

Abstract
Purpose
ALK (anaplastic lymphoma kinase) rearrangements predict for sensitivity to ALK tyrosine kinase inhibitors (TKIs); however, responses to ALK TKIs are generally short lived. Serial molecular analysis is an informative strategy used to identify genetic mediators of resistance. Although multiple studies support the clinical benefits of repeat tissue sampling, the clinical utility of longitudinal circulating tumor DNA analysis has not been established in ALK-positive lung cancer.
Patients and Methods
We used a 566-gene hybrid-capture next-generation sequencing assay to perform a longitudinal analysis of plasma specimens from 22 ALK-positive patients with acquired resistance to ALK TKIs to track the evolution of resistance during treatment. To determine tissue–plasma concordance, we compared plasma findings with the results of repeat biopsies.
Results
At disease progression, we detected an ALK fusion in plasma from 19 (86%) of 22 patients and identified ALK resistance mutations in plasma specimens from 11 patients (50%). There was 100% agreement between tissue- and plasma-detected ALK fusions. Among 16 patients for which contemporaneous plasma and tissue specimens were available, we observed 100% concordance between ALK mutation calls. ALK mutations emerged and disappeared during treatment with sequential ALK TKIs, which suggests that plasma mutation profiles were dependent on the specific TKI administered. ALK G1202R—the most frequent plasma mutation detected after progression on a second-generation TKI—was consistently suppressed during treatment with lorlatinib.
Conclusion
Plasma genotyping by next-generation sequencing is an effective method for detecting ALK fusions and ALK mutations in patients who experience disease progression on ALK TKIs. The correlation between plasma ALK mutations and the response to distinct ALK TKIs highlights the potential for plasma analysis to guide the selection of ALK-directed therapies.
INTRODUCTION
Oncogenic rearrangements that result in the constitutive activation of anaplastic lymphoma kinase (ALK) define a molecular subtype of non–small-lung cancer (NSCLC) that is characterized by sensitivity to ALK tyrosine kinase inhibitors (TKIs).1,2 Since the identification of ALK rearrangements in NSCLC in 2007, four TKIs have become standard therapies for patients with advanced disease.3-6 Although these TKIs have significantly improved clinical outcomes, most patients experience relapse within 1 to 2 years.1 Despite a shared molecular driver, the magnitude of benefit from TKI treatment varies widely between patients. Repeat biopsies upon disease progression have been instrumental in elucidating the molecular mechanisms that drive resistance to ALK inhibitors, including the distinct spectrum of ALK mutations associated with resistance to each TKI7; however, repeat biopsies may be challenging to obtain and single-site sampling may not capture the spatial heterogeneity of resistance mechanisms.
Analysis of circulating tumor DNA (ctDNA) is an emerging approach for tumor genotyping. As plasma sampling is minimally invasive and ctDNA may be derived from all metastatic sites, longitudinal monitoring of genetic alterations in ctDNA may overcome many of the limitations of tissue sampling. Numerous studies have established digital polymerase chain reaction–based plasma genotyping as a reliable method for detecting mutations in patients with EGFR-mutant NSCLC8,9; however, it is challenging to perform multiplex testing and detect fusions using digital polymerase chain reaction.10 In contrast, plasma genotyping by next-generation sequencing (NGS) can simultaneously interrogate multiple genes and identify a broad range of molecular alterations, including chromosomal rearrangements10; therefore, NGS may be optimally positioned to characterize TKI resistance in ALK-rearranged—that is, ALK-positive—NSCLC.
To date, NGS-based plasma genotyping studies have included only a small number of patients with ALK-positive NSCLC.11-13 Here, we performed longitudinal ctDNA analysis with a hybrid capture–based NGS plasma genotyping platform to assess tissue–plasma concordance and study the evolution of resistance in patients who were treated with sequential ALK TKIs.
PATIENTS AND METHODS
Study Population
This study was conducted at Massachusetts General Hospital between June 2015 and January 2017. During this period, plasma was longitudinally collected from patients with metastatic, ALK-positive NSCLC who received treatment with ALK TKIs. The institutional review board at Massachusetts General Hospital approved this study. All study participants provided written informed consent.
Data Collection
Plasma Collection
Twenty milliliters of blood was collected from patients before initiation of treatment, approximately every 8 to 12 weeks, and at disease progression. As sampling coincided with clinic visits, the sampling interval varied between patients. As a result of the timing of the study initiation, we could not collect pretreatment plasma from five patients.
Clinical Data Collection
Medical records were reviewed to extract data on clinicopathologic features and treatment histories. Follow-up data for patients was obtained through January 2017. Determination of disease progression was based on investigator assessment of imaging studies and clinical status.
