

ABSTRACT
RNA structures are increasingly recognized to be of importance during influenza A virus replication. Here, we investigated a predicted conserved hairpin in the M gene segment (nt 967-994) within the region of the vRNA 5′ packaging signal. The existence of this RNA structure and its possible role in virus replication was investigated using a compensatory mutagenesis approach. Mutations were introduced in the hairpin stem, based on natural variation. Virus replication properties were studied for the mutant viruses with disrupted and restored RNA structures. Viruses with structure-disrupting mutations had lower virus titers and a significantly reduced median plaque size when compared with the wild-type (WT) virus, while viruses with structure restoring-mutations replicated comparable to WT. Moreover, virus replication was also reduced when mutations were introduced in the hairpin loop, suggesting its involvement in RNA interactions. Northern blot and FACS experiments were performed to study differences in RNA levels as well as production of M1 and M2 proteins, expressed via alternative splicing. Stem-disruptive mutants caused lower vRNA and M2 mRNA levels and reduced M2 protein production at early time-points. When the RNA structure was restored, vRNA, M2 mRNA and M2 protein levels were increased, demonstrating a compensatory effect. Thus, this study provides evidence for functional importance of the predicted M RNA structure and suggests its role in splicing regulation.
KEYWORDS: Compensatory mutagenesis, influenza A virus, negative sense RNA virus, RNA splicing, RNA structure
Introduction
Influenza viruses belong to the Orthomyxoviridae and constitute a continuous threat for severe disease outbreaks in humans and domestic animals.1,2 The viruses have a segmented negative-sense RNA genome consisting of 8 viral RNA (vRNA) gene segments, each of which is encapsidated into a virus ribonucleoprotein (vRNP) complex. A vRNP contains multiple viral nucleoprotein (NP) molecules and the polymerase complex (PB2, PB1 and PA).3,4 Similar to other viral RNA genomes, the influenza A virus genome is expected to contain structured cis-acting elements which regulate genome functioning in virions and infected cells.5 In particular, influenza virus RNA structures are mostly suggested to be involved in mRNA splicing and vRNA packaging into virions.6-11 The locations containing the minimal packaging signals, necessary to incorporate the 8 different vRNPs into virus particles, were shown to involve the 5′- and 3′- untranslated regions (UTR) and extend into the coding parts of each gene segment (reviewed in4,11). Noda et al.12 demonstrated a highly ordered (7+1) organization of the vRNPs. Further studies revealed complex intersegment RNA-RNA interaction networks, that may vary between different virus subtypes10,13-15 and a possible role for the interactions of small vRNA hairpins (“kissing loops”) on the vRNPs.13 In segment 5 (NP), a conserved RNA pseudoknot in the region spanning the 5′ packaging signal was shown to affect virus replication.16 Conserved hairpins were predicted in the packaging signal regions of segment 4 (HA), although nucleotide covariations in these hairpins exhibited relatively weak constraints and disruption of the structures did not result in noticeable effects on virus replication.17
In the 2 gene segments with alternatively spliced pre-mRNAs, M and NS, the regions in the vicinity of splice sites are able to fold into alternative RNA structures that can regulate splicing efficiency.7,8,18,19 The M gene segment produces several types of mRNA: unspliced M1 mRNA and splice variants M2, M3 and M4 mRNA (Fig. 1A).20-22 M3 mRNA does not encode any protein, but is involved in regulating the switch to M2 mRNA production early in infection and is thereby delaying M2 protein production compared with M1.23 The ratio between M2 and M1 mRNA was shown to increase during infection, although M2 mRNA splicing is regulated in such a way that it remains a small proportion compared with M1 mRNA.24 Differences in the M2/M1 mRNA ratio were observed between different virus strains and splicing efficiency therefore seems to be strain specific.18 M4 mRNA encodes an M2 variant (M42) with a different N-terminus that can be expressed by a minority of influenza A virus strains and can complement M2 protein function through the formation of an ion channel.22
Figure 1.
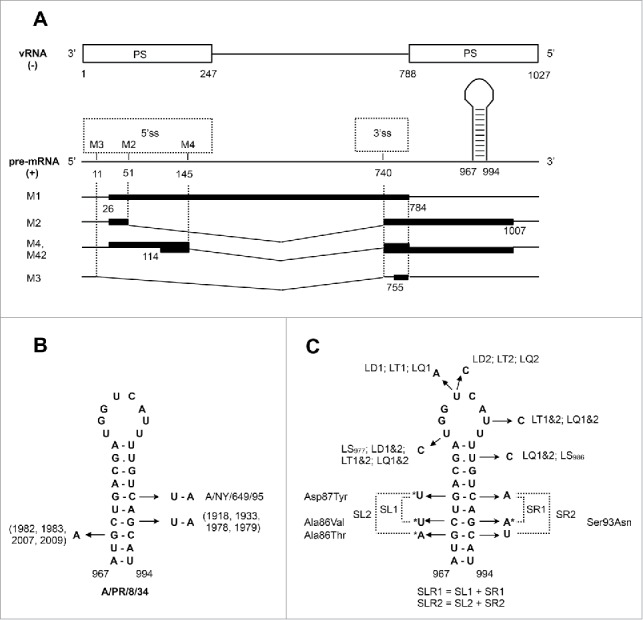
(A) Schematic overview of the main functional sites in the M gene segment. The regions of minimal packaging signals (PS) in the vRNA (negative-sense) are according to Ozawa et al.28 The base numbering starts at the vRNA 3′-end. The unspliced M1 RNA and 3 splice variants produced by alternative 5′ splice sites (5ss) contain 5 potential open reading frames,22 shown here by thick bars. M1 and M2 are important viral proteins, while M42 is probably expressed in a minority of strains; proteins M3 and M4 were never detected. The location of hairpin 967-994 is shown schematically. (B) Natural (co)variations in the 967-994 hairpin stem in human H1N1 viruses. The years of isolation of strains with these variations are indicated. A/NY/649/95 denotes A/New York/649/1995 (H1N1), a strain with unique 972/989 covariation. (C) Mutations that were introduced into the hairpin. Non-synonymous substitutions are marked with asterisks, corresponding amino acid changes in M2 protein are indicated as well.
The region containing the 5′ packaging signals in segment 7 (M) was suggested to contain a stable hairpin (at nucleotides 967-994), predicted by thermodynamics-based folding algorithms, and is conserved in all influenza A virus strains (Fig. 1A, B).5,25 This hairpin was also detected by in vitro structure probing of protein-free M vRNA.26 Several studies have investigated the region that spans this predicted M RNA structure. First, Hutchinson et al. introduced synonymous mutations in the M2 codon of amino acids 90-92 (nucleotide positions 983, 986 and 989, located in the loop and stem of the hairpin). This virus showed reduced virus replication in a plaque assay as a consequence of a packaging defect.27 Second, Ozawa et al. determined the minimal packaging regions of M and further investigated the 5′ end of the packaging region. It was shown that nucleotide composition was more important than amino acid sequence.28 Third, Kobayashi et al. introduced a combination of 3 synonymous mutations to disrupt the predicted M RNA structure and a 5-fold reduction in virus titer was observed.29 Together, these data indicate that the 5′ terminal region of the M gene segment, comprising the RNA structure as previously predicted,5,25 affects virus replication and plays a role in the influenza virus packaging mechanism.
