

ABSTRACT
While lentiviral expression vectors are widely used in many facets of molecular biology, due to their ability to stably express heterologous genes in both dividing and non-dividing cells, they suffer from the disadvantage that introns inserted into the vector genome are generally rapidly lost by splicing in packaging cell lines. The presence of an intron, if achievable, has the potential to facilitate the expression of transgene cDNAs, as splicing has been extensively shown to facilitate mRNA biogenesis and function. Moreover, if a stable intron could be introduced into a lentiviral vector, this could greatly facilitate the expression of microRNAs (miRNAs), and especially miRNA clusters, as the introduction of pri-miRNA stems into the exonic region of a lentiviral vector can strongly reduce both vector titer and the expression of any miRNA-linked indicator gene due to cleavage of the vector RNA genome by cellular Drosha. Here, we describe a novel lentiviral vector design in which transgenes and/or miRNAs are expressed using an antisense-orientated, inducible promoter driving an expression cassette bearing a functional intron. We demonstrate that this lentiviral vector, called pTREX, is able to express higher levels of both transgenes and pri-miRNA clusters when compared with a closely similar conventional lentiviral vector.
KEYWORDS: Intron, lentiviral vector, microRNA, PKR, Tet-inducible, transgene expression
Introduction
Retroviral and lentiviral vectors are widely used to express mRNAs or regulatory RNAs, such as microRNAs (miRNAs), in cultured cells or in vivo. While the titers that can be achieved using retroviral and lentiviral vectors are maximally ∼108 infectious units per ml, substantially lower than can be achieved with vectors based on adeno-associated virus (AAV), retroviral and lentiviral vectors do offer some clear advantages, including a larger packaging size (∼9 kb for lentiviral vectors versus only ∼4.8 kb for AAV) and, most importantly, the ability to efficiently integrate into the genome of transduced cells, thus potentially allowing the stable expression of the transgene of interest.1
While lentiviral vectors are particularly widely used due to their ability to infect non-dividing cells, they do suffer from one clear disadvantage. Specifically, because lentiviral vectors use RNA as their genome, any introns that are inserted into that genome are generally rapidly lost due to splicing.2 This means that lentiviral vectors, as currently designed, are only able to express cDNA copies of the gene of interest. However, it is well established that splicing can significantly boost the expression of mRNAs by promoting mRNA polyadenylation, nuclear export and accumulation.3-9 While this positive effect is often only 2 to 4-fold, some genes, including for example β-globin and preproinsulin, give rise to only very low levels of protein expression if the cognate pre-mRNA lacks all introns.6
A second, related, problem arises if the lentiviral vector is being used to express one or more miRNAs. Almost all miRNAs are initially transcribed as part of a long capped, polyadenylated pri-miRNA precursor in which the mature ∼22-nt miRNA forms one side of the upper 2-thirds of an ∼33-bp imperfect stem bearing a large terminal loop.10,11 This stem-loop is recognized by the nuclear microprocessor, consisting of the RNase III enzyme Drosha and its co-factor DGCR8, which cleaves the stem ∼22 bp away from the terminal loop to generate the pre-miRNA hairpin intermediate. This processing event cleaves the pri-miRNA into 3 fragments and, if the pri-miRNA is also an mRNA, results in a readily detectable inhibition of the expression of not only the full-length mRNA but also any encoded protein.10,12 This effect is significant when one pri-miRNA stem-loop is introduced into the 3′ untranslated region (UTR) of an mRNA and becomes severe if a miRNA cluster, containing multiple pri-miRNA stem-loops, is inserted into a 3′UTR. Perhaps as a result, miRNAs of both cellular and viral origin are usually initially expressed either as long non-coding RNAs or located within an intron.13 In the context of lentiviral expression vectors, pri-miRNA stem-loops have historically been cloned into the 3′UTR of a transgene, such as green fluorescent protein (GFP), that can be used to isolate cells expressing high levels of the miRNA by FACS for the linked GFP.14 However, the expression level of the linked transgene will again be very low if multiple pri-miRNA stems are inserted in cis and the titer of the lentiviral miRNA expression vector will be very low for the same reason, i.e., cleavage of the vector genome by the microprocessor before packaging.15,16
To address both these problems we have therefore designed a lentiviral vector, the Tet-inducible Reverse intron EXpression vector pTREX, that allows transgenes to be expressed as spliced mRNAs and also allows miRNAs to be expressed from an intronic location, thus greatly facilitating the derivation of cells expressing high levels of either heterologous transgenes or miRNAs.
Results
As noted above, it is well established that splicing facilitates the accumulation of mRNAs encoding proteins of interest, yet in the context of lentiviral vectors, genes are invariably expressed as unspliced cDNAs.2-9 We previously examined the effect of splicing on a wide range of genes expressed from transfected plasmids and observed positive effects that generally ranged from 2- to 4-fold.6 Yet, for a subset of genes, including β-globin and preproinsulin, the absence of a functional intron in the initial pre-mRNA transcript resulted in a dramatic 10–40-fold drop in the level of protein expression. Therefore, we hypothesized that a lentiviral vector that expressed mRNAs bearing a functional intron in the 5′UTR should at least modestly enhance the expression of most cDNAs and strongly enhance the expression of a subset of mRNAs.
As our strategy to retain a functional intron in a lentiviral vector, we decided to express lentiviral vector transcripts containing that intron in the antisense orientation. However, if the promoter driving antisense transcription was constitutively active, we would expect the antisense transcript to basepair with the vector genome to generate long dsRNAs in the vector packaging cell line. These dsRNAs might then activate innate antiviral immune responses, including repression of mRNA translation by the protein kinase RNA activated (PKR).17 We therefore decided to express the antisense RNA using a regulated promoter, specifically a minimal cytomegalovirus (CMV) immediate early promoter modified to include binding sites for the tetracycline transactivator (TetO/CMV), to minimize dsRNA production. As a starting point, and as the control for these studies, we used the commercially available vector pTRIPZ (GE Healthcare) shown schematically in Fig. 1A. In this lentiviral vector, the inducible TetO/CMV promoter drives the expression of a transgene, which is turboRFP in the parental pTRIPZ vector, linked to a 3′UTR that contains unique XhoI and EcoRI sites that can be used for the insertion of one or more pri-miRNA stem-loops. A 3′ constitutive promoter, derived from the ubiquitin C (UBC) gene, drives expression of the reverse tetracycline transactivator 3 (rtTA3) protein as well as a puromycin (Puro)-selectable marker linked to the rtTA3 open reading frame via an internal ribosome entry site (IRES). The pTRIPZ vector contains a self-inactivating (SIN) 3′ LTR so that transductants generated using this vector express only 2 mRNAs: a bicistronic mRNA encoding rtTA3 and Puro that is expressed constitutively and a second mRNA, initiating from the TetO/CMV promoter, that expresses a fluorescent indicator gene and any inserted pri-miRNAs, that is only expressed efficiently in the presence of the inducer doxycycline (Dox).
