

Abstract
Tumors frequently escape from immune surveillance by hijacking the natural control mechanisms that regulate normal immune responses. The programmed death-1 receptor (PD‑1) on T cells normally helps limit excessive immune activation, but it can also suppress beneficial antitumor immunity. In the clinic, blocking either PD‑1 or one of its principal counterligands, programmed death–ligand 1 (PD‑L1), can lead to dramatic responses in certain patients. Because PD‑L1 can be expressed by both the tumor cells themselves and also the host cells, including host immune cells, the actual mechanistic target of therapy has remained unclear. In the current issue of the JCI, two papers, one by Tang and colleagues and the other by Lin and colleagues, used a variety of mouse tumor models to demonstrate that the relevant target for therapy in each case was the PD‑L1 molecules expressed by host cells and not by tumor cells. If this finding is generalized to humans, then it would suggest that the tumor persuades the host to actively suppress its own attempted immune response against the tumor cells.
Success in the clinic, but against what target?
The programmed death-1 receptor (PD‑1), with its counterligands programmed death–ligand 1 (PD‑L1) and PD‑L2, forms one of the regulatory pathways used by the immune system to keep dangerous immune responses in check (1). The following discussion will focus mostly on PD‑L1, since expression of PD‑L2 is more restricted and therapeutic antibodies targeting PD-L2 are less well developed. Many tumor-infiltrating T cells upregulate PD‑1 in tumors, and sometimes the tumors themselves overexpress PD‑L1. This led to the initial hypothesis that certain tumors might upregulate PD‑L1 expression to shield themselves from attack by T cells (2). Clinical blocking antibodies against both PD‑1 and PD‑L1 have been developed for cancer immunotherapy and are approved for several indications. In a selected subset of patients, these so-called checkpoint blockade antibodies against the PD‑1 pathway can be strikingly successful (3). Ironically, however, the field is still not certain about the actual mechanism of action of these antibodies.
The PD‑1 receptor on T cells modulates the antigen-recognition signal delivered by the T cell receptor (TCR), reducing signal strength and limiting downstream pathways, such as protein kinases Akt and mTOR (1, 4). Recent reports suggest that a major effect of PD-1 may also be inhibiting the important costimulatory signal delivered via CD28 (5). PD‑1 is transiently expressed during normal T cell activation, but is usually downregulated once the activation is successful (1). However, if T cells are chronically exposed to high levels of antigen that they cannot clear, they may enter a state of constitutive PD-1 expression and become exhausted and unresponsive (6). This is thought to prevent excessive and prolonged inflammation during chronic infection.
Many tumor-infiltrating T cells show evidence of this exhausted state (6). However, exhaustion can be fully, or at least partially, reversible if the PD‑1 pathway is blocked and the T cells are restimulated (7, 8). This possibility of reinvigoration is a key point because the T cells that express PD‑1 may actually be the most desirable effector cells, i.e., they become exhausted precisely because they recognize tumor antigens (9). Thus, robust reactivation of exhausted T cells may be an important goal for clinical anti–PD‑1/PD‑L1 blockade.
To achieve this goal, however, it is important to understand how these blocking antibodies work mechanistically. Historically (2), it was first assumed that the relevant site of PD‑L1 expression was the tumor cells themselves (Figure 1A). Seemingly consistent with this model, early clinical trials suggested a higher rate of response in patients whose tumor expressed PD‑L1 prior to treatment (10). However, since then it has been shown that PD‑L1 expression can be induced in response to inflammatory cytokines such as IFN-γ from activated T cells (11). Thus, expression of PD‑L1 by tumors might simply represent a proxy marker for the level of spontaneous inflammation and T cell activation that existed prior to therapy (12). Subsequent analysis showed that patients with many activated T cells and high IFN-γ at baseline were indeed more likely to respond to PD‑1 pathway blockade (12–14). Further, more detailed immunohistochemical analysis hinted that the pattern of PD‑L1 expression that most closely predicted patient outcome was not on tumor cells, but on host stromal cells, in particular, PD‑L1 expression on DCs and macrophages (13). Thus, there has been ongoing uncertainty over whether clinical PD‑1 pathway blockade actually targeted suppression created by PD‑L1 on tumor cells or on some population of host cells.
