

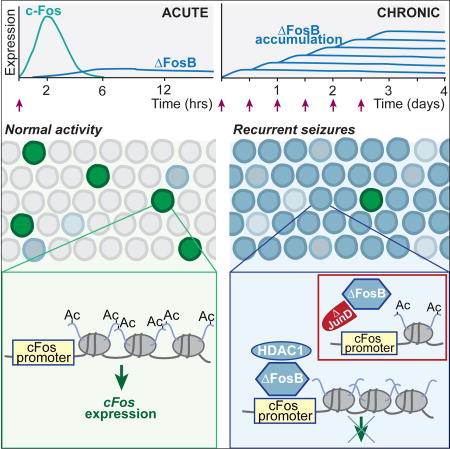
Summary
Alzheimer's disease (AD) is characterized by cognitive decline and 5–10 fold increased seizure incidence. How seizures contribute to cognitive decline in AD or other disorders is unclear. We show spontaneous seizures increase expression of ΔFosB, a highly stable Fos-family transcription factor, in the hippocampus of an AD mouse model. ΔFosB suppressed expression of the immediate early gene c-Fos, which is critical for plasticity and cognition, by binding its promoter and triggering histone deacetylation. Acute HDAC inhibition or inhibition of ΔFosB activity restored c-Fos induction and improved cognition in AD mice. Administration of seizure-inducing agents to nontransgenic mice also resulted in ΔFosB-mediated suppression of c-Fos, suggesting this mechanism is not confined to AD mice. These results explain observations that c-Fos expression increases after acute neuronal activity but decreases with chronic activity. Moreover, these results indicate a general mechanism by which seizures contribute to persistent cognitive deficits even during seizure-free periods.
Graphical abstract
Introduction
Alzheimer’s disease (AD) is the most prevalent type of dementia among the elderly, and is associated with prominent impairments in hippocampal memory (Burgess et al., 2002; Selkoe, 2002; Tulving and Markowitsch, 1998). Many current treatments are aimed at preventing disease progression in at-risk individuals or early in disease. A central component of these therapeutic efforts is the prevention of cognitive decline. Therefore, comprehensive understanding of the neuronal and network alterations associated with cognitive dysfunction in AD is critical to enable development of effective therapeutics.
It is becoming increasingly clear that network dysfunction, which can manifest as seizures and epileptiform activity, plays a contributing role in AD-related cognitive deficits. AD is associated with a 5–10 fold increased risk in seizures (Amatniek et al., 2006; Larner, 2010; Lozsadi and Larner, 2006). This rate may be underestimated due to the incidence of subclinical epileptiform activity and non-convulsive seizures, which in a majority of cases precede or coincide with the diagnosis of amnestic mild cognitive impairment or AD (Vossel et al., 2013). A recent prospective study found that 42.4% of AD patients with no clinical history of seizures exhibited subclinical epileptiform activity, and the presence of such activity was associated with faster rates of cognitive decline (Vossel et al., 2016). Furthermore, multiple mouse models of AD exhibit epileptiform activity (Chin and Scharfman, 2013; Kam et al., 2016; Minkeviciene et al., 2009; Palop et al., 2007; Palop and Mucke, 2009). Because patients with intractable epilepsy and mouse models of epilepsy display hippocampal memory deficits (Hermann et al., 2006; Hoppe et al., 2007; Stafstrom et al., 1993), the epileptiform activity associated with AD may contribute to cognitive deficits as well. Indeed, antiepileptic drug treatment reduces network dysfunction and improves hippocampus-dependent cognition in prodromal AD patients and mouse models of AD (Bakker et al., 2012; Sanchez et al., 2012). However, the mechanisms by which seizures induce persistent cognitive deficits in AD and epilepsy are poorly understood, particularly because seizure frequency is often not high.
Neuronal activity alters expression of many genes in the hippocampus, with important consequences for learning and memory. One of the most widely studied is Fos, a gene whose protein product, c-Fos, is critical for synaptic plasticity and hippocampal memory (Fleischmann et al., 2003; He et al., 2002; Tischmeyer and Grimm, 1999). Recent studies demonstrated that neuronal activity induces formation of DNA double-strand breaks (Madabhushi et al., 2015), including within the promoters of early-response genes including Fos, which facilitates their expression (Madabhushi et al., 2015). Therefore it might be expected that c-Fos expression should be high in conditions of chronic network hyperactivity. However, hippocampal c-Fos expression is biphasic, increasing after acute neuronal activity, but decreasing in chronically active neurons (Calais et al., 2013; Renthal et al., 2008; Sheng and Greenberg, 1990; Tsankova et al., 2004). The mechanisms governing such biphasic regulation of c-Fos in the hippocampus are not known. Decreased hippocampal c-Fos expression in mouse models of AD and epilepsy has been regarded as a potential contributor to hippocampus-dependent cognitive deficits (Calais et al., 2013; Palop et al., 2003, 2005; Chin et al., 2005; Le et al., 2004; Espana et al., 2010). Therefore it is critical to define the molecular mechanisms responsible for reducing c-Fos expression.
ΔFosB is an activity-dependent transcription factor in the immediate early gene family, and is an alternatively-spliced product of the FosB gene. However, unlike the products of other immediate early genes, such as c-Fos, ΔFosB has an unusually long half-life that allows it to accumulate and remain in chronically active cells for weeks (McClung et al., 2004; Nestler et al., 2001). Studies of ΔFosB function in the nucleus accumbens after chronic exposure to drugs of abuse revealed that ΔFosB epigenetically regulates a number of gene targets critical in addiction, including c-Fos, via histone modifications (Renthal et al., 2008). The term ‘epigenetic’ is used here to indicate histone modifications that alter gene expression.
We therefore hypothesized that high levels of neuronal activity induced by seizures may similarly lead to accumulation of ΔFosB in the hippocampus, where it would alter the expression of target genes such as c-Fos, causing long-term consequences for hippocampal function. Indeed the hippocampus, overexpression of ΔFosB can exert effects on learning and memory (Eagle et al., 2015). In the present study, we examined expression of ΔFosB in a transgenic mouse model of AD neuropathology that exhibits spontaneous seizures, and in a pharmacological model of epilepsy. We demonstrate that seizure-induced ΔFosB expression in the hippocampus is present and epigenetically suppresses c-Fos expression in both models. Moreover, blocking either histone deacetylation or ΔFosB activity ameliorated effects on both c-Fos expression and memory impairment. Our results reveal a molecular mechanism that explains why hippocampal c-Fos expression is chronically reduced in conditions associated with aberrant neuronal hyperactivity, and highlight potential therapeutic avenues to improve cognitive function in both AD and epilepsy.
Results
Seizure-induced ΔFosB correlates with impairments in hippocampal c-Fos expression and cognition in APP mice
Experimentally induced seizures can increase ΔFosB expression in the hippocampi of nontransgenic (NTG) wild-type mice (Chen et al., 1997; McClung et al., 2004), but the consequences of such increases on downstream gene expression are not clear. To investigate a link between seizures, ΔFosB, and c-Fos in AD mice, we first evaluated the relationship between seizures and hippocampal ΔFosB expression in a commonly used transgenic mouse model that expresses mutant human amyloid precursor protein (APP) (Line J20, Mucke et al 2000). We performed electroencephalogram (EEG) recordings in APP mice at 4–6 months of age, and found that the higher the frequency of epileptiform activity or seizures in APP mice, the greater the increase in ΔFosB expression levels in the dentate gyrus (DG) (Figure 1A,B). Overall ΔFosB expression was higher in APP mice relative to NTG controls, both by immunohistochemistry (Figure 1C) and by western blot analysis (Figure S1A,B). To confirm that ΔFosB expression in APP mice is driven by spontaneous epileptiform activity, we treated NTG and APP mice with the antiepileptic drug levetiracetam (75 mg/kg) for 14 days. Levetiracetam reduced epileptiform activity (Figure S2) consistent with previous studies (Sanchez et al., 2012), and also reduced ΔFosB expression in APP mice to levels that were no longer significantly different from NTG mice (Figure 1D).
