

Abstract
The molecular mechanism by which cancer-associated fibroblasts (CAFs) confer chemoresistance in ovarian cancer is poorly understood. The purpose of the present study was to evaluate the roles of CAFs in modulating tumor vasculature, chemoresistance, and disease progression. Here, we found that CAFs upregulated the lipoma-preferred partner (LPP) gene in microvascular endothelial cells (MECs) and that LPP expression levels in intratumoral MECs correlated with survival and chemoresistance in patients with ovarian cancer. Mechanistically, LPP increased focal adhesion and stress fiber formation to promote endothelial cell motility and permeability. siRNA-mediated LPP silencing in ovarian tumor–bearing mice improved paclitaxel delivery to cancer cells by decreasing intratumoral microvessel leakiness. Further studies showed that CAFs regulate endothelial LPP via a calcium-dependent signaling pathway involving microfibrillar-associated protein 5 (MFAP5), focal adhesion kinase (FAK), ERK, and LPP. Thus, our findings suggest that targeting endothelial LPP enhances the efficacy of chemotherapy in ovarian cancer. Our data highlight the importance of CAF–endothelial cell crosstalk signaling in cancer chemoresistance and demonstrate the improved efficacy of using LPP-targeting siRNA in combination with cytotoxic drugs.
Keywords: Cell Biology, Oncology
Keywords: Cancer
Introduction
High-grade serous ovarian cancer (HGSC) is the most common histological subtype of ovarian cancer and accounts for most ovarian cancer–related deaths. Most HGSCs are diagnosed at a late stage, and, as a result, the overall survival rate of patients with HGSC is less than 30%. The clinical biological characteristics of HGSC suggest that late diagnosis and the persistence of drug-resistant cancer cells limit our ability to cure this disease.
Tumor vasculature plays an important role in the pathogenesis and progression of HGSC and is crucial in modulating the delivery of therapeutic agents (1). Various tumor cell–derived cytokines, including VEGFs and FGFs, are involved in HGSC pathogenesis and progression. Although phase I and II trials of the VEGF-α–targeting monoclonal antibody bevacizumab in patients with ovarian cancer yielded encouraging results, phase III trials of the drug as a frontline treatment for ovarian cancer patients (Gynecologic Oncology Group 218 [GOG 218] and International Collaboration on Ovarian Neoplasms 7 [ICON7]) and recurrent ovarian cancer (Ovarian Cancer Study Comparing Efficacy and Safety of Chemotherapy and Anti-Angiogenic Therapy in Platinum-Sensitive Recurrent Disease [OCEANS] and Avastin Use in Platinum-Resistant Epithelial Ovarian Cancer [AURELIA]) have demonstrated that bevacizumab yields only a modest improvement in progression-free survival and no significant improvement in overall survival (2–5). These findings suggest that other proangiogenic mediators and pathways compensate for VEGF blockade and allow angiogenesis to occur, despite anti-VEGF therapy (1). Further research, including that aimed at identifying new proangiogenic targets and markers to optimize patient selection, is essential to maximize the potential of antiangiogenic therapy for ovarian cancer.
Cancer-associated fibroblasts (CAFs), one of the primary stromal cell types in ovarian tumor tissues (6), secrete CAF-specific proteins, cytokines, and growth factors and produce an extracellular matrix (ECM) that supports tumor cell growth and angiogenesis and confers chemoresistance (7–11). However, the mechanisms by which CAFs promote angiogenesis in ovarian cancer remain poorly understood. In addition, few studies have sought to identify CAF-derived mediator–regulated endothelial biomarkers that are associated with chemoresistance. We searched for CAF-regulated proangiogenic effector molecules in microvascular endothelial cells (MECs) and identified elevated expression of the lipoma-preferred partner (LPP) gene in MECs cocultured with CAFs. LPP is a member of a subfamily of LIM domain proteins that are characterized by an N-terminal protein–rich region and 3 C-terminal LIM domains (12, 13). It mainly localizes to the cell periphery in focal adhesion and is involved in cell-cell adhesion, cell-substrate cytoskeletal interactions, and cell motility in Madin-Darby canine kidney (MDCK) epithelial cells (14). In addition, LPP has been shown to bind to LASP1, which enhances the motility of embryonic fibroblasts (15). The roles of endothelial LPP in tumor angiogenesis and in conferring chemoresistance have not been reported to date.
The purpose of the present study was to evaluate the roles of CAFs in modulating tumor vasculature and disease progression. On the basis of our experimental results, we found elevated levels of LPP expression in MECs in the presence of CAFs and demonstrated the prognostic significance of endothelial LPP in patients with HDSC. We also delineated the molecular mechanism by which LPP increases microvascular endothelial cell motility and leakiness and decreases the delivery of paclitaxel to tumors in vivo. Furthermore, using murine models, we showed that LPP silencing inhibits ovarian tumor growth and improves paclitaxel bioavailability by reducing intratumoral microvessel leakiness. Finally, we demonstrated that CAF-derived microfibrilla–associated protein 5 (MFAP5) can upregulate LPP in MECs via a calcium-dependent MFAP5/FAK/ERK/LPP signaling pathway.
Results
CAFs upregulate LPP in MECs.
The ovarian tumor microenvironment, which is composed primarily of fibroblasts, ECM proteins, endothelial cells, and lymphocytic infiltrates, can regulate tumor growth, angiogenesis, dissemination, and chemoresistance (11, 16). CAFs have been shown to play crucial roles in cancer progression. Although increasing evidence demonstrates that CAFs have important roles in modulating the aggressive phenotypes of cancer cells, their effects on the tumor vasculature remain underexplored. We cocultured human telomerase-immortalized microvascular endothelial (TIME) cells with either primary human ovarian CAFs or normal ovarian fibroblasts (NOFs) to evaluate the effects of CAFs on endothelial cell motility and monolayer permeability. We found that TIME cells that had been cocultured with CAFs had significantly higher rates of motility and monolayer permeability than did those cocultured with NOFs (Figure 1A).
Figure 1. CAF-induced endothelial LPP expression in ovarian cancer.

