass. Proc Natl Acad Sci U S A. 2006;103(3):696–701. doi: 10.1073/pnas.0504187103. doi: 0504187103 [pii] 10.1073/pnas.0504187103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bab I, Zimmer A. Cannabinoid receptors and the regulation of bone mass. Br J Pharmacol. 2008;153(2):182–188. doi: 10.1038/sj.bjp.0707593. doi: 0707593 [pii] 10.1038/sj.bjp.0707593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Karsak M, Cohen-Solal M, Freudenberg J, Ostertag A, Morieux C, Kornak U, Essig J, Erxlebe E, Bab I, Kubisch C, de Vernejoul MC, Zimmer A. Cannabinoid receptor type 2 gene is associated with human osteoporosis. Hum Mol Genet. 2005;14(22):3389–3396. doi: 10.1093/hmg/ddi370. doi: ddi370 [pii] 10.1093/hmg/ddi370. [DOI] [PubMed] [Google Scholar]
- 177.Yamada Y, Ando F, Shimokata H. Association of candidate gene polymorphisms with bone mineral density in community-dwelling Japanese women and men. Int J Mol Med. 2007;19(5):791–801. [PubMed] [Google Scholar]
- 178.Beck TC, Gomes AC, Cyster JG, Pereira JP. CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. The Journal of experimental medicine. 2014;211(13):2567–2581. doi: 10.1084/jem.20140457. doi: jem.20140457 [pii] 10.1084/jem.20140457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10(13):4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, Boyce BF, Xing L. Tumor necrosis factor-alpha increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. The Journal of biological chemistry. 2006;281(17):11846–11855. doi: 10.1074/jbc.M512624200. [DOI] [PubMed] [Google Scholar]
- 181.Speziani C, Rivollier A, Gallois A, Coury F, Mazzorana M, Azocar O, Flacher M, Bella C, Tebib J, Jurdic P, Rabourdin-Combe C, Delprat C. Murine dendritic cell transdifferentiation into osteoclasts is differentially regulated by innate and adaptive cytokines. Eur J Immunol. 2007;37(3):747–757. doi: 10.1002/eji.200636534. [DOI] [PubMed] [Google Scholar]
- 182.Rivollier A, Mazzorana M, Tebib J, Piperno M, Aitsiselmi T, Rabourdin-Combe C, Jurdic P, Servet-Delprat C. Immature dendritic cell transdifferentiation into osteoclasts: a novel pathway sustained by the rheumatoid arthritis microenvironment. Blood. 2004;104(13):4029–4037. doi: 10.1182/blood-2004-01-0041. [DOI] [PubMed] [Google Scholar]
- 183.Fumoto T, Takeshita S, Ito M, Ikeda K. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J Bone Miner Res. 2014;29(4):830–842. doi: 10.1002/jbmr.2096. [DOI] [PubMed] [Google Scholar]
- 184.Onal M, Xiong J, Chen X, Thostenson JD, Almeida M, Manolagas SC, O’Brien CA. Receptor activator of nuclear factor kappaB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. The Journal of biological chemistry. 2012;287(35):29851–29860. doi: 10.1074/jbc.M112.377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT. Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun. 1999;265(1):144–150. doi: 10.1006/bbrc.1999.1623. [DOI] [PubMed] [Google Scholar]
- 186.Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A, Miyazaki T, Koshihara Y, Oda H, Nakamura K, Tanaka S. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis and rheumatism. 2000;43(2):259–269. doi: 10.1002/1529-0131(200002)43:2<259::AID-ANR4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 187.Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis and rheumatism. 2000;43(2):250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 188.Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, Tanaka S, Bluestone JA, Takayanagi H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. 2014;20(1):62–68. doi: 10.1038/nm.3432. [DOI] [PubMed] [Google Scholar]
- 189.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, Cua DJ, Takayanagi H. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203(12):2673–2682. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. The Journal of clinical investigation. 1999;103(9):1345–1352. doi: 10.1172/JCI5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Adamopoulos IE, Chao CC, Geissler R, Laface D, Blumenschein W, Iwakura Y, McClanahan T, Bowman EP. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis research & therapy. 2010;12(1):R29. doi: 10.1186/ar2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Adamopoulos IE, Suzuki E, Chao CC, Gorman D, Adda S, Maverakis E, Zarbalis K, Geissler R, Asio A, Blumenschein WM, McClanahan T, De Waal Malefyt R, Gershwin ME, Bowman EP. IL-17A gene transfer induces bone loss and epidermal hyperplasia associated with psoriatic arthritis. Annals of the rheumatic diseases. 