Molecular Analysis
Tumor Tissue
ALK rearrangements were identified by fluorescence in situ hybridization (FISH; n = 7) or NGS via Foundation One (n = 3), OncoPanel (n = 1), or a solid fusion assay (n = 1).14-16 In the 10 remaining patients, ALK status was determined by the combination of FISH and NGS (n = 9) or FISH and immunohistochemistry (Ventana D5F3 antibody; n = 1).17 SNaPshot NGS (Data Supplement) was employed to identify acquired genetic alterations, including ALK mutations.14
Plasma
DNA was extracted from frozen plasma specimens by using the QIAamp Circulating Nucleic Acid kit (Qiagen, Wetzlar, Germany). Libraries were constructed with Illumina TruSeq Nano DNA Library Prep kit (Illumina, San Diego, CA), enriched for a 566-gene PanCancer gene panel (Data Supplement) by using Agilent SureSelect XT Custom baits (Agilent Technologies, Santa Clara, CA), and sequenced on an Illumina HiSEquation 2500 sequencer to a median of 105 million reads, which yielded a median coverage of 1,195×.18 Sequence data were aligned to the hg19 reference genome. Variants were called with Pindel, Socrates, PureCN, and MuTect by using the default 0.5% lower threshold.19-22 Variants were then annotated, restricted to nonsynonymous mutations that involved protein-coding regions, and filtered with the assistance of common reference databases and internal controls to remove germline variants and artifacts. All ALK resistance mutations were regenotyped with the Genome Analysis Toolkit (Broad Institute, Cambridge, MA). Those with at least four alternate reads were retained on the basis of a calculated background mutation rate and false-positive rate of 1e-4 and 1e-6, respectively. The detection threshold for novel alterations in this pipeline was 1%. Called alterations were visually confirmed in Integrative Genomics Viewer (Broad Institute, Cambridge, MA).
RESULTS
Patient Characteristics
Plasma was longitudinally collected from 79 patients with metastatic, ALK-positive NSCLC (Fig 1). As this study was primarily focused on investigating the role of ctDNA analysis at disease progression, this report is limited to patients who had progressive disease and analyzable plasma specimens. Twenty-nine patients experienced disease progression during the study period. At the time of progression, we detected tumor DNA in plasma samples from 22 (76%) of 29 patients. Of the seven patients who did not have detectable ctDNA, one patient experienced CNS-only progression, five patients had intrathoracic progression, and one patient had liver oligoprogression (Fig 1). Baseline characteristics of the 22 patients and their treatment histories are summarized in Table 1, Figure 2, and the Data Supplement.
Fig 1.

Study schema depicting the incidence of progression and success rates of tissue and plasma sampling among patients who experienced disease progression during the study period. Circulating tumor DNA (ctDNA) analysis successful indicates those samples for which tumor-derived mutations were successfully identified in the plasma assessment. ctDNA insufficient for analysis indicates those samples that were technically successful, but had insufficient tumor DNA present in the plasma to reliably estimate underlying tumor genotype.
Table 1.
Baseline Clinical and Pathologic Characteristics

Fig 2.

Temporal relationship between plasma collections and administered treatments. Dark blue hash marks above boxes indicate the timing of the plasma collection. Arrows indicate that the patient continued on treatment after plasma collection. Note that some patients continued treatment beyond experiencing progression.
Plasma genotyping was performed on 88 plasma samples from 22 patients. We collected plasma at multiple time points during treatment with a single ALK TKI from eight patients and serial time points during treatment with two sequential ALK TKIs from 10 patients. For two patients, longitudinal sampling spanned treatment with three ALK TKIs. The temporal relationship between plasma collection and the treatments that were administered is depicted in the Data Supplement and Figure 2.
Concordance Between Tissue and Plasma Genotypes
We first evaluated concordance between tissue and plasma genotyping for ALK fusions, ALK mutations, and non-ALK alterations. Twenty (91%) of 22 patients underwent biopsy of a resistant site at relapse (Fig 1). Three patients underwent biopsies at two separate time points, with each biopsy representing disease progression on a distinct ALK TKI. Of 23 biopsies, tissue was adequate for NGS analysis in 16 specimens (70%) obtained from 14 patients. A biopsy was considered contemporaneous if performed within 3 weeks of plasma sampling. For two patients, tissue and plasma sampling were separated by approximately 6 weeks. As these two patients continued treatment with the same ALK TKI after experiencing progression, they were included in the analysis.
Fusion Concordance
At disease progression, an ALK fusion was detected in plasma from 19 (86%) of 22 patients. Of the three patients without a detectable ALK fusion, one patient experienced progression of intrapulmonary metastases, whereas the others experienced multisite progression (Data Supplement). We detected EML4-ALK in plasma from 18 patients (94%; Appendix Fig A1). Although we confirmed that an ALK fusion was present in the remaining patient, we were not able to determine the partner gene. Because tissue genotyping by FISH and immunohistochemistry does not identify the specific fusion partner or variant, we were unable to compare tissue and plasma fusions in eight patients. Among the 12 patients for whom the fusion partner was known and an ALK fusion was detected in the plasma, there was 100% agreement between tissue and plasma fusion calls (Fig 3A). Similarly, there was 100% concordance between tissue and plasma variant calls for the 11 patients who had an EML4-ALK variant that was identified by both plasma and tissue.
Fig 3.

Tissue–plasma concordance analysis. Gold box indicates that an alteration was detected in both plasma and tissue; blue box indicates that an alteration was present in the plasma only. (A) Concordance between fusion calls. In instances where the ALK fusion partner was not specified during tissue genotyping, patients were excluded from the concordance analysis. The fusion variant in MGH905's plasma was not identified by tissue analysis.(B) Concordance between ALK (anaplastic lymphoma kinase) mutation calls. Parentheses denote patients for whom the ALK mutation was considered the dominant resistance mechanism. For patients without paired biopsies, ALK mutations that were detected at the time of progression on an ALK tyrosine kinase inhibitor are listed. None of the patients without paired biopsies had plasma collected during progression on sequential ALK inhibitors. ALK mutations in the plasma of MGH905 occurred in cis. Because the mutations were separated by too many bases, the allelic relationship (cis v trans) could not be determined for MGH990’s ALK mutation.