Figure 1.
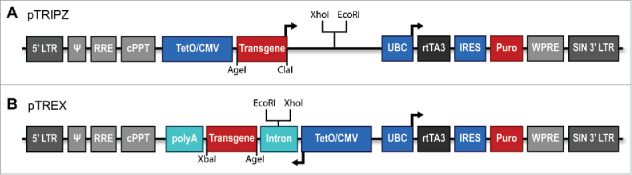
Schematic representation of control forward (pTRIPZ) and novel reverse orientation (pTREX) lentiviral expression vectors. (A) The forward orientation lentiviral vector pTRIPZ is commercially available (GE Healthcare/Dharmacon). pTRIPZ allows expression of a transgene cloned between unique AgeI and ClaI sites and miRNAs cloned between unique XhoI and EcoRI sites present in the transgene 3′UTR. Both are transcribed from a tet-inducible minimal CMV promoter (TetO/CMV). A 3′ Ubiquitin C promoter (UBC) drives the constitutive expression of the reverse tetracycline transactivator 3 (rtTA3) protein as well as a puromycin (Puro) selectable marker located 3′ to an internal ribosome entry site (IRES). (B) The pTREX lentiviral expression vector was generated from pTRIPZ by inverting sequences encompassing the TetO/CMV and inserting an intron derived from the rat preproinsulin II gene 5′ of the inserted transgene. pTREX also contains a poly(A) addition site located 3′ to the inserted transgene. Unique XhoI and EcoRI sites located in the intron are used for miRNA expression. Transgenes are cloned between the unique AgeI and XbaI sites. Additionally, both vectors contain elements necessary for the HIV-1 life cycle including the long-terminal repeats (LTRs), ψ packaging signal, Rev response element (RRE) and the central polypurine tract (cPPT) as well as a woodchuck hepatitis virus post-transcriptional regulatory element (WPRE), which enhances vector expression. The 3′ LTR bears a large deletion in U3 rendering the lentivirus self-inactivating (SIN).
To generate a vector containing an inverted transgene linked to an intron, we excised the sequence bearing the TetO/CMV promoter, the transgene and the adjacent 3′UTR from pTRIPZ and then reinserted the TetO/CMV promoter and transgene in the reverse orientation (Fig. 1B). We then inserted sequences encompassing an intron found naturally in the 5′UTR of the rat preproinsulin II gene6 into the 5′UTR of the transgene but in a modified form such that the intron now contained unique XhoI and EcoRI sites (Fig. 1B). As a final step, we inserted a genomic poly(A) addition site downstream of the transgene. The resultant pTREX vector therefore expresses precisely the same bicistronic rtTA3/Puro mRNA as pTRIPZ while a second mRNA, again controlled by the inducible TetO/CMV promoter, encodes a spliced mRNA encoding a transgene inserted in the antisense orientation that terminates at the inserted antisense poly(A) site (Fig. 1B). Importantly, the pTREX lentiviral vector bearing Thy1.1 as a transgene is, at ∼7.7 kb, the same size as the equivalent pTRIPZ-derived Thy1.1 expression vector, thus facilitating a functional comparison without concerns as to any confounding effects of vector size on packaging efficiency.
Efficient pTREX vector production requires inactivation of the pkr gene
As an initial test of the relative utility of the intron-containing pTREX lentiviral vector relative to the conventional pTRIPZ vector, we inserted enhanced green fluorescent protein (GFP) as the transgene but did not insert any pri-miRNA stem-loops. We then transfected pTREX and pTRIPZ into 293T cells along with a vector, pCMV-ΔR8.74, that encodes the HIV-1 gag, pol, rev and tat genes, and a second plasmid, pMD2.G, that expresses the vesicular stomatitis virus (VSV) glycoprotein. We then examined the production of virion particles by Western blot of the producer cells (Fig. 2A and B), by ELISA for released p24 capsid protein in the supernatant media (Fig. 2C) and finally by quantifying the number of GFP+ cells produced upon transduction of naïve 293T cells using the supernatant media from the transfected cultures (Fig. 2D). Unexpectedly, we observed 5 to 10-fold lower levels of p24 production both intracellularly in the producer cells (Fig. 2A and B) and in the extracellular media (Fig. 2C) as well as a >10-fold drop in vector titer (Fig. 2D) when pTREX was used in place of pTRIPZ. As the Gag expression vector used in these experiments, pCMV-ΔR8.74, is identical in all cultures, this result implied that pTREX was inhibiting the production of the HIV-1 proteins encoded by pCMV-ΔR8.74, including Gag, in trans. As noted above, a potential problem with a lentiviral vector that encodes an antisense transcript is the inadvertent production of dsRNAs that induce antiviral innate immune responses such as RNA interference (RNAi) and PKR-induced translational repression.17 We therefore hypothesized that the TetO/CMV promoter in pTREX was likely producing inhibitory levels of the predicted antisense transcript in the packaging cells even in the absence of added Dox. To test the hypothesis, we used 2 previously described cell lines derived by gene editing of 293T cells, called NoDice and NoDice/ΔPKR, that either lack a functional dcr gene (NoDice) or lack both dcr and pkr (NoDice/ΔPKR).18,19
Figure 2.