2015;74(6):1284–1292. doi: 10.1136/annrheumdis-2013-204782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7(9):e44552. doi: 10.1371/journal.pone.0044552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Danks L, Komatsu N, Guerrini MM, Sawa S, Armaka M, Kollias G, Nakashima T, Takayanagi H. RANKL expressed on synovial fibroblasts is primarily responsible for bone erosions during joint inflammation. Annals of the rheumatic diseases. 2015 doi: 10.1136/annrheumdis-2014-207137. [DOI] [PubMed] [Google Scholar]
- 195.Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, van der Heijde D, Zhou L, Tsuji W, Newmark R, Denosumab Rheumatoid Arthritis Study G Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis and rheumatism. 2008;58(5):1299–1309. doi: 10.1002/art.23417. [DOI] [PubMed] [Google Scholar]
- 196.Kikuta J, Wada Y, Kowada T, Wang Z, Sun-Wada GH, Nishiyama I, Mizukami S, Maiya N, Yasuda H, Kumanogoh A, Kikuchi K, Germain RN, Ishii M. Dynamic visualization of RANKL and Th17-mediated osteoclast function. The Journal of clinical investigation. 2013;123(2):866–873. doi: 10.1172/JCI65054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Adamopoulos IE, Mellins ED. Alternative pathways of osteoclastogenesis in inflammatory arthritis. Nature reviews Rheumatology. 2015;11(3):189–194. doi: 10.1038/nrrheum.2014.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin TJ, Suda T. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191(2):275–286. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yokota K, Sato K, Miyazaki T, Kitaura H, Kayama H, Miyoshi F, Araki Y, Akiyama Y, Takeda K, Mimura T. Combination of tumor necrosis factor alpha and interleukin-6 induces mouse osteoclast-like cells with bone resorption activity both in vitro and in vivo. Arthritis & rheumatology. 2014;66(1):121–129. doi: 10.1002/art.38218. [DOI] [PubMed] [Google Scholar]
- 200.Kim JH, Jin HM, Kim K, Song I, Youn BU, Matsuo K, Kim N. The mechanism of osteoclast differentiation induced by IL-1. J Immunol. 2009;183(3):1862–1870. doi: 10.4049/jimmunol.0803007. [DOI] [PubMed] [Google Scholar]
- 201.Kim N, Kadono Y, Takami M, Lee J, Lee SH, Okada F, Kim JH, Kobayashi T, Odgren PR, Nakano H, Yeh WC, Lee SK, Lorenzo JA, Choi Y. Osteoclast differentiation independent of the TRANCE-RANK-TRAF6 axis. J Exp Med. 2005;202(5):589–595. doi: 10.1084/jem.20050978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC, Baeten D, McInnes IB, Bresnihan B, Tak PP. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Annals of the rheumatic diseases. 2005;64(6):834–838. doi: 10.1136/ard.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, Jakobsson PJ, Baum W, Nimmerjahn F, Szarka E, Sarmay G, Krumbholz G, Neumann E, Toes R, Scherer HU, Catrina AI, Klareskog L, Jurdic P, Schett G. Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. The Journal of clinical investigation. 2012;122(5):1791–1802. doi: 10.1172/JCI60975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Harre U, Lang SC, Pfeifle R, Rombouts Y, Fruhbeisser S, Amara K, Bang H, Lux A, Koeleman CA, Baum W, Dietel K, Grohn F, Malmstrom V, Klareskog L, Kronke G, Kocijan R, Nimmerjahn F, Toes RE, Herrmann M, Scherer HU, Schett G. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun. 2015;6:6651. doi: 10.1038/ncomms7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 mediates TNF-induced osteoclastogenesis. The Journal of clinical investigation. 2005;115(2):282–290. doi: 10.1172/JCI23394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li P, Schwarz EM, O’Keefe RJ, Ma L, Boyce BF, Xing L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res. 2004;19(2):207–213. doi: 10.1359/JBMR.0301233. [DOI] [PubMed] [Google Scholar]
- 207.Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. The Journal of clinical investigation. 2003;111(6):821–831. doi: 10.1172/JCI16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, Burdick MD, Pope RM, Strieter RM. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. The Journal of clinical investigation. 1992;90(3):772–779. doi: 10.1172/JCI115950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Akahoshi T, Wada C, Endo H, Hirota K, Hosaka S, Takagishi K, Kondo H, Kashiwazaki S, Matsushima K. Expression of monocyte chemotactic and activating factor in rheumatoid arthritis. Regulation of its production in synovial cells by interleukin-1 and tumor necrosis factor. Arthritis and rheumatism. 1993;36(6):762–771. doi: 10.1002/art.1780360605. [DOI] [PubMed] [Google Scholar]
