

Abstract
The underlying cellular mechanisms of catatonia, an executive “psychomotor” syndrome that is observed across neuropsychiatric diseases, have remained obscure. In humans and mice, reduced expression of the structural myelin protein CNP is associated with catatonic signs in an age-dependent manner, pointing to the involvement of myelin-producing oligodendrocytes. Here, we showed that the underlying cause of catatonic signs is the low-grade inflammation of white matter tracts, which marks a final common pathway in Cnp-deficient and other mutant mice with minor myelin abnormalities. The inhibitor of CSF1 receptor kinase signaling PLX5622 depleted microglia and alleviated the catatonic symptoms of Cnp mutants. Thus, microglia and low-grade inflammation of myelinated tracts emerged as the trigger of a previously unexplained mental condition. We observed a very high (25%) prevalence of individuals with catatonic signs in a deeply phenotyped schizophrenia sample (n = 1095). Additionally, we found the loss-of-function allele of a myelin-specific gene (CNP rs2070106-AA) associated with catatonia in 2 independent schizophrenia cohorts and also associated with white matter hyperintensities in a general population sample. Since the catatonic syndrome is likely a surrogate marker for other executive function defects, we suggest that microglia-directed therapies may be considered in psychiatric disorders associated with myelin abnormalities.
Keywords: Inflammation, Neuroscience
Keywords: Demyelinating disorders, Mouse models, Schizophrenia
Introduction
White matter tracts in the CNS largely comprise long axons, associated glial cells, and the ensheathment of axons with myelin. While the role of myelin for axonal conduction and normal motor-sensory function is well known (1), the contribution of white matter integrity to cortical networks and higher cognition is just emerging. Moreover, myelin defects are increasingly linked to mental disease, but mechanistic insight is still lacking and the relationship between cause and consequence difficult to establish in humans (2, 3).
Catatonia is among the most mysterious and as yet poorly understood neuropsychiatric phenotypes. Appearing as a “psychomotor syndrome,” it reflects temporary disruption of executive control in the absence of any “classical” motor dysfunction. Catatonia is typically characterized by a fluctuating course with episodic exacerbations and has historically been associated with schizophrenia, for which it is classified as a positive symptom. Catatonia is, however, also observed in mood- and substance-induced psychotic disorders, malignant neuroleptic syndrome, most encephalitides, and even general medical conditions (4, 5). Reports on brain areas involved in catatonia are scarce. Available data point to frontal lobe regions, such as the pronounced catatonia in a case of butterfly glioma of the frontal corpus callosum (6) or frontal activation in akinetic catatonic patients detected by functional MRI (7).
The full-blown clinical picture of catatonia is dominated by immobility, catalepsy, or stupor, sometimes suddenly switching from “frozen posturing” to excessive motor activity (“movement storm”). Milder forms are more common, even though frequently missed in the diagnostic process, and addressed as catatonic signs (8). In schizophrenia, prevalence is estimated at 0.2%–3.0% (5). Treatment with benzodiazepines or electroconvulsive therapy is nonspecific and not always effective (4, 5).
Similarly to what occurs in humans, catatonia appears in mice as a state of transient immobility in which mice persist in an externally imposed abnormal posture. However, in animals, catatonia has previously been reported only upon induction by body pinch or drug exposure (9, 10). More recently, we detected catatonia in aging mice heterozygous for either Cnp or Mbp (also known as shiverer), both encoding structural proteins of the myelin sheath, 2′-3′-cyclic nucleotide 3′-phosphodiesterase (Cnp) and myelin basic protein (Mbp). Interestingly, in aging Mbp heterozygotes, Cnp expression in the forebrain is also reduced by 50% (11, 12). Correspondingly, individuals homozygous for the A allele of the SNP rs2070106 in human CNP show reduced mRNA expression (13) and association with catatonia (11). Therefore, mutant oligodendrocytes yield a predisposition to catatonia, which is not explained by any paucity of myelin; however, the responsible mechanisms have remained obscure.
The neuropathology of Cnp and Mbp heterozygous mice starts late in life and is surprisingly mild, but in either mutant accompanied by an increased number of microglial cells (11, 12, 14). We therefore hypothesized that neuroinflammation of subcortical white matter, possibly spreading into the prefrontal cortex, could be the cause of catatonic signs. Such a mechanism would be in line with the emerging role of microglia for behavioral phenotypes (15–18).
Here, we demonstrate an unexpectedly high, age-associated prevalence of catatonic signs in more than 25% of deeply phenotyped schizophrenic subjects. In 2 independent schizophrenia cohorts, we found severe catatonic signs associated with the CNP partial loss-of-function genotype rs2070106-AA. Moreover, we show by MRI that CNP rs2070106-AA carriers in the general population are more likely than G carriers (GG or AG) to display frontotemporal white matter hyperintensities (WMH) on T2-weighted images as proposed subclinical signs of vascular changes, neuroinflammation, and demyelination (19–21).
To provide proof-of-principle for microgliosis as the key disease mechanism, we studied Cnp–/– (null) mutant mice. Surprisingly, they developed neuroinflammation with catatonic signs by the early age of 8 weeks. Indeed, we show causality by depletion of microglia with the colony-stimulating factor 1 receptor (CSF1R) inhibitor PLX5622 (22, 23), which blocks a critical microglial survival pathway. The so-caused ablation of microglia prevents catatonia onset in young mutants and ameliorates existing catatonia in adult Cnp–/– mice. These findings shed light on the nature of catatonia and suggest that this striking neuropsychiatric syndrome — and possibly related executive function deficits — may be preventable as well as treatable.
Results
Catatonic signs are highly prevalent in schizophrenia and associated with the CNP partial loss-of-function genotype rs2070106-AA in independent samples.
Within the Göttingen Research Association for Schizophrenia (GRAS) population of deeply phenotyped schizophrenic subjects, 26.7% exhibited signs of catatonia. The severity of catatonic signs clearly increases with age (Figure 1A). The percentage of CNP loss-of-function SNP rs2070106-AA carriers among individuals with the highest expression (≥2) of catatonic signs is greater compared with that in noncatatonic subjects (18.2% versus 10.4%; P = 0.03; OR = 1.93; Figure 1A). This association between catatonia and rs2070106-AA is replicable in an independent sample of schizophrenic individuals (17.9% versus 7.8%; P = 0.015; OR = 2.55; Figure 1B), categorically classified for catatonia according to Leonhard (24).
Figure 1. Age–dependent association of the loss-of-function genotype CNP rs2070106-AA with catatonia in 2 independent schizophrenia cohorts and with WMH in a general population sample.

(A) Bars show mean age of schizophrenic subjects (GRAS), sorted by severity of catatonic signs. Red line denotes percentage of risk for genotype carriers (rs2070106-AA) within each severity group. Note that severity of catatonic signs increases with age. Two-sided values for Kruskal-Wallis (P = 7.6 × 10–9) and Jonckheere-Terpstra (P = 1.3 × 10–9).Mean ± SEM. Also note that CNP rs2070106-AA carriers are most frequent (18.2%) among individuals with highest expression of catatonic signs compared with noncatatonic subjects (10.4%). Two-sided P value for Mann-Whitney U test for extreme-group comparison given in the figure. (B) Distribution of CNP rs2070106 genotype in the Würzburg replication sample of schizophrenia patients based on dichotomous catatonia classification. The AA genotype is significantly more prevalent in patients with (17.9%) than without catatonia (7.8%). Two-sided P values from χ2 test given. (C) Left panel: interaction effect between age and genotype in SHIP-TREND-0 sample on overall WMH volume (minimum 10 mm3 per single WMH cluster). Shown are WMH volume residuals after correcting for intracranial volume, age (nonlinear), and gender. Genotype and age-genotype interaction term contributed 1.1% of variance to overall WMH volumes. Right panel: frequency map averaging all subjects of the general population (SHIP-TREND-0; n = 552), analyzed here. Data highlight WMH appearance predominantly in frontal regions.