(A) TIME MECs cocultured with CAFs had significantly higher motility rates and monolayer permeability compared with MECs cocultured with NOFs. P values were determined by 2-tailed Student’s t test. (B) Heatmap generated from transcriptome analyses of RNA samples isolated from TIME cells cocultured with CAFsor NOFs. A total of 1,394 genes and 2,106 genes wereup- and downregulated, respectively, in TIME cells cocultured with CAFs versus MECs cocultured with NOFs (fold change >1.5; Benjamini-Hochberg multiple testing–adjusted P < 0.05). LPP was identified as one of the significantly upregulated genes. (C) Quantitative reverse transcription PCR (qRT-PCR) analyses of endothelial cells RNA samples confirmed that endothelial LPP expression was upregulated in the presence of CAFs (#P < 0.0001, by 2-tailed Student’s t test). (D) Hematoxylin- counterstained images of immunolocalization of LPP in a normal ovary and a high-grade serous ovarian cancer showing that ovarian tumor MECs had higher LPP expression levels than did normal ovarian MECs. Scale bars: 50 μm. (E) Kaplan-Meier analysis were used to evaluate the clinical relevance of endothelial LPP expression in patients with HGSC. Elevated endothelial LPP expression was associated with lower overall and progression-free survival. The median overall survival rate of HGSC patients with high endothelial LPP levels (23 months) was significantly shorter than that of patients with low endothelial LPP levels (76 months) (n = 129; P < 0.001, by log-rank test). The median progression-free survival rate duration of HGSC patients with high endothelial LPP levels (6 months) was significantly shorter than that of patients with low endothelial LPP levels (10 months) (n = 100; P < 0.037, by log-rank test). (F) CAFs increased endothelial cell motility, and the motility-promoting effect of CAFs was attenuated in endothelial cells transfected with LPP-targeting siRNAs. Motility assays were performed using Boyden chambers. Endothelial cells in the upper chamber were allowed to migrate through the porous membrane in the presence of CAFs or NOFs in the bottom chamber (P values were determined by 2-tailed Student’s t test). (G) CAFs increased the permeability of a confluent endothelial cell monolayer, and the permeability-enhancing effect of CAFs was attenuated in endothelial cells transfected with LPP-targeting siRNAs (P values were determined by 2-tailed Student’s t test). Fluorescence-labeled dextran was added to a confluent monolayer culture of endothelial cells in the upper chamber of a Boyden chamber and the amount of dextran diffusing through the endothelial cell monolayer culture in the presence of CAFs or NOFs to the lower chamber was measured by an ELISA microplate reader. All data represent the mean ± SEM of 3 independent experiments.
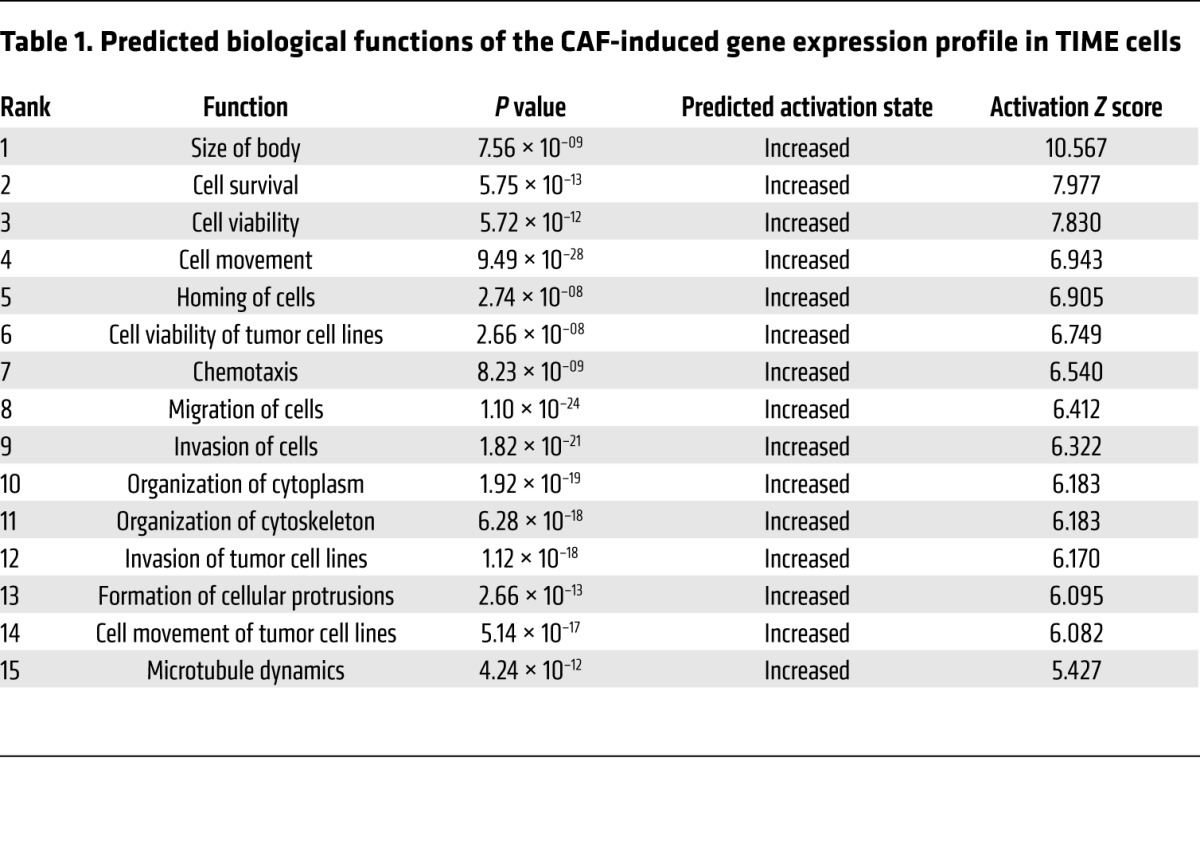
To determine the underlying molecular mechanism by which CAFs promote angiogenesis, we performed a transcriptome analysis of RNA samples isolated from TIME cells that had been cocultured with CAFs or NOFs. We identified 1,394 genes and 2,106 genes that were up- and downregulated, respectively, in TIME cells cocultured with CAFs compared with those cocultured with NOFs (fold change >1.5, Benjamini-Hochberg multiple testing–adjusted P < 0.05) (Figure 1B and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI95200DS1). To uncover the biological functions of the CAF-induced gene expression profile in TIME cells, we used Ingenuity Pathway Analysis (IPA) software to analyze the list of genes that were upregulated in TIME cells cocultured with CAFs. Among the top 15 predicted activated biological functions, 10 are related to cell motility, invasion potential, and cytoskeleton organization (Table 1), which suggests that CAFs play an important role in the mobility of endothelial cells. Since increased endothelial cell motility can facilitate angiogenesis, we examined the list of genes identified by IPA that had the highest ranked cell movement–related function (activation Z score = 6.943; P = 9.49 × 10–28). We selected LPP, a LIM domain–containing protein that interacts with the cytoskeleton, for further validation studies. As a cell motility regulatory protein, the roles of LPP in angiogenesis, chemoresistance, and tumor progression have not been investigated. We first performed a quantitative reverse transcription PCR (qRT-PCR) analysis using RNA samples isolated from endothelial cells cocultured with CAFs or NOFs and found that LPP mRNA was upregulated in TIME cells cocultured with CAFs compared with levels in those cocultured with NOFs (Figure 1C).
