

Abstract
Senescent cells (SnCs) accumulate in many vertebrate tissues with age and contribute to age-related pathologies1–3, presumably through their secretion of factors contributing to the senescence-associated secretory phenotype (SASP)4–6. Removal of SnCs delays several pathologies7–9 and increases healthy lifespan8. Aging and trauma are risk factors for the development of osteoarthritis (OA)10, a chronic disease characterized by degeneration of articular cartilage leading to pain and physical disability. Senescent chondrocytes are found in cartilage tissue isolated from patients undergoing joint replacement surgery11–14, yet their role in disease pathogenesis is unknown. To test the idea that SnCs might play a causative role in OA, we used the p16-3MR transgenic mouse, which harbors a p16INK4a (Cdkn2a) promoter driving the expression of a fusion protein containing synthetic Renilla luciferase and monomeric red fluorescent protein domains, as well as a truncated form of herpes simplex virus 1 thymidine kinase (HSV-TK)15,16. This mouse strain allowed us to selectively follow and remove SnCs after anterior cruciate ligament transection (ACLT). We found that SnCs accumulated in the articular cartilage and synovium after ACLT, and selective elimination of these cells attenuated the development of post-traumatic OA, reduced pain and increased cartilage development. Intra-articular injection of a senolytic molecule that selectively killed SnCs validated these results in transgenic, non-transgenic and aged mice. Selective removal of the SnCs from in vitro cultures of chondrocytes isolated from patients with OA undergoing total knee replacement decreased expression of senescent and inflammatory markers while also increasing expression of cartilage tissue extracellular matrix proteins. Collectively, these findings support the use of SnCs as a therapeutic target for treating degenerative joint disease.
To test whether SnCs contribute to OA pathogenesis, we first asked whether SnCs develop after articular joint injury in mice by following the expression of p16INK4a (also known as cyclin-dependent kinase inhibitor 2a; encoded by the Cdkn2a locus), a frequently used biomarker for SnCs8,17–19. We induced post-traumatic OA by ACLT in p16-3MR transgenic mice, in which p16INK4a-positive SnCs can be noninvasively monitored15,16 (Supplementary Fig. 1a). Luminescence in the articular joint region of the p16-3MR mice increased after ACLT surgery (Fig. 1a,b), peaking 2 weeks after injury and decreasing thereafter to a low-level steady state that remained above the background level (Supplementary Fig. 1b). The mRNA level of Cdkn2a increased and then decreased with kinetics similar to those of the luminescence (Fig. 1c and Supplementary Fig. 1c). The mRNA levels of Cdkn1a and Il6, encoding the SASP marker IL-6 (ref. 20), were also higher in vehicle-treated ACLT mice than in sham-operated controls 28 d after ACLT, further corroborating the development of SnCs in the injured joint (Supplementary Fig. 1d). OA disease and cartilage degeneration after ACLT were confirmed histologically and through pain assessment. ACLT-mediated injury reduced safranin O staining of proteoglycans and resulted in cartilage thinning, surface irregularities and induction of type II collagen degradation (Supplementary Fig. 1e,f). Behavioral assessment of OA-induced pain showed that ACLT surgery caused decreased weight bearing on the injured leg and an increased latency period required for the injured hindlimb to reach a pain threshold after the mouse was placed on a 55 °C platform (Supplementary Fig. 1g).
Figure 1.
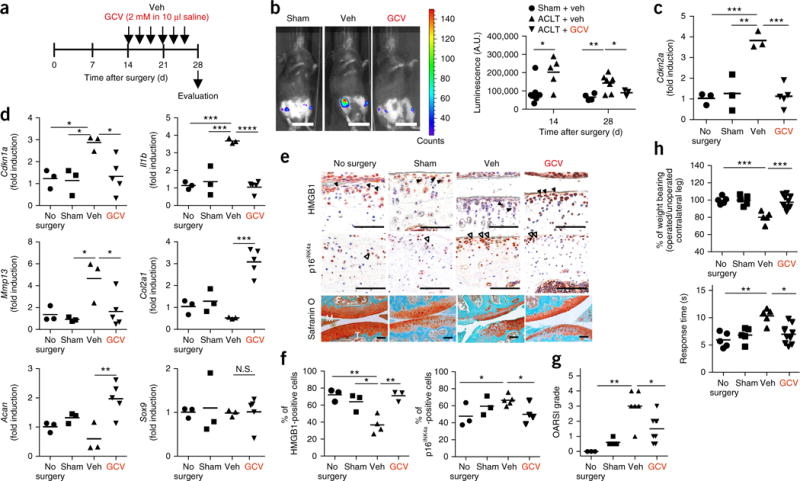
Clearance of SnCs by GCV reduces the development of post-traumatic OA. (a) Schematic of the time course for the experiments in b–h. Male p16-3MR mice undergoing ACLT were injected intra-articularly with vehicle (Veh) or ganciclovir (GCV) and evaluated as indicated. (b) Representative luminescence images (sham surgery, n = 7; Veh treated, n = 7; GCV treated, n = 3) of ACLT mice after vehicle or GCV treatment on day 28 after surgery (left) and quantification of the luminescence (right) at the indicated time s(in arbitrary units, A.U.). Scale bars, 2 cm. (c) Quantification of mRNA expression for Cdkn2a in articular joints treated on day 28 after surgery (no surgery, sham surgery and Veh treated, n = 3; GCV treated, n = 5). (d) Quantification of mRNA expression for Cdkn1a, Il1b, Mmp13, Col2a1, Acan and Sox9, normalized to Actb expression, in joints from no-surgery, sham-operated and ACLT mice that were treated as indicated (no surgery, sham surgery and Veh treated, n = 3; GCV treated, n = 5). (e) Representative images of HMGB1 (brown, closed arrowheads; no surgery, sham surgery and GCV treated, n = 3; Veh treated, n = 4) and p16INK4a (brown, open arrowheads; no surgery and sham surgery, n = 3; Veh treated, n = 5; GCV treated, n = 4) immunostaining and safranin O and methyl green (no surgery, n = 3; sham surgery, n = 5; Veh treated, n = 6; GCV treated, n = 8) in p16-3MR mice. F, femur; T, tibia; M, meniscus. Scale bars, 100 μm. (f) Quantification of non-SnCs positive for nuclear HMGB1 (no surgery and sham surgery, n = 3; Veh treated, n = 4; GCV treated, n = 3) and SnCs positive for p16INK4a (no surgery and sham surgery, n = 3; Veh treated, n = 5; GCV treated, n = 4) in articular cartilage. (g) Medial tibial plateau joint score in p16-3MR mice based on the OARSI scoring system (no surgery, n = 3; sham surgery, n = 5; Veh treated, n = 6; GCV treated, n = 8). (h) The percentage of weight placed on the operated limb versus the contralateral control (top) and the response time of mice after placement onto a 55 °C hotplate (bottom) (no surgery, sham surgery and Veh treated, n = 5; GCV treated, n = 10). All data are expressed as means, and each data point represents an individual mouse. One-way ANOVA with Tukey’s multiple-comparisons test was used for statistical analysis in c and d; a two-tailed t-test (unpaired) was used for b and f–h. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; N.S., not significant.