CNP rs2070106-AA carriers in the general population display increased age-dependent WMH in frontal and temporal brain areas.
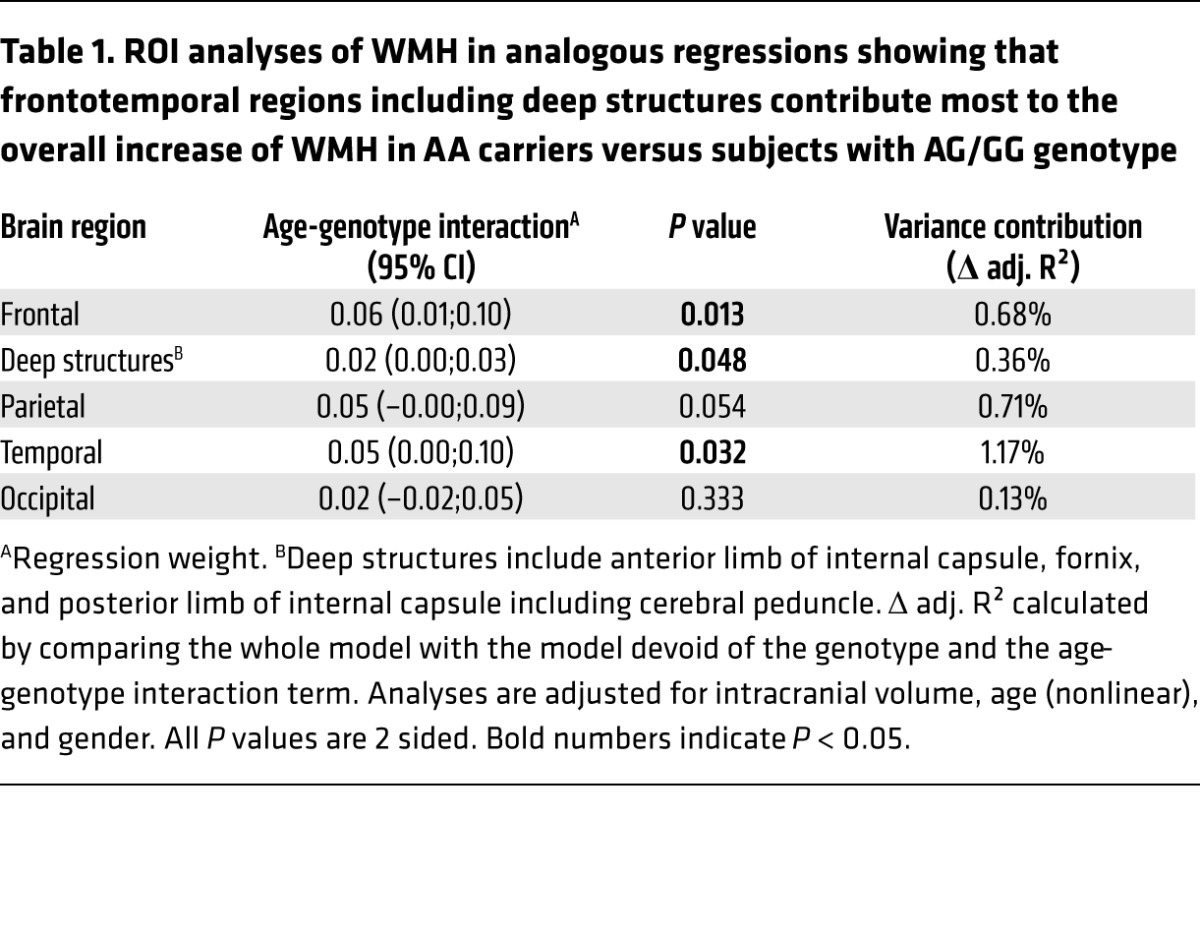
We wondered whether the CNP loss-of-function rs2070106-AA would reveal any measurable effects on suggested MRI indicators of neuroinflammation and white matter alterations. Employing a subsample of the baseline cohort of Study of Health in Pomerania (SHIP-TREND-0), namely general population subjects with MRI scans available, we quantified WMH. AA carriers showed age-dependent higher WMH volume residuals as compared with GG and AG carriers, most prominently in frontotemporal brain regions and deep brain structures, with all analyses adjusted for total intracranial volume, age (nonlinear), and gender (Figure 1C and Table 1). Control covariance analyses, stepwise including further (potentially interfering) covariates alone or together, namely education, waist circumference, serum triglycerides, and smoking, did not appreciably alter the results. Importantly, peripheral inflammation markers, namely C-reactive protein serum levels (natural logarithm) and white blood cell count were tested as covariates, but also did not substantially change the significance level (P = 0.013 and P = 0.014, respectively).
Table 1. ROI analyses of WMH in analogous regressions showing that frontotemporal regions including deep structures contribute most to the overall increase of WMH in AA carriers versus subjects with AG/GG genotype.
Catatonic signs in Cnp-null mutant mice start at around 8 weeks of age and are prevented by the CSF1R inhibitor PLX5622.
To show proof-of-principle for the causal relationship of inflammation and catatonic signs in a construct-valid experimental model, we turned to Cnp–/– mutant mice. As early as the age of 8 weeks, Cnp–/– mice developed catatonic signs (Figure 2, A and B). For illustration, Supplemental Video 1 (supplemental material available online with this article; https://doi.org/10.1172/JCI97032DS1) demonstrates a striking example of catatonia in a young Cnp–/– mutant, tested on the bar, followed by normal-appearing motor performance. This exemplifies the transiently obvious executive dysfunction in the absence of an underlying motor disturbance. Supplemental Video 2 shows excerpts of undisturbed home-cage observation in an enriched environment of another Cnp–/– mutant with normal motor performance and phases of spontaneous catatonia-like posturing (note the “manneristic” stretching of hind limbs when on the bar).
Figure 2. Early catatonia and white matter inflammation in Cnp mutant mice and their prevention by CSF1R inhibition.

(A) Catatonic signs measured by the bar test in WT, Cnp+/–, and Cnp–/– mice at the age of 8 weeks (Kruskal-Wallis, P = 0.034). (B) Image illustrating a mouse with typical catatonic posture during the bar test. (C) Schematic overview of the prevention study design, including PLX5622 (versus regular food) feeding phase (blue arrow) and time points of testing/analyses. Black arrows: bar test (results in Figure 2D) and IHC (results in Figure 2F); yellow arrows: MRS measurements (results in Figure 3). (D) Catatonic signs in WT and Cnp–/– mice (age 8 weeks) after 5-week PLX5622 or control food diet (Kruskal-Wallis, P = 0.165). (E) Schematic overview illustrating corpus callosum and neighboring cingulate cortex areas for IHC quantifications: defined ROI for quantifying APP+ swellings, Iba1+ and CD3+ cells as well as GFAP+ and CD68+ areas (densitometric analysis) shown by the yellow striped field and the blue area, respectively; cingulate area (Cg1/Cg2) for Iba1+ cell quantification displayed in rose. (F) IHC quantification within the corpus callosum of WT or Cnp–/– mice (age 12 weeks) at 4 weeks of microglia repopulation after 5 weeks of PLX5622 or control diet, as shown in C and E. Upper panels show representative images, including higher-magnification inserts, of the quantifications shown underneath: Iba1+ cells (no./mm2; 1 section/brain), APP+ swellings (yellow arrows indicating APP+ spheroids; no./mm2; 3 sections/brain), and densitometric analysis of GFAP+ area (%; 1 section/brain). Original magnification (insets), ×2 (Iba1, GFAP); ×4 (APP). All data in A, D, and F were individually tested for Gaussian distribution using the Kolmogorov-Smirnov test. Nonparametric Kruskal-Wallis test was performed for A, D and F for multiple group comparisons, followed by post hoc 1-tailed Mann-Whitney U test. All data are shown as mean ± SEM; n indicated within bars.