Figure 1. Possible locations for the immunosuppressive PD‑L1 targeted by checkpoint blockade.
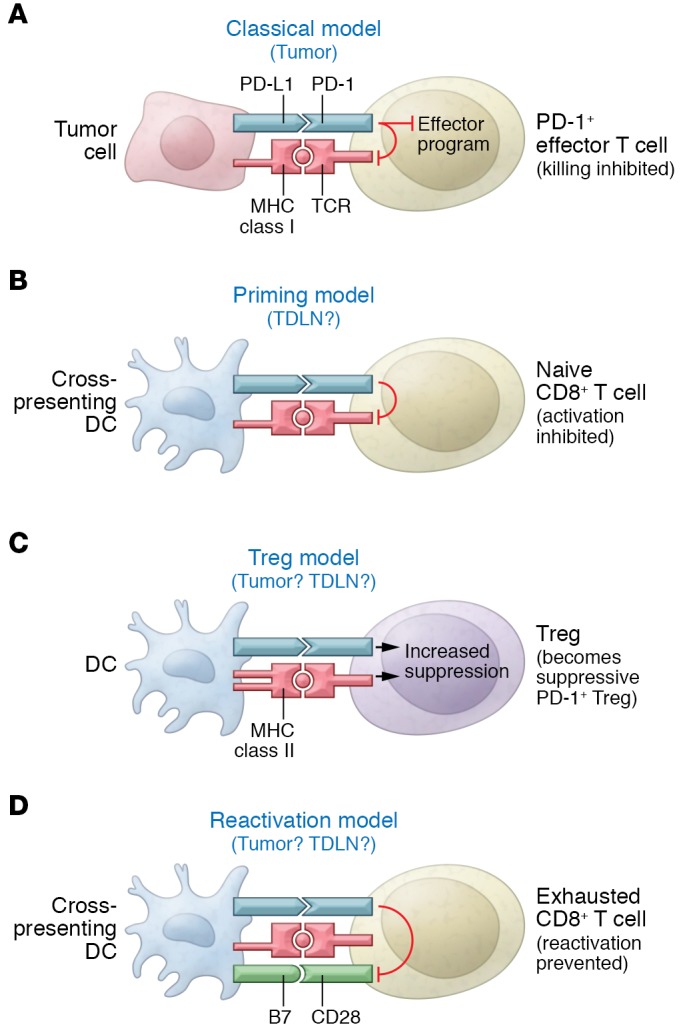
Four different hypothetical models are presented, along with the sites in which PD‑L1 might be active, either in tumor or TDLN. (A) The traditional model, in which PD‑L1 is expressed on the tumor cell itself and directly inhibits killing of the target cell by activated PD‑1+ effector T cells. (B) Model in which inhibitory PD‑L1 is expressed on DCs during the initial priming of naive tumor-specific T cells. (C) Indirect model in which PD‑L1 delivers an activating signal to Tregs via PD‑1 and the activated Tregs then mediate immune suppression. (D) Model in which DCs in tumor or TDLNs constantly interact with mature, exhausted effector T cells and PD‑L1 serves to inhibit reactivation driven by B7-CD28 costimulation. In the figure, PD‑L1 is depicted as being expressed on the same DC that could reactivate the exhausted T cell, but this interaction might also occur in trans (PD‑L1 might be expressed on a neighboring macrophage or other APC).