To localize SnCs in the articular joint, we performed immunostaining for p16INK4a protein and high-mobility-group box 1 (HMGB1)—an extracellular alarmin whose nuclear expression precedes the secretion of SASP components in cells undergoing senescence21,22. In vehicle-treated ACLT mice, p16INK4a protein was largely restricted to the superficial zone, where 66% of cells were positive for p16INK4a, and 65% of cells had little to no nuclear HMGB1 staining in articular cartilage (Fig. 1e,f). In contrast, in sham-operated controls, there were fewer cells positive for p16INK4a staining and HMGB1 was largely nuclear. We found that vehicle-treated p16-3MR ACLT mice, when compared to mice without ACLT surgery, had a greater number of p16INK4a-expressing cells in the synovium, as well as at the cartilage surface. However, there was no difference in p16INK4a-expressing SnCs in infrapatellar fat pads between the two groups (Supplementary Fig. 2a). The role of the synovium in OA has long been postulated23, and our results suggest that the synovium also contributes to the senescence burden in articular joints during OA progression. SnC localization to the superficial layer of cartilage has particular relevance for tissue regeneration, as this is where purported cartilage progenitor cells are located24. Furthermore, the superficial and deep cartilage layers interact to maintain homeostasis and increase new tissue production when needed25, further indicating a regulatory role for these superficial layer cell populations that express p16INK4a.
Next, we sought to determine whether selective removal of the SnCs that develop after ACLT would reduce or reverse the progression of OA and OA-related symptoms. To do this, we took advantage of the p16-3MR transgene, which includes HSV-TK, thereby permitting the selective killing of p16INK4a-expressing cells by ganciclovir (GCV)26. GCV treatment (Fig. 1a) reduced luminescence in the ACLT mice (Fig. 1b), indicative of the clearing of p16INK4a-positive SnCs. Clearance was confirmed by reduced injury-induced increases in the mRNA levels of Cdkn2a and Cdkn1a (Fig. 1c,d), along with increased immunostaining for nuclear HMGB1 and reduced p16INK4a protein in the cartilage (Fig. 1e,f). We also observed a reduction in p16INK4a expression in the synovium of GCV-treated p16-3MR mice (Supplementary Fig. 2a). Clearance of SnCs inhibited articular cartilage erosion such that proteoglycan staining was largely rescued and resembled that of normal tissue (Fig. 1e,g). The pain associated with ACLT injury was decreased when SnCs were removed (Fig. 1h), and the inflammatory markers matrix metalloproteinase 13 (Mmp13) and Il-1β (Il1b) (Fig. 1d), whose expression is typical of OA disease and tissue degradation27, also showed decreased mRNA levels. SnC removal increased the protein and mRNA levels of Col2a1 and Acan (corresponding to the cartilage extracellular matrix molecules type II collagen and aggrecan, respectively). The expression of Sox9, which regulates chondrogenesis in addition to having other activities, remained unchanged. However, there were pathological changes in the structure and/or appearance of the subchondral bone and the expression of bone-resorption-related genes28 was increased after ACLT surgery and OA development; these phenotypes were not rescued by GCV treatment (Supplementary Fig. 2b–d), suggesting that GCV may not reach the bone when injected intra-articularly and that systemic administration may be required16. To confirm that GCV itself did not affect the local cartilage environment but only killed SnCs in p16-3MR mice, we injected GCV into the ACLT-treated joints of nontransgenic C57BL mice, where it was found to have no influence on OA disease progression or pain (Supplementary Fig. 1). Together, these data suggest that SnCs develop after trauma in the articular cartilage and synovium of the knee joint and their removal, even after OA and symptoms develop, reduces disease impact and creates a prochondrogenic environment.
To selectively clear SnCs using a pharmacologic approach, we tested a senolytic (UBX0101) that was recently found to selectively eliminate SnCs16,29. First, we optimized the concentration of UBX0101 using p16-3MR mice and an injection regimen similar to that used to administer GCV. The development of SnCs was followed by luminescence to compare the efficacy of UBX0101 in clearing SnCs with that of GCV. An intra-articular injection with as little as 0.2 mM UBX0101 14 d after ACLT effectively cleared the SnCs induced by the surgery. The luciferase signal decreased after UBX0101 treatment (Supplementary Fig. 3a), and SnC clearance was confirmed by reduced expression of Cdkn2a, Cdkn1a, Mmp13 and Il1b (up to a 50% reduction) in UBX0101-treated ACLT mice (Supplementary Fig. 3b). The OA-related disease outcomes of pain and articular cartilage erosion were similarly reduced with UBX0101 (Supplementary Fig. 3c–e). Proteoglycan loss and the thinning and calcification of articular cartilage were attenuated in ACLT mice treated with 1 mM UBX0101. Moreover, injection of 0.2 to 5 mM UBX0101 also produced a prochondrogenic environment characterized by greater Col2a1 and Acan gene expression, indicating new cartilage growth. The expression of Sox9 and Runx2, a regulator of chondrocyte hypertrophy and bone development30, remained unchanged after treatment (Supplementary Fig. 3b). Similar to the GCV response in p16-3MR mice, abnormal subchondral bone remodeling and osteophyte formation in ACLT mice were not rescued by UBX0101 treatment (Supplementary Fig. 3f).
The half-life of UBX0101 in the articular joint was short, with concentrations falling below the half-maximal inhibitory concentration (IC50) after 1.5 h. Systemic exposure was minimal (3.3% of the intra-articular dose reached the circulation), and the IC50 was never reached (Supplementary Fig. 4). Even with this short half-life in the joint cavity, UBX0101 was effective in blocking cartilage erosion in our ACLT mouse model because UBX0101 eliminated SnCs rather than blocking their SASP secretion. Once cells have been eliminated, drug exposure is no longer necessary. Considering therapeutic translation, we next aimed to reduce the number of intra-articular injections while still achieving successful SnC removal. We thus determined the number of UBX0101 injections (one injection every 2 d) required to reduce OA progression when injections were begun 14 d after ACLT surgery (Supplementary Fig. 5). Both five and six injections of 1 mM UBX0101 resulted in lower expression of Cdkn2a, Mmp13 and Il1b, higher expression of Col2a1 and Acan, and less OA-induced pain in comparison to vehicle-treated ACLT mice.