Catatonic signs were prevented by a 5-week oral application of the CSF1R inhibitor PLX5622 via food pellets, starting at 3 weeks of age, immediately after weaning (Figure 2, C and D), consistent with a nearly complete depletion of microglia as reported earlier (22, 23) and reproduced by pilot experiments in preparation of the present study (8-week-old WT mice, treated for 5 or 8 weeks with PLX5622 versus untreated: 1–2 versus ~260 ionized calcium binding adaptor molecule-1 positive [Iba1+] cells/mm2 corpus callosum area, as delineated in Figure 2E). Intriguingly, immunohistochemical (IHC) analysis of the corpus callosum of these mice after 4 weeks of drug recovery showed still lower numbers of Iba1+ cells and amyloid precursor protein–positive (APP+) swellings compared with nontreated Cnp–/– mice, suggesting that PLX5622 treatment has a persistent antiinflammatory benefit, since it is known that microglia recover from inhibition within only 1 week (22, 23). The slightly enhanced glial fibrillary acidic protein–positive (GFAP+) area (Figure 2, E and F) as well as the mildly extended CD68+ (macrosialin) area (0.8% ± 0.04% in WT versus 6.8% ± 0.7% in Cnp–/–; P < 0.014) in young mutants is not reduced after PLX5622 treatment.
Magnetic resonance spectroscopy signs of white and gray matter inflammation in Cnp–/– mice and their prevention by CSF1R inhibition.
An independent cohort of WT and Cnp–/– mice underwent a follow-up magnetic resonance spectroscopy (MRS) study at 8 and 13 weeks of age (design shown in Figure 2C), focusing on regions of interest (ROI) in corpus callosum (white matter) and cortex (gray matter) (Figure 3A). The first MRS in 8-week-old mice was performed after 5 weeks of control versus PLX5622 diet (starting at age 3 weeks, as in the prevention study above), and the second MRS was performed in the same mice at the age of 13 weeks, i.e., after 5 weeks of PLX5622 food cessation/microglia repopulation. Brain myoinositol is seen as a global marker of glial activation including microglia that strongly correlates with neuroinflammation (12, 25–27). Quantification of myoinositol in the corpus callosum showed a distinct genotype difference and progression of inflammation over time in Cnp–/– mice (Figure 3B). Upon PLX5622, the progressive inflammatory phenotype of Cnp–/– mice was reduced to nearly the level seen in WT (Figure 3, C and D). A similar but less prominent effect of PLX5622 was observed in the cortex, where the MRS-detectable neuroinflammation in Cnp–/– mice was also less pronounced in treated mice (Figure 3, E–G).
Figure 3. MRS signs of white matter (corpus callosum) and gray matter (cortex) inflammation (myoinositol) in Cnp–/– mice and prevention by CSF1R inhibition.

(A) Representative sagittal MR images illustrating corpus callosum and cortex ROI for analysis of myoinositol levels (yellow squares). (B–D) Corpus callosum: follow-up MRS for quantification of myoinositol in WT and Cnp–/– mice at 8 and 13 weeks of age (experimental design shown in Figure 2C). Statistical comparison of the first MRS in 8-week-old mice, after 5 weeks of control (B) versus PLX5622 (C) diet (starting at age 3 weeks) and the second MRS in these same mice at the age of 13 weeks, after 5 weeks of regular food (repopulation after PLX5622). (D) Note the return to nearly WT level in PLX5622-treated Cnp–/– mice. (E–G) Cortex: same design as for corpus callosum. (E) Inflammatory phenotype of Cnp–/– mice less pronounced. (F and G) Effect of PLX5622 less prominent. All data in B–G were individually tested for Gaussian distribution using the Kolmogorov-Smirnov test. Two-way ANOVA was performed for B, E, and F, followed by post hoc 1-tailed t tests. Nonparametric Kruskal-Wallis test was used for multiple group comparisons in C, followed by post hoc 1-tailed Mann-Whitney U test. Two-way ANOVA for treatment × time interaction performed in D and G, followed by post hoc unpaired t test. P = 0.008 (D); P = 0.52 (G). All data shown as mean ± SEM; n indicated within bars.
Catatonia and IHC markers of brain inflammation and neurodegeneration in 6-month-old Cnp–/– mice are reduced by PLX5622 treatment.
The encouraging results of the prevention study made us wonder whether similar effects of microglia depletion by CSF1R inhibition could be observed at a more progressed disease state with advanced neurodegeneration (14) (Figures 4, A–F, and Figure 5, A–G). As expected, Cnp–/– mice at the age of 27 weeks were catatonic (Figure 5G). After 5 and 8 weeks of PLX5622, catatonic signs were reduced (Figure 4B). In agreement with the literature, PLX5622-treated WT mice were almost completely depleted of Iba1+ cells (22, 23), even though — compared with young mice — around 10 times more microglial cells/mm2 corpus callosum area were retained. In contrast, Cnp–/– mice showed substantially reduced microglia numbers upon PLX5622 administration to one-third of untreated Cnp–/– mice, but just reached untreated WT levels (Figure 4C). This finding may imply that upon aging and/or in situations of strong inflammatory stimulation, a considerable number of microglia lose their responsiveness to CSF1R inhibition. CD68 immunostaining, localized to the lysosomal membrane and used as an additional readout of microglia activation that is upregulated in actively phagocytic cells (28), displayed a pattern very similar to that of Iba1 (Figure 4, D–F). Interestingly, quantification of microglia in the neighboring cingulate cortex as a crucial part of the prefrontal cortex also revealed a distinct increase in Iba1+ cells in Cnp–/– mice (Figure 2E and Figure 4E). This demonstrates that white matter inflammation in mutants spreads onto gray matter areas where PLX5622 again leads to a considerable reduction in Iba1+ cells.
Figure 4. Catatonia and white matter inflammation in Cnp mutant mice and their treatment by CSF1R inhibition: Part I.

(A) Schematic overview of the treatment study design, including PLX5622 (versus regular diet) feeding phase (blue arrow) and time points/age of testing/analyses (black arrows). (B) Catatonic signs in WT and Cnp–/– mice after 5 and 8 weeks on PLX5622 or control diet. (C and D) IHC quantifications in the corpus callosum (as shown in Figure 2E) at the age of 35 weeks after 8 weeks of PLX5622 or control diet: Iba1+ cells (no./mm2; 1 section/brain) and CD68+ area (%; 1 section/brain). (E) Iba1+ cells in the cingulate cortex (no./mm2; 1 section/brain; area described in Figure 2E). (F) Representative IHC images illustrating the results in C and D. All data in B, C, D, and E were individually tested for Gaussian distribution using the Kolmogorov-Smirnov test. Nonparametric Kruskal-Wallis test was performed in B for multiple group comparisons, followed by post hoc 1-tailed Mann-Whitney U test. Two-way ANOVA was performed for C, D, and F, followed by post hoc 1-tailed unpaired t test. All data are shown as mean ± SEM; n indicated within bars.
Figure 5. Catatonia and white matter inflammation in Cnp mutant mice and their treatment by CSF1R inhibition: Part II.