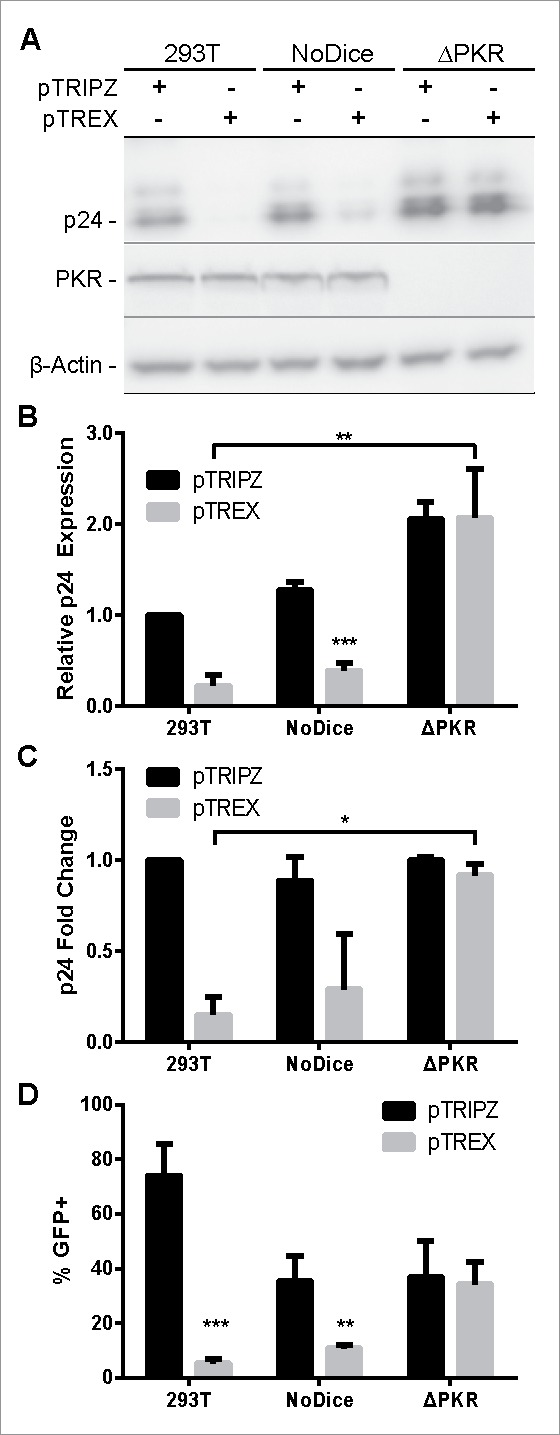
pTREX-based vectors are packaged efficiently in PKR-knockout (ΔPKR) cells. pTRIPZ and pTREX-based vectors expressing GFP were packaged in human 293T, NoDice, or NoDice/ΔPKR cells. (A) Lentiviral packaging efficiency analyzed by Western blot detection of the p24 capsid protein in packaging cells. (B) Average quantification of p24 Western blot band intensities, as performed in panel A, normalized to the β-actin internal control. (C) Amount of lentiviral particles released from each packaging line measured by ELISA detection of p24 in the supernatant media. (D) Quantification of percent GFP positivity in 293T cells 3 d post-transduction by Dox addition. Data were analyzed by a Student's t-test; *p < 0.05, **p < 0.01, ***p < 0.001. Panels B through D represent the average of 3 independent experiments. (Error bars = SD)
As shown in Fig. 2, ablation of the cellular dcr gene had at most a modest positive effect on the expression of the HIV-1 Gag protein (Fig. 2A-C) and resulted in a slight drop in the production of infectious vector particles (Fig. 2D), most probably because of the slower growth phenotype of the NoDice cell line, which lacks all cellular miRNAs, when compared with wild type 293T cells.18 In contrast, use of the NoDice/ΔPKR cells resulted in a dramatic increase in both intracellular (Fig. 2A and B) and extracellular (Fig. 2C) p24 capsid expression that was statistically significant (p < 0.05). Moreover, the level of p24 Gag production and infectious vector particle production was now identical for cells transfected with either pTREX or pTRIPZ (Fig. 2).
pTREX expresses higher levels of inserted transgenes
We next wished to examine whether the intron-containing pTREX vector would indeed express higher levels of inserted cDNAs, as predicted based on previous work.2-9 As an initial test, we packaged pTREX and pTRIPZ variants bearing gfp as the transgene and then transduced naïve 293T cells. Transduced cells were then selected using puromycin and the selected cells induced by addition of Dox or left untreated. At that point, the level of GFP protein expression was quantified by flow cytometry.
As shown in Fig. 3A and B, we noted low but significant levels of GFP expression even in the absence of Dox, which confirms that the uninduced TetO/CMV promoter is indeed leaky, as proposed above. Of note, this background GFP expression level was significantly lower with the pTREX vector than with the pTRIPZ-derived vector (compare the gray shoulder in Fig. 3A with 3B). As expected, addition of Dox did, however, result in a dramatic induction in the level of GFP expression in cells transduced by either vector, as measured by the overall pattern of GFP expression (Fig. 3A and B) and the mean fluorescent intensity (MFI) of the induced cultures. Importantly, the level of GFP expression was ∼2-fold higher in cells transduced with the pTREX vector, when compared with pTRIPZ (compare the peak fluorescence shown in panels A and B of Fig. 3 indicated by the red line). However, the total number of GFP+ cells was comparable with both pTREX and pTRIPZ, as expected given that transduced cells had been selected using puromycin resistance.
Figure 3.
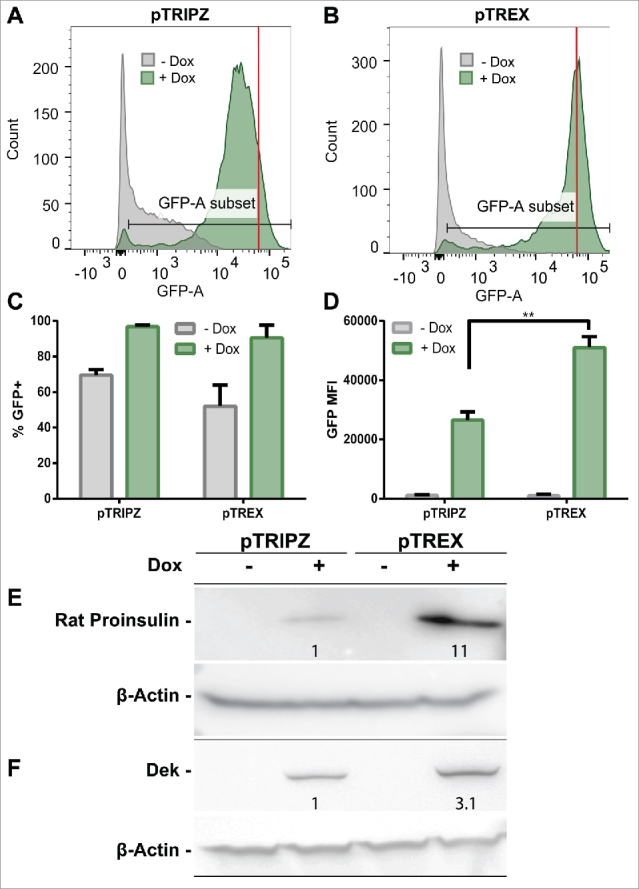
pTREX shows superior transgene expression when compared with pTRIPZ. (A-D) 293T cells were transduced with the lentiviral vectors pTRIPZ or pTREX expressing GFP as a transgene and selected with puromycin for 1 week. GFP expression was then induced by treatment with Dox for 3 d or the cells were maintained in the absence of Dox. GFP expression was then analyzed by flow cytometry. GFP signal histograms are shown for (A) pTRIPZ-transduced cells, and (B) pTREX-transduced cells, with counts of Dox-induced cells in green, and no Dox controls in gray. The average of 4 independent transductions is shown as (C) mean fluorescence intensity (MFI) of GFP+ cells or (D) percent GFP+ of the whole population. Data were analyzed by a Student's t-test; **p < 0.01. (Error bars = SD). Cells transduced with the pTREX vector showed an ∼2-fold higher MFI, as indicated by red lines in panels A and B and quantified in panel (D) Transgene expression from pTRIPZ or pTREX-transduced, puromycin-selected 293T cells were also compared via Western blot for the transgenes (E) proinsulin and (F) human Dek, which also showed significantly higher expression levels in the pTREX transduced cells, as indicated by the fold change in band intensity shown in these panels.