ALK Mutation Concordance
Point mutations in the ALK kinase domain were identified in plasma specimens from 11 patients (50%), five of whom had paired biopsies at the time of mutation detection (Fig 3B). In eight of 11 patients, ALK mutations were considered sufficient to mediate TKI resistance. In the remaining three patients, ALK mutations were detected during progression on lorlatinib, a pan-inhibitory ALK TKI that was expected to be active against a broad spectrum of mutations. ALK G1202R—present in the plasma from six patients—was the most common ALK mutation. Three patients had multiple ALK mutations.
All tissue-detected ALK mutations were also identified in corresponding plasma samples, and all plasma-detected ALK mutations were also present in paired biopsies. Overall, there was 100% concordance between detected tissue and plasma ALK mutations when temporally correlated specimens were assessed. Among 17 patients who had plasma collected during progression on a second-generation ALK inhibitor—that is, ceritinib, alectinib, or brigatinib—plasma ALK mutations were identified in eight patients (47%; Data Supplement), including I1171N (n = 1), G1202R (n = 5), I1171N/G1202R (n = 1), and E1210K (n = 1). The observed frequency of plasma ALK mutations is consistent with our previous analysis of resistance biopsies from patients who developed resistance to second-generation inhibitors, which included five patients from the current plasma cohort.7
Non-ALK Alterations
As we did not detect any differences in tissue alterations that were identified in serial specimens from the three patients who underwent multiple biopsies, we restricted the concordance assessment to one biopsy per individual. Among these 14 biopsies, we identified mutations that involved genes other than ALK in seven specimens (50%). TP53 was mutated in five resistance biopsies (36%). For three of these patients, TP53 mutation was present at the initial diagnosis. At progression, we detected a TP53 I195T mutation in a biopsy that was not detected in plasma from MGH087. Similarly, we only detected MORC I511V and WDFY4 T1039N mutations in MGH919's tissue. All other tissue mutations were also present in plasma (Data Supplement). We did not detect KRAS or EGFR mutations in plasma or tissue specimens.
We also interrogated plasma for copy number alterations. We identified high-level amplification of BCL2L1 and GNAS (> 20 copies each) in the plasma of MGH919 during treatment with lorlatinib (Data Supplement). Although we were not able to obtain tissue after the patient had developed progression on lorlatinib, amplification was not present when a prelorlatinib liver biopsy was assessed using the same 566-gene panel. We identified one case (MGH939) of MET amplification in an alectinib-resistant lung biopsy specimen. Despite high-level MET amplification (> 25 copies) in tissue, we did not detect MET amplification in the corresponding plasma specimen; however, as the maximum allelic frequency (AF) of detected alterations in the plasma of MGH939 was approximately 2%, the degree of amplification was below the limit of detection of the assay.
Evolution of Resistance During Molecular Surveillance
ALK Fusion Kinetics and Correlation With Disease Status
We hypothesized that the trend in plasma ALK fusion AF for each patient would correlate with disease status. Across patients, we observed a wide range of fusion AF in patients who had developed resistance to TKI treatment. Furthermore, the absolute change in fusion AF was modest for many patients, with peak progression values of < 0.5%. Still, changes in fusion AF reflected clinical status, with higher AF observed at disease progression than during response.
Evolution of ALK Resistance Mutations During Therapy
Preclinical models and case reports suggest that the presence of a specific ALK mutation in a TKI-resistant tissue specimen may predict response to subsequent ALK TKIs1,7,23; therefore, we analyzed plasma specimens from patients with ALK mutations who received treatment with additional ALK TKIs to evaluate the potential of using plasma findings to inform sequential therapies. As noted above, we considered ALK mutations to be the major driver of resistance for eight (36%) of 22 patients in our cohort. During the follow-up period, seven of these patients (88%) went on to receive treatment with a TKI that targeted the ALK mutation (Data Supplement). In all cases, we observed the suppression of pretreatment plasma ALK mutations when a subsequent ALK TKI with preclinical activity against the mutation was initiated (Data Supplement). For example, MGH987 developed a plasma ALK I1171N mutation on alectinib treatment that was not present during treatment with crizotinib. On the basis of studies that support its activity against ALK 11171N,23 ceritinib was initiated, which led to a radiographic and molecular response with a decrease in ALK I1171N AF from 4.5% to undetectable after 6 weeks (Fig 4A). Similarly, we detected an E1210K mutation in the plasma of MGH059 after experiencing progression on alectinib, which became undetectable during response to lorlatinib, a next-generation ALK TKI with preclinical activity against this mutation7 (Appendix Fig A2, and Data Supplement).
Fig 4.