Table 1. Predicted biological functions of the CAF-induced gene expression profile in TIME cells.
LPP overexpression is associated with poor survival rates and increased fibrosis in patients with HGSC.
Blood vessels in tumor tissue are usually poorly organized and leaky, which impairs drug delivery (17). Because LPP has been shown to be involved in cell-cell adhesion, cell-substrate cytoskeletal interactions, and cell motility (14), we hypothesized that the CAF-induced upregulation of LPP in endothelial cells in HGSC increases microvascular leakiness, thus decreasing the bioavailability of drugs such as paclitaxel to tumor cells. To test this hypothesis, we first performed immunolocalization of LPP in 10 normal ovarian and 129 HGSC tissue samples. Compared with those in normal ovarian tissue, the endothelial cells and surrounding smooth muscle cells in HGSC samples had a substantially higher LPP expression level (Figure 1D). Next, we determined the prognostic significance of endothelial LPP in HGSC. A Kaplan-Meier analysis and log-rank tests showed that high endothelial LPP expression was associated with lower overall and progression-free survival rates than was low endothelial LPP expression (Figure 1E), suggesting that endothelial LPP plays a role in ovarian cancer progression and chemoresistance.
Since the presence of CAFs is associated with tumor tissue fibrosis and our data showed that endothelial LPP expression was upregulated by coculturing MECs with CAFs, we determined whether there was a correlation between endothelial LPP expression and the degree of fibrosis. We performed Picrosirius red staining for collagen on 24 HGSC tissue samples expressing high or low levels of endothelial LPP. Collagen staining results demonstrated that HGSC patients with high expression levels of endothelial LPP had significantly higher collagen coverage and density than did patients with low expression levels of endothelial LPP (Supplemental Figure 1), suggesting an increase in fibrosis in tumor tissue with higher endothelial LPP expression.
LPP increases endothelial cell motility and monolayer permeability.
To assess the effects of CAFs in upregulating LPP to promote endothelial cell motility, we subjected TIME MECs to motility assays using Boyden chambers, in which endothelial cells in the upper chamber were allowed to migrate through the porous cell culture membrane in the presence of CAFs or NOFs in the bottom chamber. We observed that CAFs enhanced endothelial cell motility, and the motility-promoting effect of CAFs was attenuated in endothelial cells transfected with LPP-targeting siRNAs (Figure 1F). These data suggest that endothelial LPP mediates the effect of CAFs on enhancing the motility potential of endothelial cells.
To determine whether LPP mediates the effect of CAFs in modulating endothelial cell permeability, we added fluorescence-labeled dextran to a confluent monolayer culture of endothelial cells in the upper chamber of a Boyden chamber and then allowed dextran to diffuse through the culture and the porous cell membrane in the presence of CAFs or NOFs in the lower chamber. The fluorescent signal in the lower-chamber media that contained CAFs was significantly higher than that of the media that contained NOFs, suggesting that CAFs enhanced the permeability of the endothelial cell monolayer. This permeability-enhancing effect was attenuated in endothelial cells transfected with LPP-targeting siRNAs. These data suggest that LPP mediates the effect of CAFs in increasing the permeability of the endothelial cell monolayer (Figure 1G).
We compared the proliferation rates of parental and LPP-silenced endothelial cells using WST-1 cell proliferation assays. The experimental results showed that endothelial cell proliferation was not significantly affected by LPP silencing (Supplemental Figure 2), suggesting that LPP-induced endothelial cell migration in Boyden chambers and monolayer permeability are independent of cell proliferation.
LPP silencing increases paclitaxel uptake and suppresses tumor growth in vivo.
The results of our in vitro studies of LPP silencing suggest that LPP mediates the effect of CAFs in facilitating tumor angiogenesis and enhancing tumor vessel leakiness, which may subsequently reduce the uptake of chemotherapeutic agents by cancer cells. To determine the roles of LPP in tumor progression and chemoresistance in vivo, we treated OVCA432 ovarian tumor–bearing mice twice weekly with tail-vein injections of chitosan nanoparticles incorporated with 5 μg control scrambled siRNA, murine Lpp–targeting siRNA 1, or murine Lpp–targeting siRNA 2 in combination with weekly i.p. injections of either sterile PBS or paclitaxel (3.5 mg/kg) for 6 weeks. All mice in all treatment groups were euthanized at the experimental endpoint. We harvested and weighed the i.p. tumor nodules and found that endothelial Lpp expression in tumor tissues from mice treated with Lpp-targeting siRNAs was markedly lower than that in tumor tissues from mice treated with scrambled siRNA (Figure 2A). Furthermore, mice treated with Lpp-targeting siRNA 1 or siRNA 2 had significantly smaller tumor burdens than did mice treated with scrambled siRNA (P = 0.0048 and P = 0.0008, respectively) (Figure 2B). Immunolocalization of tumor vessels by CD31 staining revealed that the microvessel densities in the Lpp-silenced groups were lower than those in the control group (Figure 2C), suggesting that Lpp silencing suppresses tumor angiogenesis and cancer progression.
Figure 2. LPP silencing increases paclitaxel uptake and suppresses tumor growth in vivo.