We next confirmed the efficacy of intra-articular UBX0101 injections for treating ACLT-induced OA in C57BL mice. On the basis of the short half-life of the drug in the articular space and the dosing experiments, six intra-articular injections (1 mM, once every 2 d over 2 weeks starting on day 14 after ACLT) were performed (Fig. 2a). Similar to the effects of GCV in p16-3MR mice, UBX0101 treatment selectively killed SnCs that were induced by ACLT surgery in articular cartilage. This SnC clearance was confirmed by (i) a reduced number of cells without nuclear HMGB1 and higher numbers of non-SnCs positive for nuclear HMGB1 (Fig. 2b,c); (ii) a higher number of non-SnCs expressing, Ki-67 and proliferating cell nuclear antigen (PCNA) (Fig. 2b,c and Supplementary Fig. 6a); (iii) fewer p16INK4a- and MMP13-positive chondrocytes (Fig. 2b,c); and (iv) reduced levels of Cdkn2a, Cdkn1a, Il6 and Mmp13 mRNAs 28 d after injury (Fig. 2d). We also observed fewer Ki-67-expressing cells in the synovium of vehicle-treated C57BL mice, a finding consistent with the presence of synovial SnCs, which was abrogated by UBX0101 treatment (Supplementary Fig. 6b). This correlates with the reduction in expression of p16INK4a observed in the synovium of GCV-treated p16-3MR mice.
Figure 2.
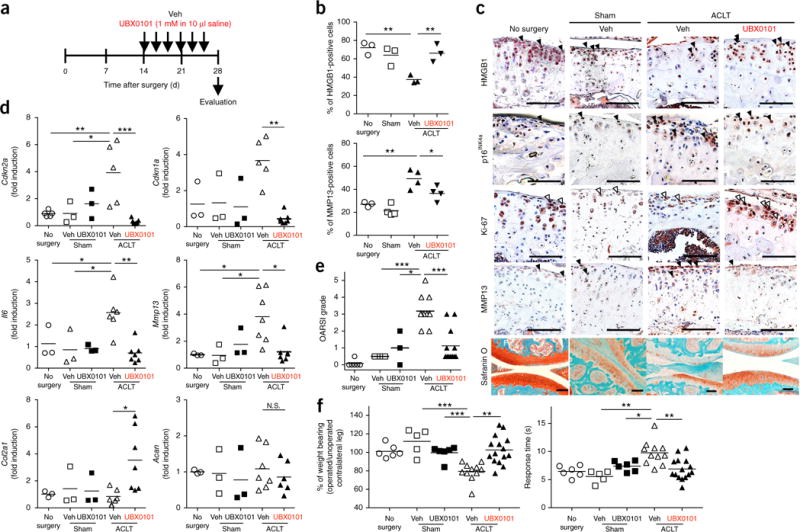
SnC clearance by UBX0101 attenuates post-traumatic OA and creates a prochondrogenic environment. (a) Schematic of the time course for the experiments in b–f. Male C57BL mice that underwent ACLT were injected intra-articularly every other day with vehicle (Veh) or UBX0101 (UBX) and evaluated as indicated. (b,c) Quantification of non-SnCs positive for nuclear HMGB1 and MMP13 immunostaining (b) and representative immunostaining images of HMGB1 (brown, closed arrowheads; n = 3 for each group), p16INK4a (brown, closed arrowheads), n = 3, Ki-67 (brown, open arrowheads; no surgery, n = 3; sham surgery, Veh treated and UBX0101 treated, n = 4) and MMP13 (brown, closed arrowheads; no surgery, n = 2; sham surgery, n = 3; Veh and UBX0101 treated, n = 4) and the medial tibial plateau stained with safranin O and methyl green in articular cartilage from no-surgery (n = 6), sham-operated (n = 5) and ACLT mice treated with vehicle (n = 11) or UBX0101 (n = 12) (c). Scale bars, 100 μm. (d) Quantification of mRNA expression for Cdkn2a (no surgery, n = 6; sham operated with vehicle or UBX0101, n = 3; ACLT with vehicle, n = 5; ACLT with UBX, n = 7), Cdkn1a (no surgery, sham operated with vehicle or UBX0101, n = 3; ACLT with vehicle, n = 6; ACLT with UBX, n = 7), Il6 (no surgery, sham operated with vehicle or UBX0101, n = 3; ACLT with vehicle, n = 5; ACLT with UBX, n = 7), Mmp13 (no surgery, sham operated with vehicle or UBX0101, n = 3; ACLT with vehicle, n = 7; ACLT with UBX, n = 7), Col2a1 (no surgery, sham operated with vehicle or UBX0101, n = 3; ACLT with vehicle, n = 6, ACLT with UBX; n = 7) and Acan (no surgery, sham operated with vehicle or UBX0101, n = 3; ACLT with vehicle, n = 7; ACLT with UBX, n = 6) normalized to Actb levels in joints. (e) OARSI scores of articular joints (no surgery, n = 6; sham operated with vehicle, n = 5; sham operated with UBX, n = 3; ACLT with vehicle, n = 11; ACLT with UBX, n = 12). (f) The percentage of weight placed on the operated limb versus the contralateral control limb (left) and the response time after placement onto a 55 °C platform (right) (no surgery, n = 6; sham operated with vehicle, n = 5; sham operated with UBX, n = 6; ACLT with vehicle, n = 11; ACLT with UBX, n = 15). All data are expressed as means, and each data point represents an individual mouse. One-way ANOVA with Tukey’s multiple-comparisons test was used for statistical analysis in d; a two-tailed t-test (unpaired) was used for b, e and f. *P < 0.05, **P < 0.01, ***P < 0.001.
The long-term durability of a single UBX0101 treatment course and its ability to attenuate articular cartilage degeneration and ameliorate OA symptoms were evaluated over 84 d (Fig. 2c–f and Supplementary Figs. 7 and 8). Reduction of proteoglycan and OA-related pain was maintained between 28 and 56 d after surgery and the gene and protein levels of the prochondrogenic factors Col2a1 and Acan remained upregulated in comparison to the untreated controls (Supplementary Figs. 7a and 8a–d). Subchondral bone alterations induced by ACLT surgery did not change over the 84-d study (Supplementary Figs. 7b–d and 8d,e). However, OA disease (cartilage degradation and subchondral bone sclerosis) and OA-induced pain returned at 84 d after ACLT or 4 weeks after the last drug injection (Supplementary Fig. 8b,c,e), suggesting that SnCs may return by week 12 after ACLT. In mouse ACLT surgery, the ligament cannot be repaired, so continued damage may occur in the joint in addition to age-related accumulation of SnCs over time. Thus, repeated treatment may be required to remove SnCs that accumulate owing to injury and aging to prevent disease development and to achieve long-term disease modification.