While GFP expression from the intron-containing pTREX vector was only ∼2-fold higher than seen with the pTRIPZ vector lacking any introns, there are a significant number of genes that have been reported to be highly or moderately dependent on splicing for high transgene expression, including, respectively, the rat preproinsulin II gene and the human proto-oncogene dek.2-9 We indeed observed a more significant phenotype using these 2 cDNAs. In the case of the preproinsulin gene, levels of proinsulin present in puromycin-selected cells transduced with pTREX were ∼11-fold higher than seen with pTRIPZ-transduced cells (Fig. 3E) while, in the case of dek, the level of protein production was ∼3-fold higher when pTREX was used (Fig. 3F). We therefore conclude that pTREX does indeed allow the more efficient expression of inserted transgenes and that this positive effect can be highly significant for a subset of cDNAs.
The pTREX vector permits enhanced microRNA expression
While we anticipated that the intron-containing pTREX might prove capable of expressing higher levels of inserted protein-coding genes, our primary goal in developing pTREX was in the hope that this vector would permit the generation of high titers of lentiviral vector particles that express significantly increased levels of an inserted miRNA, or miRNA cluster, in transduced cells. To achieve this goal, we first inserted the murine Thy1.1 gene as the transgene in pTREX and pTRIPZ and then inserted either a single pri-miRNA stem-loop, encoding human miR-15518; a pri-miRNA cluster derived from Epstein-Barr virus (EBV) encoding the 3 viral miRNAs miR-BHRF1–1, 1–2 and 1–320; or finally, a large pri-miRNA cluster derived from Kaposi's sarcoma-associated herpesvirus (KSHV) that encodes the 5 KSHV miRNA miR-K1, miR-K2, miR-K3, miR-K4 and miR-K5.21 These pri-miRNAs were inserted between the XhoI and EcoRI sites present in the 3′UTR of the Thy1.1 transgene in pTRIPZ or between the XhoI and EcoRI sites found in the intron located in the 5′UTR of the Thy1.1 transgene in pTREX. The 3 resultant vectors, as well as an empty vector lacking any inserted pri-miRNAs, were then packaged in NoDice/ΔPKR cells and used to transduce HeLa cells. After puromycin selection and expansion, the HeLa transductants were induced by Dox treatment of 3 d and then harvested for flow cytometric analysis of the level of expression of the Thy1.1 protein (Fig. 4 A to F), for RT-PCR analysis of the expression of the Thy1.1 transgene mRNA (Fig. 4G and H) and finally for qRT-PCR analysis of mature miRNA expression (Fig. 5).
Figure 4.

pTREX expresses a higher level of a marker gene when pri-miRNAs are present in cis. pTREX and pTRIPZ vectors bearing a Thy1.1 transgene were packaged in NoDice/ΔPKR cells and transduced into HeLa cells. Transduced cells were selected for puromycin resistance, and then treated with Dox for 3 d. Thy1.1 positivity was measured with an APC-conjugated antibody by flow cytometry. Thy1.1 histograms are shown for (A) empty vector, (B) miR-155, (C) miR-BHRF1–123, and (D) miR-K1–5-transduced HeLa cells. pTREX is shown in blue and pTRIPZ is shown in red. The average of 3 independent experiments is shown as (E) percent Thy1.1+ cells and (F) MFI of positive cells. Student's t-test, ***p < 0.001. (Error bars = SD). Thy1.1 transcript abundance, and the level of intron removal for pTREX, were determined by RT-PCR using total RNA from (G) pTRIPZ and (H) pTREX transduced cells. PCR was also performed using the input plasmid DNA, as a control for primer function and for unspliced RNA expression. RT-PCR was performed for GAPDH as a loading control. Green arrows indicate primer locations.
Figure 5.
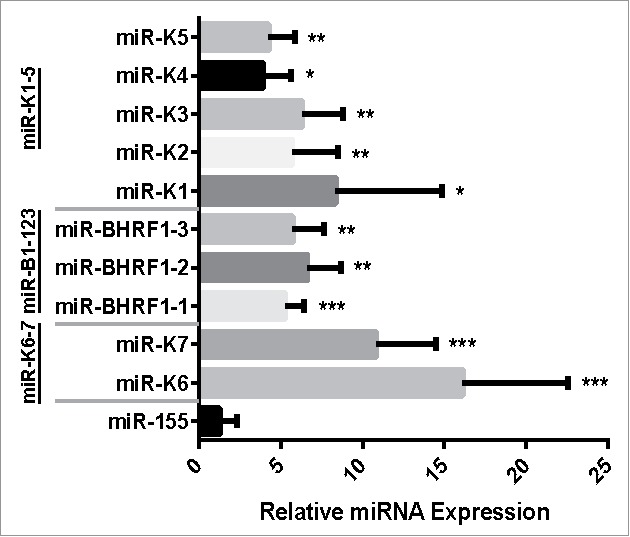
pTREX expresses miRNA clusters at a significantly higher level than does pTRIPZ. HeLa cells were transduced with miRNA expression vectors derived from pTREX and pTRIPZ, selected with puromycin, and induced with Dox for 3 d before RNA was harvested. miRNA expression levels were measured by TaqMan RT-qPCR and fold change calculated using the ΔΔCt method, normalizing to the miRNA expression level observed in pTRIPZ, which was set at 1. Data shown represent the average of 4 independent replicates analyzed using a one-sample t-test; *p < 0.05, **p < 0.01, ***p < 0.001 (Error bars = SD).