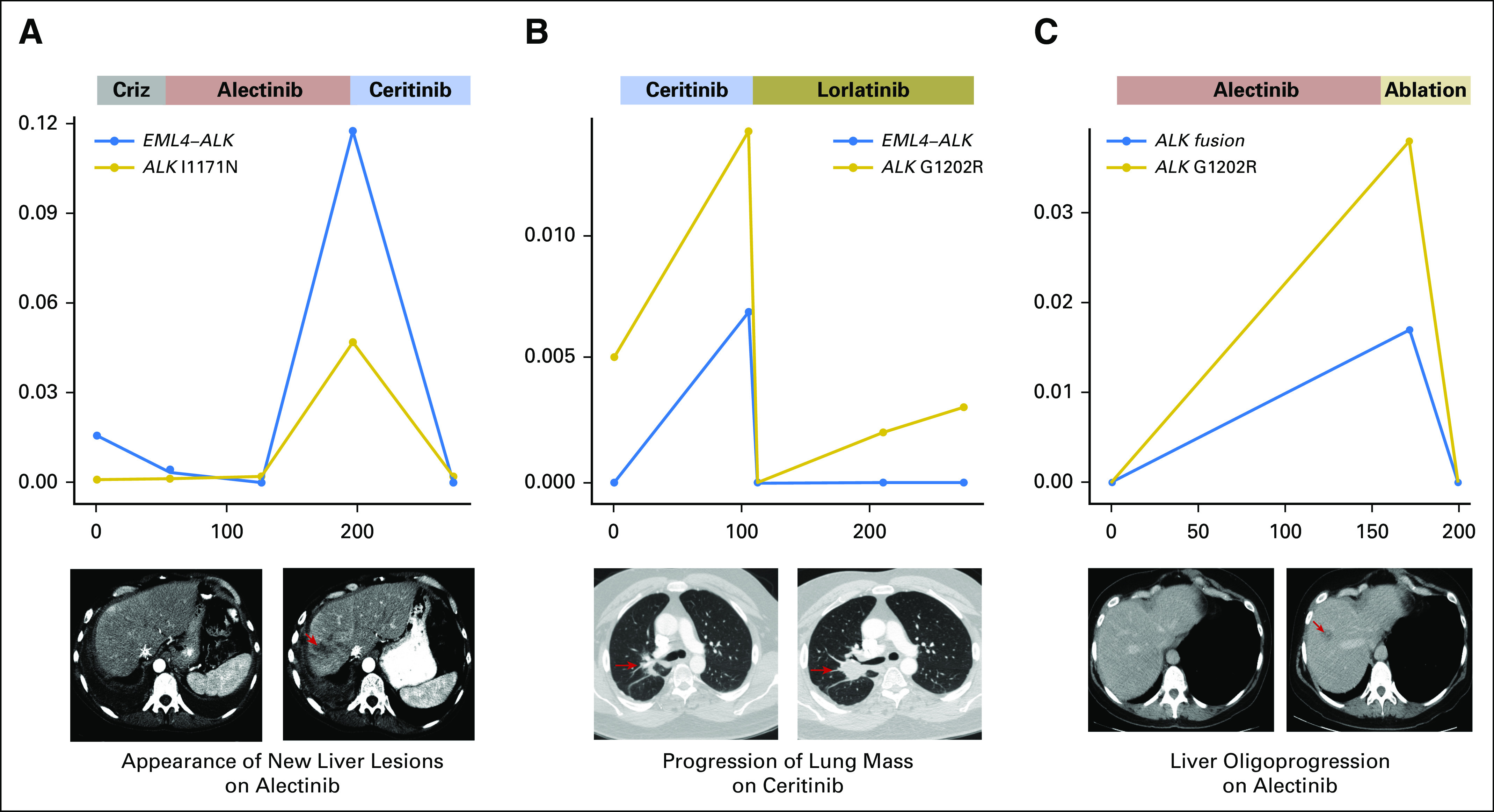
Plasma ALK (anaplastic lymphoma kinase) mutation kinetics during treatment. Figure illustrates the change in the allelic fraction of EML4-ALK and ALK mutations during sequential treatment with next-generation ALK inhibitors for (A) MGH987 and (B) MGH087. (C) ALK fusion and mutation kinetics before and after ablation of an oligometastasis (MGH989). The x-axis depicts days from initial plasma collection, whereas the y-axis reports allelic fraction. Criz, crizotinib.
Among ALK resistance mutations that were identified in patients who experienced progression on second-generation ALK TKIs, the solvent front G1202R mutation is the most common.7 Of note, lorlatinib has been shown to retain activity against cancers that harbor G1202R.7,24 In our cohort of patients, six (35%) of 17 patients who had plasma collected during treatment with a second-generation ALK TKI acquired the G1202R mutation when experiencing disease progression. One patient (MGH989) underwent microwave ablation to treat a liver lesion with a biopsy-proven G1202R mutation and continued treatment with alectinib for an additional 6 months. After ablation, G1202R AF decreased from 4% to undetectable (Fig 4C). The remaining five patients were treated with lorlatinib. Two of these patients—MGH087, depicted in Figure 4B and MGH936, depicted in Appendix Figure A2—achieved a response to treatment and plasma clearance of G1202R. MGH990 had a radiographic response to treatment, but we could not evaluate the molecular response as we were only able to collect a single plasma specimen.
Although the G1202R mutation was suppressed in plasma during the treatment with lorlatinib, two of the four patients with plasma-detected G1202R developed progressive disease soon after initiating lorlatinib (Appendix Fig A3). MGH947 experienced symptomatic progression of a brain metastasis, which prompted a craniotomy. G1202R was not detected in the resected lesion. MGH919 developed multisite progression and rapidly declined. We were not able to obtain a biopsy, but plasma analysis demonstrated an increase in copy number of GNAS and BCL2L1 and a marked increase in the AF of mutations that involved other genes during treatment (Fig 5). In both cases, EML4-ALK fusion AF increased at progression despite the suppression of ALK mutations, which supports the notion that resistance was independent of genetic ALK alterations.
Fig 5.