(A) APP+ swellings (indicating APP+ spheroids; no./mm2; 3 sections/brain) and (B) densitometric analysis of GFAP+ area (%; indicating astrogliosis; 1 section/brain). (C) CD3+ cells (no./mm2; indicating T lymphocyte invasion; 1 section/brain). (D and E) Representative IHC images illustrating CD3 and PDGFRα staining. (F) PDGFRα+ cells (no./mm2; indicating oligodendrocyte precursors; 1 section/brain). (G) Catatonic signs in WT, Cnp–/– (age of onset at 8 weeks of age, shown in Figure 2A), and Cnp+/– mice compared with mice with mutations in other myelin-related genes (Mbp+/–, Plp–/y). Age of onset of catatonia in heterozygous mice (Cnp+/–, Mbp+/–) seen at around 60–72 weeks and in Plp–/y mice at 25 weeks. All data in A, B, C, F, and G were individually tested for Gaussian distribution using the Kolmogorov-Smirnov test. Nonparametric Kruskal-Wallis test was performed for B, C, and G for multiple group comparisons, followed by post hoc 1-tailed Mann-Whitney U test. Two-way ANOVA was performed for A and F, followed by post hoc 1-tailed unpaired t test. All data are shown as mean ± SEM; n indicated within bars.
Both the remarkably increased axonal swellings (APP+) and the strongly enhanced GFAP+ area seen in untreated Cnp–/– mice were diminished under PLX5622, but remained greater than that seen in WT (Figure 5, A and B). The same holds true for CD3+ T lymphocytes, which are attracted by chemokines and cytokines into the inflammatory brain and were also considerably reduced upon PLX5622 treatment (Figure 5, C and D). Since the abundance of oligodendrocyte precursor cells is known to be influenced by microglia and CSF1R inhibition during postnatal development was shown to decrease their numbers (29), PDGFRα staining was also performed. Indeed, Cnp–/– mice, with their elevated numbers of PDGFRα+ oligodendrocyte precursors, as well as WT mice respond to PLX5622 with a reduction of PDGFRα+ cells to nearly 50% (Figure 5, E and F).
Other myelin mutants with neuroinflammation also develop catatonia.
Finally, we asked whether and when other mouse mutants exhibiting subtle myelin abnormalities and mild neuroinflammation later in life would likewise display a catatonic phenotype. This included mice heterozygous for Mbp (12) and Plp-null mutant mice (30). Behavioral testing began at 8 weeks of age and was repeated every 4 to 6 weeks. Indeed, we detected signs of catatonia in heterozygous Mbp+/– mice at ages 60 to 72 weeks, similar to those seen in the aging phenotype of Cnp+/– mice (11). Hemizygous Plp–/y mice displayed catatonic signs already at 25 weeks of age (Figure 5G). We note, however, that in all of these mouse lines, including Cnp mutants, and similarly across the different age groups, catatonic signs show some variability regarding time on the bar. While as a group, catatonic mice clearly differ from WT mice, the severity range is sometimes considerable.
To test for specificity of catatonic signs as a white matter problem, the bar test was performed at various ages in a wide range of CNS mutants with reported behavioral phenotypes relevant for other facets of neuropsychiatric disease, e.g., autistic, cognitive, or metabolic syndromes. This included mutations of synaptic genes Nlgn4–/– (31), Caps1–/– (32), Psd93–/–, and Psd95–/– (33), autophagy dysfunction in Ambra1+/– mutants (34), and ApoE–/– mice with a disturbed blood-brain barrier (35), yet none of these control mice revealed catatonic signs (all P ≥ 0.2 compared with respective WT). In addition, normal WT (C57BL/6N) mice were tested at 1, 6, 10, or 14 weeks after a single injection of high-dose LPS (5 mg/kg i.p. given at the age of 4 months), known to induce persistent microgliosis (36). Also here, no catatonic signs were observed (all P ≥ 0.2 compared with respective untreated controls). Interestingly, even catatonic Plp–/y mice did not reveal any further increase in their catatonic signs upon LPS (5 mg/kg i.p.) when tested at 1, 4, 8, or 12 weeks after a single injection (all P ≥ 0.8 compared with PBS-injected controls), suggesting that just the myelin-associated inflammation is critical for catatonic signs.
Discussion
We have identified altered myelin gene expression and minor structural abnormalities of CNS myelin as the trigger of an inflammatory response predominantly in white matter tracts and an underlying cause of catatonic signs in the behavior of mice and humans. This provides a mechanistic insight into a previously enigmatic neuropsychiatric phenotype and expands our view on the role of white matter integrity in cognitive and executive functions in general. Importantly, we have discovered a potential therapy by targeting microglial cells, which emerge as mediators of this neuropsychiatric syndrome.
Our study was based on the previously reported “catatonia-depression” phenotype of aged mice heterozygous for Cnp (11), which led us to a translational approach from mice to humans and back. Studying the deeply phenotyped GRAS sample of schizophrenic patients (37, 38), we first demonstrated an age-dependent, unexpectedly high prevalence of more than 25% of catatonic signs, exceeding by far the current estimates of approximately 0.2%–3.0% catatonia in schizophrenic subjects (5). This major discrepancy is likely explained by the often-missed clinical diagnosis, in particular of the milder forms that are much more common and classified as neurological soft signs (8).
Importantly, we noticed that the more severe catatonic signs of GRAS patients are associated with rs2070106-AA, a CNP partial loss-of-function genotype (13, 39, 40), a finding that we replicated in an independent schizophrenia cohort. We could further show by MRI in a general population sample that CNP rs2070106-AA subjects were more likely than G carriers (GG or AG) to display WMH in frontal and temporal brain areas as well as in deep brain structures. These subclinical findings are not unusual in healthy individuals, where they have been associated with vascular changes, demyelination, and activated microglia (19–21). Even though this literature is suggestive, we have of course no direct proof (e.g., brain biopsies) that inflammation is increased in white matter tracts of live human AA carriers. We note, however, that diffusion tensor imaging identified higher axial diffusivity and a higher apparent diffusion coefficient in the frontal part of the corpus callosum of AA as compared with GG subjects, consistent with a more progressed axonal loss/degeneration (11). This finding further supports the presence of at least low-grade inflammation in AA individuals.
While WMH in humans may be an indirect indicator of white matter inflammation, presence of the latter in Cnp–/– mice and its reduction upon microglia depletion were directly shown in the present study. In fact, since microgliosis is a feature of Cnp mutant mice (14) and aged Cnp heterozygotes exhibit a catatonia-depression phenotype together with late-onset brain inflammation (11), we tested our hypothesis that neuroinflammation itself is causal for the catatonic signs of myelin mutant mice. We chose to treat Cnp-null mutant mice at an age at which they were still free of motor impairments, and indeed, we could completely prevent catatonia onset in these young animals by depleting microglia via administration of the CSF1R inhibitor PLX5622 (22, 23, 41) immediately after weaning. The CSF1R inhibitor also proved effective for treatment of existing catatonia in older Cnp–/– mutants, in which it caused a reduction (but not prevention) of axonal degeneration. This is in line with CNP deficiency as a “driver” of neurodegeneration following traumatic brain injury (42). However, the complete prevention of catatonic signs in the young Cnp-null mutants strongly suggests that catatonia is not caused by axonal degeneration but is primarily a “microglial disease” induced by mild myelin perturbations. This conclusion is also supported by the catatonic phenotype in aged heterozygous Mbp mice that exhibit microglial activation (12), but will not develop the axonal degeneration phenotype of Plp- and Cnp-null mutant mice with motor impairments (14, 43).
We note that catatonic signs in mice are presently measured by the bar test only, which requires an experienced examiner to yield reproducible results. Having to build on a single readout may explain the cohort-to-cohort variation in the expression severity of this phenotype, which does not show consistent worsening over time (compared with WT controls). Thus, catatonic signs constitute a dichotomous variable in mice (yes/no) rather than a continuous one, as in humans. In fact, several readouts underlie the severity rating used here for humans (9 subtests of Cambridge Neurological Inventory [CNI], including gait mannerisms, gegenhalten, mitgehen, imposed posture, abrupt, or exaggerated spontaneous movements, iterative movements, automatic obedience, and echopraxia; ref. 8). To obtain a similarly robust continuous measure for mice, more catatonia tests are presently being developed in our laboratory, but these tests still require replications in independent mouse cohorts and extensive crossvalidation with bar test results. We hope to ultimately provide a catatonia severity composite score for mice — as we previously established for autistic phenotypes (44) — which will then help diminish sample heterogeneity as typically obtained for single tests.