Figure 1. Epileptiform activity and seizures in APP mice were associated with increased ΔFosB and decreased c-Fos in the hippocampus.

(A,B) EEG and immunohistochemical staining for ΔFosB in 4–6 month old APP mice reveal that increased hippocampal ΔFosB immunoreactivity (IR) is associated with epileptiform activity and frequency of seizures (APP, n = 14). A representative epileptiform discharge in the shaded region in the middle set of traces in A is magnified on the right side of the panel. Electrodes were in left and right frontal cortex (L/RFC), hippocampus (HIP) and parietal cortex (PC). (C) Increased ΔFosB IR in the DG of APP vs. NTG mice (n = 12/genotype). (D) 14-day treatment of APP mice with levetiracetam reduces ΔFosB IR (n = 4–5 per treatment/genotype, 2-way RM ANOVA with Bonferroni posthoc tests). (E,F) Reduced number of c-Fos-IR cells in the DG of APP vs. NTG mice (n = 16–18/genotype). (G, H) Expression of ΔFosB and c-Fos is inversely related in the DG of APP mice (n = 19 mice) and in individual cells in DG (n = 23 cells). Arrows indicate cells expressing primarily ΔFosB (green) or c-Fos (red), arrowheads indicate cells expressing both ΔFosB and c-Fos (yellow). (I) Average path length to find the hidden platform in the Morris water maze is associated with hippocampal ΔFosB expression (n = 21–22/genotype). (J–L) Increased ΔFosB IR was detected in the DG of Tg2576 (n = 9–10/genotype) and PSAPP mice (n = 5/genotype), but not in I5A mice (n = 7–9/genotype). AU, arbitrary units. Data were normalized to NTG control values (rel to NTG). *p<0.05, **p<0.01, ****p<0.0001. Error bars indicate SEM. See also Figures S1–S2 and Tables S3–S4.
To determine whether such increased ΔFosB expression in the DG is related to c-Fos expression, we confirmed our previous findings of reduced c-Fos expression in the DG of APP mice (Chin et al., 2005; Palop et al., 2005) (Figure 1E,F). In addition, we observed that APP mice with the highest ΔFosB expression had the lowest numbers of c-Fos-immunoreactive (IR) cells in the DG, and that the magnitude of ΔFosB and c-Fos expression were inversely correlated in APP mice on both a mouse-by-mouse and a cell-by-cell basis (Figure 1G,H). Because c-Fos expression is tightly linked to spatial memory (Fleischmann et al., 2003; He et al., 2002; Tischmeyer and Grimm, 1999), we examined whether ΔFosB expression levels in APP mice were related to performance in the Morris water maze. Indeed, the magnitude of ΔFosB expression correlated with the distance to find the hidden platform in the water maze (Figure 1I). We also observed some variability in the water maze performance of NTG mice (Figure 1I), which can sometimes be randomly affected even in control conditions. There was no obvious systematic relationship with ΔFosB levels, which did not vary greatly in NTG mice.
The increase in DG ΔFosB expression is not restricted to the particular line of APP mice used in our studies (line J20), as Tg2576 and PSAPP mice also exhibited increased ΔFosB-IR in the DG (Figures 1J,K and S1E,F). All of these lines of mice express human APP with mutations linked to autosomal dominant AD, produce high levels of Aβ (Chin, 2011), and exhibit epileptiform activity (Chin and Scharfman, 2013; Corbett et al., 2013; Kam et al., 2016; Minkeviciene et al., 2009; Palop et al., 2007). Notably, similarly aged I5A mice, which overexpress wild-type human APP and do not produce high levels of Aβ (Mucke et al., 2000), do not exhibit increased ΔFosB (Figure 1L, Figure S1G).
Epigenetic mechanism of c-Fos suppression in APP mice
ΔFosB suppresses c-Fos expression in nucleus accumbens by directly binding to the c-Fos promoter and recruiting HDAC1, thereby promoting deacetylation of histone H4 (Renthal et al., 2008). We hypothesized that this mechanism could also be responsible for the decreased c-Fos expression observed in DG of APP mice. Indeed, chromatin immunoprecipitation (ChIP) experiments demonstrated increased binding of ΔFosB to the c-Fos promoter in the hippocampus of APP mice (Figure 2A). Interactions between ΔFosB and HDAC1 have been characterized in nucleus accumbens (Renthal et al., 2008), and co-immunoprecipitation experiments showed ΔFosB also binds HDAC1 in the hippocampus (Figure 2B). Consistent with this, a target of HDAC1, histone H4, was hypoacetylated at the c-Fos promoter in the hippocampus of APP mice (Figure 2C). No significant differences in histone H3 acetylation at the c-Fos promoter were observed between NTG and APP mice (Figure 2D,E), indicating relative specificity. Because histone H4 acetylation is important for gene transcription (Grunstein, 1997; Lee et al., 1993; Struhl, 1998; Tsankova et al., 2004), we measured c-Fos mRNA in the hippocampus of NTG and APP mice and confirmed that APP mice exhibited decreased levels of hippocampal c-Fos mRNA compared to NTG controls (Figure 2F). Together, these data support the hypothesis that increased hippocampal ΔFosB suppresses c-Fos expression in APP mice.
Figure 2. Epigenetic suppression of the c-Fos gene in the hippocampus of APP mice.

(A) Increased ΔFosB binding to the c-Fos promoter in the hippocampus APP vs. NTG mice (n = 8/genotype). (B) ΔFosB co-immunoprecipitates (IP) with HDAC1 in the hippocampus of NTG and APP mice. Two exposures of the same ΔFosB blot are shown. (C) c-Fos promoter is hypoacetylated on histone H4 in APP vs. NTG mice (n = 7/genotype). (D,E) No changes in tetra-acetyl histone H3 (n = 8/genotype) or di-acetyl histone H3 (n = 8/genotype) were detected in APP vs. NTG mice. (F) Reduced c-Fos mRNA expression in APP vs. NTG mice (n = 12/genotype). Data were normalized to NTG control values (rel to NTG). *p<0.05, ***p<0.001. Error bars indicate SEM. See also Tables S3–S4.
Pharmacologically-induced seizures are sufficient to increase ΔFosB and suppress c-Fos in NTG mice within days
We next investigated whether seizures alone, outside of the context of other AD-related factors, were sufficient to drive ΔFosB-mediated c-Fos suppression and hippocampal memory deficits in NTG mice. We assessed ΔFosB and c-Fos IR in the DG of NTG mice given a single convulsant injection of kainic acid (KA) or saline control, and examined mice either 2 hrs later as seizures were occurring or 3 days later when seizures were over. Mice injected with KA exhibited dose-dependent behavioral seizures that were observed in the first two hours post-injection (Figure S3A). For mice that were sacrificed 2 hrs after KA injection, high doses (15 and 25 mg/kg) of KA caused severe behavioral seizures, and increased levels of both ΔFosB and c-Fos compared to saline-treated mice (Figure 3A,B, and Figure S1A,C), whereas no changes were observed in mice receiving a low dose (5 mg/kg) of KA that did not elicit behavioral seizures.