Importance of host-derived PD‑L1
In this issue, Tang and colleagues (15) used PD‑L1–deficient host mice and transplantable tumor cell lines with CRISPR/Cas9-mediated deletion of PD-L1 to determine whether the host or tumor was the relevant molecular target for PD‑L1 antibody blockade. The results from three different tumor models indicated that the PD-L1 expressed by host cells was crucial, while expression by tumor cells was essentially irrelevant. Since PD‑L1 can be expressed on a variety of host cell types, Tang et al. used bone marrow chimera studies to show that the relevant site of host PD‑L1 expression appeared to be a myeloid-derived cell population expressing the marker CD11b.
In a second paper in this issue (16), Lin and colleagues used the MC38 tumor mouse model and B16F10 and ID8 tumor lines to address the question analogous to the one addressed in Tang et al. Lin et al. used CRISPR technology to delete PD‑L1 in tumors and compared these tumors to those in PD‑L1–deficient hosts. As with the Tang study, this study also found that host cells and not tumor cells were the relevant sites of expression for PD‑L1. Adoptive-transfer studies further suggested that either DCs or macrophages were the relevant cell types expressing PD‑L1. Using human tumor biopsies, the authors found that expression of PD‑L1 on DCs and macrophages was a better predictor of clinical response to checkpoint blockade than expression by tumor cells. Although this study used only a small number of clinical samples, the results are consistent with those of previous reports from larger studies (13).
Thus, taken together, both studies reached the same conclusion, that PD‑L1 expression on host cells, and probably host myeloid cells, may be the relevant mechanistic target for PD‑1/PD‑L1 checkpoint blockade. If this finding can be generalized to human tumors, then it has important implications, as discussed below. However, several caveats need to be considered. First, both studies used transplantable mouse cell lines, which may behave differently than the spontaneous, autochthonous tumors found in humans. Second, there might be substantial variation in the role of PD‑L1 on the tumor cells, depending on the specific stage of the tumor (17, 18). For example, the relative importance of tumor PD‑L1 may be affected by the inherent immunogenicity of the tumor, and it may also depend on whether the tumor is at an early stage and just beginning to escape immune surveillance or is an established tumor already full of immunosuppressive host cells (19). Thus, it seems likely that both tumor and host have the potential to contribute to suppression. However, with these caveats in mind, the implied role for host-derived PD‑L1 raises several interesting questions, with potential implications for therapy.
New answers raise new questions
Under the earlier paradigm, in which PD‑L1 was assumed to be expressed by the tumor itself, PD‑L1 was presumed to engage PD‑1 on mature effector T cells and inhibit the final killing step (Figure 1A). However, both Tang et al. and Lin et al. found that in all of their models, tumors lacking PD‑L1 still responded to PD‑L1 checkpoint blockade, indicating that PD‑L1 expression on tumor cells was not relevant. Yet if the relevant site of PD‑L1 is actually on host cells, then how does this host PD‑L1 create immunosuppression? Additionally, what makes PD‑L1 so important on professional antigen-presenting cells (APCs) such as DCs or macrophages? This seems paradoxical because, at least in the traditional understanding of T cell activation, by the time the CD8+ T cells reach the late effector stage in the tumor, they should no longer be dependent on interaction with APCs.
Three possibilities suggest themselves. First, the PD‑L1 on host APCs might not be regulating the effector phase at all, but rather controlling the initial priming step (Figure 1B). The interaction between host APC and naive T cell is obligatory for priming, which can be influenced by PD‑L1 or PD‑L2 (1, 20). Perhaps PD‑1 ligation during priming might contribute to the state of fixed unresponsiveness that can be seen in tumor-reactive T cells (21). However, a fixed unresponsive state, established during initial priming, does not explain the prompt reactivation of at least some exhausted T cells under PD‑L1 blockade. Rather, this suggests that the PD‑L1 on host APCs creates a tonic, ongoing suppressive signal via an unknown mechanism and keeps effector T cells unresponsive.