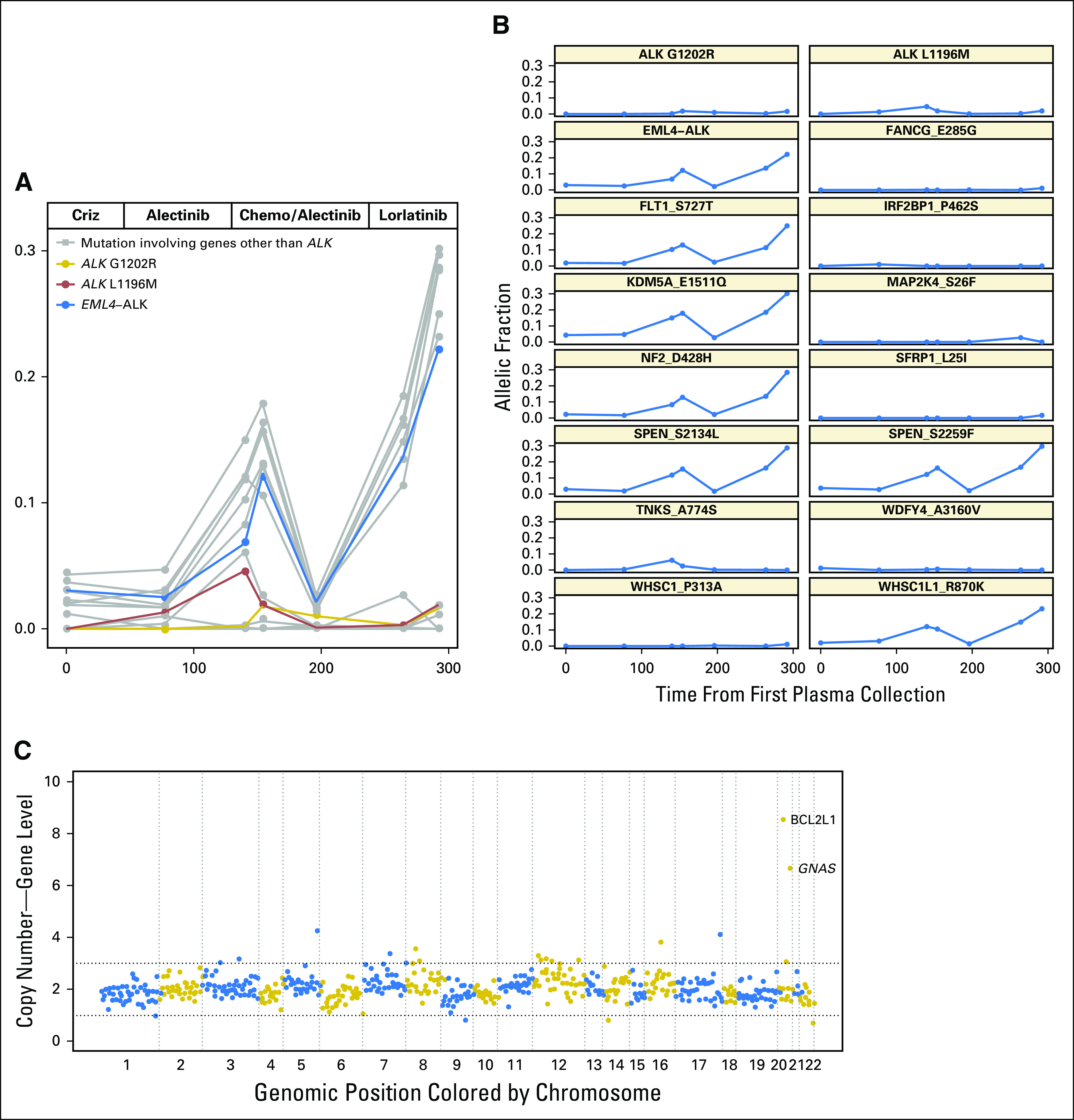
Longitudinal analysis of the plasma of MGH919. (A) Allelic fraction of ALK (anaplastic lymphoma kinase) fusion (blue), ALK mutations (red and gold), and non-ALK alterations (gray) during treatment with sequential ALK inhibitors. The x-axis represents days from initial plasma collection, whereas the y-axis represents allelic fraction. (B) Individual mutation kinetics during treatment. (C) Plasma copy number plot from a specimen collected during treatment with lorlatinib shows focal amplification of GNAS and BCL2L1. ALK L1196M and G1202R mutations were in trans. Chemo, chemotherapy; Criz, crizotinib.
DISCUSSION
Despite a shared molecular driver, the clinical course is variable for patients with ALK-positive NSCLC treated with ALK TKIs. Personalized treatment approaches currently rely on serial tissue sampling to guide subsequent management; however, tissue sampling often fails to capture the dynamic and heterogeneous nature of resistance. To our knowledge, we present here the largest study of longitudinal plasma genotyping in ALK-positive NSCLC to date. Our findings highlight the potential clinical utility of hybrid-capture NGS for improving our understanding of the molecular drivers of resistance.
In our cohort of patients, 76% of plasma samples contained sufficient tumor-derived DNA for molecular analysis compared with 65% of biopsy specimens, which confirms that both are reliable approaches. We identified plasma ALK mutations in one half of our patients at disease progression, many of whom had been exposed to a second-generation ALK inhibitor. Overall, there was a high degree of concordance between plasma and tissue alterations. In addition to demonstrating agreement between plasma and tissue at specific time points, we observed changes in plasma ALK mutation status and mutation AF during longitudinal analysis. These results highlight the advantages of noninvasive genotyping and the potential of serial plasma analysis to elucidate the impact of therapeutic selective pressure on clonal dynamics. Furthermore, our observation that the presence of specific plasma ALK mutations correlated with the response and resistance to distinct ALK TKIs lends support to the emerging practice of using ALK mutations to inform the selection of ALK TKIs.