Here, we studied the functioning of this RNA structure using a compensatory mutagenesis strategy. The results demonstrated that hairpin disruptive mutations had a negative effect on virus replication, which can be reverted by structure-restoring mutations. Surprisingly our data indicate involvement of the RNA structure in regulating M mRNA splicing, despite its location in the packaging signal region at a distance more than 200 nucleotides away from the end of the M gene intron (Fig. 1A).
Results
Strategy of the stem compensatory mutagenesis
The best way to obtain convincing experimental evidence of functional importance of a double-stranded RNA structure is to demonstrate compensatory effects in mutants containing double substitutions at the paired nucleotide positions. In case the RNA structure is located in the protein-coding RNA region, the introduced mutations should minimally affect the functions of the encoded protein(s) to avoid protein function modifications and misinterpretation of the data. The hairpin at position 967-994 is located downstream of the M1 stop-codon. However, each of the base pairs in the stem involves at least one nucleotide at the first or second position in M2 codons, making it impossible to design compensatory mutants in this structure with only silent substitutions. Therefore it was decided to explore the natural variation in the stem nucleotides of the hairpin to select mutations yielding functional M2 protein. In particular, variation observed in the human H1N1 strains was explored, as the experimental system used was based on the A/PR/8/34 (H1N1) reverse genetics system.
First, substitutions that characterize the previously identified natural covariation at base pair 970-9915,25 (Fig. 1B) were selected. At this position there is a CG pair in the majority of human strains (including A/PR/8/34), in contrast to UA in some of the early human H1N1 viruses and in avian strains. In addition, the G-C/U-A covariation at position 972-989, observed in one of the human H1N1 strains (A/New York/649/1995 (H1N1); accession CY011281), was selected. It involved 2 transversions (Fig. 1), one silent, of nucleotides that are invariant in all other M segments from human strains available in the database (in total 9163 sequences), apparently indicating the importance of pairing at this position. Apart from these 2 natural covariations, it was noted that several human strains of different subtypes, including some human H1N1 viruses, have the G969A substitution (Ala86Thr in M2), which is easy to combine in experiments with a silent C992U substitution of the nucleotide, paired in the suggested structure model. Thus, in total 3 pairs of point mutations that can be used for compensatory mutagenesis of the A/PR/8/34 M gene segment were selected (Fig. 1C). To enhance the structure-destabilizing effects of substitutions, they were combined in 2 sets (SL1-SR1-SLR1 and SL2-SR2-SLR2), each dealing with disruption and reversion of 2 base pairs. The mutations were introduced in the stem (S) of the predicted RNA structure and were located either on the left side (L), right side (R) or both sides (LR). Thus SLR1 included all mutations of SL1 and SR1, and SLR2 included all mutations of SL2 and SR2. In this way, SL1, SR1, SL2 and SR2 mutants should destabilize hairpins, each with 2 mismatches, whereas the perfect RNA duplex should be restored in SLR1 and SLR2 despite the 4 nucleotide substitutions compared with the WT sequence.
Stem mutant viruses and the effect on viral replication
Mutations were introduced into the stem of the predicted RNA structure as described above (Fig. 1C and 2A). Mutant viruses, including an A/PR/8/34 WT control, were produced by reverse genetics and the M gene segment of the recombinant viruses was sequenced. All mutants contained the desired mutations and no additional co-mutations were detected. Subsequently, virus titers of the 2 sets of mutant viruses (SL1-SR1-SLR1 and SL2-SR2-SLR2) were determined (Fig. 2A). Recombinant viruses with mutations at one side of the stem (SL1, SR1, SL2 and SR2) had lower virus titers (107.3, 107.3, 107.1 and 107.1 TCID50/ml, respectively) than the WT virus (108.4). However, recombinant viruses that contained mutations at both sides of the stem (SLR1 and SLR2), predicted to re-stabilize the RNA structure, had virus titers of 108.3 and 108.1 respectively, which was similar to the WT virus (108.4). When mutant viruses SL1, SR1, SLR1 and the WT virus were passaged 6 times in MDCK cells, at each passage, a 1-2 log lower virus titer was observed between disruptive mutants SL1/SR1 and the compensatory mutant SLR1 and WT virus. At each passage the MA gene segment was sequenced to confirm the presence of the desired mutations and absence of undesired co-mutations (data not shown).
Figure 2.
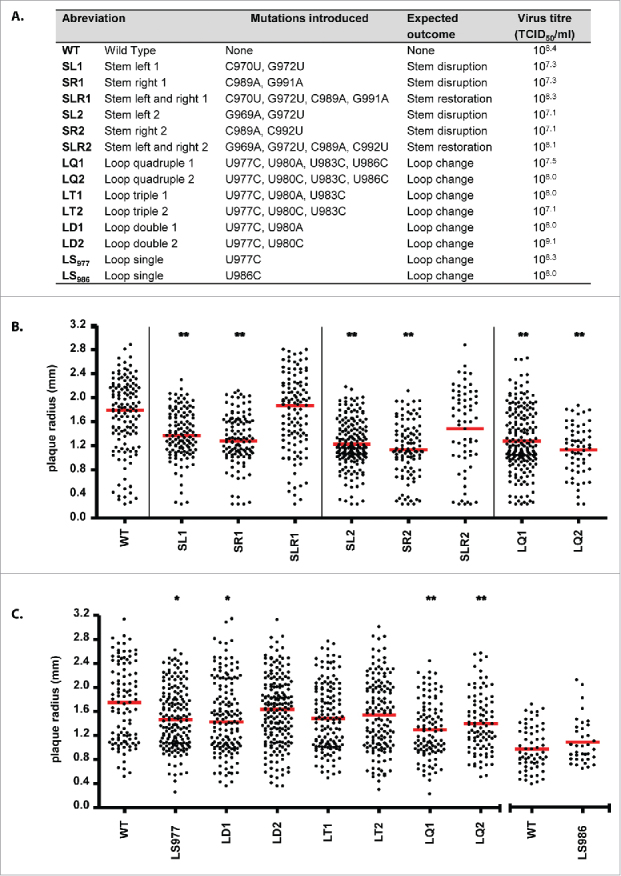
(A) Overview of the mutations that were introduced into the M gene segment of the 967-994 RNA structure showing the expected outcome and the effect on virus replication. (B) Investigation of the effect of the stem (SL1, SR1, SLR1, SL2, SR2 and SLR2) and quadruple loop mutants (LQ1 and LQ2) in a plaque assay compared with the WT control. The plaque radius (in mm) is shown. Statistical significance was determined by using the Kruskal-Wallis and Dunn's multiple comparison test and is depicted by one (p < 0.05) or 2 (p < 0.01) asterisks. The red bars represent the median. (C) The effect of single, double, triple and quadruple loop mutants were compared in a plaque assay. The x-axis was disconnected to identify a separately performed plaque assay where mutant SL986 was compared with the WT virus. Statistical significance was determined by using the Kruskal-Wallis and Dunn's multiple comparison test or the Mann-Whitney t-test. The red bars represent the median.