(A) Hematoxylin- counterstained micrographs showing that endothelial Lpp expression in tumor tissues collected from mice treated with Lpp-targeting siRNAs was markedly lower than that in tumor tissues collected from control mice treated with scrambled siRNA (arrowheads indicate tumor microvessels). Scale bars: 50 μm. (B) Mice treated with Lpp-targeting siRNA 1or siRNA 2 had significantly smaller tumor burdens than did scrambled siRNA–treated mice (P = 0.0048 and P = 0.0008, respectively). In addition, paclitaxel-treated mice injected with Lpp-targeting siRNA 1 or Lpp-targeting siRNA 2 had significantly lower tumor weights than did scrambled siRNA–injected mice (n = 10/group; mean ± SD; P = 0.0055 and P = 0.0005, respectively, by Mann-Whitney U test). (C) Mice treated with Lpp-targeting siRNA 1 or siRNA 2 had significantly lower microvessel densities than did control group mice (n = 10/group; mean ± SD; P = 0.019 and P = 0.003, respectively, by Mann-Whitney U test). Microvessel densities were determined by immunolocalization of CD31-positive microvessels in harvested tumor nodules. (D) Fluorescence micrographs showing that the FITC-dextran signals in ovarian tumor tissues harvested from mice treated with Lpp-targeting siRNA 1 and from mice treated with Lpp-targeting siRNA 2 were significantly lower than those in ovarian tumor tissues from control mice, indicating reduced vessel leakiness in tumors from mice treated with Lpp-targeting siRNAs. Mice were injected with FITC-dextran via the tail vein 1 hour before sacrifice. Tumor vessel leakiness was evaluated by fluorescence microscopic quantification of tumor tissue FITC-dextran signals. Green: FITC-dextran; red: CD31. (E) Fluorescence-labeled paclitaxel signals in ovarian tumor tissues harvested from mice treated with Lpp-targeting siRNA 1 and from mice treated with Lpp-targeting siRNA 2 were significantly higher than those in control tumor tissues, suggesting increased drug delivery to the tumors via circulation in mice treated with Lpp-targeting siRNAs. Mice were injected with Oregon Green 488 fluorescence–labeled paclitaxel via the tail vein 1 hour before sacrifice. Drug delivery was evaluated by quantifying the green fluorescence signals in the tumor tissue. Green: Oregon Green 488–paclitaxel; red: CD3. (D and E) Scale bars: 100 μm (top), 50 μm (bottom).
Next, we determined whether Lpp silencing can increase paclitaxel delivery to ovarian cancer cells through tumor vessel normalization and promote the treatment efficacy of paclitaxel in ovarian tumor–bearing mice. For each of the aforementioned siRNA treatment groups, we injected half the mice with FITC-dextran via the tail vein 1 hour before euthanasia to evaluate tumor vessel leakiness and injected the other half with Oregon Green 488 fluorescence–labeled paclitaxel via the tail vein 1 hour before euthanasia to evaluate drug delivery within the tumor tissue. As expected, among the mice injected with the scrambled siRNA, the tumor burden in mice treated with paclitaxel was significantly smaller than that in mice treated with PBS (P = 0.0107). In addition, the tumor weights in the paclitaxel-treated mice injected with either Lpp-targeting siRNA was significantly smaller than tumor weights in mice injected with scrambled siRNA (P = 0.0055 and P = 0.0005) (Figure 2B), suggesting that Lpp confers paclitaxel resistance in these mice.
Fluorescence microscopy was used to visualize FITC-dextran and Oregon Green 488 green fluorescence–labeled paclitaxel in frozen tissue sections prepared from tumor nodules harvested from the different treatment groups. Compared with those from control mice, ovarian tumor tissues from mice treated with Lpp-targeting siRNA had a markedly lower FITC-dextran signal (Figure 2D). Because dextran, which has a molecular weight of 70,000 kDa, can pass through only the endothelial cell layer of leaky tumor vessels, the lower FITC-dextran signal in the tumors from mice treated with Lpp-targeting siRNA suggests that Lpp silencing decreases vessel leakiness in the tumor tissue of these mice. The fluorescence-labeled paclitaxel signal in ovarian tumor tissues harvested from mice treated with Lpp-targeting siRNA was substantially higher than that in tumor tissues from control mice (Figure 2E), suggesting that Lpp silencing promotes the delivery of paclitaxel via blood vessels to cancer cells and subsequently increases the bioavailability of the agent to cancer cells in these mice.
CAF-derived MFAP5 upregulates endothelial LPP expression.
To identify CAF-derived mediators that modulate LPP expression in MECs, we first examined the promoter sequence of LPP. We found that this sequence has multiple AP1-binding sites, which suggests that LPP expression can be regulated by the c-Fos/c-Jun transcriptional complex (Supplemental Figure 3). By querying the IPA database, we obtained a list of upstream ligands that have been shown to activate c-Fos/c-Jun signaling pathways. By comparing the IPA ligand list with our information on upregulated secretory ligands identified in CAFs (8), we generated a list of secretory ligands that were overexpressed in CAFs compared with expression in NOFs and that have been shown to activity c-Fos/c-Jun (Supplemental Table 2). Among them, MFAP5 was selected for further validation studies, because MFAP5 has recently been shown to be a CAF-derived mediator that can promote ovarian cancer cell motility through the c-Jun signaling cascade and because stromal MFAP5 overexpression is associated with poor clinical outcomes in patients with HGSC (18).
To determine whether CAF-derived MFAP5 in the tumor microenvironment can upregulate LPP in endothelial cells, we performed a correlative study of MFAP5 expression levels in CAFs and LPP expression levels in MECs in 96 HGSC tissue samples. We found that CAF-derived MFAP5 expression was significantly correlated with endothelial LPP expression (Figure 3A). To determine whether CAF-derived MFAP5 upregulates LPP in endothelial cells, we treated TIME and human MEC-1 (hMEC-1) cells with recombinant MFAP5 (recMFAP5) or PBS. qRT-PCR and Western blot analyses showed that cells treated with recMFAP5 had significantly higher LPP levels than did those treated with PBS (Figure 3, B and C).
Figure 3. CAF-derived MFAP5 modulates endothelial LPP expression and tumor vasculature.