Although tissue sampling is the gold standard for molecular analysis, sampling a single site of disease may overestimate the contribution of a particular alteration to the resistant phenotype. For example, we detected an ALK G1202R mutation at 50% AF in a progressing liver lesion, which suggested that it was a major contributor to alectinib resistance in the case of MGH919. In contrast, the plasma G1202R AF of 1% was less than the AF of other plasma-detected molecular alterations, including multiple non-ALK alterations (Fig 5). The patient’s primary disease progression on lorlatinib supports the conclusion that ALK G1202R was likely not the major driver of resistance. Moreover, this case reinforces the notion that the analysis of liquid biopsies may be more informative than tissue-based genotyping in certain situations.
To date, the study of resistance to ALK therapeutics has primarily focused on ALK mutations. As a significant proportion of patients may not acquire ALK mutations, we evaluated whether tracking plasma fusion AF might be used to anticipate disease progression. Our results indicate that even when fusions can be detected longitudinally, the dynamic range of the fusion AF during response and progression may be limited. These findings suggest that trending plasma ALK fusions as a surrogate for disease status may not be uniformly applicable. Indeed, we did not detect an ALK fusion in the plasma from three of our patients during disease progression. The ratio of the resistance mutation AF to the activating mutation AF can offer insight into tumor heterogeneity and predict clinical outcomes in patients with EGFR-mutant NSCLC8,25; however, the differing efficiencies of capturing point mutations and structural variants may result in an ALK mutation AF that exceeds fusion AF, which suggests that similar calculations may be challenging for ALK-positive NSCLC. Despite these shortcomings, our data suggest that the quantitative assessment of structural variants may be clinically useful and complementary to radiographic assessment in some patients.
Overall, our data confirm that plasma genotyping by using hybrid-capture NGS technology can reliably detect ALK fusions and ALK resistance mutations in patients with ALK-positive NSCLC; however, there are several limitations of this study, including the small size of our cohort, inconsistent sampling intervals that led to variation in the duration of follow-up after and before disease progression, and the inability to obtain pretreatment plasma and paired biopsies for all patients. Furthermore, as a result of the small contribution of tumor DNA to total cell-free DNA in most patients, we cannot definitively exclude the presence of undetected amplification events at resistance. Finally, as the assay only analyzed genetic alterations in 566 genes, the current analysis does not account for resistance as a result of alterations in genes not covered by the panel and nongenetic mediators of drug resistance.
The optimal strategy for integrating plasma genotyping into clinical practice remains to be established. For patients with EGFR-mutant NSCLC, there are two US Food and Drug Administration–approved liquid biopsies that may be used to select initial and subsequent targeted therapies. As identifying ALK resistance mutations is emerging as an important consideration for the management of ALK-positive NSCLC, we anticipate that genotyping plasma to characterize resistance to ALK TKIs will also become a routine part of patient care. Although tissue remains the gold standard, larger ALK-specific concordance studies may ultimately pave the way for ctDNA analysis to become an essential component of the evaluation of ALK TKI resistance. Beyond identifying alterations at resistance, one of the major advantages of plasma genotyping is its ability to track response kinetics throughout the treatment course. Additional studies are indicated to determine whether plasma mutation clearance and subclinical molecular relapse are predictive of clinical outcomes.
ACKNOWLEDGMENT
We also thank Jeremy Decker for assistance with plasma preparation and circulating tumor DNA processing.
Appendix
Fig A1.

ALK (anaplastic lymphoma kinase) fusions detected in plasma. Figure illustrates the spectrum of ALK fusion variants that were detected in plasma specimens. Blue indicates the presence of a fusion or fusion variant, whereas white indicates the absence of a detectable fusion. Numbers denote the exon breakpoints for each gene. In two of the three patients for whom plasma analysis failed to detect a fusion, an EML4-ALK fusion (variants 3 and 5) was identified in matching tissue. The remaining patient did not undergo a repeat biopsy, but was ALK positive by fluorescence in situ hybridization at diagnosis. E, EML4; A, ALK.
Fig A2.

Longitudinal monitoring during sequential treatment with ALK (anaplastic lymphoma kinase) inhibitors. Figure illustrates ALK fusion (MGH059 only; left) and ALK mutation plasma allelic fraction during progression on alectinib and the subsequent response to lorlatinib. The assay did not detect an ALK fusion in the plasma of MGH936 during longitudinal assessment. The y-axis indicates the allelic fraction of the ALK alterations.
Fig A3.

Plasma ALK (anaplastic lymphoma kinase) fusion and ALK mutation allelic fractions of MGH919 (left) and MGH947 (right) during treatment with ALK inhibitors. The y-axis indicates the allelic fraction of the ALK fusion and ALK mutations. Chemo, chemotherapy; Criz, crizotinib.
Footnotes
Funded by the National Cancer Institute Grant No. 5R01CA164273 (A.T.S.), Be a Piece of the Solution, Novartis, and LungStrong. Supported in part by an ASCO/Conquer Cancer Young Investigator Award (I.D.-J.).