Next, plaque assays were performed to determine virus replication efficiency in a more sensitive way with larger plaque sizes corresponding to more efficient virus replication. Viruses containing the SL1 and SR1 mutations showed smaller median plaque sizes (1.37 and 1.28 mm respectively) when compared with the WT (1.79 mm) and SLR1 (1.87 mm) virus (Fig. 2B). This was statistically significant (p < 0.001) for the SL1 and SR1 mutant compared with the WT virus. The second set of mutations (SL2-SR2-SLR2) showed a similar pattern (Fig. 2B), with median plaque sizes of 1.17 and 1.11 mm for mutant SL2 and SR2, respectively when compared with WT (1.79 mm) and SLR2 (1.60 mm), which was statistically significant (p < 0.001) compared with the WT virus. Together, these data provide experimental support for the existence and functional importance of the predicted RNA hairpin structure, shown by the compensatory effect of the stabilizing mutants (SLR1 and SLR2) on virus replication.
Loop mutant viruses and the effect on viral replication
Next, recombinant viruses containing 4 silent mutations in the hairpin loop (L) at position 977, 980, 983 and 986 (quadruple mutant; LQ) of the predicted RNA structure were produced to investigate if the loop may be involved in RNA interactions (e.g. via “kissing loops”) that affect virus replication (Fig. 1C and 2A). It should be noted that although nucleotide 986 is suggested to be paired in the M mRNA hairpin stem (Fig. 1B), it is likely to be located in the larger loop of the vRNA (negative sense) counterpart hairpin5,26 due to the 986–975 AC mismatch that also makes the formation of the isolated neighboring base pair 985–976 unlikely. At nucleotide position 980 2 nucleotide changes were possible that resulted in silent mutations; consequently both U980A (LQ1) and U980C (LQ2) mutations were introduced. Loop mutants LQ1 and LQ2 had a virus titer of 107.5 and 108.0 respectively, compared with 108.4 for the WT virus (Fig. 2A). Both LQ1 and LQ2 mutant viruses were tested in a plaque assay (Fig. 2B) and showed a reduced median plaque size (1.19 mm for LQ1 and 1.17 mm for LQ2) when compared with the WT virus (1.79 mm), which was statistically significant (p < 0.001). Subsequently, the influence of different mutations in the loop were determined by introducing mutations from the left (nt position 977) to the right (nt position 986) side of the hairpin loop. Single (LS977), double (LD1/LD2), triple (LT1/LT2) and quadruple (LQ1/LQ2) mutant viruses, where 1 represents an A at position 980 and 2 a C at this position, were produced. Subsequently, the virus titer was determined (Fig. 2A). In general, the different loop mutants had virus titers that were similar to the WT virus (108.4), except LT2 (107.1) and LQ1 (107.5). Next, these mutant viruses were tested in a plaque assay (Fig. 2C). For the single loop mutant (LS977) a small decrease in median plaque size (1.47 mm) was observed compared with the WT virus (1.75 mm), which was statistically significant (p < 0.05). Both double mutants (LD1 and LD2) showed a decreased median plaque size (1.43 mm and 1.64 mm respectively) compared with the WT (1.75 mm); this difference was only statistically significant (p < 0.05) for mutant LD1. Both triple mutants (LT1 and LT2) showed a small decrease in median plaque size (1.48 mm and 1.56 mm respectively) compared with the WT (1.75 mm), although this difference was not statistically significant. Overall, the biggest and most significant (p < 0.001) decrease in median plaque size was observed for the quadruple mutants (1.20 mm and 1.34 mm compared with 1.75 mm for the WT). Therefore, a recombinant virus containing only the U986C (LS986) mutation was produced and no difference in virus titer (Fig. 2A) was observed. In the plaque assay (Fig. 2C) the mutant had even a slightly higher (0.99 mm) median plaque size compared with the WT (0.88 mm), but this difference was not statistically significant. Therefore, the isolated mutation at nucleotide position 986 was not responsible for the decrease in median plaque size, observed in the quadruple mutants.
Functional effects of stem-loop mutations
To gain more insight into the possible function of the M RNA structure, northern blot assays were performed to determine RNA levels. Mutants SL1, SR1, SLR1 and LQ1 were selected (as a representative set), together with WT virus, a negative control (NC) consisting of RNA extracted from mock infected MDCK cells and a positive control consisting of RNA extracted from 293T cells transfected with expression plasmids. Total RNA was extracted from MDCK cells that were inoculated at a multiplicity of infection (MOI) of 1 with the selected set of viruses and run on a 1% agarose gel. After Northern blot, the membranes were hybridized with probes against (-)RNA, (+)RNA and M2 mRNA (Fig. 3). To determine the volume (intensity) of the bands the area equivalent to the widest band of the blot was selected for analyses. The volume (intensity) of each band was then compared with the WT and the relative quantity (RQ) calculated.
Figure 3.
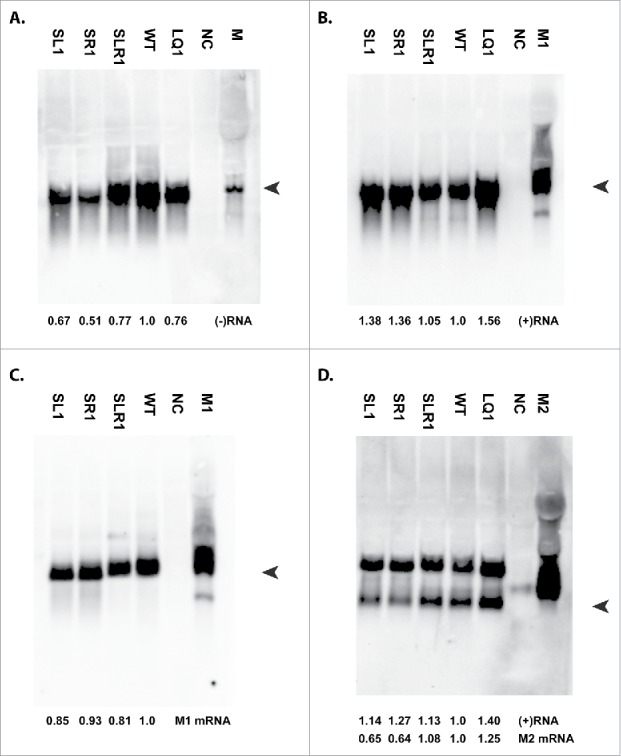
Northern blot experiments. Total RNA was extracted from MDCK cells inoculated with SL1, SR1, SLR1, WT, LQ1 and mock (NC). As a positive control for (-)RNA a unidirectional pSP72-PhuTmu plasmid containing the M gene segment was transfected into 293T cells, which produces only negative sense vRNA. Additionally, pCAGGS-M1 (M1) and pCAGGS-M2 (M2) plasmids were transfected into 293T cells to serve as positive mRNA controls. Approximately 40 hours after transfection, total RNA was extracted and used to produce Northern blot. Arrow heads indicate the region of interest. A representative blot of 3 experiments is shown for (-)RNA (A), (+)RNA (B) and M2 mRNA (D). The 2 bands in panel D correspond to unspliced M1 and spliced (M2) mRNA. (C) Total RNA from SL1, SR1, SLR1 and WT was used for polyA purification to isolate mRNA and this was used in a Northern blot with the (+)RNA probe.