In the case of the empty vector transduced cells, we observed a bimodal expression pattern for Thy1.1, with both pTREX and pTRIPZ giving rise to both a peak of high expressing cells and a peak with low or negative Thy1.1 expression (Fig. 4A). Curiously, even though all of these cells had been selected for puromycin resistance, and therefore had been transduced, this was a particular problem for pTRIPZ. Specifically, while ∼80% of pTREX transduced cells were strongly Thy1.1 positive, only ∼15% of the pTRIPZ transductants were scored as Thy1.1 positive (Fig. 4E). The reasons for this presumably epigenetic silencing effect, which was largely restricted to pTRIPZ transduced cells, is unclear but this was seen with all pTRIPZ-derived vectors expressing Thy1.1 (Fig. 4E), but not GFP (Fig. 3A), and was unaffected by whether the pTRIPZ vector had also been designed to express miRNAs (Fig. 4E).
As noted above, it is anticipated that insertion of pri-miRNA stem-loops into the 3′UTR of the Thy1.1 indicator gene, as is done in pTRIPZ, will reduce expression of that gene at both the mRNA and protein level, and that this inhibitory effect will increase when more pri-miRNA stem loops are inserted.10,12,15,16 Conversely, we hypothesized that insertion of pri-miRNA stem-loops into the 5′UTR intron present in pTREX would have no effect on the expression of the linked Thy1.1 indicator gene at either the protein or mRNA level.
As shown in Fig. 4B to D, we indeed observed a reduction in the level of cell surface Thy1.1 expression from the pTRIPZ vector if pri-miRNAs were introduced into the 3′UTR of the Thy1.1 transgene, and this effect was profound when 3 (miR-BHRF1–1, 2 and 3) or 5 (miR-K1 to K5) pri-miRNAs were introduced, though this effect was statistically significant even with the single miR-155 pri-miRNA (Fig. 4F). In contrast, introduction of up to 5 pri-miRNA stem loops into the 5′UTR intron present in pTREX had no detectable effect on Thy1.1 expression from this vector (Fig. 4B to D and 4F).
Similarly, analysis of Thy1.1 mRNA expression from the pTRIPZ-based miRNA expression vectors using RT-PCR showed that while the predicted Thy1.1 mRNA was detectable for the empty pTRIPZ Thy1.1 expression vector, and barely detectable with the pTRIPZ vector expressing miR-155, the Thy1.1 mRNA became undetectable when multiple pri-miRNA stems were present in the Thy1.1 3′UTR present in pTRIPZ (Fig. 4G). This does not reflect any problem with the PCR primers used, as shown by the plasmid DNA control lanes. Conversely, when expression of the Thy1.1 mRNA was examined in cells transduced by the pTREX-based miRNA expression vectors, the level of Thy1.1 mRNA was uniform across all cultures regardless of the number of pri-miRNAs inserted into the Thy1.1 5′UTR intron. Moreover, as the PCR primers used in this analysis amplify across this inserted intron, these data also show that the intron, as well as any inserted pri-miRNA stem-loops, had all been removed from the mature Thy1.1 mRNAs expressed in the pTREX transduced cells due to splicing, even though these were clearly present in the starting plasmid DNAs used here as controls (Fig. 4G). In conclusion, these data confirm that excision of the intron present in pTREX is efficient and that splicing is not affected by the insertion of pri-miRNA stems into this intron.
While the data presented in Fig. 4 confirm that pTREX-based miRNA expression vectors allow a much higher level of expression of the encoded indicator Thy1.1 transgene due to avoidance of both Thy1.1 mRNA and vector RNA cleavage by Drosha, these data do not address the key question of whether the observed higher mRNA expression level also correlates with an increase in the mature miRNA expression level in transduced cells. To determine whether this is indeed the case, we transduced HeLa cells with the 3 pTREX and pTRIPZ-based miRNA expression vectors described in Fig. 4, as well as a fourth set of vectors encoding a 2 pri-miRNA cluster derived from KSHV, consisting of miR-K6 and miR-K7.21 Transduced cells were selected for puromycin resistance and then induced with Dox for 3 d before harvesting total RNA and performing TaqMan qRT-PCR for each encoded miRNA as well as for endogenous U6 snRNA, as a normalization control. Note that this protocol does not take advantage of the higher vector titers seen with pTREX relative to pTRIPZ and also does not involve the selection of “Thy1.1 high” cells by FACS, which would be predicted to increase the expression of the linked miRNA(s), but rather analyzes a mixed population of transduced, puromycin resistant cells.
As shown in Fig. 5, we did not, in fact, see a significant increase in mature miRNA expression in pTREX relative to pTRIPZ if the single pri-miR-155 stem-loop was expressed. However, in the case of the vectors expressing the 2 KSHV miRNAs miR-K6 and K7, the 3 EBV miRNAs miR-BHRF1–1, 1–2 and 1–3, or the 5 KSHV miRNAs miR-K1 through miR-K5, we observed a statistically significant (p < 0.05) ∼5-fold or greater increase in the level of expression for all 10 viral miRNAs. We therefore conclude that pTREX offers substantial advantages over conventional miRNA expression vectors, such as pTRIPZ, when the simultaneous expression of 2 or more miRNAs from a single lentiviral vector is desired.
Discussion
The goal of this project was to design and test an inducible lentiviral vector, here called pTREX, that expresses an intron-containing transgene in the antisense orientation with the goal of increasing transgene expression, increasing the expression of encoded miRNAs and, finally, increasing the generally low infectious titer of lentiviral miRNA expression vectors. The latter problem arises because pri-miRNA stem-loops inserted into the 3′ UTR of a linked indicator transgene, the method used previously to express pri-miRNAs using lentiviral vectors (Fig. 1),14 results in the cleavage of both the transgene mRNA and the vector genomic RNA by the nuclear microprocessor complex. This issue has been reported previously,15,16 and one suggested means of avoiding this problem is to co-transfect the packaging cells with siRNAs specific for Drosha, which can reduce the amount of pri-miRNA expression vector cleavage. However, by inserting the pri-miRNA stem-loops into an intron transcribed in the antisense orientation (Fig. 1) we were able to not only avoid all vector RNA cleavage (Fig. 4H), thus resulting in a higher level of vector and transgene expression (Fig. 4F), but also generate transduced cells that express an ∼5 to 15-fold higher level of all members of pri-miRNA clusters cloned into the pTREX vector intron, when compared with the similar pTRIPZ control vector (Fig. 5). We note that others have described previously plasmid-based miRNA expression vectors in which the pri-miRNAs were inserted into an intron and these plasmids were also reported to express higher levels of not only the encoded miRNAs but also of the linked indicator genes.22,23
Because it is well established that introns can also facilitate the expression of cloned cDNAs,2-9 we also were hopeful that the pTREX vector would also allow increased transgene expression. In fact, this positive effect, while readily detectable, was generally a modest 2–3-fold, as seen with cDNAs encoding GFP and human Dek (Fig. 3). However, in the case of the preproinsulin gene, which we have previously shown to be highly intron-dependent, protein expression was found to be ∼11-fold higher with the pTREX vector than seen with the control pTRIPZ vector (Fig. 3). As several genes have been reported to be highly dependent on splicing for effective protein expression,3-9 we believe the pTREX vector has the potential to be generally useful, especially given that even a modest ∼2–3-fold enhancement in protein expression, as previously reported for most cDNAs when introns are introduced in cis,6 may prove experimentally important.