(A) Plot shows a significant correlation between LPP expression in endothelial cells and MFAP5 expression in CAFs (n = 96; R = 0.652, P < 0.001, by Spearman rank correlation). Hematoxylin-counterstained images of immunolocalization of MFAP5 and LPP in 2 HGSC tissue samples showing that high levels of endothelial LPP expression were associated with high levels of stromal MFAP5 (Case 815) and that low levels of endothelial LPP expression were associated with low levels of stromal MFAP5 (Case 1265). Scale bars: 50 μm. (B) qRT-PCR analyses show that TIME and hMEC-1 MECs treated with recMFAP5 had significantly higher levels of LPP mRNA than did PBS-treated MECs (mean ± SEM of 3 independent experiments; P values were determined by 2-tailed Student’s t test). (C) Western blots show that TIME and hMEC-1 MECs treated with recMFAP5 had markedly increased LPP protein expression levels compared with PBS-treated MECs. (D) Murine fibroblasts transfected with 3 different Mfap5-specific siRNAs had significantly lower levels of Mfap5 mRNA expression than did those transfected with the scrambled siRNA or the vehicle (mean ± SEM of 3 independent experiments; †P < 0.001, by 2-tailed Student’s t test). (E) Bioluminescence images showing markedly decreased luciferase signals in A224 ovarian tumor–bearing mice treated with chitosan nanoparticles incorporated with Mfap5-targeting siRNAs compared with mice injected with chitosan nanoparticles incorporated with the scrambled siRNA. Tumor growth was monitored using the IVIS 200 Bioluminescence and Fluorescence Imaging System. (F) Box and whisker plot showing significantly lower luminescence signal intensities in mice treated with chitosan nanoparticles incorporated with Mfap5-targeting siRNA 68 and Mfap5-targeting siRNA 69 than signals in mice injected with chitosan nanoparticles incorporated with the scrambled siRNA. Boxes represent the interquartile range of the records, and the lines across the boxes indicate the median. Whiskers indicate the highest and lowest values that were no greater than 1.5 times the interquartile range (n = 10 per group; *P < 0.01, by Mann-Whitney U test). (G) Box and whisker plot showing that the tumor weights in mice treated with Mfap5-targeting siRNA were significantly lower than tumor weightsin mice treated with scrambled siRNA at the experimental endpoint (n = 10/group; *P < 0.01, by Mann-Whitney U test). (H) Hematoxylin-counterstained images of immunolocalization of murine Mfap5 and CD34 show that tumors from Mfap5-targeting siRNA– treated mice had markedly lower stromal Mfap5 expression and lower CD34-positive microvessel densities than did tumors from control mice (n = 5 per group; mean ± SD; *P < 0.01, by Mann-Whitney U test). Tumor cells were injected i.p. Scale bars: 50 μm. (I) Hematoxylin-counterstained images of immunolocalization of murine Mfap5 and CD34 show that tumors from Mfap5-targeting siRNA– treated mice had markedly lower stromal Mfap5 expression and lower CD34-positive microvessel densities than did tumors from control mice (n = 5 per group; mean ± SD; *P < 0.01, by Mann-Whitney U test). Tumor cells were delivered by intraovarian injection. Scale bars: 50 μm. S, Stroma; T, Tumor. (J) Hematoxylin-counterstained images of immunolocalization of Lpp show that tumors from mice treated with Mfap5-targeting siRNAs had significantly lower endothelial Lpp expression levels than did those treated with scrambled siRNA. Arrowheads indicate microvessels in the tumor tissue. Scale bars: 50 μm.
Silencing of Mfap5 downregulates endothelial Lpp expression and reduces intratumoral microvessel densities and tumor progression in vivo.
To determine the roles of MFAP5 in regulating endothelial LPP expression and modulating tumor progression and angiogenesis in vivo, we first injected mice i.p. with A224 ovarian cancer cells. Two weeks after tumor cell injection, ovarian cancer–bearing mice were injected via the tail veins with chitosan nanoparticles with one of two different murine Mfap5-targeting siRNAs or control scrambled siRNA (Figure 3D). Using the IVIS 200 Bioluminescence and Fluorescence Imaging System (Caliper Life Sciences), we detected markedly lower luciferase activity in the Mfap5-targeting siRNA groups than in the control group (Figure 3, E and F). By week 6, we euthanized the mice and resected their tumors; the tumor weights in the Mfap5-targeting siRNA groups were significantly lower than were tumor weights in the scrambled siRNA-treated group (P < 0.001) (Figure 3G). Immunolocalization of murine Mfap5 and CD34 on paraffin-embedded sections of ovarian tumors from mice showed markedly lower stromal Mfap5 expression and lower CD34-positive microvessel densities in the Mfap5-targeting siRNA groups than in the control group, confirming that nanoparticle-delivered Mfap5-targeting siRNAs knocked down Mfap5 expression and reduced intratumoral microvessel densities (Figure 3H).
We further confirmed that CAF-derived Mfap5 regulates endothelial Lpp expression using a mouse model in which ovarian cancer cells were directly injected into the ovaries, and the aforementioned chitosan nanoparticle treatment schedule was used. Tumors from mice with stromal Mfap5 silencing had markedly lower CD34-positive microvessel densities than did tumors from mice without stromal Mfap5 silencing (Figure 3I). Immunostaining analysis revealed that the tumor tissue samples harvested from mice treated with Mfap5-targeting siRNAs had significantly lower endothelial Lpp expression than did those from mice treated with the scrambled siRNA, confirming that knockdown of Mfap5 downregulates endothelial Lpp expression (Figure 3J).
Fibroblast-derived MFAP5 enhances intratumoral microvessel formation.
To confirm the role of fibroblast-derived MFAP5 in the regulation of endothelial LPP and tumor angiogenesis in vivo, we s.c. coinjected nude mice with A224 ovarian cancer cells and ovarian fibroblasts, which had been transfected with MFAP5 full-length cDNA or a mock transfectant. Compared with those from mice injected with control fibroblasts, the tumors from mice injected with MFAP5-transfected fibroblasts showed a marked increase in progression, as demonstrated by increased cancer cell bioluminescence, dry tumor weights (Supplemental Figure 4, A and B), and higher microvessel density (Supplemental Figure 4, C and D). These data suggest that fibroblast-derived MFAP5 facilitates tumor angiogenesis and increases tumor growth rates in vivo. Furthermore, immunolocalization of Lpp on tissue sections revealed that endothelial Lpp expression in tumors formed from MFAP5-transfected, fibroblast-injected cells was substantially higher than that in tumors formed from control fibroblast–injected cells, suggesting that fibroblast-derived MFAP5 upregulates endothelial LPP expression (Supplemental Figure 4E).
recMFAP5 upregulates endothelial LPP expression and promotes angiogenesis in vivo.
To determine the extent to which MFAP5 protein promotes endothelial LPP expression, tumor progression, and angiogenesis in vivo, mice were implanted i.p. with Matrigel plugs reconstituted in recMFAP5 or control buffer. A histological analysis revealed that recMFAP5-containing Matrigel implants had more CD31-positive endothelial cells than did those containing PBS (Figure 4A). In addition, using the angiogenesis module of the MetaMorph imaging analysis software program (Molecular Devices) to determine the phenotype of infiltrated endothelial cells, we found that the recMFAP5-containing Matrigel implants had significantly longer total tube lengths, higher total tube areas, more segments, and more nodes than did the PBS-containing Matrigel implants (Figure 4B). To determine whether recMfap5 directly upregulates endothelial Lpp in vivo, we performed transcriptome profiling and qRT-PCR analyses, which showed significantly higher levels of Lpp mRNA in endothelial cells isolated from Matrigel plugs reconstituted in recMfap5 than in endothelial cells isolated from Matrigel plugs reconstituted in PBS (Figure 4, C and D). Upregulation of endothelial Lpp protein expression by recMfap5 in these i.p. implants was confirmed by immunostaining (Figure 4E). These data suggest that MFAP5 indeed upregulates LPP in MECs in vivo.