To test the potential of UBX0101 in treating later-stage disease, we injected the drug into mice 42 d after ACLT (Supplementary Fig. 9a). After the standard intra-articular injections over 2 weeks, UBX0101 treatment increased the number of non-SnCs with nuclear HMGB1 (Supplementary Fig. 9b), reduced OA-induced pain (Supplementary Fig. 9c) and reduced the levels of Cdkn2a, Cdkn1a and Mmp13 mRNAs (Supplementary Fig. 9d). Further, the expression of chon-drogenic genes (Col2a1 and Acan) and proteoglycan staining in articular cartilage increased even with the late UBX0101 treatment (Supplementary Fig. 9d,e). ACLT mice treated with UBX0101 had a reduction in subchondral bone sclerosis but similar osteophyte formation relative to vehicle-treated controls (Supplementary Fig. 9f). Together, these results indicate that UBX0101 can clear SnCs from articular cartilage to reduce OA symptoms and modify the disease by creating a pro-regenerative environment even in later-stage disease.
Aging in mice contributes to the development of spontaneous and injury-induced OA, similar to what occurs in humans31,32. We therefore explored the consequences of selective removal of naturally occurring age-related SnCs on arthritis development using the previously reported INK-ATTAC transgenic mouse model, which expresses the FK506 binding protein–caspase-8 fusion protein under the control of the p16INK4a promoter7,8. Starting at 12 months of age, AP20187, a molecule that induces apoptosis in p16INK4a-expressing cells8, was injected intraperitoneally into female mice twice a week until animals became moribund. In the articular joints of these mice, elimination of naturally occurring p16INK4a-positive SnCs markedly reduced age-related cartilage degeneration in comparison to vehicle treatment alone, as demonstrated by increased safranin O staining for proteoglycans, cartilage thickness and normalized Osteoarthritis Research Society International (OARSI) scores (Fig. 3a–c and Supplementary Fig. 10a).
Figure 3.
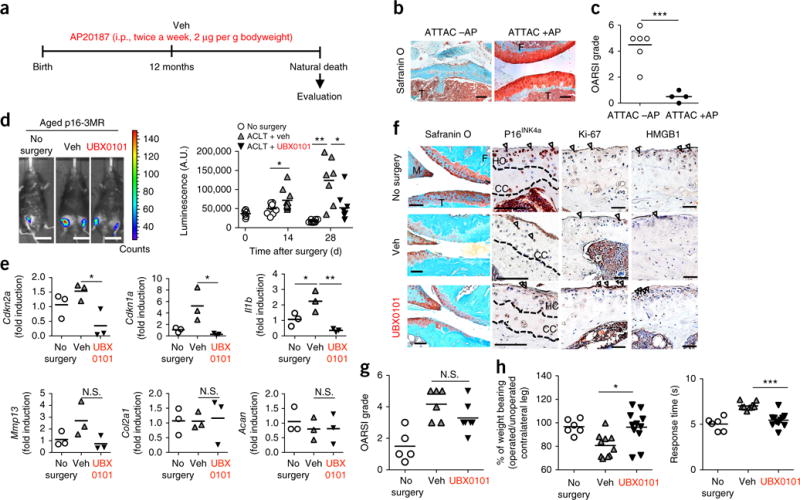
Clearance of SnCs slows the development of naturally occurring OA and post-traumatic OA in aged mice. (a) Study design for clearance of naturally occurring p16INK4a-positive SnCs in female INK-ATTAC mice. AP20187 (AP) was administrated intraperitoneally (i.p.) twice a week starting at 12 months of age. (b) Representative images of safranin O and methyl green staining from vehicle-treated (−AP; n = 6) and AP20187-treated (+AP; n = 4) mice. F, femur; T, tibia. Scale bars, 100 μm. (c) OARSI grade for vehicle-treated (n = 6 mice aged 20, 21, 25, 28, 28 and 32 months) and AP20187-treated (n = 4 mice aged 21, 28, 28 and 34 months) mice. (d) Representative whole-body luminescence images on day 28 after ACLT surgery (no surgery, n = 8; Veh treated, n = 7; UBX0101 treated, n = 8) (left) and quantification of luminescence (in arbitrary units, A.U.) at the indicated times after the surgery of male p16-3MR mice aged 19–20 months treated as indicated in Figure 2a (right). Scale bars, 2 cm (no surgery, n = 8; Veh treated, n = 7; UBX treated, n = 8). (e) Quantification of mRNA levels for Cdkn2a, Cdkn1a, Mmp13, Col2a1 and Acan normalized to Actb levels in joints 28 d after ACLT (n = 3 for all groups). (f) Representative images of safranin O and methyl green staining and immunostaining for p16INK4a (brown, arrows; no surgery, n = 4; Veh treated, n = 3; UBX0101 treated, n = 4), Ki-67 (no surgery and Veh treated, n = 3; UBX0101 treated, n = 4) and HMGB1 (brown, arrowheads; no surgery, n = 5; Veh treated and UBX0101 treated, n = 3) on articular cartilage. HC, hyaline cartilage; CC, calcified cartilage. Scale bars, 100 μm. (g) OARSI grade for no-surgery, vehicle-treated and UBX0101-treated ACLT mice (no surgery, n = 5; Veh treated, n = 6; UBX treated, n = 7). (h) The percentage of weight placed on the operated limb versus the contralateral control (left) and the response time of mice after placement onto a 55 °C platform (right) (no surgery, n = 6; Veh treated, n = 10; UBX treated, n = 11). All data are expressed as means, and each data point represents an individual mouse. One-way ANOVA with Tukey’s multiple-comparisons test was used for statistical analysis in e; a two-tailed t-test (unpaired) was used for c, d, g and h. *P < 0.05, **P < 0.01, ***P < 0.001.
The synergistic impact of age and trauma may further exacerbate disease. To address this possibility, we performed ACLT surgery on p16-3MR mice aged 19 months and evaluated the effect of selective SnC removal on OA disease. In contrast with young animals, luminescence in the articular joint region of vehicle-treated aged p16-3MR mice increased continually over 28 d after ACLT surgery (Fig. 3d). This SnC development in ACLT mice was corroborated by higher mRNA levels of Cdkn2a, Cdkn1a and the SASP factor Il1b and fewer of Ki-67-positive non-SnCs as compared to controls that did not undergo surgery (Fig. 3e and Supplementary Fig. 10b). Moreover, the distribution of the SnCs in aged mice after ACLT was different from that in young mice after ACLT. In particular, 40% of p16INK4a-positive SnCs were localized throughout the articular cartilage of vehicle-treated ACLT mice instead of only in the superficial layer (Fig. 3f and Supplementary Fig. 10b).