Lower vRNA levels were detected for all mutant viruses when compared with the WT virus (Fig. 3A). The reduction in RQ vRNA levels were more pronounced for SL1 (0.67) and SR1 (0.51) than for SLR1 (0.77) and LQ1 (0.76). The vRNA levels correlated with the virus titers, as both SL1 and SR1 showed a reduction of ∼1 log in virus titer compared with the WT virus. Mutant SLR1 replicated similar to the WT virus in a plaque assay, but the vRNA level was slightly reduced compared with that of the WT virus.
The Northern blot detecting (+)RNA is shown in Fig. 3B. Here, higher RQ (+)RNA levels were detected for the SL1 (1.38), SR1 (1.36) and LQ1 (1.56) mutants compared with SLR1 (1.05) and WT (1.0). Both cRNA and unspliced M1 mRNA are detected by the (+)RNA probe and this will result in bands of approximately the same size. Therefore, poly A+ mRNA was isolated from total RNA and this was subsequently used in a Northern blot experiment (Fig. 3C) with the (+)RNA probe to measure M1 mRNA levels for the stem mutants. Similar RQ levels of M1 mRNA were detected for SL1 (0.85), SR1 (0.93), SLR1 (0.81) compared with the WT (1.0).
The RQ M2 mRNA levels were lower (Fig. 3D) for the structure-disrupting mutants SL1 (0.65) and SR1 (0.64) as compared with WT (1.0). A compensatory effect for structure-restoring mutant SLR1 was observed with near-WT RQ levels of 1.08. In contrast, the quadruple loop mutant LQ1 resulted in higher RQ M2 mRNA levels (1.25) compared with the WT virus. In the M2 mRNA Northern blot a second (larger) band was observed, as the region of the M2 mRNA probe is also present in cRNA and unspliced M1 mRNA. Both the structure-disrupting and the loop mutants showed higher RQ (+)RNA levels (1.14 for SL1, 1.27 for SR1 and 1.40 for LQ1), although a larger increase was observed with the other (+)RNA probe (1.38 for SL1, 1.36 for SR1 and 1.56 for LQ1 (Fig. 3B). Structure-restoring mutant SLR1 showed a minor increase in RQ level with both probes of 1.05 with the (+)RNA probe and 1.13 with the M2 mRNA probe. A similar (+)RNA pattern was observed in 3 independent experiments with both probes. Based on these data a slightly higher level of cRNA in the structure disrupting mutants is suggested, as well as lower vRNA and M2 mRNA levels.
Detection of MA protein levels
As the Northern blot data showed a relatively strong reduction in M2 mRNA production for stem disrupting mutants, the kinetics of M1 and M2 protein levels at early time-points were studied by FACS analysis. MDCK cells were inoculated at a MOI of 3 with mutant (SL1, SR1, SLR1, LQ1) and WT viruses and samples were collected with a 30 minute time interval. Cells were harvested and stained for NP and the M1 or M2 protein. Within the virus infected cell population (NP positive cells), the percentage M1 or M2 positive cells and the mean fluorescence (MF) levels of M1 or M2 were determined. The percentage of M positive cells was multiplied by the MF to calculate M1 and M2 protein levels. Similar results were obtained in 3 independent experiments with a slightly different experimental setup. A representative experiment is shown in Fig. 4.
Figure 4.
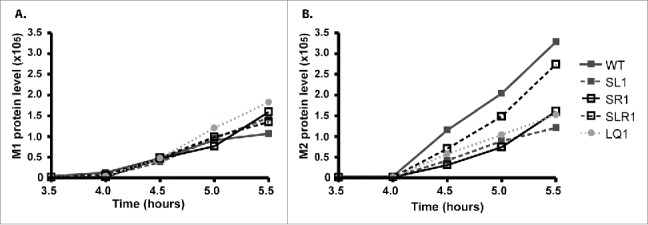
Time-lapse FACS experiment to determine M1 and M2 protein levels in influenza A virus infected MDCK cells. MDCK cells were inoculated with mutant (SL1, SR1, SLR1, LQ1) and WT viruses at MOI 3. From 2½ hours post-inoculation onwards, cells were harvested at a 30 minute time interval and used to stain for NP protein as a marker for virus infected cells and, simultaneously, for either M1 or M2 protein. Data are shown from the moment of first detection (3½) to 5½ hours post-inoculation to emphasize the differences at early time-points. (A) M1 protein levels within the virus infected (NP-positive) population and (B) M2 protein levels are shown. M1 and M2 protein levels were calculated by multiplying MF levels with the percentage of M1 or M2 infected cells.
Similar levels of total M1 protein were observed for all mutant viruses (Fig. 4A). For M2, negligible levels of total M2 protein were observed until 4 hours post-inoculation (Fig. 4B). From 4 hours onwards M2 production increased, albeit at a slower rate for the stem-disruptive mutants SL1 and SR1. The structure-restoring mutant SLR1 resembled the pattern of the WT although the values at the different time-points were slightly lower. The loop mutant LQ1 showed an intermediate pattern at 4 .5 hours but resembled the pattern of the structure disrupting mutants at 5 and 5.5 hours. Overall, the patterns of total protein corresponded to the Northern blot data, except for loop mutant LQ1, where a higher level of M2 protein production was expected.
Together, these data showed that the structure disruptive mutations in the stem (SL1 and SR1) negatively influenced virus replication, produced similar amounts of M1 mRNA and less M2 mRNA and vRNA. Additionally, a consistent pattern for mRNA and protein levels was observed. The structure-restoring mutant (SLR1) replicated like the WT virus and had M1/M2 mRNA and protein levels that were comparable to the WT, although still a small reduction in vRNA level was observed.
Discussion
Previous identification of RNA structures in the influenza virus genome13,16,17,26,29-31 and the hypotheses that vRNA-vRNA interactions play a role in the enigmatic influenza A virus packaging mechanism9,32,33 make them an interesting subject for experimental studies. Here, we studied a previously predicted RNA hairpin structure in the packaging signal region of the M gene segment (nt 967-994)5,25,29 using a compensatory mutagenesis strategy with A/PR/8/34 influenza A virus to determine the effect of RNA folding on virus replication. The mutant viruses were further used to study possible functions of the hairpin structure during virus replication by determining differences in RNA and protein levels.
In several previous studies mutations were introduced into the region spanning this hairpin27,28 or a mutagenesis approach was based upon the predicted RNA hairpin structure29; in all studies a (small) effect on virus replication was observed and the 5′-terminal region in the M gene segment (983–1027) was shown to play a role in packaging.27,28 Based on these data and the location of the hairpin within the packaging region28 a role in influenza virus genome packaging was therefore implied. The hairpin structure may be involved in packaging by interacting with another gene segment based on loop-loop “kissing” complex contacts between gene segments.9,13 A pronounced influence of the hairpin on pre-mRNA splicing was less expected, bearing in mind that the distance to the intron of the M gene segment (52–739) is more than 200 nucleotides (Fig. 1).