Figure 4. CAF-derived MFAP5 upregulates endothelial LPP expression and promotes angiogenesis in vivo.

(A) Micrographs showing that recMFAP5-containing Matrigel plugs implanted i.p. into mice had significantly more CD31-positive endothelial cells than did PBS-containing Matrigel implants. Matrigel reconstituted with basic FGF (bFGF), a known proangiogenic protein, was used as a positive control. Scale bars: 100 μm. (B) Box and whisker plots showing the effect of recMFAP5 on total tube length, total tube area, and segment and node numbers in Matrigel plugs reconstituted with recMFAP5. The phenotypes of the endothelial cell networks in the Matrigel implants were analyzed using MetaMorph software. Compared with that reconstituted with PBS, the Matrigel reconstituted with recMFAP5 had significantly longer total tube lengths, larger total tube areas, more segments, and more nodes. The boxes in the box plot represent the interquartile range, and the lines across the boxes indicate the median. The whiskers indicate the highest and lowest values that were no greater than 1.5 times the interquartile range (n = 10/group; P values were determined by Mann-Whitney U test). (C) Heatmap showing differentially expressed genes that were up- or downregulated in endothelial cells isolated from recMFAP5-containing Matrigel implants compared with cells from PBS-containing Matrigel implants in mice. Transcriptome profiling of endothelial cells isolated from Matrigel implants revealed that 394 genes were expressed at significantly higher levels and 449 genes were expressed at significantly lower levels in recMFAP5-containing Matrigel implants compared with cells from PBS-containing Matrigel implants (P < 0.05, by moderated t test and Benjamini-Hochberg multiple testing correction). Expression of Lpp was increased by 2-fold in endothelial cells isolated from recMFAP5-containing Matrigel implants compared with cells from PBS-containing Matrigel implants. (D) qRT-PCR analyses showing that endothelial cells isolated from recMFAP5-containing Matrigel implants had significantly higher levels of Lpp mRNA than did PBS-containing Matrigel implants (mean ± SEM of 3 independent experiments; 2-tailed Student’s t test). (E) Hematoxylin-counterstained micrographs showing that endothelial cells from recMFAP5-containing Matrigel implants had markedly higher Lpp protein levels than did cells from PBS-containing Matrigel implants. Scale bars: 10 μm.
LPP mediates the effect of MFAP5 on endothelial cell motility and monolayer permeability.
To determine whether LPP mediates the effect of MFAP5 on endothelial cell motility, we treated hMEC-1 and TIME human MECs transfected with LPP-targeting siRNAs or control scrambled siRNA with recMFAP5 or control buffer. Cells treated with MFAP5 had markedly increased motility potential, which was abrogated in cells transfected with LPP-targeting siRNAs but not in cells transfected with scrambled siRNA (Figure 5A), suggesting that LPP mediated the effects of MFAP5 on endothelial cell motility. In addition, we found that 3 times as many hMEC-1 and TIME cells invaded through porous cell culture inserts coated with Matrigel in recMFAP5-treated wells compared with that observed in control wells. Again, cells transfected with scrambled siRNA were significantly more invasive than were those transfected with LPP-targeting siRNAs (Figure 5B). These data suggest that LPP mediates the effect of MFAP5 on the invasive potential of these cells.
Figure 5. LPP mediates the effect of MFAP5 on endothelial cell motility and monolayer permeability.

(A) hMEC-1 and TIME endothelial cells treated with MFAP5 had markedly increased motility potential compared with control cells. This increase in motility induction was abrogated in cells transfected with LPP-targeting siRNA but not in cells transfected with control scrambled siRNA, which suggests that LPP mediates the effect of MFAP5 on endothelial cell motility (mean ± SEM of 3 independent experiments; *P < 0.01, by 2-tailed Student’s t test). (B) A significantly greater number of hMEC-1 and TIME cells invaded through porous Matrigel-coated cell culture inserts in the presence of recMFAP5 than in the absence of recMFAP5. The effect of MFAP5 on promoting invasive potential was abrogated in endothelial cells transfected with LPP-targeting siRNAs (mean ± SEM of 3 independent experiments; P values were determined by 2-tailed Student’s t test). (C) Micrographs show that recMFAP5 enhanced the tubular network formation of hMEC-1 and TIME cells seeded on Matrigel in a dose-dependent manner. Scale bars: 50 μm. (D) Image analyses showed dose-dependent increases in tube length, tube area, number of segments, and number of branch points for tubes formed from hMEC-1 and TIME cells seeded onto MFAP5-containing Matrigel compared with those formed from cells seeded onto control Matrigel (mean ± SEM of 3 independent experiments; P values were determined by 2-tailed Student’s t test). (E) Monolayer permeability analyses using the xCELLigence system show that MFAP5-treated, confluent endothelial cell monolayer hMEC-1 and TIME cultures had a marked decrease in impedance compared with PBS-treated cells (mean ± SEM of 4 independent experiments). (F) Effect of MFAP5 on the permeability of endothelial cell monolayers. hMEC-1 and TIME monolayers treated with recMFAP5 had a significantly greater amount of fluorescence-labeled dextran in the bottom wells of Transwells than did those treated with PBS (mean ± SEM of 3 independent experiments; P values were determined by 2-tailed Student’s t test). (G) Effect of LPP silencing on MFAP5-enhanced endothelial cell permeability. hMEC-1 and TIME monolayers treated with recMFAP5 had a significantly greater amount of fluorescence-labeled dextran in the bottom wells of Transwells than did those treated with PBS, and the effect was abrogated when hMEC-1 and TIME were transfected with LPP-targeting siRNA (mean ± SEM of 3 independent experiments; *P < 0.01, by 2-tailed Student’s t test).