More severe OA developed after ACLT surgery in aged than in young p16-3MR mice, as demonstrated by histology and OARSI scoring of vehicle-treated animals (Fig. 3f,g). The baseline morphology of the aged animals without surgery showed thinning cartilage with reduced safranin O staining for proteoglycans and higher OARSI scores. Removal of SnCs from aged p16-3MR animals using the standard regimen of UBX0101 (Fig. 2a) reduced OA-induced pain, similar to what was observed in young mice (Fig. 3h). Furthermore, expression of Cdkn2a, Cdkn1a, Il6 and Mmp13 was reduced with SnC clearance. Moreover, Cdkn1a and Mmp13 gene expression levels in ACLT mice treated with UBX0101 were lower than those in animals that did not receive any surgery, suggesting that there were higher baseline levels of SnCs in the aged mice (Fig. 3e). However, in contrast with the young animals, there was not higher expression of prochondrogenic genes (Col2a1 and Acan) in the older animals after treatment at 28 d (Fig. 3e). Histologically, there was a small increase in safranin O staining of the articular cartilage in ACLT mice treated with UBX0101, but the OARSI scores were not significantly lower than those of the vehicle-treated ACLT mice, given the small number of animals in this study (Fig. 3f,g). ACLT- and age-induced changes in bone did not significantly change with UBX0101 treatment (Supplementary Fig. 10c,d). However, SnCs found in the synovium were cleared with UBX0101 treatment, and those in the subchondral bone marrow were slightly reduced in number (Supplementary Fig. 10e). These findings suggest that UBX0101 can clear SnCs from aged articular cartilage, but the age-related decline in the proliferative and synthetic capacities of articular chondrocytes may reduce subsequent tissue regeneration33. However, based on the drastic articular joint improvement, in terms of OARSI scores, in the older animals with naturally occurring OA, it is likely that additional treatments, and potentially systemic treatment, might be required to reduce disease and rebuild cartilage tissue.
The relevance of SnCs and their clearance in clinical OA was evaluated in explant and in vitro cultures from patients undergoing total knee arthroplasty. To validate the presence of SnCs in human disease tissues, we performed senescence-associated β-galactosidase (SA-β-gal) staining and HMGB1 immunohistochemistry on arthritic and healthy human cartilage explants. SA-β-gal-positive SnCs were observed throughout the depth of osteoarthritic cartilage, and there was little to no difference in nuclear HMGB1 staining in OA tissues as compared to tissues from healthy controls (Supplementary Fig. 11). However, staining was variable in cartilage tissue isolated from different regions of the joint within an individual, suggesting variability across the articular surface. To avoid bias from this variability in SnCs across the articular joint, we isolated cells from the tissue for further testing.
Exposure to the senolytic UBX0101 effectively cleared SnCs isolated from human OA cartilage grown in culture as a monolayer. Preliminary screening showed that treatment with 43 μM UBX0101 killed senescent chondrocytes isolated from human OA tissue, as determined by the SA-β-gal assay (Supplementary Fig. 12). To further understand the effects of UBX0101 on OA, human OA chondrocytes were exposed to UBX0101 (43 μM) for 2 d (Fig. 4a); this regimen killed 20% of the SnCs and no more than 5% of non-SnCs, as determined by staining for SA-β-gal activity and nuclear HMGB1, Annexin V–propidium iodide (PI) live–dead cell staining, and the protein levels of p21 (Fig. 4b,c and Supplementary Fig. 12d). Increased levels of activated caspase-3 suggest that UBX0101 kills SnCs through the apoptotic pathway in treated human OA chondrocytes (but not healthy chondrocytes; Fig. 4d and Supplementary Fig. 12e). After human OA chondrocytes were exposed to UBX0101 for 2 d followed by drug-free culture for 8 d, the cultures showed a decline in expression of the senescence-related genes CDKN2A (without statistical significance), MMP3 and IL6. Expression levels of the OA-related genes MMP13 and IL1B were also decreased (Fig. 4e). Furthermore, selective removal of SnCs from the OA chondrocyte cultures by UBX0101 increased the proliferation rate of the remaining chondrocytes by up to 15%, as determined by labeling of individual cells with 5-ethynyl-20-deoxyuridine (EdU) (Fig. 4f). These data suggest that transient UBX0101 treatment may cause a reversible cessation of cell growth (quiescence) in normal cells, which resume proliferation after removal of UBX0101.
Figure 4.
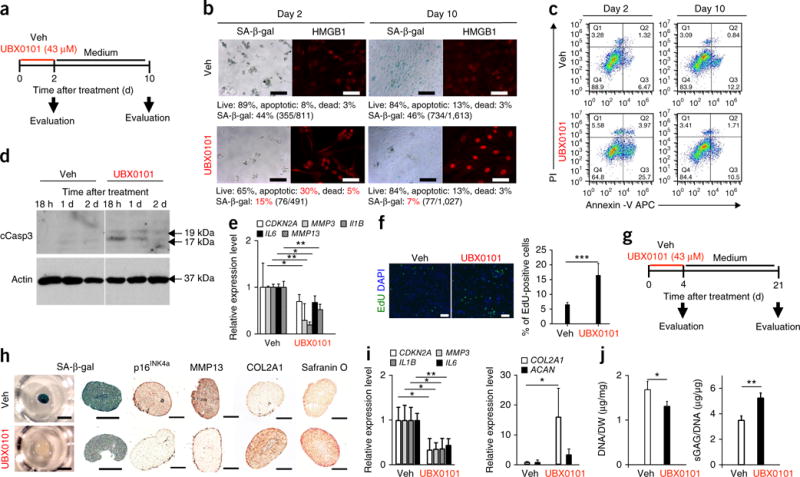
UBX0101 clears SnCs by inducing apoptosis and improves the cartilage-forming ability of chondrocytes from human OA tissue. (a) Schematic of the monolayer experiments in b–e. (b) Quantification of SA-β-gal-positive cells and the percentage of live (gate Q2, PI−Annexin V−), apoptotic (gate Q3, PI−Annexin V+ and gate Q4, PI+Annexin V+), and dead (gate Q1, PI+Annexin V−) SnCs in monolayer-cultured human OA chondrocytes treated with vehicle or 43 μM UBX0101. Representative images are shown of cells with nuclear HMGB1 (red) visualized by immunostaining (n = 10 images per group). Scale bars, 100 μm. (c) Representative flow cytometric plots measuring apoptosis from three independent experiments. (d) Immunoblot analysis of cleaved caspase-3 (cCasp3) and actin in human OA chondrocytes at 18 h, 1 d and 2 d after incubation with vehicle or 43 μM UBX0101. The experiment was performed two independent times. See Supplementary Figure 13 for uncropped immunoblots. (e) Quantification of mRNA levels for CDKN2A, MMP3, IL1B, IL6 and MMP13 normalized to ACTB levels; n = 3 for each group. (f) Representative EdU staining (left; green) and quantification of the percentage of EdU-positive cells remaining (right) in human OA chondrocyte cultures after removing SnCs using UBX0101; n = 5 for each group. Scale bars, 100 μm. (g) Schematic of the 3D pellet culture experiments in h–j. (h) SA-β-gal staining (left; scale bars, 1 mm) and sectioned images immunostained for p16INK4a, MMP13 and COL2A1 and stained for safranin O (right; scale bars, 100 μm) in 3D pellet cultures of human OA chondrocytes treated with vehicle or 43 μM UBX0101; n = 3 for each group. (i) Quantification of mRNA levels for CDKN2A, MMP3, IL1B, IL6, COL2A1 and ACAN normalized to ACTB levels; n = 3 for each group. (j) DNA content normalized to dry weight (DW) and sGAGs normalized to DNA content; n = 3 for each group. In e, f, i and j, data are shown as averages ± s.d. Statistical analysis was performed using two-tailed t-tests (unpaired). *P < 0.05, **P < 0.01, ***P < 0.001.