Interestingly, treating older Cnp mutants with PLX5622 was less effective in eliminating microglial cells. Thus, at least a subpopulation of microglia seems to become unresponsive to CSF1R inhibition in the course of neurodegeneration. This represents a therapeutically relevant observation that may reflect the activation status of these cells and deserves further investigation. Based on our data, we cannot exclude that some of these resilient cells are invading peripheral macrophages, known to be Iba1+ and perhaps resistant to PLX5622, or that a higher dose of PLX5622 would have eliminated even those seemingly less responsive cells. The fact, however, that in our pilot experiments with older WT mice, a higher cumulative dose (8-week treatment versus 5-week treatment) had not resulted in any stronger depletion may point against this interpretation.
We point out that CSF1R inhibition also affects cells other than microglia, which may have contributed to the catatonic phenotype (45). Upon CSF1R inhibition, we saw not only a decline in CD3+ T cells that are attracted to the brain by the inflammatory milieu and may influence microglial behavior, but also a decrease in the GFAP+ area as a measure of robust astrogliosis. In addition, we noted a diminished number of PDGFRα+ oligodendrocyte precursors following CSF1R inhibition, similar to what was shown earlier during brain development (29). We thus have to assume that indirect effects could add to the therapeutic benefit of PLX5622.
A large number of studies in mouse models of neuropsychiatric disease have analyzed the contributions of single genes and developmental defects on cognitive dysfunction, autistic traits, signs of depression, and other mental disease–relevant phenotypes (46–49). However, this research has mainly focused on negative and cognitive symptoms, which are easier to model in mice than any of the positive symptoms (delusions, hallucinations), most of which are considered human specific. Catatonia, defined in DSM-5 as a disease specifier for schizophrenia and major mood disorders (5), emerges as an intriguing exception, a positive symptom, and quantifiable readout that can be studied across species.
Our study is, to our knowledge, the first molecular-genetic approach to catatonia and catatonic signs. However, we have to assume that the etiology of these conditions might be heterogeneous. Myelin perturbations may be just one of several possible causes. Further studies are needed to determine whether secondary neuroinflammation is always essential in the “final common pathway” to catatonia. The inefficiency, however, of LPS-mediated brain inflammation to induce catatonic signs in WT mice or to further enhance them in catatonic Plp–/y mutants supports the concept of primary myelin alterations (or other underlying causes) being of critical importance for the development of a catatonic phenotype. In fact, peripheral LPS injection most likely acts by causing the liver to produce high levels of circulating TNF-α, which enters the brain at specific sites to incite the abnormal stimulation of endogenous microglia and trigger a vicious and long-lasting circle of events that may even lead to neurodegeneration (50, 51). Here, gray matter areas, such as dopaminergic nuclei, seem to be at higher risk than white matter, whereas myelin-associated inflammation is a unique feature of Cnp–/– and other myelin mutant mice.
The sudden loss of motor control in catatonia, followed by an equally sudden regaining of control — often within seconds — ultimately suggests a dysfunction of synaptic circuitry. Importantly, we could show that chronic neuroinflammation in the subcortical white matter progresses into the cingulate cortex, where activated microglia are known to perturb normal synaptic function (16). The production of nitric oxide and axonal conduction blocks, for instance, constitute well-established links between activated microglia and neurodegeneration (52). Within the cortex, the inflammation associated with myelinated fibers may also affect nonmyelinated axons and dendrites (as bystanders), but is probably rather transient or fluctuating because neuronal somata (unlike axons) and the synaptic circuitry are more likely to recover from acute mitochondrial perturbations caused by reactive oxygen species such as NO, as seen in chronic progressive multiple sclerosis (53).
Inflammation, identified in the present study as a major mechanism of catatonic signs, might also affect other phenotypes of mental disease, which could explain the frequently observed fluctuations in their clinical presentation (“episodes”). Alteration of the body’s inflammatory state, as encountered in infectious diseases or even during the normal estrous cycle (luteolysis), may have an additional amplifying impact on inflammation within the CNS and thus contribute to the still poorly explained episodic course of many neuropsychiatric diseases. Also, the most severe acute form of catatonia, the life-threatening febrile pernicious catatonia (4), may represent a fulminant inflammation of white matter tracts.
Catatonia in Cnp mutant mice was prevented by depletion of microglia at a young age and was even treatable in older animals. While this is important as a proof-of-principle, more research will be needed to define effects of repeated treatment cycles with CSF1R inhibitors as well as to delineate more specific pharmacological targets in activated microglia. In fact, repeated treatment cycles with treatment-free intervals may still be effective, but reduce the risk of side effects, potentially resembling hereditary diffuse leukoencephalopathy with spheroids (HDLS), a CNS white matter disease described in individuals with loss-of-function mutations of the CSF1R gene (54). Catatonic signs in patients with schizophrenia are generally mild and per se may not even require specific treatments. However, they likely constitute a surrogate marker for disturbance of broader executive functions and for cognitive deficits, which are severely disabling and currently untreatable, but may also be responsive to specific microglia-targeting antiinflammatory therapies.
Methods
Human studies
GRAS sample of schizophrenic subjects.
The GRAS data collection (37, 38) involved deeply phenotyped patients (n = 1095, age 39.1 ± 12.7 years; 66.8% men) diagnosed with schizophrenia or schizoaffective disorder according to DSM-IV-TR (55). Catatonic signs as the present study’s target phenotype are from the CNI (8). Genotyping of CNP SNP rs2070106 was performed using a semi-custom Axiom MyDesign Genotyping Array (Affymetrix) as reported previously (35, 56).
Würzburg replication sample.
Schizophrenic subjects (n = 288, age 41.4 ± 13.5 years; 54.5% men) were assessed categorically into catatonia versus no catatonia according to Leonhard’s classification (24), as described earlier (57). Genotyping of CNP SNP rs2070106 was performed by means of a quantitative reverse-transcriptase PCR–based (qRT-PCR–based) system using a custom-made primer (KASP assay, LGC Genomics).
SHIP general population sample.
A subsample (n = 552, age 46.2 ± 11.4 years; 42.6% men) of the baseline cohort of SHIP-TREND-0 (accessible via application at www.community-medicine.com) was analyzed (58). Only individuals with available SNP information, valid brain MRI scans, complete covariate data, and no neurological conditions were included. Genotyping was performed using the Illumina HumanOmni2.5-Quad and imputation of genotypes via IMPUTE v.2.1.2.3 against the HapMap II (CUv22, Build36) reference panel. The call rate was very high for the CNP SNP rs2070106 (1.00).
MRI acquisition.
T1- and T2-weighted MRI were used to measure regional patterns of WMH. All images were obtained using a 1.5T Siemens MRI scanner (Magnetom Avanto, Siemens Medical Systems) with an axial T1-weighted MPRAGE sequence and the following parameters: 1 mm isotropic voxels (flip-angle 15°); 3.37 ms echo time; 1900 ms repetition time, and 1100 ms inversion time. Axial T2-FLAIR sequence had the following parameters: 0.9 × 0.9 mm in-plane spatial resolution; 3.0 mm slice thickness (flip-angle 15°); 325 ms echo time; 5,000 ms repetition time. An automated multimodal segmentation algorithm for WMH determination produced a probabilistic map, thresholded to generate a binary image. Thresholding was based on the default threshold value obtained from algorithm training data. To calculate WMH volume within specific ROI, we applied a multiatlas segmentation method. This included nonlinear registration of multiple atlases with ground-truth labels for every individual scan. Finally, WMH was determined for every region of the brain by masking WMH from all other regions (20).