- 210.Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J Exp Med. 1997;186(1):131–137. doi: 10.1084/jem.186.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Plater-Zyberk C, Hoogewerf AJ, Proudfoot AE, Power CA, Wells TN. Effect of a CC chemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunology letters. 1997;57(1–3):117–120. doi: 10.1016/s0165-2478(97)00075-8. [DOI] [PubMed] [Google Scholar]
- 212.Bruhl H, Cihak J, Schneider MA, Plachy J, Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M, Schlondorff D, Mack M. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: evidence for regulatory activity of CCR2+ T cells. J Immunol. 2004;172(2):890–898. doi: 10.4049/jimmunol.172.2.890. [DOI] [PubMed] [Google Scholar]
- 213.Rampersad RR, Tarrant TK, Vallanat CT, Quintero-Matthews T, Weeks MF, Esserman DA, Clark J, Di Padova F, Patel DD, Fong AM, Liu P. Enhanced Th17-cell responses render CCR2-deficient mice more susceptible for autoimmune arthritis. PLoS One. 2011;6(10):e25833. doi: 10.1371/journal.pone.0025833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Koch AE, Kunkel SL, Harlow LA, Mazarakis DD, Haines GK, Burdick MD, Pope RM, Strieter RM. Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis. The Journal of clinical investigation. 1994;93(3):921–928. doi: 10.1172/JCI117097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Volin MV, Shah MR, Tokuhira M, Haines GK, Woods JM, Koch AE. RANTES expression and contribution to monocyte chemotaxis in arthritis. Clinical immunology and immunopathology. 1998;89(1):44–53. doi: 10.1006/clin.1998.4590. [DOI] [PubMed] [Google Scholar]
- 216.Lebre MC, Vergunst CE, Choi IY, Aarrass S, Oliveira AS, Wyant T, Horuk R, Reedquist KA, Tak PP. Why CCR2 and CCR5 blockade failed and why CCR1 blockade might still be effective in the treatment of rheumatoid arthritis. PLoS One. 2011;6(7):e21772. doi: 10.1371/journal.pone.0021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Vallet S, Raje N, Ishitsuka K, Hideshima T, Podar K, Chhetri S, Pozzi S, Breitkreutz I, Kiziltepe T, Yasui H, Ocio EM, Shiraishi N, Jin J, Okawa Y, Ikeda H, Mukherjee S, Vaghela N, Cirstea D, Ladetto M, Boccadoro M, Anderson KC. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood. 2007;110(10):3744–3752. doi: 10.1182/blood-2007-05-093294. doi: blood-2007-05-093294 [pii] 10.1182/blood-2007-05-093294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Vergunst CE, Gerlag DM, Lopatinskaya L, Klareskog L, Smith MD, van den Bosch F, Dinant HJ, Lee Y, Wyant T, Jacobson EW, Baeten D, Tak PP. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis and rheumatism. 2008;58(7):1931–1939. doi: 10.1002/art.23591. [DOI] [PubMed] [Google Scholar]
- 219.Vergunst CE, Gerlag DM, von Moltke L, Karol M, Wyant T, Chi X, Matzkin E, Leach T, Tak PP. MLN3897 plus methotrexate in patients with rheumatoid arthritis: safety, efficacy, pharmacokinetics, and pharmacodynamics of an oral CCR1 antagonist in a phase IIa, double-blind, placebo-controlled, randomized, proof-of-concept study. Arthritis and rheumatism. 2009;60(12):3572–3581. doi: 10.1002/art.24978. [DOI] [PubMed] [Google Scholar]
- 220.Gerlag DM, Hollis S, Layton M, Vencovsky J, Szekanecz Z, Braddock M, Tak PP, Group ES Preclinical and clinical investigation of a CCR5 antagonist, AZD5672, in patients with rheumatoid arthritis receiving methotrexate. Arthritis and rheumatism. 2010;62(11):3154–3160. doi: 10.1002/art.27652. [DOI] [PubMed] [Google Scholar]
- 221.van Kuijk AW, Vergunst CE, Gerlag DM, Bresnihan B, Gomez-Reino JJ, Rouzier R, Verschueren PC, van der Leij C, Maas M, Kraan MC, Tak PP. CCR5 blockade in rheumatoid arthritis: a randomised, double-blind, placebo-controlled clinical trial. Annals of the rheumatic diseases. 2010;69(11):2013–2016. doi: 10.1136/ard.2010.131235. [DOI] [PubMed] [Google Scholar]