We next evaluated UBX0101 treatment of human OA chondrocytes in a 3D pellet culture system that supports maintenance of the chondrocyte phenotype and chondrogenesis. After 21 d of growth in culture, human OA chondrocyte pellet cultures that were then transiently exposed to 43 μM UBX0101 for 4 d had dramatically reduced levels of SnCs (Fig. 4g), as determined by reduced SA-β-gal activity (Fig. 4h). UBX0101 also reduced CDKN2A, MMP3, IL1B and IL6 mRNA levels and increased COL2A1 and ACAN mRNA levels (Fig. 4i). Similarly, UBX0101 decreased p16INK4a and MMP13 protein levels and increased the levels of COL2A1, proteoglycan and sulfated glycosaminoglycans (sGAGs), confirming new cartilage growth (Fig. 4i,j).
In summary, we report the development of SnCs after articular joint injury in the superficial layer of cartilage and the synovium in mice. These cells express a SASP that includes inflammatory molecules and degradative enzymes. The removal of SnCs, through transgenic mouse models or pharmaceutical intervention, reduced the development of post-traumatic OA and related pain and created a prochondrogenic environment. Aged mice had a higher senescence burden and developed more severe OA after injury. The relevance of our findings to human disease was validated using chondrocytes isolated from arthritic patients. These findings provide new insights into therapies targeting SnCs for the treatment of trauma and age-related degenerative joint disease.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
Mice generation and drug treatment
We purchased young (aged 10 weeks) male C57BL mice from Charles River (Germantown, MD, USA). p16-3MR transgenic mice (a C57BL/6J strain) were generated as previously described15 and were bred in the animal facility of the Johns Hopkins University School of Medicine. All studies described using INK-ATTAC mice (a C57BL/6J strain) were performed and the mice were generated as previously described8. The experimental protocol was approved by and performed in accordance with protocols from the Institutional Animal Care and Use Committee at Johns Hopkins University and the Mayo Clinic Institutional Animal Care and Use Committee. All mice were housed under pathogen-free conditions with five or fewer mice per cage. Mice had free access to food and water. The mice used for all experiments were randomly assigned to control or treatment groups and to those used in OA evaluation. For drug treatments, p16-3MR and normal C57BL mice were injected intra-articularly with 1 or 2 mM GCV (Sigma-Aldrich, G2036) in 10 μl of saline or 0.2, 1 or 5 mM UBX0101 in 10 μl of saline. INK-ATTAC mice were intraperitoneally administrated 2.0 μg of AP20187 (B/B homodimerizer; Clontech) per gram bodyweight or vehicle from 12 months until the end of life.
Cell isolation and culture
Human articular cartilage samples explanted from patients with OA undergoing total knee arthroplasty were received from the National Disease Resource Institution (Philadelphia, PA, USA) according to an IRB-approved protocol. Patients in the registry in NDRI (National Disease Research Interchange) gave informed consent for the donation of organs and tissues for research and education. Cartilage tissue was cut into 1 mm3 pieces, washed three times with PBS supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin (Pen/Strep; Invitrogen, 15140-122) and digested on an orbital shaker for 16 h at 37 °C with 0.17% (w/v) type II collagenase (Worthington Biochemical, 4176) in high-glucose DMEM (Gibco, 11965-092) supplemented with 10% FBS (HyClone, SH30070.03). After digestion, the filtrate was passed through a 70-μm strainer, and cells were rinsed three times with DMEM supplemented with 1% Pen/Strep and 10% FBS. For 3D pellet culture of human OA chondrocytes34, 400,000 cells were seeded in 96-well MicroWell round-bottom plates (Thermo Fisher Scientific). Pellets were formed in the bottom by centrifugation at 150g for 10 min. The human OA chondrocyte pellets were maintained at 37 °C with 5% CO2 in 200 μl of chondrocyte growth medium consisting of high-glucose DMEM, 1% Pen/Strep, 10% FBS, 1% nonessential amino acids (NEAA) (Gibco, 11140-050), 1 M HEPES (Gibco, 15630-080), 100 mM sodium pyruvate (Gibco, 11360-070), 0.2 M l-proline (Sigma, P5607) and 25 mg/ml ascorbic acid (Sigma, A5960). Pellets were harvested after 21 d for evaluation. Media were changed three times per week until the end of the experiment.
Surgically induced OA mouse model
Anterior cruciate ligament transection (ACLT) and sham surgery were performed on male p16-3MR mice aged 10 weeks or 19 months and male C57BL mice aged 10 weeks. Animals were placed under general anesthesia with 3% isoflurane, and hindlimbs were shaved and prepared for aseptic surgery. For the sham operation, the knee joint was exposed following a medial capsular incision, which cut the patellar tendon, and the surgical skin incision was closed with sutures. For the ACLT surgery, after opening the joint capsule, the ACL was transected with microscissors under a surgical microscope. After irrigation with saline to remove tissue debris, the skin incision was closed. All procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine. On week 4, 8 or 12, the mice were euthanized and the joint was collected for mRNA quantification and histological assessment of the medial tibial plateau joint through blinded graded observations by two observers who followed the Osteoarthritis Research Society International (OARSI) scoring system35.
Bioluminescence
For in vivo luminescence imaging, male p16-3MR mice aged 10 weeks or 19 months were injected intra-articularly with 10 μl (150 μg/ml) of Xenolight RediJect Coelenterazine h (Calipers). Twenty-five minutes later, the mice were anesthetized with isoflurane and luminescence was measured with a PerkinElmer IVIS SpectrumCT in vivo imaging system (Caliper Life Sciences; 5 min, medium binning).
Real-time RT–PCR
To extract RNA, the hindlimbs that underwent surgery and no-surgery controls were dissected and immediately transferred to liquid nitrogen and were then pulverized with a mortar and pestle. Total RNA was extracted from knee joints using TRIzol and was reverse transcribed to cDNA using SuperScript II reverse transcriptase following the manufacturer’s protocol (Invitrogen). Real-time PCR for Cdkn2a, Cdkn1a, Il1b, Mmp3, Mmp13, Col2a1, Acan, Sox9, Runx2, Tnfrsf11b (Opg), Bglap, Rankl and Ctsk was performed using the StepOnePlus Real-Time PCR System with SYBR Green PCR Master Mix (Applied Biosystems by Life Technologies, 4367659). All signals were normalized to that for Actb. Relative gene expression was calculated by the ∆∆CT method, in which ∆CT was calculated using the Actb reference gene. ∆∆CT was calculated relative to the unoperated control group in the in vivo studies and relative to control samples in the in vitro studies. See Supplementary Table 1 for a list of primers.