Figure 3. Pharmacologically-induced seizures are sufficient to increase ΔFosB and suppress c-Fos in NTG mice within days.

(A,B) At 2 hrs after single administration of 15 or 25 mg/kg KA, there were increases in ΔFosB IR (n = 4/group) and c-Fos IR (n = 4/group). (C) ΔFosB IR is increased 3 days after single administration 15 or 25 mg/kg KA (n = 4/group). (D) c-Fos IR is increased 3 days after single administration of 5 or 10 mg/kg KA, but decreased 3 days after 15 or 25 mg/kg KA (n = 4/group). (E) ΔFosB and c-Fos expression are positively correlated with increasing doses of KA when assessed 2 hrs after KA injection. (F) ΔFosB and c-Fos expression are inversely correlated with increasing doses of KA when assessed 3 days after KA injection. (G) Summary of results from A–D. “0” indicates no change from control, whereas “+” and “−“ indicate increase and decrease from control, respectively. (H) Increased ΔFosB binding to the c-Fos promoter in mice 3 days after 15 mg/kg KA (n = 5/group). (I) Decreased Fos mRNA expression in mice 3 days after 15 mg/kg KA (n = 8–9/group). (J) NTG mice treated with 3 weekly doses of saline, but not 15 mg/kg KA, show preference for the displaced object in the test phase of the object location memory task (n = 6/group). Data were normalized to saline-treated control values (rel to Sal). *p<0.05, **p<0.01, ***p<0.001. ns, non-significant. Error bars indicate SEM. See also Figures S1 and S3 and Tables S3–S4.
Three days after KA injection, ΔFosB expression remained increased in mice that received high doses of KA, consistent with its long half-life, and was unchanged in mice that received low doses of KA (Figure 3C and Figure S1A,D). ΔFosB expression persisted for at least 4 weeks after KA injection (Figure S3B). At 3 days post-injection, low doses of KA increased c-Fos expression, whereas high doses of KA decreased c-Fos expression (Figure 3D). The dose-dependent relationship between ΔFosB and c-Fos expression was positively correlated 2 hrs after KA (Figure 3E), but inversely correlated 3 days after KA (Figure 3F). These results suggest that at 2 hrs post-KA, ΔFosB had not yet suppressed c-Fos expression; however, by 3 days post-KA it had. Therefore, induction of seizures in NTG mice appears to be sufficient, after 3 days, to simulate the increased ΔFosB/decreased c-Fos signature observed in the DG of APP mice. A summary of the cellular changes following different seizure severities is provided in Figure 3G.
To determine whether the decreased c-Fos levels observed in the DG of mice exposed to high doses of KA were also due to ΔFosB-mediated epigenetic suppression, we performed ChIP experiments. Three days after mice were administered 15 mg/kg KA, there was increased binding of ΔFosB to the Fos promoter compared to saline-treated mice (Figure 3H), and also decreased hippocampal Fos mRNA expression (Figure 3I).
We investigated whether the increased ΔFosB expression in KA-treated mice was also associated with hippocampal memory deficits. We used the object location memory task, a hippocampus-dependent spatial memory task (Faust et al., 2013; Scharfman and Binder, 2013). Mice received weekly injections of either saline or KA (15 mg/kg) for 3 weeks to ensure maximal ΔFosB expression and persistent decreases in c-Fos prior to behavioral testing. Mice that received saline injections displayed intact memory, but mice that received KA injections exhibited impaired memory (Figure 3J). Together, these data support the hypothesis that seizures are sufficient to drive ΔFosB-mediated suppression of c-Fos and contribute to hippocampal memory deficits.
Persistent ΔFosB expression leads to desensitization of c-Fos expression with repetitive seizures
c-Fos mRNA induction in the cortex is attenuated after repeated neuronal stimulation by either brain injury or seizures (Calais et al., 2013; Ivkovic et al., 1994; Renthal et al., 2008; Winston et al., 1990). We hypothesized that similar to the effect of chronic exposure to drugs of abuse (Renthal et al., 2008), repeated seizures may lead to desensitization of c-Fos expression due to the up-regulation and persistent presence of ΔFosB in the hippocampus. We therefore examined whether increased ΔFosB expression coincided with desensitization of c-Fos mRNA induction in the hippocampus following multiple KA injections. We first verified the expression time course of c-Fos mRNA by injecting NTG mice with a single dose of 15 mg/kg of KA and then assessing levels of hippocampal c-Fos mRNA at various time points. As expected, c-Fos mRNA was robustly increased 1 hr post-KA and returned to baseline levels 4.5 hrs post-KA (Figure 4A). We then injected NTG mice with KA (15 mg/kg) either 1, 2, 3, 4, or 5 times, with injections spaced 4.5 hrs apart for the first 4 injections and 12 hrs apart for the 4th and 5th injections. Mice were sacrificed 1 h following their final injection, and hippocampal ΔFosB protein and c-Fos mRNA expression were assessed. Increased expression of ΔFosB protein was first detected after the second injection, and remained high throughout the remainder of the protocol (Figure 4B), and coincided with desensitization of c-Fos mRNA expression (Figure 4C). These results support the hypothesis that persistent ΔFosB up-regulation can desensitize c-Fos expression to repeated stimulation. The mean maximum seizure score in mice injected multiple times with KA was lower than in mice injected only once (Supplemental Fig S4A), so other factors such as modification of glutamate receptors (Borbely et al., 2009; Vilagi et al., 2009), may also be involved in attenuation of c-Fos induction and seizures scores. The multiple KA injections did not result in any obvious cell death (Supplemental Fig S4B).
Figure 4. Persistent ΔFosB expression leads to desensitization of c-Fos expression with repetitive seizures.

(A) Hippocampal c-Fos mRNA expression in NTG mice is increased 1–2 hrs after single administration of 15 mg/kg KA, but returns to baseline at 4.5 hrs (n = 3–4/group). (B) DG ΔFosB IR was increased in mice that received multiple administrations of 15 mg/kg KA. (C) The increase in c-Fos mRNA expression after each KA injection is attenuated with multiple injections, which is first evident when ΔFosB expression increases as shown in panel B. Data were normalized to saline-treated control values (rel to Sal). *p<0.05, **p<0.01, ***p<0.001. Error bars indicate SEM. See also Figure S4 and Tables S3–S4.
HDAC1 contributes to the suppression of physiological c-Fos induction in the hippocampus of APP mice
Given that ΔFosB suppresses c-Fos expression during seizures, we hypothesized that prolonged levels of ΔFosB might serve as a persistent brake that limits c-Fos expression during physiological neuronal activity. Indeed, APP mice exhibit impaired induction of c-Fos expression after exploration of a novel environment (NE) (Palop et al., 2003), but the mechanisms were not clear. Because ΔFosB binds HDAC1 in the hippocampus, and suppression of c-Fos in the nucleus accumbens is dependent on ΔFosB-mediated recruitment of HDAC1 to the c-Fos promoter (Kennedy et al., 2013; Renthal et al., 2008), we predicted that acute inhibition of HDAC1 would improve physiological c-Fos induction.