Mouse studies
In all experiments, the experimenter was unaware of mouse genotypes and treatments (fully blinded).
Mouse maintenance and genotyping.
Male and female mice were group-housed separately in ventilated cabinets (Scantainers; Scanbur Karlslunde) unless otherwise indicated for experiments requiring single housing. Mice were maintained on a 12-hour light/12-hour dark cycle (lights off at 7 pm) at 20–22°C, with access to food and water ad libitum, woodchip bedding, and paper tissue as nesting material. Mutant mice (all C56BL/6J background) were genotyped as previously described for Cnp (11, 14), Mbp (12), and Plp (30). Only males were used for experiments with Mbp and Plp mutants, while both females and males were used for experiments with Cnp mutants (housing and testing were always separated). We did not observe any gender differences in catatonic signs. The following neuropsychiatric phenotype-relevant mouse lines, with mutations affecting synapses, blood-brain barrier function or autophagy, were used as controls: Psd93–/– (Dlg2–/–), Psd95–/– (Dlg4–/–) (33), Nlgn4–/– (31), Cadps1–/– (32), ApoE–/– (35), and Ambra1+/– (34).
Treatments.
PLX5622 (formulated in AIN-76A standard chow by Research Diets; 1,200 ppm) and control food (AIN-76A) were provided by Plexxikon Inc. (22, 23). For tracking of potential batch-to-batch variations in future studies, PLX5622 lot numbers are given: 17032710A5TT1.0i; 16010809A9TT1.0i; 17010309A7TT1.0i; and 16092608A1TT1.0i. LPS was injected intraperitoneally (5 mg/kg LPS; O111:B4; Sigma-Aldrich).
Bar test for catatonia.
All mice were transferred to the experimental room 30 minutes prior to testing for habituation. The bar test was performed as previously described (11, 59). Briefly, the mouse was gently carried by the tail to a horizontal bar made of stainless steel (12 cm length, 2.5 mm diameter). Upon grasping the bar with both forepaws, the mouse was moved downwards so that its hind paws had contact with the floor before its tail was released. All experiments were recorded using a high-resolution camcorder (Sony HDR-CX405, Sony Europe Limited). Catatonic signs were scored manually from video recordings as the duration of uninterrupted time a mouse stood nonmoving with at least 1 forepaw on the bar and both hind paws on the ground. Scoring was performed by trained observers blinded to treatments and genotypes (for illustration, see Supplemental Video 1).
Immunohistochemistry.
Anesthetized mice were perfused with Ringer’s solution (Braun) followed by 4% formaldehyde. Brains were collected, postfixed overnight in 4% formaldehyde, cryoprotected in 30% sucrose, and stored at –80°C. Whole mouse brains were cut into 30-μm–thick coronal sections on a cryostat (Leica, CM1950) and kept in storage solution (25% ethylene glycol/25% glycerol in PBS) at –20°C. For APP and CD3 staining, sections were microwaved in citrate buffer (1 mM, pH 6.0) and for APP detection further pretreated in 3% H2O2. All sections were permeabilized and blocked with 5% normal horse serum (NHS)/0.5% Triton X-100 in PBS for 1 hour at room temperature. Sections at a bregma level between +1.15 and +0.5 mm were immunostained for Iba1 (rabbit, 1:1,000, catalog 019-19741, Wako), GFAP (mouse, 1:500, catalog NCL-GFAP-GA5, Novocastra-Leica), CD3 (rat, clone CD3-12, 1:100, catalog MCA1477, Bio-Rad), CD68 (rat, 1:400, catalog MCA1957GA, Bio-Rad), PDGFRα (rabbit, 1:300, catalog 3174, Cell Signaling), or APP (mouse, 1:850, catalog MAB348, Chemicon-Millipore) in 3% NHS/0.5% Triton X-100 in PBS over 2 nights at 4°C. For fluorescent microscopy, 1 hour incubation at room temperature with donkey anti-rabbit Alexa Fluor 647 (catalog A-31573), donkey anti-mouse Alexa Fluor 488 (catalog A-21202), goat anti-rat Alexa Fluor 647 (catalog A-21247), or goat anti-rabbit Alexa Fluor 555 (catalog A-21428) antibodies (1:1,000; Thermo Fisher Scientific-Life Technologies) in 3% NHS/0.5% Triton X-100 in PBS was used. For DAB-based immunostaining, biotinylated horse anti-mouse antibody (1:200; Vector Laboratories) in 3% NHS/0.5% Triton X-100 in PBS and subsequent Vectastain Elite ABC Kit (Vector Laboratories) were used according to the manufacturer’s instructions. Cell nuclei were counterstained with DAPI (1:5,000, Sigma-Aldrich) or Mayer’s hemalum solution (Merck).
Morphometry.
For the analysis of Iba1, GFAP, CD3, CD68, and PDGFRα fluorescent staining, brain slices were scanned using an inverted epifluorescent microscope with a 20×/NA0.4 or 40×/NA0.6 objective lens (Leica, DMI6000B) and quantified using Fiji software (http://fiji.sc/Fiji). Corpus callosum or cingulate cortex (Cg1 and Cg2) of each brain was defined on a DAPI channel as ROI. Iba1+DAPI+, CD3+DAPI+, and PDGFRα+DAPI+ cells were manually counted and density calculated with normalization to ROI. GFAP+ and CD68+ regions were quantified upon uniform thresholding with the respective area expressed as percentage of corpus callosum. APP+ swellings were manually counted using a light microscope (Olympus BX-50) connected to a computer-driven motorized stage, z-axis position encoder (microcator), and a microfire video camera interfaced to a PC using Stereo Investigator 6.55 software (MicroBrightfield Inc.). Representative images of APP+ swellings were taken with a light microscope with a 100×/NA 1.30 oil objective lens (Zeiss Imager Z1).
MRI and 1H-MRS.
Mice were anesthetized with 5% isoflurane, intubated, and kept at 1.75% isoflurane by active ventilation with a constant respiratory frequency of 85 breaths/min (Animal Respirator Advanced, TSE Systems). MRI and localized 1H-MRS were performed at a magnetic field strength of 9.4T (Bruker BioSpin). MRI consisted of T2-weighted images (2D-FSE, TR/TE = 2800/11 ms, 100 × 100 × 300 μm3) based on which respective volumes of interest for localized proton-MR spectra were positioned. MR spectra (STEAM, TR/TE/TM = 6,000/10/10 ms) were obtained from a volume of interest in the cortex (3.9 × 0.7 × 3.2 mm3) and corpus callosum (3.9 × 0.7 × 1.7 mm3). Metabolite quantification was completed with spectral evaluation by LCModel (Version 6.3-1L). Results with Cramer-Rao lower bounds greater than 20% were excluded from further analysis.
Statistics
Group differences for continuous variables in human samples were assessed using the Kruskal-Wallis and Jonckheere-Terpstra trend tests. Genotype comparisons used the χ2 test. Multivariate linear regression models were run with WMH volume as a dependent variable and rs2070106 genotype-age interaction term as a predictor of interest. WMH volumes were transformed via cubic root due to their highly skewed distributions (20). For whole brain, total WMH volume was calculated summing all clusters greater than 10 mm3 to reduce noise, followed by hypothesis-driven (11) post hoc testing of WMH from 5 predefined ROI (frontal, temporal, parietal and, as control, occipital lobes, as well as deep structures). For sensitivity analyses, all models described above were rerun using bootstrap methodology (2,000 replications) to derive SEM and CI independently of parametric assumptions such as Gaussian distribution or homoscedasticity. No major differences in standard ordinary least squares results were found. For mouse statistics, data distribution and variance homogeneity were determined by Kolmogorov-Smirnov test and outliers via the Grubbs test (https://graphpad.com/quickcalcs/Grubbs1.cfm). Two-way ANOVA with/without repeated measures was used for normally distributed data. Kruskal-Wallis test was used for data without normal distribution. Between-group comparisons were performed by Student’s t test for dependent/independent samples or Mann-Whitney U test. P ≤ 0.05 was considered significant. All statistical analyses were performed using SPSS (v 17.0; IBM-Deutschland GmbH), STATA14/MP (Stata Inc.), or Prism 5 software (GraphPad Software).