Hotplate analysis
Mice were placed on the hotplate analgesia meter at 55 °C (Columbus Instruments). The latency period for a hindlimb response (for example, shaking, jumping or licking) was recorded as the response time before and at different time points after surgery36. At least three measurements were taken per mouse. The observer was blinded to the genotype and treatment of the mice.
Weight bearing
Static incapacitance measurements were performed using the Incapacitance Tester (Columbus Instruments). Mice were first acclimated to the chamber at least three times before measurement. After acclimatization, mice were maneuvered inside the chamber to stand with one paw on each scale. The weight placed on each hindlimb was measured over a 3-s interval for at least three separate measurements. Results are expressed as a percentage of the weight placed on the operated limb versus the weight placed on the contralateral control limb. The observer was blinded to the genotype and treatment of the mice.
Pharmacokinetic half-life determination
Local and blood pharmacokinetics analyses were performed after a single intra-articular administration of 1 mM UBX0101 in saline to determine the half-life, and measurements were generated by Seventh Wave laboratories (MO, USA).
Biochemical analysis of sGAG content
Pellets (n = 3 per group) were lyophilized for 2 d and then digested overnight in 125 mg/ml papainase (Worthington, 3126) for 16 h at 60 °C. The sGAG content was determined by 1,9-dimethylmethylene blue (DMMB) dye assay, which measured absorbance at 525 nm and used chondroitin sulfate as a standard. DNA content was determined by Hoechst Dye 33342 DNA assay, using calf thymus DNA as a standard. sGAG content was expressed as micrograms of sGAG per microgram of DNA.
Histology
Pellets and mouse joints were fixed in 4% paraformaldehyde overnight, dehydrated in increasing concentrations of ethanol and embedded in paraffin. Sections (5 μm) were cut from the paraffin blocks and applied to glass slides. The sections were stained for proteoglycans with aqueous safranin O (0.1%) for 5 min, and the specimens were then mounted.
Quantification of subchondral bone damage
Osteophyte thickness (a distance on the marginal zone of the medial tibial plateau starting at an original edge of the tibial articular cartilage) was measured with AxioVision SE64 software. Medial tibial bone sclerosis (the extent of subchondral bone sclerosis and reduction in bone marrow area) was scored by measuring the subchondral trabecular bone to marrow ratio. On each safranin O–stained joint section, the decrease or increase in trabecular bone area was assigned a score from −5 to 5, where 0 represents no increase or decrease, −5 represents severe bone loss and 5 represents severe bone sclerosis.
Immunohistochemistry
For immunohistochemical staining, the endogenous peroxidase activity of the sections was quenched using 2.5% (v/v) hydrogen peroxide in methanol and sections were then incubated at 3 °C with 0.25% (w/v) hyaluronidase for 1 h. The AEC Broad-Spectrum Histostain-SP Kit (Invitrogen, 959944) was used following the manufacturer’s instructions. Primary antibodies for p16INK4a (1:500; Abcam, ab54210), MMP-13 (1:200; Abcam, ab39012), type I collagen (1:100; Abcam, ab34710), type II collagen (1:300; Abcam, ab34712) and type X collagen (1:50; Abcam, ab58632), osteocalcin (1:100; Millipore, AB10911), HMGB1 (1:500; Abcam, ab18256), Ki-67 (1:100; Novusbio, NB110-89717) and PCNA (1:100; Thermo Scientific, MA511358) were used and diluted in 4% BSA dissolved in PBST 2.5% Triton X-100 in PBS.
Immunofluorescence
Monolayer cultures of human OA chondrocytes were fixed in 4% paraformaldehyde, permeabilized with PBS containing 0.25% Triton X-100 for 10 min and then blocked with 4% BSA containing 0.25% Triton X-100 for 30 min at room temperature. The cells were incubated with primary antibodies against HMGB1 (1:100; Abcam) overnight at 4 °C. The cells were rinsed three times with PBS and incubated with goat anti-rabbit IgG (H&L) conjugated with Alexa Fluor 594 (1:1,000; Life Technologies, A-11012). After washing, the nuclei were counterstained with DAPI for 5 min. Nuclear HMGB1 was imaged by fluorescence microscopy (Carl Zeiss, USA). Cell proliferation was detected using Click-iT EdU Alexa Fluor 488 imaging kits (Promega, C10337). Total cells were counted following nuclear DAPI counterstaining in ten random fields per culture dish to determine the percentage of EdU-positive cells.
SA-β-galactosidase staining
SA-β-gal staining was done using a kit (Biovision, K320-250) according to the manufacturer’s instructions. SnCs were identified as blue-stained cells under light microscopy. Total cells were counted using a nuclear DAPI counterstain in ten random fields per culture dish to determine the percentage of SA-ß-gal-positive cells.
Flow cytometry analysis for apoptosis detection
For Annexin V/PI analysis, live trypsinized and floating cells were collected, centrifuged and rinsed with PBS. Cells were stained with the Annexin V apoptosis detection kit APC according to the manufacturer’s instructions (eBioscience, 88-8007). Data were analyzed with the BD Accuri C6 flow cytometer and FlowJo X 10.0 software (Becton Dickinson).
Immunoblotting
To assay the cleaved caspase-3 and p16INK4a proteins, chondrocytes were harvested, washed twice with ice-cold PBS and lysed with RIPA buffer (Thermo Scientific, 89900) containing protease (Sigma, P8340) and phosphatase (Sigma, P5726) inhibitor mixture according to the manufacturer’s instructions. Total protein concentration was determined using the BCA assay (Pierce, 23227). Proteins were separated by 10% SDS–PAGE and then transferred onto nitrocellulose membranes. The membranes were blocked for 1 h with 5% BSA in TBST and incubated overnight at 4 °C with primary antibodies specific for cleaved caspase-3 (1:1,000; Cell Signaling, 9661), p21 (1:1,000; Santa Cruz Biotechnology, sc-417), ß-actin (1:200; Santa Cruz Biotechnology, sc-47778) and GAPDH (1:1,000; Santa Cruz Biotechnology, sc-1694). After washing away unbound primary antibodies, the membranes were incubated with horseradish peroxidase–conjugated anti-rabbit secondary antibodies (1:5,000; Bio-Rad, 1706515) and anti-mouse secondary antibodies (1:5,000; Cell Signaling, 7076) and protein bands were detected using an ECL detection system with X-ray film (Fujifilm).