To test this hypothesis, we treated mice with the class I HDAC inhibitor 4-PBA one hr prior to exposure to NE. After exploring the NE for two hrs, mice were sacrificed and their brains processed to assess c-Fos-IR cells in the DG. Mice treated with saline and/or left in their home cages were used as controls. As expected, saline-treated NTG but not APP mice exhibited a greater number of total c-Fos-IR cells when exposed to the NE than when left in their home cages (Figure 5A). Surprisingly, neither NTG nor APP mice treated with 4-PBA 1 hr prior to placement in the NE exhibited increased total numbers of c-Fos-IR cells compared to their home cage controls (Figure 5B). We initially thought that the lack of effect might be due to non-specific HDAC blockade, since 4-PBA inhibits all class I HDACs (HDACs 1–3, 8). To inhibit HDAC1 more specifically, we treated NTG and APP mice with MS-275, an inhibitor of HDAC1-3 that has an IC50 that is 8–30 fold lower for HDAC1 than for HDACs 2 and 3 (Bahari-Javan et al., 2012; Hu et al., 2003; Khan et al., 2008). The brain penetration of MS-275 has also been characterized (Hooker et al., 2010; Simonini et al., 2006), allowing us to administer a dose predicted to be near the IC50 for HDAC1. However, similar to 4-PBA treatment, treatment with MS-275 2 hrs before placement in the NE did not increase the total number of c-Fos-IR cells of NTG or APP mice compared to home cage controls (Figure 5C).
Figure 5. Acute HDAC inhibition partially restores c-Fos induction in APP mice.

(A) NTG but not APP mice exhibit increased total c-Fos IR in the hippocampus after exploration of a novel environment for 2 hrs (NE +) relative to home cage controls (NE −) (n = 9–10/group). (B,C) Neither NTG nor APP mice exhibit significant increases in the total number of c-Fos IR cells when exposed to NE 1 hr after 4-PBA treatment (n = 6/group) or 2 hrs after MS-275 treatment (n = 8–10/group). (D) In control conditions, NTG but not APP mice exhibit increased numbers of c-Fos-IR cells above optical threshold when exposed to NE vs. home cage (n = 9–10/group). (E,F) Both NTG and APP mice exhibit increased number of c-Fos-IR cells above optical threshold when exposed to NE 1 hr after 4-PBA treatment (n = 6/group) or 2 hrs after MS-275 treatment (n = 8–10/group). (G,H) Histone H4 acetylation on the Fos promoter in APP mice is normalized after treatment with 4-PBA (n = 6/group) or MS-275 (n = 6/group). Data in panels D-F were normalized to NTG home cage control values (rel to NTG/–). *p<0.05, ***p<0.001. Error bars indicate SEM. See also Figure S5–S6, and Tables S3–S4.
Because c-Fos is only expressed in a sparse population of DG cells at baseline, we hypothesized that increases in physiologically-induced c-Fos following HDAC inhibition might be detectable only in select granule cells that were highly active. Furthermore, because HDAC inhibition appears to increase the baseline number of DG c-Fos-IR cells in home cage controls (Figure 5A–C), it was possible that the specific population of c-Fos-IR cells induced during NE exploration was difficult to detect by total cell counts. Therefore, we investigated whether acute HDAC inhibitor treatment increased c-Fos IR in a specific population of DG cells capable of inducing c-Fos at high levels. To do this, we set an optical threshold during quantification so that only DG cells with high c-Fos-IR were counted (Figure S5). Using this quantification method, we found that saline-treated NTG mice exposed to the NE exhibited higher numbers of c-Fos-IR cells above threshold compared to home cage controls while saline-treated APP mice did not demonstrate this effect (Figure 5D). However, when treated with either 4-PBA or MS-275 prior to placement in the NE, both NTG and APP mice that explored the NE exhibited increased numbers of c-Fos-IR cells above threshold compared to home cage controls (Figure 5E,F). To verify that treatment with 4-PBA and MS-275 inhibited HDAC1, we confirmed that there were no significant differences in hippocampal histone H4 acetylation at the c-Fos promoter among any groups treated with 4-PBA or MS-275 (Figure 5G,H). Moreover, no differences in the number of object interactions during NE exploration were observed between NTG and APP mice regardless of treatment (Figure S6A–C), indicating that the induction of c-Fos was not due to behavioral alterations. We also confirmed that APP mice exhibited increased ΔFosB-IR in the DG compared to NTG controls regardless of treatment or cage condition (Figure S6D–F). ΔFosB-IR was inversely related to the number of c-Fos-IR cells above threshold in saline and MS-275-treated APP mice, and trended toward an inverse relation in 4-PBA-treated APP mice (Figure S6G–I). Moreover, acute HDAC inhibitor treatment did not affect the number of spikes observed by EEG recording, indicating that the alterations in c-Fos expression were not due to gross changes in EEG activity (Figure S6J). Together, these results demonstrate that HDAC1 contributes to the suppression of physiological c-Fos induction in the DG of APP mice.
Acute HDAC1 inhibition improves hippocampus-dependent memory deficits in APP mice
Physiological c-Fos induction is critical for hippocampal memory (Fleischmann et al., 2003; He et al., 2002). Therefore, we hypothesized that acute HDAC inhibition would also improve hippocampal memory in APP mice if administered prior to training. We assessed hippocampal memory using the object location memory task because it could be completed within the same 2 hr timeframe as the NE paradigm. We treated mice with saline, 4-PBA, or MS-275 prior to training in the object location memory task. Saline-treated NTG mice, but not APP mice, spent an increased percentage of time interacting with the displaced object during testing compared to training. When treated with 4-PBA or MS-275, both NTG and APP mice spent an increased percentage of time interacting with the displaced object during the testing phase (Figure 6A,B). We hypothesize that ΔFosB recruits HDAC1 to deacetylate its gene targets, and thus HDAC1 functions downstream of ΔFosB. Therefore, inhibition of HDACs with either 4-PBA or MS-275 should not affect ΔFosB levels, especially with the acute timeframe of treatment given that the half-life of ΔFosB is 8 days. Consistent with our hypothesis, APP mice displayed increased levels of ΔFosB in the DG compared to NTG controls regardless of treatment (Figure 6C, D). Moreover, acute treatment with HDAC inhibitors did not non-specifically alter spiking frequency in APP mice (Figure S6J). These results support the hypothesis that HDAC1 actively and acutely suppresses the expression of genes important for hippocampal memory and contributes to cognitive deficits in APP mice.
Figure 6. Acute HDAC inhibition improves hippocampus-dependent learning and memory in APP mice.

(A,B) APP mice exhibited increased time interacting with the displaced object during the test phase of the task when, prior to training, they were treated with either 4-PBA (n = 5–6/group) or MS-275 (n = 6/group) relative to treatment with vehicle (control). (C, D) Increased ΔFosB expression in APP vs. NTG mice is unaffected by 4-PBA (n = 5–6/group) or MS-275 (n = 7/group). Data in panels C–D were normalized to vehicle-treated NTG values (rel to NTG/Ctrl). *p<0.05, **p<0.01, ***p<0.001, ns, non-significant. Error bars indicate SEM. See also Tables S3–S4.
Direct inhibition of ΔFosB improves c-Fos induction and hippocampus-dependent memory in APP mice
Our results suggest that direct inhibition of ΔFosB should also improve DG c-Fos induction and spatial memory deficits in APP mice. To test whether this is true, we used ΔJunD, an N-terminally truncated mutant of JunD, the normal binding partner of ΔFosB. When expressed in neurons, ΔJunD serves as a dominant-negative inhibitor of ΔFosB (Berton et al., 2007; Vialou et al., 2010).