The compensatory mutagenesis study described here, revealed that introduction of mutations, based on natural variation, at either side of the stem in the hairpin structure resulted in decreased viral replication both in virus titration and plaque assay experiments. This is also consistent with the reduced infectivity of mutants containing stem mismatches as studied by Kobayashi et al.29 Introduction of silent mutations in the loop of the predicted hairpin structure also resulted in reduced plaque sizes, with the largest effect observed with quadruple mutant LQ1. The fact that virus replication was affected by mutations in the loop supports the hypotheses that the loop of the hairpin structure is involved in a functional interaction (e.g., with another M gene segment domain or another molecule). Unfortunately, without knowing the interacting partner, further experimental studies are difficult.
In Northern blot experiments, disruptive stem mutants (SR1 and SL1) showed reduced levels of vRNA and M2 mRNA compared with WT. Differences in M1 mRNA levels were relatively small. M2 mRNA levels were restored completely and vRNA levels partly in compensatory mutant virus SLR1. For loop mutant (LQ1) lower vRNA and higher M2 mRNA levels than the WT were observed. It should be noted that the different splice events in the M gene segment result in overlapping mRNA regions for M2, M3 and M4 (Fig. 1A) which might potentially complicate the interpretation of the Northern blot data.22 However, cross-reactivity with M3 and M4 mRNA is not expected as Shih et al.23 showed that M3 mRNA is only produced early in infection and Wise et al.22 detected no M3 and M4 mRNA in cells infected with A/PR/8/34 at 6 hours post-inoculation. A FACS time-course experiment demonstrated that M2 protein was produced at a slower rate for the structure-disrupting mutants (SL1 and SR1), the compensatory mutant SLR1 showed a pattern that resembled that of the WT. M1 protein production was similar for all the mutants and WT virus, and this pattern was consistent with the Northern blot data. However, after 5.5 hours post-inoculation these differences became smaller and thus no difference in M2 protein levels were observed as with the Northern blot at later time-points. These data suggest that M2 mRNA production is slightly delayed (this may be caused by a delayed switch from M3 to M2 mRNA23) as the differences in M2 protein production were only detectable at early time-points.
Interestingly, mutant virus M2 H90-V92 (amino acid position 90-92), described by Hutchinson et al.27 contained a combination of our loop mutations U983C and U986C, together with substitution C989A in the stem. This M2 mutant resulted in lower virus titers, smaller plaques and a ∼5-fold reduction in vRNA accumulation between 8 and 22 hours and was therefore suggested to cause a defect in packaging, with possible pleiotropic effects. However, no effect on mRNA levels was detected. On the other hand, our quadruple loop mutant (containing U983C and U986C) showed equal amounts of M2 mRNA and stem mutants SL1 and SR1 (containing C989A) both had reduced M2 mRNA levels. The differences may be due to the different combination of mutations that were tested.
Our results indicate that the predicted RNA structure is involved in splicing. This splicing regulation can be realized by long-distance interactions with M mRNA splice site regions that change their structures and/or accessibility to the splicing machinery (Fig. 5), or mediated by binding to other splicing factors. In NS mRNA, a 85-nucleotide-long cis-acting region, located 147 nucleotides downstream of the NS intron, was shown to inhibit splicing.34 Although here the opposite effect is observed, the similarity in the location of M and NS exonic regulatory elements is intriguing. It is also possible to suggest that the folding of the M 967-994 hairpin operates as a splicing enhancer by simply preventing its sequence to exert an inhibitory effect. On the other hand, the splicing regulation systems of NS and M mRNAs differ considerably, in particular, M1 mRNA is spliced in nuclear speckles while NS1 mRNA is not.35 Splicing and nuclear export of M1 mRNA is characterized by a complex regulation with the presence of alternative 5′ splice sites and several splicing factors.22,35-38
Figure 5.
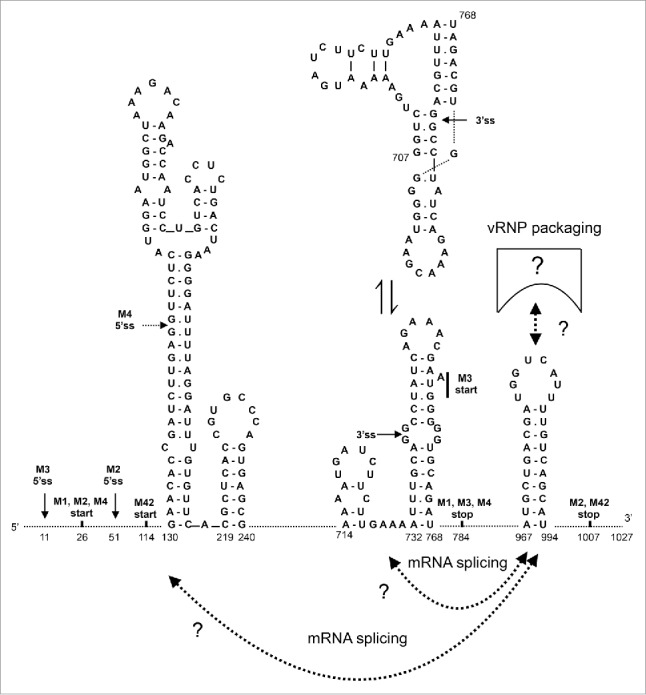
Putative functional interactions of the 967-994 hairpin of the M gene segment. The interactions of the 967-994 hairpin with some of the structures suggested to fold in the splice site regions30,42-45 may regulate RNA splicing. An equilibrium between alternative structures in the 3′ splice site region (707-768) was previously suggested.42,45 The apical part of the stem loop domain 130-217 may also undergo a conformational transition, the structure shown here is more likely for the A/PR/8/34 M segment sequence.44 The hairpin and/or its complement in the negative-sense vRNA may be also involved in other interactions and functions such as RNA packaging. All known M RNA splice sites are indicated here, but the A/PR/8/34 M4 5′ splice site is not expected to be used, because its sequence does not correspond to the optimal consensus.22 Thus M42 protein expression should not occur in this strain, while expression of M3 and M4 ORFs in influenza A viruses is purely hypothetical.22,37 5ss, 5′ splice site; 3ss, 3′ splice site.
The restoration of viral fitness in the compensatory mutants showed that changes in both hairpin nucleotide sequence and encoded amino acids in this region can be tolerated more than distortions of the helical stem structure. Furthermore, in addition to the 2 covariations in H1N1 strains at position 970/991 and 972/989 described above (Fig. 1B), the covariations of the 971/990 pair between them were also identified in the GenBank sequences of M segments from viruses of other subtypes: in 3 American avian strains of 1994 and 1995 (H7N3, H7N2 and H11N2) and in the group of 29 avian H5N1 viruses of the Egypt 2009/2010 outbreak. Strong structural constraints in the M gene segment 967-994 hairpin are consistent with the multifunctional role, suggested here, in virus replication.