AUTHOR CONTRIBUTIONS
Conception and design: Ibiayi Dagogo-Jack, A. Rose Brannon, Jeffrey A. Engelman, Rebecca J. Leary, Alice T. Shaw, Justin F. Gainor
Administrative support: Lorin A. Ferris
Provision of study material or patients: Ibiayi Dagogo-Jack, Jennifer Ackil, Lecia V. Sequist, Inga T. Lennes, Rebecca S. Heist, Christopher G. Azzoli, Anna F. Farago, Jochen K. Lennerz, Alice T. Shaw, Justin F. Gainor
Collection and assembly of data: Ibiayi Dagogo-Jack, A. Rose Brannon, Lorin A. Ferris, Catarina D. Campbell, Jessica J. Lin, Katherine R. Schultz, Jennifer Ackil, Sara Stevens, Satoshi Yoda, Lecia V. Sequist, Anthony John Iafrate, Rebecca S. Heist, Christopher G. Azzoli, Jochen K. Lennerz, Rebecca J. Leary, Alice T. Shaw, Justin F. Gainor
Data analysis and interpretation: Ibiayi Dagogo-Jack, A. Rose Brannon, Catarina D. Campbell, Jessica J. Lin, Leila Dardaei, Satoshi Yoda, Harper Hubbeling, Subba R. Digumarthy, Markus Riester, Aaron N. Hata, Inga T. Lennes, Anthony John Iafrate, Anna F. Farago, Jeffrey A. Engelman, Jochen K. Lennerz, Cyril H. Benes, Rebecca J. Leary, Alice T. Shaw, Justin F. Gainor
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Ibiayi Dagogo-Jack
Consulting or Advisory Role: Boehringer Ingelheim
Honoraria: Foundation Medicine
A. Rose Brannon
Employment: Novartis Institutes of BioMedical Research
Stock and Other Ownership Interests: Johnson & Johnson
Lorin A. Ferris
No relationship to disclose
Catarina D. Campbell
Employment: Novartis, Pfizer (I)
Stock and Other Ownership Interests: Novartis
Jessica J. Lin
Honoraria: Chugai Pharma
Consulting or Advisory Role: Boehringer Ingelheim
Katherine R. Schultz
No relationship to disclose
Jennifer Ackil
No relationship to disclose
Sara Stevens
No relationship to disclose
Leila Dardaei
No relationship to disclose
Satoshi Yoda
No relationship to disclose
Harper Hubbeling
No relationship to disclose
Subba R. Digumarthy
No relationship to disclose
Markus Riester
Employment: Novartis
Aaron N. Hata
Research Funding: Amgen, Novartis, Relay Therapeutics
Lecia V. Sequist
Honoraria: AstraZeneca
Consulting or Advisory Role: AstraZeneca, Genentech, Bristol-Myers Squibb, Pfizer
Research Funding: Boehringer Ingelheim (Inst), Clovis Oncology (Inst), Genentech (Inst), Merrimack Pharmaceuticals (Inst), Novartis (Inst), AstraZeneca (Inst), Johnson & Johnson (Inst), Merck (Inst), Pfizer (Inst)
Inga T. Lennes
Honoraria: Blue Cross and Blue Shield of Massachusetts
Consulting or Advisory Role: Kyruus
Anthony John Iafrate
Stock and Other Ownership Interests: Archer Biosciences
Consulting or Advisory Role: Debiopharm Group, Constellation Pharmaceuticals, Chugai Pharma, Roche
Research Funding: Blueprint Medicines
Patents, Royalties, Other Intellectual Property: ArcherDx exclusive license to AMP Technology
Rebecca S. Heist
Consulting or Advisory Role: Boehringer Ingelheim
Research Funding: GlaxoSmithKline, Sanofi, AbbVie, Novartis, Roche, Incyte, Celgene, Mirati Therapeutics, Peregrine Pharmaceuticals, Exelixis, Millennium Pharmaceuticals, Debiopharm Group
Christopher G. Azzoli
Consulting or Advisory Role: Merck, ARIAD Pharmaceuticals, Takeda
Anna F. Farago
Honoraria: Foundation Medicine
Consulting or Advisory Role: PharmaMar, Merrimack Pharmaceuticals, Takeda, AbbVie, Intervention Insights
Research Funding: PharmaMar (Inst), AbbVie (Inst), AstraZeneca (Inst), Bristol-Myers Squibb (Inst), Merck (Inst), Loxo (Inst), Ignyta (Inst)
Travel, Accommodations, Expenses: PharmaMar, AbbVie, Stemcentrx
Jeffrey A. Engelman
Employment: Novartis
Stock and Other Ownership Interests: Loxo, Agios, Kura, Gatekeeper Pharmaceuticals, Novartis
Consulting or Advisory Role: Novartis, Agios, Loxo, Clovis Oncology, Ventana Medical Systems, G1 Therapeutics, Sanofi, Chugai Pharma, Warp Drive Bio, Asana Biosciences, EMD Serono, Synta, Allostery, Genentech, Aveo, Biodesix, Merck, Cell Signaling Technology, Endo Pharmaceuticals, AstraZeneca, GlaxoSmithKline, Amgen, Bristol-Myers Squibb, Cancer Progress
Research Funding: Novartis, Jounce Therapeutics, Sanofi, Araxes, AstraZeneca, Amgen (Inst)
Patents, Royalties, Other Intellectual Property: Coinventor on a patent application that has been licensed to Ventana Medical System/Roche
Other Relationship: Third Rock Ventures
Jochen K. Lennerz
No relationship to disclose
Cyril H. Benes
Honoraria: Araxes
Research Funding: Amgen, Novartis
Rebecca J. Leary
Employment: Novartis
Stock and Other Ownership Interests: Novartis
Patents, Royalties, Other Intellectual Property: Patents, pending patents, and a royalty-sharing agreement with Johns Hopkins University
Alice T. Shaw
Honoraria: Pfizer, Novartis, Genentech, Foundation Medicine
Consulting or Advisory Role: Pfizer, Novartis, Genentech, Roche, ARIAD Pharmaceuticals, Ignyta, Blueprint Medicines, Daiichi Sankyo, EMD Serono, Taiho Pharmaceutical, KSQ Therapeutics