To achieve ΔJunD overexpression in the DG, we utilized an adeno-associated virus serotype 2 (AAV2) delivery system (Vialou et al., 2010). We confirmed ΔJunD overexpression in the DG by stereotaxically infusing AAV2 carrying GFP alone (AAV-GFP) into the left DG and AAV2 carrying both GFP and ΔJunD (AAV-GFP/ΔJunD) into the right DG of NTG mice, and assessed ΔJunD expression at 28 days. Using an antibody that recognizes both ΔJunD and endogenous JunD, we verified that AAV-GFP/ΔJunD overexpressed ΔJunD by nearly three-fold above endogenous JunD levels (Figures 7A and S7).
Figure 7. Direct inhibition of ΔFosB signaling improves c-Fos expression and hippocampus-dependent memory in APP mice.

(A) Unilateral AAV-GFP/ΔJunD infusion into the right DG achieves robust ΔJunD overexpression in comparison with AAV-GFP infused into the contralateral side. (B) Bilateral AAV-ΔJunD infusion into the DG of APP mice improves performance in the object location memory task. (C) Bilateral AAV-GFP/ΔJunD (ΔJunD) infusion into the DG of NTG and APP mice achieves robust ΔJunD overexpression vs. AAV-GFP (GFP) control. (D) ΔJunD overexpression does not impact ΔFosB expression in APP mice. (E) ΔJunD overexpression restores c-Fos expression in APP mice. JunD/ΔJunD and ΔFosB quantification data were normalized to AAV-GFP-treated NTG values (rel to NTG/GFP). n = 5–8/group for panels B-E. *p<0.05, **p<0.01, ***p<0.001, ns, non-significant. Error bars indicate SEM. See also Figure S7 and Tables S3–S4.
To test whether ΔJunD overexpression improves hippocampal memory and increases c-Fos expression in the DG of APP mice, we infused either AAV-GFP or AAV-GFP/ΔJunD bilaterally into the DG of NTG and APP mice, and tested them in the object location memory task 28 days later. NTG mice spent more time interacting with the displaced object in the testing phase of the task regardless of treatment (Figure 7B). APP mice that received AAV-GFP spent similar amounts of time with the displaced object in both training and testing phases, demonstrating impaired hippocampal memory (Figure 7B). However, APP mice that received AAV-GFP/ΔJunD spent an increased percentage of time interacting with the displaced object in the testing phase (Figure 7B), indicating that ΔJunD overexpression improves hippocampal memory in APP mice.
After behavioral testing, we verified that JunD-IR was increased in NTG and APP mice receiving AAV-GFP/ΔJunD infusion (Figure 7C) and that ΔFosB expression was increased in APP mice regardless of the type of virus infused (Figure 7D). We then quantified the total number of c-Fos-IR cells in the DG of NTG and APP mice receiving AAV infusions. ΔJunD overexpression did not affect c-Fos expression in the DG of NTG mice. APP mice that received AAV-GFP/ΔJunD exhibited increased numbers of c-Fos-IR cells in the DG compared to APP mice that received AAV-GFP (Figure 7E). This result indicates that ΔFosB directly mediates the suppression of c-Fos expression in the DG of APP mice.
Discussion
A growing body of evidence suggests that network dysfunction in the form of epileptiform activity contributes to cognitive deficits in AD, even in early stages of the disease (Bakker et al., 2012; Vossel et al., 2013; Vossel et al., 2016). Understanding how specific neuronal alterations contribute to cognitive deficits may therefore highlight additional therapeutic avenues to improve the quality of life for AD patients. Moreover, because long-term treatment with antiepileptic drugs is associated with many adverse effects, targeting more specific mechanisms may enable development of treatments with fewer side effects.
Our studies reveal a molecular mechanism by which epileptiform activity might contribute to cognitive deficits in AD and epilepsy. This mechanism highlights seizure-induced ΔFosB as a transcription factor that critically regulates hippocampal memory, although other effects of seizures and/or APP/Aβ may also play a role. Due to the unusually long half-life of ΔFosB, even infrequent epileptiform activity would be sufficient to maintain persistent high levels of ΔFosB. Therefore, ΔFosB is well-poised to modulate gene expression over extended periods of time, which may be part of why cognitive deficits persist even during seizure-free periods in AD and epilepsy patients, as well as in relevant mouse models.
A critical implication of ΔFosB-mediated c-Fos suppression is that it also explains the biphasic expression curve of c-Fos expression after sustained high levels of neuronal activity. Hippocampal c-Fos expression is increased by acute neuronal activity, but decreased by chronic activity (McClung et al., 2004; Renthal et al., 2008; Sheng and Greenberg, 1990; Tsankova et al., 2004). The gradual but persistent nature of activity-induced ΔFosB expression directly accounts for the biphasic expression profile of c-Fos. Acute neuronal activity induces c-Fos expression prior to the accumulation of ΔFosB to high enough levels in the nucleus to regulate gene expression. Following chronic neuronal activity, persistent increases in ΔFosB allow it to reach a threshold that enables regulation of genes such as c-Fos. Therefore, although c-Fos expression is frequently used as a marker of neuronal activity, it may only accurately reflect activity under acute conditions. However, the simultaneous use of both ΔFosB and c-Fos as activity markers provides a more accurate readout of neuronal activity.
We focused on ΔFosB-mediated suppression of hippocampal c-Fos expression as a mechanism for hippocampal memory deficits. However, the extent to which the normalization of other ΔFosB-targeted genes contribute to cognitive improvement was not investigated. We chose to investigate ΔFosB-mediated c-Fos suppression because the role of c-Fos in synaptic plasticity and hippocampal memory is well-established (Fleischmann et al., 2003; He et al., 2002; Tischmeyer and Grimm, 1999). Furthermore, ΔFosB may not be the sole mechanism underlying hippocampal c-Fos suppression in APP mice. Other factors, such as altered regulation of CREB-regulated transcription coactivator-1 (Espana et al., 2010; Saura, 2012; Yiu et al., 2011) and increased levels of miR-181 (Rodriguez-Ortiz et al., 2014) have also been implicated in reduced c-Fos expression exhibited by mouse models of AD.
We hypothesized that ΔFosB actively suppresses the expression of genes important for cognition in the hippocampus and that even a temporary relief of this suppression would improve hippocampal memory. Indeed, acute HDAC inhibition improved hippocampal c-Fos induction and hippocampal memory in APP mice. Although acute HDAC inhibition may have improved memory in APP mice by altering the expression of many genes important for cognition in addition to c-Fos, the increase in c-Fos is likely to have contributed strongly to the efficacy of acute HDAC inhibition. Chronic treatment with class 1 HDAC inhibitors including 4-PBA improved learning and memory in APP mice (Kilgore et al., 2010; Ricobaraza et al., 2009), but our results demonstrate that even a temporary relief of gene suppression can improve cognition when administered prior to training in a memory paradigm. For AD patients, acute treatment options may minimize side effects by allowing for an infrequent, but well-timed schedule of administration (e.g. during family visitation), thus improving overall quality of life.
Acute HDAC inhibition did not increase the total number of c-Fos-IR cells in the DG of APP mice following exploration in the NE paradigm; however, it did increase c-Fos expression in a certain population of neurons. As such, it is possible that acute HDAC inhibition is unable to affect more severe and/or chronic epigenetic mechanisms that may have occurred, such as histone or DNA methylation. Overexpression of ΔJunD, a more chronic treatment that might alter multiple modes of epigenetic modification, was able to improve the total number of c-Fos-IR neurons in the DG of APP mice, lending support for this hypothesis.