Here, we showed that mutants with a destabilized hairpin stem produced less vRNA, spliced M2 mRNA and affected virus replication. When mutations were introduced on both sides of the hairpin structure, the replication defect was restored, together with a complete restoration of wild type M2 splice levels and partial restoration of vRNA synthesis. Our data are consistent with a role of this hairpin in influenza virus genome packaging and suggest its involvement in regulating the M splicing mechanism.
Materials and methods
Cells
293T cells were cultured in DMEM (Lonza, Breda, the Netherlands) supplemented with 10% FCS, 100 IU/ml penicillin (Lonza), 100 µg/µl streptomycin (Lonza), 2 mM glutamine (Lonza), 1 mM sodiumpyruvate (Gibco, Leusden, the Netherlands) and non-essential amino acids (Lonza). Madin-Darby Canine Kidney (MDCK) cells were cultured in EMEM (Lonza) supplemented with 10% FCS, 100 IU/ml penicillin, 100 µg/µl streptomycin, 2 mM glutamine, 1,5 mg/ml sodiumbicarbonate (Lonza), 10 mM hepes and non-essential amino acids.
Plasmids and mutagenesis
Plasmids containing the gene segments of A/PR/8/34 were described previously.39 The M gene segment was used to introduce mutations into the region of the predicted RNA structure using the Quickchange Multi Site Directed Mutagenesis kit (Agilent, Amstelveen, the Netherlands) according to the manufacturer's instructions. The presence of the desired mutations in the plasmids were confirmed using a 3130XL genetic analyzer (Applied Biosystems, Bleiswijk, the Netherlands). All primer sequences are available upon request. The A/PR/8/34 M unidirectional pSP72-PhuTmu plasmid was described previously.39 The coding regions of A/PR/8/34 M1 and M2 were cloned into the EcoRI-XhoI restriction sites of pCAGGS, the pCAGGS plasmid was kindly provided by Dr. A. Garcia-Sastre (Icahn School of Medicine, New York, USA).
Production of recombinant virus
Recombinant viruses were produced as described previously.16 In brief, 293T cells were transfected with 5 µg of plasmid DNA containing the desired M mutant and 5 µg of the remaining A/PR/8/34 bidirectional plasmids each. The 293T supernatant was harvested and used to inoculate MDCK cells. The MDCK supernatant containing recombinant virus was further used to assess the 50 percent tissue culture infectious dose (TCID50) in virus stocks as described previously.16
Plaque assay
The plaque assay was performed as described previously.16,40 Data were analyzed using ImageQuant TL colony counting software version 8.1 (GE Healthcare, Life sciences, Eindhoven, the Netherlands). Plaque size was plotted as the plaque radius (in mm) and displayed using Graphpad Prism 4 (Graphpad Software, La Jolla, USA).
Northern blot
Unidirectional M, pCAGGS-M1 and pCAGGS-M2 plasmids were transfected in 293T cells to serve as positive controls in the Northern blot experiments. Samples for Northern blot analysis were prepared by inoculating MDCK cells with the desired mutant viruses at MOI 1. One hour after inoculation the cells were washed with PBS once and fresh infection medium was added. Approximately 40 hours after transfection or 8 hours after inoculation, cells were washed with PBS once. Trizol (Invitrogen, Life Technologies, Bleiswijk, the Netherlands) was added to the cells and RNA was isolated according to the manufacturer's instructions. Total RNA (90 µg) was also used to extract mRNA using the MicroPoly(A)Purist kit (Ambion, Life Technologies, Bleiswijk, the Netherlands) according to the instructions of the manufacturer. Samples for the Northern blot were prepared by mixing RNA (7.5 µg) and RNA millennium marker (2 µl; Ambion) 1:1 with Glyoxal loading dye (Ambion), this was then incubated at 50 °C for 30 minutes. Samples were loaded on a 1% agarose gel prepared with 1x Gel/Running buffer (Ambion) and run at 68 Volts. RNA was subsequently transferred from the agarose gel to a positively charged nylon membrane (Brightstar-Plus, Ambion) by downward transfer for ∼3½ hours. The RNA was then cross-linked for 15 minutes at 80 °C and the blot was pre-hybridized in 10 ml of ULTRAhyb (Ambion) buffer for 30 minutes at 68 °C using a hybridization (roller) oven (GE Healthcare, Eindhoven, the Netherlands). Biotinylated probes to detect positive sense RNA ((+)RNA), M2 mRNA and negative sense vRNA ((-)RNA) were generated using the MAXIscript Kit (Ambion) according to the manufacturer's instructions. PCR products that contained a T7 promoter sequence were used as a template. The (+)RNA probe was located in the M1 region (nucleotide 232-632), the M2 mRNA probe was located at the 3′ proximal region of M2 (nucleotide 787-969) and the (-)RNA probe contained the M1/M2 overlapping “splice acceptor” region (nucleotide 501-800). Northern blotting was performed using the NorthernMax-Gly kit (Ambion) according to the manufacturer's instructions. After pre-hybridization, the probe was added to a final concentration of 0.1 nM and hybridized at 68 °C overnight. The next day, the blot was washed once with low stringency wash solution (2x SSC, 0.1% SDS) for 10 minutes at room temperature, followed by 2 washes with high stringency wash solution (0.1X SSC, 0.1% SDS) for 15 minutes at 68 °C. The biotin signal was detected using the AP chemiluminescent blotting kit (KPL, Gaithersburg, USA) according to the manufacturer's instructions. The blot was imaged with a Chemidoc MP imaging system (BioRad, Veenendaal, the Netherlands) and analyzed using Image Lab software (version 4.0.1, BioRad).
M1 and M2 protein expression in infected cells
MDCK cells were inoculated with the desired mutant viruses at a MOI of 3 and incubated at 37 °C and 5% CO2 for the appropriate time. From 2½ hours onwards a plate containing the set of desired viruses was removed from the incubator with a 30 minute time interval. The cells were washed with PBS once and 500 µl of 0.05% trypsin-EDTA (Life Technologies, Bleiswijk, the Netherlands) was added to the cells and incubated at 37 °C. After 5-10 minutes of incubation, the cells were transferred to a tube containing PBS-1% FCS and centrifuged for 5 minutes at 1500 rpm. Next, the cells were resuspended in 400 µl PBS-1% FCS and 2 × 200 µl (for M1 and M2 staining) was transferred to a 96-well v-bottom plate (Corning life sciences, Amsterdam, the Netherlands). The plate was incubated at 4 °C until all samples were harvested. At the end of the day, the plate containing all the samples was centrifuged for 5 minutes at 1500 rpm and 4 °C. The cells were resuspended in 100 µl of cytofix/cytoperm (BD Biosciences, Breda, the Netherlands) and incubated on ice for 20 minutes. Subsequently, the plate was centrifuged for 5 minutes at 500 × g and 4 °C and the cells were resuspended in 100 µl perm-wash buffer (BD Biosciences). The plates were stored overnight at 4 °C in the dark. The next day, the cells were stained for NP and M1 or NP and M2 essentially as described previously.41 Primary antibodies against NP (IgG2A, Clone Hb65; ATCC), M1 (IgG1, Clone Hb64; ATCC) and a monoclonal mouse antibody against M2 (IgG1, clone 14C2; Abcam, Cambridge, UK) were used at a 1/100 dilution in perm/wash. Secondary goat-anti-mouse antibody against IgG1 (APC; Thermo Fisher Scientific, Bleiswijk, the Netherlands) and goat-anti-mouse antibody against IgG2A (Alexa Fluor 488; Invitrogen, Life Technologies) were diluted 1:100 in perm/wash. The cells were resuspended in PBS and the percentage of M1 and M2 positive cells, as well as the M1 and M2 mean fluorescence were determined within the NP positive population by FACS on a FACS Canto (BD Biosciences, Breda, the Netherlands). The data were analyzed using FACSDiva software (BD Biosciences).