Research Funding: Pfizer, Novartis, Genentech
Justin F. Gainor
Honoraria: Merck, Incyte, ARIAD Pharmaceuticals
Consulting or Advisory Role: Novartis, Boehringer Ingelheim, Clovis Oncology, Genentech, Bristol-Myers Squibb, Theravance, Loxo
Research Funding: Merck, Novartis, Genentech, Bristol-Myers Squibb, Adaptimmune, AstraZeneca, ARIAD Pharmaceuticals
Travel, Accommodations, Expenses: Affymetrix
REFERENCES
- 1.Lin JJ, Riely GJ, Shaw AT. Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov. 2017;7:137–155. doi: 10.1158/2159-8290.CD-16-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non–small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 4.Kim DW, Mehra R, Tan DS, et al. Activity and safety of ceritinib in patients with ALK-rearranged non–small-cell lung cancer (ASCEND-1): Updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol. 2016;17:452–463. doi: 10.1016/S1470-2045(15)00614-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw AT, Gandhi L, Gadgeel S, et al. Alectinib in ALK-positive, crizotinib-resistant, non–small-cell lung cancer: A single-group, multicentre, phase 2 trial. Lancet Oncol. 2016;17:234–242. doi: 10.1016/S1470-2045(15)00488-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim DW, Tiseo M, Ahn MJ, et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non–small-cell lung cancer: A randomized, multicenter phase II trial. J Clin Oncol. 2017;35:2490–2498. doi: 10.1200/JCO.2016.71.5904. [DOI] [PubMed] [Google Scholar]
- 7.Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6:1118–1133. doi: 10.1158/2159-8290.CD-16-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non–small-cell lung cancer. J Clin Oncol. 2016;34:3375–3382. doi: 10.1200/JCO.2016.66.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2016;2:1014–1022. doi: 10.1001/jamaoncol.2016.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oxnard GR, Paweletz CP, Sholl LM. Genomic analysis of plasma cell-free DNA in patients with cancer. JAMA Oncol. 2017;3:740–741. doi: 10.1001/jamaoncol.2016.2835. [DOI] [PubMed] [Google Scholar]
- 11.Paweletz CP, Sacher AG, Raymond CK, et al. Bias-corrected targeted next-generation sequencing for rapid, multiplexed detection of actionable alterations in cell-free DNA from advanced lung cancer patients. Clin Cancer Res. 2016;22:915–922. doi: 10.1158/1078-0432.CCR-15-1627-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwaederlé MC, Patel SP, Husain H, et al. Utility of genomic assessment of blood-derived circulating tumor DNA (ctDNA) in patients with advanced lung adenocarcinoma. Clin Cancer Res. 2017;23:5101–5111. doi: 10.1158/1078-0432.CCR-16-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next-generation sequencing of cell-free circulating tumor DNA. Clin Cancer Res. 2016;22:5772–5782. doi: 10.1158/1078-0432.CCR-16-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med. 2014;20:1479–1484. doi: 10.1038/nm.3729. [DOI] [PubMed] [Google Scholar]
- 15.Sholl LM, Do K, Shivdasani P, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016;1:e87062. doi: 10.1172/jci.insight.87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conde E, Hernandez S, Prieto M, et al. Profile of Ventana ALK (D5F3) companion diagnostic assay for non–small-cell lung carcinomas. Expert Rev Mol Diagn. 2016;16:707–713. doi: 10.1586/14737159.2016.1172963. [DOI] [PubMed] [Google Scholar]
- 18.Curigliano G, Gómez Pardo P, Meric-Bernstam F, et al. Ribociclib plus letrozole in early breast cancer: A presurgical, window-of-opportunity study. Breast. 2016;28:191–198. doi: 10.1016/j.breast.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ye K, Schulz MH, Long Q, et al. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schröder J, Hsu A, Boyle SE, et al. Socrates: Identification of genomic rearrangements in tumour genomes by re-aligning soft clipped reads. Bioinformatics. 2014;30:1064–1072. doi: 10.1093/bioinformatics/btt767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riester M, Singh AP, Brannon AR, et al. PureCN: Copy number calling and SNV classification using targeted short read sequencing. Source Code Biol Med. 2016;11:13. doi: 10.1186/s13029-016-0060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katayama R, Friboulet L, Koike S, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;20:5686–5696. doi: 10.1158/1078-0432.CCR-14-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solomon B, Bauer TM, Felip E, et al. Safety and efficacy of lorlatinib (PF-06463922) from the dose-escalation component of a study in patients with advanced ALK+ or ROS1+ non-small cell lung cancer (NSCLC) J Clin Oncol. 2016;34(suppl 15; abstr 9009) [Google Scholar]
- 25.Chabon JJ, Simmons AD, Lovejoy AF, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016;7:11815. doi: 10.1038/ncomms11815. [DOI] [PMC free article] [PubMed] [Google Scholar]