Understanding how network dysfunction induces neuronal alterations that cause cognitive deficits emphasizes the importance of the role that seizures play in AD. Furthermore, many diseases including schizophrenia (Hyde and Weinberger, 1997), autism (Viscidi et al., 2013), Down syndrome (Lott et al., 2012), Fragile X syndrome (Hecht, 1991), Rett Syndrome (Jian et al., 2006), and Angelman Syndrome (Pelc et al., 2008) are all accompanied by both seizures and cognitive deficits. Therefore, it is possible that seizures and network dysfunction may be common denominators contributing to cognitive deficits in many brain disorders. In addition, seizure-induced increases in ΔFosB may contribute to deficits in cognition or other behaviors via alteration of downstream gene targets that are already vulnerable due to compromised genetic backgrounds.
Experimental Procedures
Mice
4- to 6-month-old heterozygous transgenic and NTG mice from lines hAPP-J20, Tg2576, and PSAPP were used in these experiments. Male and female mice were used unless otherwise specified, and sex was balanced within a cohort. All mice referred to as APP mice were of the line hAPP-J20, which express a human APP (hAPP) gene carrying Swedish (K670N, M671L) and Indiana (V717F) familial AD mutations under control of the platelet-derived growth factor β–chain promoter, on a C57Bl/6 background (Mucke et al., 2000). Tg2576 mice express hAPP carrying the Swedish FAD mutation (Hsiao et al., 1996). PSAPP mice express hAPP carrying the Swedish mutation and presenilin-1 with FAD mutation (M146L) (Holcomb et al., 1998). For brain harvesting, mice were flush-perfused transcardially with DEPC-treated saline. Hemibrains were frozen on dry ice for biochemistry or postfixed in 4% paraformaldehyde for immunohistochemistry. All procedures were approved by the Thomas Jefferson University and Baylor College of Medicine Institutional Animal Care and Use Committees.
Drug Treatment
Levetiracetam was dissolved in saline at 15 mg/ml and administered intraperitoneally (IP) at a dose of 75 mg/kg every 8 h for 14 d. Kainic acid (Sigma) was dissolved in saline at 2 mg/ml and administered IP at doses described in Results. 4-PBA (Aldrich) was dissolved with equimolar NaOH in ddH2O to 0.125 M and injected IP at a dose of 200 mg/kg 1 h prior to NE or object placement paradigms. A stock solution of 50 mg/ml MS-275 in dimethyl sulfoxide (DMSO) was dissolved into a working solution of 1.5 mg/ml MS-275 in saline containing 20% DMSO, which was administered at 6 mg/kg via subcutaneous injection 2 h prior to NE or object placement paradigms.
Kainic Acid Injections and Seizure Severity
Scoring NTG mice were injected with kainic acid IP at specified doses and monitored for 2 h. Seizure severity and elapsed times since injection were recorded using a modified Racine scale (Racine, 1972; Roberson et al., 2007): 0, normal behavior; 1, immobility; 2, generalized spasm, tremble, or twitch; 3, tail extension; 4, forelimb clonus; 5, generalized clonic activity with loss of posture; 6, bouncing or running seizures; 7, full tonic extension; 8, death. Acute KA groups were sacrificed 2 h following a single KA injection (15 mg/kg). Subchronic KA groups were sacrificed 3 d following a single KA injection (15 mg/kg). For mice receiving multiple kainic acid injections, all injections were separated by 4.5 h except for the 4th and 5th injections, which were separated by 12 h. Unless otherwise specified, mice were sacrificed 0.5 h following their final injection.
Video-EEG Monitoring
Electrodes were implanted as described in Supplemental Methods. EEG recordings were performed on at least 2 different days for a minimum of 4 h each using a Stellate Harmonie acquisition system. EEG signal processing was performed using native Stellate software and Labchart Pro (AD Instruments). The frequency and total number of spikes for each mouse were analyzed.
AAV-mediated gene transfer
AAV2 carrying CMV-ΔJunD-IRES2-eGFP (AAV-GFP/ΔJunD) or CMV-eGFP (AAV-GFP) was previously described (Berton et al., 2007; Vialou et al., 2010), demonstrating that these constructs are neurotropic and achieve stable neuronal gene expression within 18–22 days of infusion into the brain. One µL of virus solution was stereotaxically infused unilaterally or bilaterally into the hippocampus at rostral (−1.7 mm A/P, 1.2 mm M/L, 2 mm D/V from bregma) and caudal (−2.7 mm A/P, 2 mm M/L, 2.1 mm D/V from bregma) coordinates. Approximately 2×108 infectious particles were infused into each hippocampus. Mice were allowed to recover for 28 days post-surgery before behavior testing/sacrifice.
Morris Water Maze
Female NTG and APP mice (4–6 months old, 20–24/genotype) were tested as described in detail in the Supplemental Methods. Mice were first tested in cued platform sessions, 2 trials per day for 2 days. Hidden platform training involved 6 trials/d over 7 d. After the last training trial, the platform was removed and mice were given a 60 s probe trial test. Performance was videotracked and analyzed using TopScan (CleverSys, Inc). The experimenter was blinded to genotypes during testing.
Object Location Memory Task
The object location memory task was based on previously described protocols (Scharfman and Binder, 2013) and is described in detail in the Supplemental Methods. Briefly, mice were trained in 3 trials to recognize set locations of 2 identical objects in an empty mouse cage, and the time spent exploring each object during each of the 3 trials was recorded. After a 2 hr delay, the mouse was placed back in the test cage, in which one of the objects had been displaced. The time spent exploring the displaced versus the non-displaced object was recorded. For HDAC inhibitor treatments, training began 1 h following IP injection with saline or 4-PBA, or 2 h following subcutaneous injection with 20% DMSO in saline or MS-275. For kainic acid experiments, mice were injected with saline or 15 mg/kg kainic acid once a week for 3 weeks and trained/tested 3 d following the third injection.
Novel Environment
Female NTG and APP mice were placed in a NE either without treatment, 1 h following 4-PBA treatment, or 2 h following MS-275 treatment. Home cage control mice for each group received the same treatment in close temporal proximity. The home cages of mice contained only bedding, a water bottle, and a food hopper. The NE cage included a novel bedding material, a banana slice, the grid of a pipette tip rack, a 50 ml conical vial cap, and a repeat pipetter tip wrapped in red tape. The top of the cage lid was removed for observation. Mice explored the NE for 2 h while being video monitored, and were then sacrificed. The number of object interactions was quantified for each mouse in 10-min bins at 1, 50, and 110 min into the recording.
Immunohistochemistry
Tissue preparation and immunohistochemistry were performed as described (Chin et al., 2005; Corbett et al., 2013). Brains were sectioned at 30 µm. Antibodies are listed in Supplemental Table S1. For staining with 3,3’-diaminobenzidine (DAB), further amplification was accomplished using Avidin-Biotin Complex (Vectastain). DAB (Sigma) was used as a chromagen. ΔFosB-IR quantification was performed by assessing the mean pixel intensity using ImageJ software. Quantification of c-Fos was performed by total cell count or by using the threshold function provided by Metamorph. A pixel intensity threshold was set such that approximately 40–50 (approximately 30% of total) cells were above threshold in the DG of NTG mice from the homecage group. This threshold was used for all mice and the number of c-Fos-IR cells above threshold was recorded. For immunofluorescence, Metamorph software was used to quantify optical density throughout the rostral-caudal extent of the DG.