Statistics
The data displayed normal variance. No statistical method was used to predetermine sample size. Pilot experiments were performed with a small number of animals (Supplementary Fig. 1) to determine average and s.d. in the model and characterization methods. From this information, a power analysis was performed to determine adequate animal numbers to achieve statistical significance. The experiments were not randomized, except for the in vivo animal studies with mice, as described above. No samples or animals were excluded from the analysis except mice who died early as a result of fighting (we performed 12 in vivo experiments, and 1 or 2 animals were excluded per experiment, each of which usually entailed 40–60 animals; so, at most, 1 animal out of 40 animals was excluded from analysis). All in vivo data are expressed as the mean and each data point represents an individual mouse. All in vitro data are expressed as averages ± s.d. Statistical significance (P values) was determined by an unpaired, two-tailed Student’s t-test with Welch’s correction (we do not assume an equal s.d. in each group) or one-way ANOVA (Tukey’s multiple-comparisons test). All analyses were performed with Prism 6 software from GraphPad (San Diego, CA, USA). Statistical significance was determined to be P < 0.05.
Data availability
The data from this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank J. Xu (F.M. Kirby Research Center at Johns Hopkins University) for in vivo luminescence imaging, A. Bendele (Bolder Biopath, Inc.) for the subchondral bone damage analysis and Y. Oh (Johns Hopkins University) for immunoblotting. This work was supported by Unity Biotechnology, Inc. (J.H.E., A.P.V., Y.P., N.D.), the Bloomberg-Kimmel Institute for Cancer Immunotherapy (J.H.E.), the Morton Goldberg Professorship (J.H.E.), National Institute on Aging (NIA) grant AG017242 (J.C.), AG009909 (M.D.), National Cancer Institute (NCI) grant R01CA96985 (J.M.v.D.), a grant from the Paul F. Glenn Foundation (J.M.v.D. and D.J.B.) and a Fulbright scholarship from the Institute of International Education (O.H.J.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
O.H.J. and C.K. designed, carried out and analyzed data from most of the experiments and wrote the manuscript with input from all co-authors; S.R. performed experiments; R.-M.L. and M.D. designed experiments and interpreted data; A.P.V. designed and analyzed data from experiments; J.C. provided mice, designed experiments, analyzed and interpreted data, and revised the manuscript; J.W.C. and D.H.K. performed experiments; Y.P. and N.D. conceived the application of senescence removal to OA treatment and participated in in vivo experimental design; D.J.B. and J.M.v.D. carried out experiments on naturally occurring OA; J.H.E. conceived, designed and supervised the study, analyzed and interpreted data, and wrote the manuscript. All authors discussed the results and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS J.C., R.-M.L., Y.P., D.J.B., J.M.v.D., M.D., N.D. and J.H.E. own equity in Unity Biotechnology. Johns Hopkins University and Unity Biotechnology own intellectual property related to the research. O.H.J., C.K. and J.H.E. are inventors of Johns Hopkins University intellectual property licensed to Unity Biotechnology.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
EDITORIAL SUMMARY
AOP: Pharmacological or genetic depletion of senescent cells in the joint ameliorates osteoarthritis in mice.
References
- 1.Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Coppé JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campisi J. Cancer, aging and cellular senescence. In Vivo. 2000;14:183–188. [PubMed] [Google Scholar]
- 6.Nelson G, et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker DJ, et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker DJ, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wieland HA, Michaelis M, Kirschbaum BJ, Rudolphi KA. Osteoarthritis—an untreatable disease? Nat Rev Drug Discov. 2005;4:331–344. doi: 10.1038/nrd1693. [DOI] [PubMed] [Google Scholar]
- 11.Martin JA, Brown T, Heiner A, Buckwalter JA. Post-traumatic osteoarthritis: the role of accelerated chondrocyte senescence. Biorheology. 2004;41:479–491. [PubMed] [Google Scholar]
- 12.Price JS, et al. The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002;1:57–65. doi: 10.1046/j.1474-9728.2002.00008.x. [DOI] [PubMed] [Google Scholar]
- 13.Philipot D, et al. p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation–associated matrix remodeling in osteoarthritis. Arthritis Res Ther. 2014;16:R58. doi: 10.1186/ar4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCulloch K, Litherland GJ, Rai TS. Cellular senescence in osteoarthritis pathology. Aging Cell. 2017;16:210–218. doi: 10.1111/acel.12562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demaria M, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014;31:722–733. doi: 10.1016/j.devcel.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. doi: 10.1016/j.molcel.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 18.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- 19.Ohtani N, Yamakoshi K, Takahashi A, Hara E. The p16INK4a–RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest. 2004;51:146–153. doi: 10.2152/jmi.51.146. [DOI] [PubMed] [Google Scholar]
- 20.Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi: 10.1101/gad.235184.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holmlund U, et al. The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology. 2007;122:430–437. doi: 10.1111/j.1365-2567.2007.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davalos AR, et al. p53-dependent release of alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013;201:613–629. doi: 10.1083/jcb.201206006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6:625–635. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- 24.Dowthwaite GP, et al. The surface of articular cartilage contains a progenitor cell population. J Cell Sci. 2004;117:889–897. doi: 10.1242/jcs.00912. [DOI] [PubMed] [Google Scholar]
- 25.Sharma B, et al. Human cartilage repair with a photoreactive adhesive–hydrogel composite. Sci Transl Med. 2013;5:167ra6. doi: 10.1126/scitranslmed.3004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laberge RM, et al. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis. 2013;4:e727. doi: 10.1038/cddis.2013.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldring MB, Otero M. Inflammation in osteoarthritis. Curr Opin Rheumatol. 2011;23:471–478. doi: 10.1097/BOR.0b013e328349c2b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burr DB, Gallant MA. Bone remodelling in osteoarthritis. Nat Rev Rheumatol. 2012;8:665–673. doi: 10.1038/nrrheum.2012.130. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Y, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016;15:428–435. doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Komori T. Signaling networks in RUNX2-dependent bone development. J Cell Biochem. 2011;112:750–755. doi: 10.1002/jcb.22994. [DOI] [PubMed] [Google Scholar]
- 31.Ruan MZ, et al. Proteoglycan 4 expression protects against the development of osteoarthritis. Sci Transl Med. 2013;5:176ra34. doi: 10.1126/scitranslmed.3005409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loeser RF, et al. Microarray analysis reveals age-related differences in gene expression during the development of osteoarthritis in mice. Arthritis Rheum. 2012;64:705–717. doi: 10.1002/art.33388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loeser RF. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthritis Cartilage. 2009;17:971–979. doi: 10.1016/j.joca.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekiya I, Vuoristo JT, Larson BL, Prockop DJ. In vitro cartilage formation by human adult stem cells from bone marrow stroma defines the sequence of cellular and molecular events during chondrogenesis. Proc Natl Acad Sci USA. 2002;99:4397–4402. doi: 10.1073/pnas.052716199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative—recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18(Suppl 3):S17–S23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber S, Backer MM, Yanai J, Pick CG. The antinociceptive effect of fluvoxamine. Eur Neuropsychopharmacol. 1996;6:281–284. doi: 10.1016/s0924-977x(96)00031-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data from this study are available from the corresponding author upon reasonable request.