Statistics
Statistical significance of the plaque assay data was performed using the nonparametric Kruskal-Wallis test with a Dunns post-test, when more than 2 groups were compared. A p-value <0.05 was considered to be significant. Statistical significance was compared with the median plaque size of the wild-type (WT) virus. If 2 groups were compared, the nonparametric Mann-Whitney t-test was performed where p < 0.05 was considered significant. All statistical analyses were performed using GraphPad Prism 4.02 for Windows (GraphPad Software, San Diego, CA, USA).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Garcia-Sastre for the pCAGGS plasmid. We thank Ramona Mögling and Miranda de Graaf for excellent technical assistance.
Funding
This work was financially supported by NIAID/NIH contract HSN272201400008C.
References
- 1.Horimoto T, Kawaoka Y. Influenza: Lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol 2005; 3(8):591-600; PMID:16064053; https://doi.org/ 10.1038/nrmicro1208 [DOI] [PubMed] [Google Scholar]
- 2.Nelson MI, Holmes EC. The evolution of epidemic influenza. Nat Rev Genet 2007; 8(3):196-205; PMID:17262054; https://doi.org/ 10.1038/nrg2053 [DOI] [PubMed] [Google Scholar]
- 3.Martin-Benito J, Ortin J. Influenza virus transcription and replication. Adv Virus Res 2013; 87:113-37; PMID:23809922; https://doi.org/ 10.1016/B978-0-12-407698-3.00004-1 [DOI] [PubMed] [Google Scholar]
- 4.Eisfeld AJ, Neumann G, Kawaoka Y. At the centre: Influenza A virus ribonucleoproteins. Nat Rev Microbiol 2015; 13(1):28-41; PMID:25417656; https://doi.org/ 10.1038/nrmicro3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gultyaev AP, Fouchier RA, Olsthoorn RC. Influenza virus RNA structure: Unique and common features. Int Rev Immunol 2010; 29(6):533-56; PMID:20923332; https://doi.org/ 10.3109/08830185.2010.507828 [DOI] [PubMed] [Google Scholar]
- 6.Gultyaev AP, Olsthoorn RC. A family of non-classical pseudoknots in influenza A and B viruses. RNA Biol 2010; 7(2):125-9; PMID:20200490; https://doi.org/ 10.4161/rna.7.2.11287 [DOI] [PubMed] [Google Scholar]
- 7.Ward AC, Azad AA, McKimm-Breschkin JL. Changes in the NS gene of neurovirulent strains of influenza affect splicing. Virus Genes 1995; 10(1):91-4; PMID:7483294; https://doi.org/ 10.1007/BF01724301 [DOI] [PubMed] [Google Scholar]
- 8.Gultyaev AP, Heus HA, Olsthoorn RC. An RNA conformational shift in recent H5N1 influenza A viruses. Bioinformatics 2007; 23(3):272-6; PMID:17090581; https://doi.org/ 10.1093/bioinformatics/btl559 [DOI] [PubMed] [Google Scholar]
- 9.Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. Genome packaging in influenza A virus. J Gen Virol 2010; 91(Pt 2):313-28; PMID:19955561; https://doi.org/ 10.1099/vir.0.017608-0 [DOI] [PubMed] [Google Scholar]
- 10.Fournier E, Moules V, Essere B, Paillart JC, Sirbat JD, Isel C, Cavalier A, Rolland JP, Thomas D, Lina B, et al.. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res 2012; 40(5):2197-209; PMID:22075989; https://doi.org/ 10.1093/nar/gkr985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerber M, Isel C, Moules V, Marquet R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol 2014; 22(8):446-55; PMID:24798745; https://doi.org/ 10.1016/j.tim.2014.04.001 [DOI] [PubMed] [Google Scholar]
- 12.Noda T, Sagara H, Yen A, Takada A, Kida H, Cheng RH, Kawaoka Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006; 439(7075):490-2; PMID:16437116; https://doi.org/ 10.1038/nature04378 [DOI] [PubMed] [Google Scholar]
- 13.Gavazzi C, Yver M, Isel C, Smyth RP, Rosa-Calatrava M, Lina B, Moulès V, Marquet R. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc Natl Acad Sci U S A 2013; 110(41):16604-9; PMID:24067651; https://doi.org/ 10.1073/pnas.1314419110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Essere B, Yver M, Gavazzi C, Terrier O, Isel C, Fournier E, Giroux F, Textoris J, Julien T, Socratous C, et al.. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc Natl Acad Sci U S A 2013; 110(40):E3840-8; PMID:24043788; https://doi.org/ 10.1073/pnas.1308649110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fournier E, Moules V, Essere B, Paillart JC, Sirbat JD, Cavalier A, Rolland JP, Thomas D, Lina B, Isel C, et al.. Interaction network linking the human H3N2 influenza A virus genomic RNA segments. Vaccine 2012; 30(51):7359-67; PMID:23063835; https://doi.org/ 10.1016/j.vaccine.2012.09.079 [DOI] [PubMed] [Google Scholar]
- 16.Gultyaev AP, Tsyganov-Bodounov A, Spronken MI, van der Kooij S, Fouchier RA, Olsthoorn RC. RNA structural constraints in the evolution of the influenza A virus genome NP segment. RNA Biol 2014; 11(7):942-52; PMID:25180940; https://doi.org/ 10.4161/rna.29730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gultyaev AP, Spronken MI, Richard M, Schrauwen EJ, Olsthoorn RC, Fouchier RA. Subtype-specific structural constraints in the evolution of influenza A virus hemagglutinin genes. Sci Rep 2016; 6:38892; PMID:27966593; https://doi.org/ 10.1038/srep38892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dubois J, Terrier O, Rosa-Calatrava M. Influenza viruses and mRNA splicing: Doing more with less. MBio 2014; 5(3):e00070-14; PMID:24825008; https://doi.org/ 10.1128/mBio.00070-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang T, Nogales A, Baker SF, Martinez-Sobrido L, Turner DH. Mutations designed by ensemble defect to misfold conserved RNA structures of influenza A segments 7 and 8 affect splicing and attenuate viral replication in cell culture. PLoS One 2016; 11(6):e0156906; PMID:27272307; https://doi.org/ 10.1371/journal.pone.0156906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamb RA, Lai CJ, Choppin PW. Sequences of mRNAs derived from genome RNA segment 7 of influenza virus: Colinear and interrupted mRNAs code for overlapping proteins. Proc Natl Acad Sci U S A 1981; 78(7):4170-4; PMID:6945577; https://doi.org/ 10.1073/pnas.78.7.4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inglis SC, Brown CM. Spliced and unspliced RNAs encoded by virion RNA segment 7 of influenza virus. Nucleic Acids Res 1981; 9(12):2727-40; PMID:6169001; https://doi.org/ 10.1093/nar/9.12.2727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, et al.. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog 2012; 8(11):e1002998; PMID:23133386; https://doi.org/ 10.1371/journal.ppat.1002998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shih SR, Nemeroff ME, Krug RM. The choice of alternative 5′ splice sites in influenza virus M1 mRNA is regulated by the viral polymerase complex. Proc Natl Acad Sci U S A 1995; 92(14):6324-8; PMID:7541537; https://doi.org/ 10.1073/pnas.92.14.6324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valcarcel J, Portela A, Ortin J. Regulated M1 mRNA splicing in influenza virus-infected cells. J Gen Virol 1991; 72(Pt 6):1301-8; PMID:1710647; https://doi.org/ 10.1099/0022-1317-72-6-1301 [DOI] [PubMed] [Google Scholar]