Western Blot
Western blot analysis was performed as described (Corbett et al., 2013). The hippocampus from one hemibrain of each mouse was isolated and homogenized with a Polytron tissue homogenizer in ice-cold RIPA buffer. Equal amounts of protein were resolved by SDS-PAGE on 4–12% gels and transferred to nitrocellulose, and probed with primary antibodies listed in Supplemental Table S1. IR-dye-conjugated secondary antibodies were used for detection and quantification using a LI-COR Odyssey infrared imaging system.
Co-immunopreciptation
Hippocampi were lysed in immunoprecipitation buffer (320 mM sucrose, 10 mM Tris-HCl pH 7.4, 10 mM EDTA, 1% sodium deoxycholate) and diluted 1:5 in binding buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 0.5 mM EDTA, 0.5% Triton-X). 4 µg of rabbit anti-HDAC1 (Abcam) was added to immunoprecipitate HDAC1 prior to Western blot analysis. HDAC1 and FosB were immunoblotted.
RNA Extraction
RNA extraction was adapted from instructions from the Qiagen RNeasy kit. Briefly, hippocampi were homogenized in RLT/β-mercaptoethanol buffer by passing the lysate through an 18G needle followed by a 25G needle. After centrifugation, RNA was extracted, and eluted with nuclease-free water. Final RNA concentration was measured using a NanoDrop 2000 spectrophotometer.
qRT-PCR
Reverse transcription was performed using the Taqman Gold RT-PCR kit (ABI). Quantitative PCR was performed with an ABI 7500 PCR machine using SYBR Green as a fluorophore. Primers used to amplify cDNA are listed in Supplemental Table S2.
Chromatin Immunoprecipitation
ChIP was performed similarly to described methods (Renthal et al., 2008; Tsankova et al., 2004). Hippocampi were subdissected and fixed in 1% formaldehyde. Samples were sonicated to generate genomic fragments 200–1000 bp in length and pre-cleared with Protein A beads (Millipore) prior to incubation with antibodies listed in Supplemental Table 1 at 4°C overnight. The antibody-chromatin complex was immunoprecipitated with Protein A beads and then washed with a series of buffers (Millipore), then chromatin was then eluted and reverse cross-linking was performed with Proteinase K. DNA was purified via phenol-chloroform extraction. Final DNA concentration was measured using the NanoDrop 2000. Quantitative PCR was performed with an ABI 7500 PCR machine using SYBR green as a fluorophore. Primers used to amplify the c-fos promoter and β-actin promoter are listed in Supplemental Table S2.
Statistical Analysis
Statistical analyses were performed using Prism 5. Raw values for data presented as “relative to NTG controls” are listed in Supplemental Table S3. Differences between means were assessed using a two-tailed, unpaired Student’s t-test unless otherwise indicated. Differences among 3 or more means were assessed using one-way ANOVA and Tukey post hoc tests. Two-way ANOVA using Bonferonni or Student-Newman-Keuls post hoc tests were used to assess differences in analyses with two variables. Repeated measures two-way ANOVA with Bonferonni post hoc tests were used for analyzing object location memory data. Regression analysis was used to detect correlations. To assess the relationship between c-Fos expressing cells and ΔFosB expression, c-Fos cell counts were log transformed in order to perform a linear regression and test for statistical significance. Detailed results of all statistical analyses are listed in Supplemental Table S4.
Supplementary Material
Acknowledgments
This work was supported by the Margaret Q. Landenberger Research Foundation (JC), the Hassel Family Foundation (JC), and National Institutes of Health Grants NS085171 (JC) and F30-AG048710 (JCY).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
B.F.C. and J.C. conceived the project. B.F.C., X.Z., E.N. and J.C. designed the experiments. B.F.C., X.Z, M.S.P., U.T., J.C.Y., D.M.I., I.P. A.H., C-H.F., G.S.S., A.A.A., S.A., L.Z., and J.C. performed the experiments and analyzed the data. All authors discussed the results and B.F.C., J.C.Y., and J.C. wrote the manuscript.
References
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Bahari-Javan S, Maddalena A, Kerimoglu C, Wittnam J, Held T, Bahr M, Burkhardt S, Delalle I, Kugler S, Fischer A, et al. HDAC1 regulates fear extinction in mice. J Neurosci. 2012;32:5062–5073. doi: 10.1523/JNEUROSCI.0079-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, Speck CL, Jones LR, Stark CE, Yassa MA, Bassett SS, Shelton AL, Gallagher M. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, Covington HE, Ebner K, 3rd, Tsankova NM, Carle TL, Ulery P, Bhonsle A, Barrot M, Krishnan V, Singewald GM, et al. Induction of deltaFosB in the periaqueductal gray by stress promotes active coping responses. Neuron. 2007;55:289–300. doi: 10.1016/j.neuron.2007.06.033. [DOI] [PubMed] [Google Scholar]
- Burgess N, Maguire EA, O'Keefe J. The human hippocampus and spatial and episodic memory. Neuron. 2002;35:625–641. doi: 10.1016/s0896-6273(02)00830-9. [DOI] [PubMed] [Google Scholar]
- Calais JB, Valvassori SS, Resende WR, Feier G, Athie MC, Ribeiro S, Gattaz WF, Quevedo J, Ojopi EB. Long-term decrease in immediate early gene expression after electroconvulsive seizures. J Neural Transm. 2013;120:259–266. doi: 10.1007/s00702-012-0861-4. [DOI] [PubMed] [Google Scholar]
- Chen J, Kelz MB, Hope BT, Nakabeppu Y, Nestler EJ. Chronic Fos-related antigens: stable variants of deltaFosB induced in brain by chronic treatments. J Neurosci. 1997;17:4933–4941. doi: 10.1523/JNEUROSCI.17-13-04933.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J. Selecting a mouse model of Alzheimer's disease. Methods Mol Biol. 2011;670:169–189. doi: 10.1007/978-1-60761-744-0_13. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Scharfman HE. Shared cognitive and behavioral impairments in epilepsy and Alzheimer's disease and potential underlying mechanisms. Epilepsy Behav. 2013;26:343–351. doi: 10.1016/j.yebeh.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett BF, Leiser SC, Ling HP, Nagy R, Breysse N, Zhang X, Hazra A, Brown JT, Randall AD, Wood A, et al. Sodium channel cleavage is associated with aberrant neuronal activity and cognitive deficits in a mouse model of Alzheimer's disease. J Neurosci. 2013;33:7020–7026. doi: 10.1523/JNEUROSCI.2325-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle AL, Gajewski PA, Yang M, Kechner ME, Al Masraf BS, Kennedy PJ, Wang H, Mazei-Robison MS, Robison AJ. Experience-Dependent Induction of Hippocampal DeltaFosB Controls Learning. J Neurosci. 2015;35:13773–13783. doi: 10.1523/JNEUROSCI.2083-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espana J, Valero J, Minano-Molina AJ, Masgrau R, Martin E, Guardia-Laguarta C, Lleo A, Gimenez-Llort L, Rodriguez-Alvarez J, Saura CA. beta-Amyloid disrupts activity-dependent gene transcription required for memory through the CREB coactivator CRTC1. J Neurosci. 2010;30:9402–9410. doi: 10.1523/JNEUROSCI.2154-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust TW, Robbiati S, Huerta TS, Huerta PT. Dynamic NMDAR-mediated properties of place cells during the object place memory task. Front Behav Neurosci. 2013;7:202. doi: 10.3389/fnbeh.2013.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann A, Hvalby O, Jensen V, Strekalova T, Zacher C, Layer LE, Kvello A, Reschke M, Spanagel R, Sprengel R, et al. Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J Neurosci. 2003;23:9116–9122. doi: 10.1523/JNEUROSCI.23-27-09116.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- He J, Yamada K, Nabeshima T. A role of Fos expression in the CA3 region of the hippocampus in spatial memory formation in rats. Neuropsychopharmacology. 2002;26:259–268. doi: 10.1016/S0893-133X(01)00332-3. [DOI] [PubMed] [Google Scholar]