Despite the advantages presented by the pTREX vector in terms of both transgene and miRNA expression, pTREX does have the disadvantage that it generates low levels of dsRNAs in transfected packaging cells, resulting in inhibition of protein expression presumably due to activation of PKR (Fig. 2). Thus, the titers achieved using pTREX in wildtype 293T cells, which are the workhorse packaging cell line for lentiviral vector production, are low. This result may explain why we were only able to find a small number of published papers describing retroviral or lentiviral vectors that express their transgene using an antisense promoter. One report, by Karlsson et al.,24 described a vector based on murine leukemia virus in which the human genomic β-globin gene, including both introns and the genomic promoter, was inserted in an antisense orientation and gave rise to regulated expression of β-globin in transduced erythroleukemia cells. Holehonnur et al.25 described a lentiviral vector that contains an antisense expression cassette that includes an intron under the control of the neurotropic 1,3αCaMKII promoter and showed that this vector could successfully transduce neurons in vivo. Finally, Cooper et al.2 describe a SIN lentiviral vector in which the UBC promoter, together with a UBC-derived intron, were used to express an indicator gene in the antisense orientation in transduced cells. Of note, none of these papers discussed how to optimize vector particle production in the transfected packaging cell line, which for both lentiviral vector papers was the same 293T cell line used here, so how the problem of PKR activation by dsRNAs was prevented is unclear. As noted in Fig. 2, we found that the induction of PKR activity by dsRNAs produced by pTREX, even using the inducible TetO/CMV promoter in the absence of Dox, still resulted in the potent inhibition of vector particle production. Fortunately, with the advent of facile methods for gene editing in cultured human cells, it proved simple to mutationally inactivate the pkr gene in the 293T packaging cell line 19 and thereby fully rescue the efficient production of pTREX-derived lentiviral particles (Fig. 2). We note that because the pTREX vector bears a 3′ LTR that is deleted for all enhancer and promoter sequences present in the LTR “U3” region, pTREX is a self-inactivating (SIN) vector that is incapable of initiating transcription in the 5′ LTR in transduced cells. Therefore, we did not anticipate that pTREX would produce any dsRNAs in transduced cells and in fact no evidence of toxicity was observed. We anticipate that we will be able to distribute the pTREX vector, as well as the 293T-based NoDice/ΔPKR packaging cell line, to basic researchers interested in testing the ability of pTREX to give enhanced transgene and/or miRNA expression in their system upon request.
Materials and Methods
Cell culture
293T and HeLa cells were grown in Dulbecco's Modified Eagle Medium (DMEM, Sigma Aldrich) supplemented with 5% fetal bovine serum (FBS) and 10 µg/mL gentamicin (Gibco, 15710064). NoDice and NoDice/ΔPKR cells18,19 were grown in DMEM with 10% FBS and 1x Antibiotic-Antimycotic (Gibco, 15240062). 293T cells were switched to 10% FBS media when used in experiments comparing with NoDice or NoDice/ΔPKR cells.
Plasmids
pTREX was created by PCR amplification of the Tet-inducible minimal TetO/CMV promoter and transgene TurboRFP from pTRIPZ (RHS4696, Dharmacon) followed by cloning in the reverse orientation at the same location in pTRIPZ. The 5′ rat preproinsulin II intron,6 modified to contain internal XhoI and EcoRI cloning sites, was PCR amplified and cloned 5′ to the transgene. A bovine growth hormone genomic polyadenylation site was then PCR amplified and cloned 3′ to the transgene. Primers used for cloning can be found in Table S1 (1–8). The integrity of all PCR amplified DNA sequences was confirmed.
Transgenes were inserted into the AgeI/ClaI or AgeI/XhoI sites of pTRIPZ (replacing TurboRFP). In pTREX, transgenes were inserted between the AgeI/XbaI sites (Fig. 1). The enhanced GFP (gfp) gene was cloned from pLCE.26 Flag-tagged rat preproinsulin and Dek cDNAs were derived from the plasmids pCMV/Δ1+2/INS and pCMV/DEK, respectively.6 Mouse Thy1.1 (CD90.1) was cloned from a MSCV-Thy1.1 expression vector (a gift from the Qi-Jing Li laboratory). Primers used for PCR amplification of these sequences can be found in Table S1 (9–17).
All lentiviral miRNA expression vectors analyzed contained Thy1.1 as their transgene. All pri-miRNAs were cloned between the XhoI and EcoRI sites present in both pTREX and pTRIPZ. Human miR-155 was cloned using the XhoI/EcoRI sites present in the pL-SIN-CMV-AcGFP-miR-155 vector.27 miR-BHRF1–1 and 1–3 were expressed from artificial miR-30-based expression cassettes.28 Sequences are given in Supplemental Table S1 (18, 19). miR-BHRF1–2 was PCR cloned from the EBV genome and inserted between the miR-BHRF1–1 and 1–3 expression cassettes. KHSV miRNAs miR-K1 through miR-K5, or miR-K6 and miR-K7, were PCR amplified from the KSHV genome present in the BC1 cell line21 using primers listed in Table S1 (20–25).
Lentiviral packaging plasmids pCMV-ΔR8.74 and pMD2.G were gifts from Didier Trono, (Addgene plasmids #22036 and 12259). pCMV-ΔR8.74 expresses all HIV-1 proteins except for env, vpr, vif, vpu, and nef. pMD2.G expresses the VSV-G envelope glycoprotein.
Lentivirus transgene packaging and transduction
All transgene expression vectors were packaged by transfecting 5 × 106 293T, NoDice, or NoDice/ΔPKR cells in 10 cm plates with 5.5 µg lentiviral vector, 6 µg pCMV-ΔR8.74 helper plasmid, 2.5 µg pMD2.G, and 36.3 µl of polyethylenimine (PEI). Plasmids along with PEI were pre-mixed in 1 ml of OPTI-MEM then added to cells 15 minutes later. Culture media were changed the next day, and supernatants collected 3 d post-transfection. Lentiviral particles harvested from each 10-cm plate were passed through a 0.45 µm filter and then used to transduce 2 wells of a 6-well plate of 293T cells at 8 × 105 cells per well.