- 25.Gultyaev AP, Olsthoorn RCL, Spronken MIJ, Fouchier RAM.. Detection of structural constraints and conformational transitions in the influenza virus RNA genome using structure predictions and mutual information calculations. Proceedings Iwbbio 2014: International Work-Conference on Bioinformatics and Biomedical Engineering, 1:273-84. [Google Scholar]
- 26.Ruszkowska A, Lenartowicz E, Moss WN, Kierzek R, Kierzek E. Secondary structure model of the naked segment 7 influenza A virus genomic RNA. Biochem J 2016; 473(23):4327-48; PMID:27694388; https://doi.org/ 10.1042/BCJ20160651 [DOI] [PubMed] [Google Scholar]
- 27.Hutchinson EC, Curran MD, Read EK, Gog JR, Digard P. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J Virol 2008; 82(23):11869-79; PMID:18815307; https://doi.org/ 10.1128/JVI.01634-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ozawa M, Maeda J, Iwatsuki-Horimoto K, Watanabe S, Goto H, Horimoto T, Kawaoka Y. Nucleotide sequence requirements at the 5′ end of the influenza A virus M RNA segment for efficient virus replication. J Virol 2009; 83(7):3384-8; PMID:19158245; https://doi.org/ 10.1128/JVI.02513-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi Y, Dadonaite B, van Doremalen N, Suzuki Y, Barclay WS, Pybus OG. Computational and molecular analysis of conserved influenza A virus RNA secondary structures involved in infectious virion production. RNA Biol 2016; 13(9):883-94; PMID:27399914; https://doi.org/ 10.1080/15476286.2016.1208331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moss WN, Priore SF, Turner DH. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 2011; 17(6):991-1011; PMID:21536710; https://doi.org/ 10.1261/rna.2619511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hsu MT, Parvin JD, Gupta S, Krystal M, Palese P. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc Natl Acad Sci U S A 1987; 84(22):8140-4; PMID:2446318; https://doi.org/ 10.1073/pnas.84.22.8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Isel C, Munier S, Naffakh N. Experimental approaches to study genome packaging of influenza A viruses. Viruses 2016; 8(8):218; PMID:27517951; https://doi.org/ 10.3390/v8080218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giese S, Bolte H, Schwemmle M. The feat of packaging eight unique genome segments. Viruses 2016; 8(6):165; PMID:27322310; https://doi.org/ 10.3390/v8060165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nemeroff ME, Utans U, Krämer A, Krug RM. Identification of cis-acting intron and exon regions in influenza virus NS1 mRNA that inhibit splicing and cause the formation of aberrantly sedimenting presplicing complexes. Mol Cell Biol 1992; 12(3):962-70; PMID:1532050; https://doi.org/ 10.1128/MCB.12.3.962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mor A, White A, Zhang K, Thompson M, Esparza M, Muñoz-Moreno R, Koide K, Lynch KW, García-Sastre A, Fontoura BM. Influenza virus mRNA trafficking through host nuclear speckles. Nat Microbiol 2016; 1(7):16069; PMID:27572970; https://doi.org/ 10.1038/nmicrobiol.2016.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shih SR, Krug RM. Novel exploitation of a nuclear function by influenza virus: The cellular SF2/ASF splicing factor controls the amount of the essential viral M2 ion channel protein in infected cells. EMBO J 1996; 15(19):5415-27; PMID:8895585. [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson D, Lamb RA. The influenza A virus spliced messenger RNA M mRNA3 is not required for viral replication in tissue culture. J Gen Virol 2008; 89(Pt 12):3097-101; PMID:19008398; https://doi.org/ 10.1099/vir.0.2008/004739-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsai PL, Chiou NT, Kuss S, García-Sastre A, Lynch KW, Fontoura BM. Cellular RNA binding proteins NS1-BP and hnRNP K regulate influenza A virus RNA splicing. PLoS Pathog 2013; 9(6):e1003460; PMID:23825951; https://doi.org/ 10.1371/journal.ppat.1003460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Wit E, Spronken MI, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. Efficient generation and growth of influenza virus A/PR/8/34 from eight cDNA fragments. Virus Res 2004; 103(1-2):155-61; PMID:15163504; https://doi.org/ 10.1016/j.virusres.2004.02.028 [DOI] [PubMed] [Google Scholar]
- 40.Matrosovich M, Matrosovich T, Garten W, Klenk HD. New low-viscosity overlay medium for viral plaque assays. Virol J 2006; 3:63; PMID:16945126; https://doi.org/ 10.1186/1743-422X-3-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spronken MI, Short KR, Herfst S, Bestebroer TM, Vaes VP, van der Hoeven B, Koster AJ, Kremers GJ, Scott DP, Gultyaev AP, et al.. Optimisations and challenges involved in the creation of various bioluminescent and fluorescent influenza A virus strains for in vitro and in vivo applications. PLoS One 2015; 10(8):e0133888; PMID:26241861; https://doi.org/ 10.1371/journal.pone.0133888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moss WN, Dela-Moss LI, Kierzek E, Kierzek R, Priore SF, Turner DH. The 3′ splice site of influenza A segment 7 mRNA can exist in two conformations: A pseudoknot and a hairpin. PLoS One 2012; 7(6):e38323; PMID:22685560; https://doi.org/ 10.1371/journal.pone.0038323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen JL, Kennedy SD, Turner DH. Structural features of a 3′ splice site in influenza a. Biochemistry 2015; 54(21):3269-85; PMID:25909229; https://doi.org/ 10.1021/acs.biochem.5b00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang T, Kennedy SD, Moss WN, Kierzek E, Turner DH. Secondary structure of a conserved domain in an intron of influenza A M1 mRNA. Biochemistry 2014; 53(32):5236-48; PMID:25026548; https://doi.org/ 10.1021/bi500611j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moss WN, Dela-Moss LI, Priore SF, Turner DH. The influenza A segment 7 mRNA 3′ splice site pseudoknot/hairpin family. RNA Biol 2012; 9(11):1305-10; PMID:23064116; https://doi.org/ 10.4161/rna.22343 [DOI] [PMC free article] [PubMed] [Google Scholar]