- Hecht F. Seizure disorders in the fragile X chromosome syndrome. Am J Med Genet. 1991;38:509. doi: 10.1002/ajmg.1320380274. [DOI] [PubMed] [Google Scholar]
- Hermann BP, Seidenberg M, Dow C, Jones J, Rutecki P, Bhattacharya A, Bell B. Cognitive prognosis in chronic temporal lobe epilepsy. Ann Neurol. 2006;60:80–87. doi: 10.1002/ana.20872. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hooker JM, Kim SW, Alexoff D, Xu Y, Shea C, Reid A, Volkow N, Fowler JS. Histone deacetylase inhibitor, MS-275, exhibits poor brain penetration: PK studies of [C]MS-275 using Positron Emission Tomography. ACS Chem Neurosci. 2010;1:65–73. doi: 10.1021/cn9000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe C, Elger CE, Helmstaedter C. Long-term memory impairment in patients with focal epilepsy. Epilepsia. 2007;48(Suppl 9):26–29. doi: 10.1111/j.1528-1167.2007.01397.x. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, et al. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–728. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- Hyde TM, Weinberger DR. Seizures and schizophrenia. Schizophr Bull. 1997;23:611–622. doi: 10.1093/schbul/23.4.611. [DOI] [PubMed] [Google Scholar]
- Ivkovic S, Kanazir S, Stojiljkovic M, Rakic L, Ruzdijic S. Desensitization of c-fos mRNA expression in rat brain following cortical lesions. Mol Cell Neurosci. 1994;5:11–22. doi: 10.1006/mcne.1994.1002. [DOI] [PubMed] [Google Scholar]
- Jian L, Nagarajan L, de Klerk N, Ravine D, Bower C, Anderson A, Williamson S, Christodoulou J, Leonard H. Predictors of seizure onset in Rett syndrome. J Pediatr. 2006;149:542–547. doi: 10.1016/j.jpeds.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Kam K, Duffy AM, Moretto J, LaFrancois JJ, Scharfman HE. Interictal spikes during sleep are an early defect in the Tg2576 mouse model of beta-amyloid neuropathology. Sci Rep. 2016;6:20119. doi: 10.1038/srep20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, Chaudhury D, Damez-Werno DM, Haggarty SJ, Han MH, et al. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat Neurosci. 2013;16:434–440. doi: 10.1038/nn.3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larner AJ. Epileptic seizures in AD patients. Neuromolecular Med. 2010;12:71–77. doi: 10.1007/s12017-009-8076-z. [DOI] [PubMed] [Google Scholar]
- Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Lott IT, Doran E, Nguyen VQ, Tournay A, Movsesyan N, Gillen DL. Down syndrome and dementia: seizures and cognitive decline. J Alzheimers Dis. 2012;29:177–185. doi: 10.3233/JAD-2012-111613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozsadi DA, Larner AJ. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer's disease. Dement Geriatr Cogn Disord. 2006;22:121–124. doi: 10.1159/000093664. [DOI] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015;161:1592–1605. doi: 10.1016/j.cell.2015.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, Nestler EJ. DeltaFosB: a molecular switch for long-term adaptation in the brain. Brain Res Mol Brain Res. 2004;132:146–154. doi: 10.1016/j.molbrainres.2004.05.014. [DOI] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, Penke B, Zilberter Y, Harkany T, Pitkanen A, et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, Self DW. DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci U S A. 2001;98:11042–11046. doi: 10.1073/pnas.191352698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu GQ, Mucke L. Vulnerability of dentate granule cells to disruption of arc expression in human amyloid precursor protein transgenic mice. J Neurosci. 2005;25:9686–9693. doi: 10.1523/JNEUROSCI.2829-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelc K, Boyd SG, Cheron G, Dan B. Epilepsy in Angelman syndrome. Seizure. 2008;17:211–217. doi: 10.1016/j.seizure.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Renthal W, Carle TL, Maze I, Covington HE, Truong HT, 3rd, Alibhai I, Kumar A, Montgomery RL, Olson EN, Nestler EJ. Delta FosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ortiz CJ, Baglietto-Vargas D, Martinez-Coria H, LaFerla FM, Kitazawa M. Upregulation of miR-181 decreases c-Fos and SIRT-1 in the hippocampus of 3xTg-AD mice. J Alzheimers Dis. 2014;42:1229–1238. doi: 10.3233/JAD-140204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA. CREB-regulated transcription coactivator 1-dependent transcription in Alzheimer's disease mice. Neurodegener Dis. 2012;10:250–252. doi: 10.1159/000333341. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Binder DK. Aquaporin-4 water channels and synaptic plasticity in the hippocampus. Neurochem Int. 2013;63:702–711. doi: 10.1016/j.neuint.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Sheng M, Greenberg ME. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron. 1990;4:477–485. doi: 10.1016/0896-6273(90)90106-p. [DOI] [PubMed] [Google Scholar]
- Simonini MV, Camargo LM, Dong E, Maloku E, Veldic M, Costa E, Guidotti A. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc Natl Acad Sci U S A. 2006;103:1587–1592. doi: 10.1073/pnas.0510341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, Chronopoulos A, Thurber S, Thompson JL, Holmes GL. Age-dependent cognitive and behavioral deficits after kainic acid seizures. Epilepsia. 1993;34:420–432. doi: 10.1111/j.1528-1157.1993.tb02582.x. [DOI] [PubMed] [Google Scholar]
- Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- Tischmeyer W, Grimm R. Activation of immediate early genes and memory formation. Cell Mol Life Sci. 1999;55:564–574. doi: 10.1007/s000180050315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulving E, Markowitsch HJ. Episodic and declarative memory: role of the hippocampus. Hippocampus. 1998;8:198–204. doi: 10.1002/(SICI)1098-1063(1998)8:3<198::AID-HIPO2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Vialou V, Robison AJ, Laplant QC, Covington HE, Dietz DM, 3rd, Ohnishi YN, Mouzon E, Rush AJ, Watts EL, 3rd, Wallace DL, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci. 2010;13:745–752. doi: 10.1038/nn.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscidi EW, Triche EW, Pescosolido MF, McLean RL, Joseph RM, Spence SJ, Morrow EM. Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS One. 2013;8:e67797. doi: 10.1371/journal.pone.0067797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, et al. Incidence and impact of subclinical epileptiform activity in Alzheimer's disease. Ann Neurol. 2016;80:858–870. doi: 10.1002/ana.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston SM, Hayward MD, Nestler EJ, Duman RS. Chronic electroconvulsive seizures down-regulate expression of the immediate-early genes c-fos and c-jun in rat cerebral cortex. J Neurochem. 1990;54:1920–1925. doi: 10.1111/j.1471-4159.1990.tb04892.x. [DOI] [PubMed] [Google Scholar]
- Yiu AP, Rashid AJ, Josselyn SA. Increasing CREB function in the CA1 region of dorsal hippocampus rescues the spatial memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2011;36:2169–2186. doi: 10.1038/npp.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.