Lentivirus packaging efficiency
pTRIPZ and pTREX vectors expressing GFP were packaged in 293T, NoDice, and NoDice/ΔPKR cells. The packaging cells and supernatant media were harvested 3 d post-transfection. Packaging cells were subjected to Western blot analysis probing for PKR (Abcam ab32506), β-Actin (Santa Cruz sc-47778), and HIV-1 p24 (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: HIV-1 p24 Gag Monoclonal (#24–3) from Dr. Michael H. Malim).29-31 Western blot bands were quantified using ImageJ (Rasband WS. ImageJ, US. National Institutes of Health, Bethesda, Maryland, USA, imagej.nih.gov/ij/, 1997–2012). To assess vector packaging efficiency, supernatants containing the 2 vectors packaged in 293T, NoDice, and NoDice/ΔPKR cells were subjected to HIV-1 p24 ELISA (Advanced Bioscience Laboratories, HIV-1 p24 antigen capture assay #5421); and used to transduce 293T cells. Transduced cells were treated with doxycycline shortly after transduction. Transduction and transgene expression efficiency were measured by flow cytometric analysis of cellular GFP expression 3 d post-transduction.
Post-selection transgene expression
pTRIPZ or pTREX vectors expressing GFP, preproinsulin, and Dek were packaged in NoDice/ΔPKR cells. Lentiviral particles were harvested 3 d post-transfection and used to transduce 293T cells. Lentivirus-transduced cells were subjected to puromycin selection (0.2–0.5 µg/ml) starting 3 d post-transduction, and kept in puromycin-containing media for 2 weeks with frequent media changes until cell death could no longer be detected. Selected cells were treated with doxycycline for at least 2 d before harvest. GFP expression was analyzed by flow cytometry using a BDFACSCanto II (BD Biosciences 338962) and proinsulin and Dek expression measured by Western blot using a Flag antibody (Sigma-Aldrich F3165).
microRNA and Thy1.1 expression
All lentiviral miRNA expression vectors were packaged by transfecting 3 × 105 NoDice/ΔPKR cells in a 6-well plate with 1.2 µg lentiviral plasmid, 1.2 µg pCMV-ΔR8.74 helper plasmid and 0.5 µg pMD2.G envelope plasmid. Plasmid DNA and 7.3 µL PEI were mixed in 150 µL OPTI-MEM and allowed to sit for 15 minutes before being added to cells. The culture media were exchanged for 2 ml of DMEM supplemented with 10% FBS and gentamicin the following day. Supernatant media were collected 3 d post-transfection and filtered through a 0.45 µm filter onto HeLa cells plated at 3 × 106 cells per 10-cm plate on the previous day. Two days post-transduction, 1 µg/ml puromycin was added and the cells cultured until colonies were visible. Cells were then collected and re-plated into 2 wells of a 6-well plate (one for flow cytometry and one for RNA) and Thy1.1/miRNA expression induced by treating with 1 µg/ml doxycycline for 3 d.
Thy1.1 expression was tested in Dox-induced cells by staining for Thy1.1 positivity and measuring expression by flow cytometry. Briefly, cells were collected by centrifugation and then resuspended in 100 µl PBS with 5% FBS and 1 µL APC conjugated Thy1.1 antibody (Biolegend 202526). Cells were allowed to sit in the dark for 20 minutes and then collected and resuspended in PBS with 5% FBS. Cells were then subjected to flow cytometry on a BDFACSCanto II.
Transcript abundance and splicing was analyzed by RT-qPCR. Total RNA was harvested from transduced, puromycin selected, Dox-induced cultures using TRIzol and 4 µg total RNA then reverse transcribed using an oligo-dT primer and SuperScript III Reverse Transcriptase according to the manufacturer's protocol (ThermoFisher Scientific, 18080093). PCR was performed on the original plasmids and cDNA using primers 26–29 listed in Table S1. cDNA was also analyzed for GAPDH mRNA expression using primers 30 and 31 listed in Table S1.
miRNA expression levels were analyzed by TaqMan qRT-PCR. Total RNA was harvested from Dox induced cells using TRIzol according to the manufacturer's protocol (Ambion, 15596018). 10 ng total RNA was reverse transcribed using the TaqMan microRNA reverse transcription kit (Applied Biosystems, 4366596) and miRNA specific stem-loop primers (ThermoFisher Scientific). cDNA was diluted 1:3 and TaqMan qPCR was performed using TaqMan Universal PCR Master Mix, no AmpErase UNG (Applied Biosystems, 4324018) and a miRNA specific probe (ThermoFisher Scientific). Endogenous U6 snRNA levels were determined by the same protocol as an internal control. The relative miRNA expression levels were calculated by first normalizing to U6 then the pTREX expression level was normalized to the expression level seen using pTRIPZ, which was set at 1.0.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The research reported in this manuscript was supported by National Institutes of Health grants R01-AI067968 and R01-AI117780. K.T. was supported by National Institutes of Health T32-CA009111. The authors thank Michael Malim, Qi-Jing Li and Didier Trono for the gift of reagents used in this research. The authors would also like to thank the Duke Cancer Institute Flow Cytometry Shared Resource Core for their help with collecting flow cytometry data.