Study approval
The GRAS study was approved by the ethics committees of Georg-August-University and participating centers across Germany, complying with the Helsinki Declaration. The Würzburg replication sample was approved by the Würzburg University Ethics Committee. The baseline cohort of SHIP-TREND-O, conducted in Northeast Germany, was approved by the Greifswald University Ethics Committee. All subjects (and/or legal representatives) gave written, informed consent. All animal tests were approved by the local Animal Care and Use Committee (LAVES, Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany) in accordance with the German Animal Protection Law.
Author contributions
HE and KAN created the concept and designed and supervised the study. MM, JH, MH, RAB, MB, SL, SKS, GH, CD, HV, AR, HJG, and HE acquired, analyzed, and interpreted data for human studies. HJ, SA, EB, HP, BLW, KAN, SB, and HE acquired, analyzed, and interpreted data for mouse phenotyping. HJ, SA, EB, SG, UCG, HBW, KAN, and HE acquired, analyzed, and interpreted data for mouse genetics. HE and KAN drafted the manuscript. HE, KAN, MM, HJ, SA, and EB drafted display items. All authors read and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
This work was supported by the Max Planck Society, the Max Planck Förderstiftung, the Deutsche Forschungsgemeinschaft (DFG) (CNMPB and SPP1757), EXTRABRAIN EU-FP7, and the Niedersachsen-Research Network on Neuroinfectiology (N-RENNT). SHIP is part of the Community Medicine Research Net of the University Medicine Greifswald, which is funded by the Federal State of Mecklenburg–West Pomerania. Genome-wide data in SHIP have been supported by a joint grant from Siemens Healthineers, Erlangen, and the Federal State of Mecklenburg–West Pomerania. KAN holds a European Research Council Advanced Investigator grant. The authors thank all subjects for participating in the study, and all colleagues who have contributed over the past decade to GRAS data collection.
Version 1. 12/18/2017
Electronic publication
Version 2. 02/01/2018
Print issue publication
Footnotes
Conflict of interest: Brian West is an employee of Plexxikon Inc.
Reference information: J Clin Invest. 2018;128(2):734–745.https://doi.org/10.1172/JCI97032.
See the related Commentary at Micro(glial)-managing executive function: white matter inflammation drives catatonia.
Contributor Information
Hana Janova, Email: janova@em.mpg.de.
Sahab Arinrad, Email: arinrad@em.mpg.de.
Evan Balmuth, Email: balmuth@em.mpg.de.
Marina Mitjans, Email: mitjans@em.mpg.de.
Johannes Hertel, Email: hertelj@uni-greifswald.de.
Mohamad Habes, Email: Mohamad.Habes@uphs.upenn.edu.
Robert A. Bittner, Email: Robert.Bittner@kgu.de.
Hong Pan, Email: pan@em.mpg.de.
Sandra Goebbels, Email: sgoebbels@em.mpg.de.
Martin Begemann, Email: begemann@em.mpg.de.
Sönke Langner, Email: soenke.langner@uni-greifswald.de.
Sarah Kittel-Schneider, Email: sarah.kittel-schneider@kgu.de.
Georg Homuth, Email: georg.homuth@uni-greifswald.de.
Christos Davatzikos, Email: Christos.Davatzikos@uphs.upenn.edu.
Henry Völzke, Email: voelzke@uni-greifswald.de.
Andreas Reif, Email: andreas.reif@kgu.de.
Hans Jörgen Grabe, Email: grabeh@uni-greifswald.de.
Susann Boretius, Email: SBoretius@dpz.eu.
Hannelore Ehrenreich, Email: ehrenreich@em.mpg.de.
References
- 1.Nave KA, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. 2014;30:503–533. doi: 10.1146/annurev-cellbio-100913-013101. [DOI] [PubMed] [Google Scholar]
- 2.Filley CM, Fields RD. White matter and cognition: making the connection. J Neurophysiol. 2016;116(5):2093–2104. doi: 10.1152/jn.00221.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nave KA, Ehrenreich H. Myelination and oligodendrocyte functions in psychiatric diseases. JAMA Psychiatry. 2014;71(5):582–584. doi: 10.1001/jamapsychiatry.2014.189. [DOI] [PubMed] [Google Scholar]
- 4. Fink M, Taylor MA. Catatonia: a clinician’s guide to diagnosis and treatment. Cambridge, UK: Cambridge University Press; 2003. [Google Scholar]
- 5.Tandon R, et al. Catatonia in DSM-5. Schizophr Res. 2013;150(1):26–30. doi: 10.1016/j.schres.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 6.Arora M, Praharaj SK. Butterfly glioma of corpus callosum presenting as catatonia. World J Biol Psychiatry. 2007;8(1):54–55. doi: 10.1080/15622970600960116. [DOI] [PubMed] [Google Scholar]
- 7.Northoff G, et al. Orbitofrontal cortical dysfunction in akinetic catatonia: a functional magnetic resonance imaging study during negative emotional stimulation. Schizophr Bull. 2004;30(2):405–427. doi: 10.1093/oxfordjournals.schbul.a007088. [DOI] [PubMed] [Google Scholar]
- 8.Chen EY, et al. The Cambridge Neurological Inventory: a clinical instrument for assessment of soft neurological signs in psychiatric patients. Psychiatry Res. 1995;56(2):183–204. doi: 10.1016/0165-1781(95)02535-2. [DOI] [PubMed] [Google Scholar]
- 9.Amir S. Catalepsy induced by body pinch: relation to stress-induced analgesia. Ann N Y Acad Sci. 1986;467:226–237. doi: 10.1111/j.1749-6632.1986.tb14631.x. [DOI] [PubMed] [Google Scholar]
- 10.Chaperon F, Thiébot MH. Behavioral effects of cannabinoid agents in animals. Crit Rev Neurobiol. 1999;13(3):243–281. doi: 10.1615/CritRevNeurobiol.v13.i3.20. [DOI] [PubMed] [Google Scholar]
- 11.Hagemeyer N, et al. A myelin gene causative of a catatonia-depression syndrome upon aging. EMBO Mol Med. 2012;4(6):528–539. doi: 10.1002/emmm.201200230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poggi G, et al. Cortical network dysfunction caused by a subtle defect of myelination. Glia. 2016;64(11):2025–2040. doi: 10.1002/glia.23039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peirce TR, et al. Convergent evidence for 2’,3’-cyclic nucleotide 3’-phosphodiesterase as a possible susceptibility gene for schizophrenia. Arch Gen Psychiatry. 2006;63(1):18–24. doi: 10.1001/archpsyc.63.1.18. [DOI] [PubMed] [Google Scholar]
- 14.Lappe-Siefke C, et al. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33(3):366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- 15.Chen SK, et al. Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell. 2010;141(5):775–785. doi: 10.1016/j.cell.2010.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–134. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parkhurst CN, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15(5):300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 19.Fazekas F, et al. The morphologic correlate of incidental punctate white matter hyperintensities on MR images. AJNR Am J Neuroradiol. 1991;12(5):915–921. [PMC free article] [PubMed] [Google Scholar]
- 20.Habes M, et al. White matter hyperintensities and imaging patterns of brain ageing in the general population. Brain. 2016;139(Pt 4):1164–1179. doi: 10.1093/brain/aww008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson JE, et al. Microglial activation in white matter lesions and nonlesional white matter of ageing brains. Neuropathol Appl Neurobiol. 2007;33(6):670–683. doi: 10.1111/j.1365-2990.2007.00890.x. [DOI] [PubMed] [Google Scholar]