Furthermore, a tube formation assay demonstrated that hMEC-1 and TIME cells seeded on Matrigel containing recMFAP5 had a dose-dependent tubular network formation that was enhanced compared with that in cells seeded on control Matrigel (Figure 5C). Further analysis using the angiogenesis module of MetaMorph imaging analysis software revealed that the total tube lengths, total tube areas, number of segments, and number of branch points of tubes formed from hMEC-1 and TIME cells seeded onto MFAP5-containing Matrigel were significantly and dose-dependently greater than those of tubes formed from cells seeded onto control Matrigel (P < 0.05) (Figure 5D). In addition, the effect of recMFAP5 on tube formation was abrogated in endothelial cells transfected with LPP-targeting siRNAs but not in cells transfected with scrambled siRNA. These data further support the notion that LPP mediates the proangiogenic roles of MFAP5 (Supplemental Figure 5, A and B).
To evaluate the effect of MFAP5 on endothelial cell monolayer permeability in vitro, we plated hMEC-1 and TIME cells onto the E-plate of an xCELLigence system (ACEA Biosciences) to create confluent monolayer cultures and used a real-time cell analyzer to measure impedance in the presence or absence of recMFAP5. Endothelial cell monolayer cultures treated with recMFAP5 had markedly lower impedance than did those without recMFAP5 treatment, suggesting a disruption of the endothelial monolayer barrier by MFAP5 (Figure 5E). To validate this observation, we performed an in vitro permeability assay by measuring the traversal of FITC-dextran probes (molecular mass, 70,000 kDa) through hMEC-1 and TIME cell monolayers to the bottom of a Transwell in the presence or absence of recMFAP5. The amount of fluorescence-labeled dextran in the recMFAP5-containing bottom wells was larger than that in the bottom wells that did not contain recMFAP5 (Figure 5F). To determine whether LPP mediates the effect of MFAP5 on endothelial cell monolayer permeability, we repeated the above experiments using endothelial cells transfected with LPP-targeting siRNAs or scrambled siRNA and observed that silencing LPP in endothelial cells abrogated the effects of MFAP5 on endothelial cell monolayer permeability (Figure 5G).
While MFAP5 mediated the motility and monolayer permeability of endothelial cells via upregulation of LPP expression, proliferation assay results showed that endothelial cell proliferation was not significantly affected by MFAP5 (Supplemental Figure 6).
LPP mediates the effect of MFAP5 on focal adhesions and stress fiber formation.
Capillary endothelium permeability and endothelial cell motility are modulated by mechanical forces that are conveyed by the ECM and focal adhesion formation (19–24). To determine the mechanism by which LPP modulates endothelial cell motility and microvessel permeability, we used immunofluorescence microscopy to assess the colocalization of LPP and key proteins associated with focal adhesions, including paxillin, FAK, and vinculin. LPP colocalized with all 3 molecules in the focal adhesions located at the cell membrane of the endothelial cells (Figure 6A), suggesting that LPP is a key component of the focal adhesions of endothelial cells. To determine the roles of LPP in focal adhesion formation, we silenced LPP in TIME and hMEC-1 MECs and used vinculin/LPP staining to determine the number of focal adhesions. Cells transfected with LPP-targeting siRNAs had significantly fewer focal adhesions than did those transfected with control scrambled siRNA (Figure 6B and Supplemental Figure 7A), suggesting that LPP plays a role in focal adhesion formation. The role of LPP in stress fiber formation was also determined by F-actin staining. TIME cells transfected with LPP-targeting siRNAs had markedly less stress fiber formation than did cells transfected with scrambled siRNA (Figure 6B and Supplemental Figure 7A).
Figure 6. LPP mediates the effect of MFAP5 on focal adhesions and stress fiber formation.

(A) Immunofluorescence micrographs showing that LPP colocalized with key focal adhesion proteins including paxillin, FAK, and vinculin in the focal adhesions located at the cell membrane of the 2 MEC lines hMEC-1 and TIME, suggesting that LPP is a key component of the focal adhesions of endothelial cells. Red: focal adhesion proteins; green: LPP; blue: nuclei. (B) Immunofluorescence micrographs showing that TIME MECs transfected with LPP-targeting siRNA had fewer F-actin stress fibers and focal adhesions than did cells transfected with control scrambled siRNA, suggesting that LPP plays important roles in stress fiber and focal adhesion formation. Dot plot summarizes the data (mean ± SD; n = 10/treatment group; P values were determined by 2-tailed Student’s t test). Red: F-actin/vinculin; green: LPP; blue: nuclei. (C) Immunofluorescence micrographs showing that hMEC-1 and TIME MECs treated with 200 ng/ml recMFAP5 had more F-actin stress fibers and focal adhesions than did cells treated with PBS, suggesting that MFAP5 could increase stress fiber formation in endothelial cells. Red: F-actin; blue: nuclei. (D) Immunofluorescence micrographs showing that hMEC-1 and TIME MECs treated with 200 ng/ml recMFAP5 had more focal adhesions than did cells treated with PBS, suggesting that MFAP5 could increase focal adhesions in endothelial cells. Green: vinculin; blue: nuclei. (E) Immunofluorescence micrographs showing that recMFAP5-treated cells had markedly more stress fibers (red) attached to upregulated LPP (green) in focal adhesions on the cell membrane than did control cells. (F) Immunofluorescence micrographs showing that MFAP5-induced stress fiber formation and that focal adhesions were abrogated in TIME cells transfected with LPP-targeting siRNAs but not with the control scrambled siRNA, suggesting that LPP mediates the effect of MFAP5 in increasing stress fiber formation and focal adhesions. Red: F-actin/vinculin; green: LPP; blue: nuclei. Data are summarized in the dot plot (mean ± SD of 10 independent experiments; P values were determined by 2-tailed Student’s t test). Scale bars: 5 μm (A–F).
Because we found that MFAP5 upregulates LPP in MECs, we determined whether MFAP5 increased focal adhesions and stress fiber formation in MECs. We treated hMEC-1 and TIME cells with recMFAP5 or PBS and assessed the number of focal adhesions and amount of stress fiber formation. Compared with cells treated with PBS, hMEC-1 and TIME cells treated with recMFAP5 had markedly increased focal adhesions and stress fiber formation (Figure 6, C and D). Immunofluorescence microscopy to assess the colocalization of LPP and F-actin revealed that MFAP5-treated cells also had markedly more stress fibers attached to upregulated LPP in focal adhesions on the cell membrane (Figure 6E).
To confirm that LPP mediates the effect of MFAP5 on increasing stress fiber formation and focal adhesions, we transfected TIME and hMEC-1 cells with LPP-targeting siRNAs or scrambled siRNA and then treated the cells with recMFAP5 or PBS. Stress fiber formation and focal adhesions were determined by F-actin and vinculin LPP staining, respectively. Compared with cells treated with PBS, those treated with recMFAP5 had markedly increased stress fiber formation and focal adhesions, and these effects were abrogated in cells transfected with LPP-targeting siRNAs but not in cells transfected with scrambled siRNA (Figure 6F and Supplemental Figure 7B).