This ongoing suppression of effector cells is somewhat difficult to explain because professional APCs in tumors and tumor-draining lymph nodes (TDLNs) make up a fairly small population and cannot be in sustained contact with every effector T cell at all times. This ongoing suppression might be accounted for by the possibility (Figure 1C) of PD‑L1 on host APCs sending an activating signal to a population of suppressive T cells such as CD4+ Tregs. Tregs are very potent (22), but it is unclear whether PD‑L1 expression on host APCs plays a role in creating or maintaining the Treg population in tumors. While this is not the traditional role for PD‑L1, such an effect on Tregs has been suggested in some models (23–25). Thus, under this scenario, therapeutically blocking the PD‑1/PD‑L1 pathway might help to destabilize the intratumoral Treg population.
Finally, a third possibility (Figure 1D) might be that host APCs form a critical bottleneck for reactivation or reversal of unresponsiveness in exhausted T cells. Recent studies suggest that reversal exhaustion by PD‑1 blockade is strictly dependent on CD28 signaling in the exhausted T cells (7). This is an important finding because engagement of CD28 presumably would occur only via B7 molecules on professional APCs. This implies that reengagement with host APCs may be an obligatory step in order for exhausted T cells to become functional again. If, however, these APCs expressed high levels of PD‑L1, either in an autocrine or paracrine fashion, then PD‑1 signaling might crossinhibit the CD28 signal in the T cells (5), thus preventing reactivation.
All of these possibilities are speculative at present, and they are not mutually exclusive. Host PD‑L1 could well play a role at multiple points. But the two studies in this issue suggest an import mechanistic contribution by PD‑L1 expressed on host APCs, thus opening up a number of new avenues for further investigation.
Acknowledgments
DHM is supported by NIH R01 CA103320 and CA21229.
Version 1. 01/16/2018
Electronic publication
Version 2. 02/01/2018
Print issue publication
Footnotes
Conflict of interest: D.H. Munn holds intellectual property interests in the therapeutic use of IDO-inhibitor drugs for immuno-oncology, receives royalties and consulting income from NewLink Genetics, Inc., and owns stock and stock options in NewLink Genetics.
Reference information: J Clin Invest. 2018;128(2):570–572. https://doi.org/10.1172/JCI99047.
References
- 1.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol. doi: 10.1038/nri.2017. [published online ahead of print November 13, 2017]. https://doi.org/10.1038/nri.2017.108. [DOI] [PubMed] [Google Scholar]
- 2.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 3.Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity. 2016;44(5):1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 4.Bengsch B, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. 2016;45(2):358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hui E, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamphorst AO, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355(6332):1423–1427. doi: 10.1126/science.aaf0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pauken KE, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354(6316):1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gros A, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. doi: 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spranger S, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med. 2013;5(200):200ra116. doi: 10.1126/scitranslmed.3006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayers M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daud AI, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest. 2016;126(9):3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang H, et al. PD-L1 on host cells is essential for PD-L1 blockade–mediated tumor regression. J Clin Invest. 2018;128(2):580–588. doi: 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin H, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade–mediated tumor regression. J Clin Invest. 2018;128(2):805–815. doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burr ML, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549(7670):101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juneja VR, et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214(4):895–904. doi: 10.1084/jem.20160801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noguchi T, et al. Temporally distinct PD-L1 expression by tumor and host cells contributes to immune escape. Cancer Immunol Res. 2017;5(2):106–117. doi: 10.1158/2326-6066.CIR-16-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767–1778. doi: 10.1056/NEJMra1514296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schietinger A, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. 2016;45(2):389–401. doi: 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Pino-Lagos K, de Vries VC, Guleria I, Sayegh MH, Noelle RJ. Programmed death 1 ligand signaling regulates the generation of adaptive Foxp3+CD4+ regulatory T cells. Proc Natl Acad Sci U S A. 2008;105(27):9331–9336. doi: 10.1073/pnas.0710441105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma MD, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv. 2015;1(10):e1500845. doi: 10.1126/sciadv.1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francisco LM, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]