- 22.Dagher NN, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation. 2015;12:139. doi: 10.1186/s12974-015-0366-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elmore MR, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82(2):380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leonhard K. Classification of Endogenous Psychoses and Their Differentiated Etiology. Wien, Austria: Springer-Verlag Wien; 1999. [Google Scholar]
- 25.Badar-Goffer RS, Ben-Yoseph O, Bachelard HS, Morris PG. Neuronal-glial metabolism under depolarizing conditions. A 13C-n.m.r. study. Biochem J. 1992;282(Pt 1):225–230. doi: 10.1042/bj2820225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang L, Munsaka SM, Kraft-Terry S, Ernst T. Magnetic resonance spectroscopy to assess neuroinflammation and neuropathic pain. J Neuroimmune Pharmacol. 2013;8(3):576–593. doi: 10.1007/s11481-013-9460-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ross BD, Bluml S, Cowan R, Danielsen E, Farrow N, Gruetter R. In vivo magnetic resonance spectroscopy of human brain: the biophysical basis of dementia. Biophys Chem. 1997;68(1–3):161–172. doi: 10.1016/s0301-4622(97)00032-x. [DOI] [PubMed] [Google Scholar]
- 28.Zotova E, et al. Inflammatory components in human Alzheimer’s disease and after active amyloid-β42 immunization. Brain. 2013;136(Pt 9):2677–2696. doi: 10.1093/brain/awt210. [DOI] [PubMed] [Google Scholar]
- 29.Hagemeyer N, et al. Microglia contribute to normal myelinogenesis to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017;134(3):441–458. doi: 10.1007/s00401-017-1747-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klugmann M, et al. Assembly of CNS myelin in the absence of proteolipid protein. Neuron. 1997;18(1):59–70. doi: 10.1016/S0896-6273(01)80046-5. [DOI] [PubMed] [Google Scholar]
- 31.Jamain S, et al. Reduced social interaction and ultrasonic communication in a mouse model of monogenic heritable autism. Proc Natl Acad Sci U S A. 2008;105(5):1710–1715. doi: 10.1073/pnas.0711555105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jockusch WJ, et al. CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell. 2007;131(4):796–808. doi: 10.1016/j.cell.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Winkler D, et al. Hypersocial behavior biological redundancy in mice with reduced expression of PSD95 or PSD93. Behav Brain Res. doi: 10.1016/j.bbr.2017.02.011. [published online ahead of print February 9, 2017 ]. https://doi.org/10.1016/j.bbr.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 34.Dere E, et al. Heterozygous ambra1 deficiency in mice: a genetic trait with autism-like behavior restricted to the female gender. Front Behav Neurosci. 2014;8:181. doi: 10.3389/fnbeh.2014.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hammer C, et al. Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol Psychiatry. 2014;19(10):1143–1149. doi: 10.1038/mp.2013.110. [DOI] [PubMed] [Google Scholar]
- 36.Qin L, Liu Y, Hong JS, Crews FT. NADPH oxidase and aging drive microglial activation, oxidative stress, and dopaminergic neurodegeneration following systemic LPS administration. Glia. 2013;61(6):855–868. doi: 10.1002/glia.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Begemann M, et al. Modification of cognitive performance in schizophrenia by complexin 2 gene polymorphisms. Arch Gen Psychiatry. 2010;67(9):879–888. doi: 10.1001/archgenpsychiatry.2010.107. [DOI] [PubMed] [Google Scholar]
- 38.Ribbe K, et al. The cross-sectional GRAS sample: a comprehensive phenotypical data collection of schizophrenic patients. BMC Psychiatry. 2010;10:91. doi: 10.1186/1471-244X-10-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwamoto K, Ueda J, Bundo M, Nakano Y, Kato T. Effect of a functional single nucleotide polymorphism in the 2’,3’-cyclic nucleotide 3’-phosphodiesterase gene on the expression of oligodendrocyte-related genes in schizophrenia. Psychiatry Clin Neurosci. 2008;62(1):103–108. doi: 10.1111/j.1440-1819.2007.01786.x. [DOI] [PubMed] [Google Scholar]
- 40.Mitkus SN, et al. Expression of oligodendrocyte-associated genes in dorsolateral prefrontal cortex of patients with schizophrenia. Schizophr Res. 2008;98(1–3):129–138. doi: 10.1016/j.schres.2007.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waisman A, Ginhoux F, Greter M, Bruttger J. Homeostasis of microglia in the adult brain: review of novel microglia depletion systems. Trends Immunol. 2015;36(10):625–636. doi: 10.1016/j.it.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Wieser GL, et al. Neuroinflammation in white matter tracts of Cnp1 mutant mice amplified by a minor brain injury. Glia. 2013;61(6):869–880. doi: 10.1002/glia.22480. [DOI] [PubMed] [Google Scholar]
- 43.Griffiths I, et al. Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science. 1998;280(5369):1610–1613. doi: 10.1126/science.280.5369.1610. [DOI] [PubMed] [Google Scholar]
- 44.El-Kordi A, et al. Development of an autism severity score for mice using Nlgn4 null mutants as a construct-valid model of heritable monogenic autism. Behav Brain Res. 2013;251:41–49. doi: 10.1016/j.bbr.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 45.Chitu V, Gokhan Ş, Nandi S, Mehler MF, Stanley ER. Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. 2016;39(6):378–393. doi: 10.1016/j.tins.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10(2):126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13(10):1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schroeder JC, Reim D, Boeckers TM, Schmeisser MJ. Genetic animal models for autism spectrum disorder. Curr Top Behav Neurosci. 2017;30:311–324. doi: 10.1007/7854_2015_407. [DOI] [PubMed] [Google Scholar]
- 49.Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11(7):490–502. doi: 10.1038/nrn2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeong HK, Jou I, Joe EH. Systemic LPS administration induces brain inflammation but not dopaminergic neuronal death in the substantia nigra. Exp Mol Med. 2010;42(12):823–832. doi: 10.3858/emm.2010.42.12.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin L, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55(5):453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 53.Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol. 2012;8(11):647–656. doi: 10.1038/nrneurol.2012.168. [DOI] [PubMed] [Google Scholar]
- 54.Rademakers R, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200–205. doi: 10.1038/ng.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. American Psychiatric Association. Diagnostic statistical manual of mental disorders: DSM-IV-TR. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- 56.Stepniak B, et al. Accumulated environmental risk determining age at schizophrenia onset: a deep phenotyping-based study. Lancet Psychiatry. 2014;1(6):444–453. doi: 10.1016/S2215-0366(14)70379-7. [DOI] [PubMed] [Google Scholar]
- 57.Reif A, et al. A neuronal nitric oxide synthase (NOS-I) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol Psychiatry. 2006;11(3):286–300. doi: 10.1038/sj.mp.4001779. [DOI] [PubMed] [Google Scholar]
- 58.Völzke H, et al. Cohort profile: the study of health in Pomerania. Int J Epidemiol. 2011;40(2):294–307. doi: 10.1093/ije/dyp394. [DOI] [PubMed] [Google Scholar]
- 59.Kuschinsky K, Hornykiewicz O. Morphine catalepsy in the rat: relation to striatal dopamine metabolism. Eur J Pharmacol. 1972;19(1):119–122. doi: 10.1016/0014-2999(72)90086-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.