- 222.Hayashida K, Nanki T, Girschick H, Yavuz S, Ochi T, Lipsky PE. Synovial stromal cells from rheumatoid arthritis patients attract monocytes by producing MCP-1 and IL-8. Arthritis research. 2001;3(2):118–126. doi: 10.1186/ar149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Fukuda S, Kohsaka H, Takayasu A, Yokoyama W, Miyabe C, Miyabe Y, Harigai M, Miyasaka N, Nanki T. Cannabinoid receptor 2 as a potential therapeutic target in rheumatoid arthritis. BMC musculoskeletal disorders. 2014;15:275. doi: 10.1186/1471-2474-15-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Ha J, Choi HS, Lee Y, Kwon HJ, Song YW, Kim HH. CXC chemokine ligand 2 induced by receptor activator of NF-kappa B ligand enhances osteoclastogenesis. J Immunol. 2010;184(9):4717–4724. doi: 10.4049/jimmunol.0902444. [DOI] [PubMed] [Google Scholar]
- 225.Coelho FM, Pinho V, Amaral FA, Sachs D, Costa VV, Rodrigues DH, Vieira AT, Silva TA, Souza DG, Bertini R, Teixeira AL, Teixeira MM. The chemokine receptors CXCR1/CXCR2 modulate antigen-induced arthritis by regulating adhesion of neutrophils to the synovial microvasculature. Arthritis and rheumatism. 2008;58(8):2329–2337. doi: 10.1002/art.23622. [DOI] [PubMed] [Google Scholar]
- 226.Kasama T, Strieter RM, Lukacs NW, Lincoln PM, Burdick MD, Kunkel SL. Interleukin-10 expression and chemokine regulation during the evolution of murine type II collagen-induced arthritis. The Journal of clinical investigation. 1995;95(6):2868–2876. doi: 10.1172/JCI117993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Heinig M, Petretto E, Wallace C, Bottolo L, Rotival M, Lu H, Li Y, Sarwar R, Langley SR, Bauerfeind A, Hummel O, Lee YA, Paskas S, Rintisch C, Saar K, Cooper J, Buchan R, Gray EE, Cyster JG, Erdmann J, Hengstenberg C, Maouche S, Ouwehand WH, Rice CM, Samani NJ, Schunkert H, Goodall AH, Schulz H, Roider HG, Vingron M, Blankenberg S, Munzel T, Zeller T, Szymczak S, Ziegler A, Tiret L, Smyth DJ, Pravenec M, Aitman TJ, Cambien F, Clayton D, Todd JA, Hubner N, Cook SA. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature. 2010;467(7314):460–464. doi: 10.1038/nature09386. doi: nature09386 [pii] 10.1038/nature09386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, International IBDGC. Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wallace C, Rotival M, Cooper JD, Rice CM, Yang JH, McNeill M, Smyth DJ, Niblett D, Cambien F, Cardiogenics C, Tiret L, Todd JA, Clayton DG, Blankenberg S. Statistical colocalization of monocyte gene expression and genetic risk variants for type 1 diabetes. Hum Mol Genet. 2012;21(12):2815–2824. doi: 10.1093/hmg/dds098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci U S A. 2012;109(11):4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic Cytokines Can Instruct Lineage Choice. Science. 2009;325(5937):217–218. doi: 10.1126/science.1171461. [DOI] [PubMed] [Google Scholar]
- 232.Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-gamma in response to chronic infection. Nature. 2010;465(7299):793–797. doi: 10.1038/nature09135. doi: nature09135 [pii]10.1038/nature09135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, Trumpp A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458(7240):904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 234.Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, Lio P, Macdonald HR, Trumpp A. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 235.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. The Journal of experimental medicine. 2005;201(11):1771–1780. doi: 10.1084/jem.20041419. doi: jem.20041419 [pii] 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Ueda Y, Yang K, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXCL12 expression. The Journal of experimental medicine. 2004;199(1):47–58. doi: 10.1084/jem.20031104. 199/1/47 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Li P, Schwarz EM, O’Keefe RJ, Ma L, Looney RJ, Ritchlin CT, Boyce BF, Xing L. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50(1):265–276. doi: 10.1002/art.11419. [DOI] [PubMed] [Google Scholar]
- 238.Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol. 2014;14(5):302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 239.Lawlor KE, Campbell IK, Metcalf D, O’Donnell K, van Nieuwenhuijze A, Roberts AW, Wicks IP. Critical role for granulocyte colony-stimulating factor in inflammatory arthritis. Proc Natl Acad Sci U S A. 2004;101(31):11398–11403. doi: 10.1073/pnas.0404328101. [DOI] [PMC free article] [PubMed] [Google Scholar]