References
- 1.Kotterman MA, Chalberg TW, Schaffer DV. Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu Rev Biomed Eng 2015; 17:63-89; PMID:26643018; https://doi.org/ 10.1146/annurev-bioeng-071813-104938 [DOI] [PubMed] [Google Scholar]
- 2.Cooper AR, Lill GR, Gschweng EH, Kohn DB. Rescue of splicing-mediated intron loss maximizes expression in lentiviral vectors containing the human ubiquitin C promoter. Nucleic Acids Res 2015; 43(1): 682-90; PMID:25520191; https://doi.org/ 10.1093/nar/gku1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le Hir H, Nott A, Moore MJ. How introns influence and enhance eukaryotic gene expression. Trends Biochem Sci 2003; 28(4): 215-20; PMID:12713906; https://doi.org/ 10.1016/S0968-0004(03)00052-5 [DOI] [PubMed] [Google Scholar]
- 4.Nott A, Meislin SH, Moore MJ. A quantitative analysis of intron effects on mammalian gene expression. RNA 2003; 9(5): 607-17; PMID:12702819; https://doi.org/ 10.1261/rna.5250403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valencia P, Dias AP, Reed R. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc Natl Acad Sci U S A 2008; 105(9): 3386-91; PMID:18287003; https://doi.org/ 10.1073/pnas.0800250105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu S, Cullen BR. Analysis of the stimulatory effect of splicing on mRNA production and utilization in mammalian cells. RNA 2003; 9(5): 618-30; PMID:12702820; https://doi.org/ 10.1261/rna.5260303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vagner S, Vagner C, Mattaj IW. The carboxyl terminus of vertebrate poly(A) polymerase interacts with U2AF 65 to couple 3′-end processing and splicing. Genes Dev 2000; 14(4): 403-13; PMID:10691733; https://doi.org/ 10.1101/gad.14.4.403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ryu WS, Mertz JE. Simian virus 40 late transcripts lacking excisable intervening sequences are defective in both stability in the nucleus and transport to the cytoplasm. J Virol 1989; 63(10): 4386-94. PMID:25506721620616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonsson JJ, Foresman MD, Wilson N, McIvor RS. Intron requirement for expression of the human purine nucleoside phosphorylase gene. Nucleic Acids Res 1992; 20(12): 3191-8; PMID:1620616; https://doi.org/ 10.1093/nar/20.12.3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004; 10(12): 1957-66; PMID:15525708; https://doi.org/ 10.1261/rna.7135204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006; 125(5): 887-901; PMID:16751099; https://doi.org/ 10.1016/j.cell.2006.03.043 [DOI] [PubMed] [Google Scholar]
- 12.Lin YT, Sullivan CS. Expanding the role of Drosha to the regulation of viral gene expression. Proc Natl Acad Sci U S A 2011; 108(27): 11229-34; PMID:21690333; https://doi.org/ 10.1073/pnas.1105799108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cullen BR. Transcription and processing of human microRNA precursors. Mol Cell 2004; 16(6): 861-5; PMID:15610730; https://doi.org/ 10.1016/j.molcel.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 14.Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ. A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A. 2005; 102(37): 13212-7; PMID:16141338; https://doi.org/ 10.1073/pnas.0506306102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu YP, Vink MA, Westerink JT, Ramirez de Arellano E, Konstantinova P, Ter Brake O, Berkhout B. Titers of lentiviral vectors encoding shRNAs and miRNAs are reduced by different mechanisms that require distinct repair strategies. RNA 2010; 16(7): 1328-39; PMID:20498457; https://doi.org/ 10.1261/rna.1887910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brandl A, Wittmann J, Jack HM. A facile method to increase titers of miRNA-encoding retroviruses by inhibition of the RNaseIII enzyme Drosha. Eur J Immunol 2011; 41(2): 549-51; PMID:21268023; https://doi.org/ 10.1002/eji.201040960 [DOI] [PubMed] [Google Scholar]
- 17.Hartmann G. Nucleic Acid Immunity. Adv Immunol 2017; 133: 121-169; PMID:28215278; https://doi.org/ 10.1016/bs.ai.2016.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogerd HP, Whisnant AW, Kennedy EM, Flores O, Cullen BR. Derivation and characterization of Dicer- and microRNA-deficient human cells. RNA 2014; 20(6): 923-37; PMID:4024645; https://doi.org/ 10.1261/rna.044545.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kennedy EM, Whisnant AW, Kornepati AV, Marshall JB, Bogerd HP, Cullen BR. Production of functional small interfering RNAs by an amino-terminal deletion mutant of human Dicer. Proc Natl Acad Sci U S A 2015; 112(50): E6945-54; PMID:26621737; https://doi.org/ 10.1073/pnas.1513421112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai X, Schäfer A, Lu S, Bilello JP, Desrosiers RC, Edwards R, Raab-Traub N, Cullen BR. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog 2006; 2(3): e23; PMID:16557291; https://doi.org/ 10.1371/journal.ppat.0020023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai X, Lu S, Zhang Z, Gonzalez CM, Damania B, Cullen BR. Kaposi's sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci U S A 2005; 102(15): 5570-5; PMID:15800047; https://doi.org/ 10.1073/pnas.0408192102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du G, Yonekubo J, Zeng Y, Osisami M, Frohman MA. Design of expression vectors for RNA interference based on miRNAs and RNA splicing. FEBS J 2006; 273(23): 5421-7; PMID:17076699; https://doi.org/ 10.1111/j.1742-4658.2006.05534.x [DOI] [PubMed] [Google Scholar]
- 23.Hu T, Chen P, Fu Q, Liu Y, Ishaq M, Li J, Ma L, Guo D. Comparative studies of various artificial microRNA expression vectors for RNAi in mammalian cells. Mol Biotechnol 2010; 46(1): 34-40; PMID:20300885; https://doi.org/ 10.1007/s12033-010-9264-7 [DOI] [PubMed] [Google Scholar]
- 24.Karlsson S, Papayannopoulou T, Schweiger SG, Stamatoyannopoulos G, Nienhuis AW. Retroviral-mediated transfer of genomic globin genes leads to regulated production of RNA and protein. Proc Natl Acad Sci U S A 1987; 84(8): 2411-5; PMID:3470803; https://doi.org/ 10.1073/pnas.84.8.2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holehonnur R, Lella SK, Ho A, Luong JA, Ploski JE. The production of viral vectors designed to express large and difficult to express transgenes within neurons. Mol Brain 2015; 8:12; PMID:25887710; https://doi.org/ 10.1186/s13041-015-0100-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottwein E, Cullen BR. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol 2010; 84(10): 5229-37; PMID:20219912; https://doi.org/ 10.1128/JVI.00202-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gottwein E, Mukherjee N, Sachse C, Frenzel C, Majoros WH, Chi J-tA, Braich R, Manoharan M, Soutschek J, Ohler U, et al.. A viral microRNA functions as an ortholog of cellular miR-155. Nature 2007; 450: 1096-99; PMID:18075594; https://doi.org/ 10.1038/nature05992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell 2002; 9(6): 1327-33; PMID:12086629; https://doi.org/ 10.1016/S1097-2765(02)00541-5 [DOI] [PubMed] [Google Scholar]
- 29.Fouchier RA, Meyer BE, Simon JH, Fischer U, Malim MH. HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J 1997; 16(15): 4531-9; PMID:9303297; https://doi.org/ 10.1093/emboj/16.15.4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simon JH, Fouchier RA, Southerling TE, Guerra CB, Grant CK, Malim MH. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J Virol 1997; 71(7): 5259-67; PMID:9188594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simon JH, Carpenter EA, Fouchier RA, Malim MH. Vif and the p55(Gag) polyprotein of human immunodeficiency virus type 1 are present in colocalizing membrane-free cytoplasmic complexes. J Virol. 1999; 73(4): 2667-74; PMID:10074112 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.