Taken together, our data demonstrate that LPP mediates the effect of MFAP5 in the enhancement of focal adhesion and stress fiber formation, which may lead to increased endothelial cell motility and increased contractile forces within the cells, thus increasing microvessel permeability.
CAF-derived MFAP5 increases paclitaxel uptake and suppresses tumor growth in vivo.
Since our data showed that CAF-derived MFAP5 upregulates LPP expression in endothelial cells and our in vitro studies suggest that LPP silencing promotes the delivery of paclitaxel via blood vessels to cancer cells, increasing the bioavailability of the agent to cancer cells in mice, we hereby determined the effects of MFAP5 on paclitaxel resistance in ovarian tumor–bearing mice.
In this experiment, nude mice were s.c. coinjected with OVCA432 ovarian cancer cells with control ovarian fibroblasts or MFAP5-overexpressing ovarian fibroblasts. One week after the initial cancer cell and fibroblast injection, tumor-bearing mice were given weekly paclitaxel (3.5 mg/kg) injections via the tail vein for 2 weeks. One hour prior to euthanasia at the experimental endpoint, half the mice were injected with FITC-dextran via the tail vein for the evaluation of tumor vessel leakiness, and the remaining mice were injected with Oregon Green 488 green fluorescence–labeled paclitaxel via the tail vein for the evaluation of drug delivery within the tumor tissue. After euthanasia, s.c. tumor nodules were harvested and weighed. The experimental results showed that mice injected with a mixture of OVCA432 ovarian cells and MFAP5-overexpressing fibroblasts had significantly larger tumor burdens than did mice injected with a mixture of OVCA432 ovarian cancer cells and control fibroblasts on the basis of bioluminescence and tumor weights (P = 0.0138 and P < 0.0001, respectively) (Supplemental Figure 8A), suggesting that MFAP5 confers paclitaxel resistance to OVCA432 ovarian cancer cells.
To determine whether the presence of MFAP5 promotes tumor vessel leakiness and decreases paclitaxel delivery to ovarian cancer cells, we examined FITC-dextran and Oregon Green 488 green fluorescence–labeled paclitaxel on frozen tissue sections prepared from the harvested tumor nodules. Compared with tissues from mice injected with control fibroblasts, ovarian tumor tissues from mice injected with MFAP5-overexpressing fibroblasts had a significantly higher FITC-dextran signal (Supplemental Figure 8B), suggesting that MFAP5 increases vessel leakiness in the tumor tissue of these mice. The fluorescence-labeled paclitaxel signal in ovarian tumor tissues harvested from mice injected with MFAP5-overexpressing fibroblasts was markedly lower than that in tumor tissues from control mice (Supplemental Figure 8C), suggesting that MFAP5 reduces the delivery of paclitaxel via blood vessels to cancer cells and subsequently decreases the bioavailability of the agent to cancer cells in these mice.
CAF-derived MFAP5 activates LPP through the calcium-dependent MFAP5/FAK/ERK/LPP signaling pathway.
Our data indicated that LPP is a key downstream effector molecule that plays a role in modulating the effect of MFAP5 on endothelial cell motility and permeability. Previous studies showed that αVβ3 integrin is a major receptor for MFAP5 and that MFAP5 plays a role in αVβ3 integrin–mediated angiogenesis (25). In addition, Ca2+ mobilization is involved in integrin signaling and cell migration (26–28). We therefore hypothesized that the binding of MFAP5 to αVβ3 integrin activates calcium-dependent signaling pathways that transcriptionally upregulate LPP expression and subsequently increase the motility and permeability of endothelial cells.
To test these hypotheses, we first determined whether the effect of MFAP5 on LPP expression in endothelial cells is Ca2+ dependent. The stimulatory effect of MFAP5 on endothelial cell motility (Figure 7A) and stress fiber formation (Figure 7B) was abrogated in cells preloaded with the cell-permeant calcium chelator BAPTA/AM, suggesting that calcium signaling is involved in the modulation of MFAP5 function. Moreover, using the calcium dye Fluo-4 AM and confocal fluorescence microscopy, we found that exogenous recMFAP5 mobilized intracellular Ca2+ in hMEC-1 cells (Figure 7C). recMFAP5-induced calcium mobilization was attenuated in cells treated with the inositol 1,4,5-triphosphate receptor inhibitor xestospongin C but not the ryanodine receptor blocker (Figure 7, D and E), suggesting that MFAP5 induces calcium release via the inositol 1,4,5-triphosphate receptor instead of the ryanodine receptor. Furthermore, we also detected store-operated calcium entry into hMEC-1 cells (Figure 7F), which might also contribute to MFAP5-induced calcium mobilization after emptying of inositol 1,4,5-triphosphate–sensitive intracellular calcium stores.
Figure 7. CAF-derived MFAP5 activates LPP through the calcium-dependent MFAP5/FAK/ERK/LPP signaling pathway.

(A) hMEC-1 and TIME MECs treated with recMFAP5 had significantly higher motility rates than did MECs treated with the control buffer, and the stimulatory effect of MFAP5 on cell motility was abrogated in cells preloaded with the cell-permeant calcium chelator BAPTA-AM (mean ± SEM of 3 independent experiments; P < 0.01, by 2-tailed Student’s t test). (B) Fluorescence micrographs show that MFAP5-induced stress fiber formation was abrogated in MECs that had been preloaded with BAPTA-AM, suggesting that calcium signaling is involved in modulating MFAP5 function. Red: F-actin; blue: nuclei. Scale bars: 5 μm. (C–E) Mean normalized time courses of calcium mobilization induced by treating hMEC-1 cells with recMFAP5 in the absence and presence of calcium channel blockers. Calcium influx was monitored with confocal fluorescence microscopy. recMFAP5 was added to the imaging chamber at t0. Blue lines indicate the mean; red lines indicate the SEM. The inositol 1,4,5-triphosphate receptor inhibitor xestospongin C abrogated calcium mobilization, while inhibition of ryanodine receptor with ryanodine did not prevent calcium mobilization. (F) Mean normalized time courses of store-operated calcium entry. Thapsigargin was used to empty intracellular Ca2+ stores in the absence of extracellular Ca2+. Addition of Ca2+ to the medium at t0 resulted in rapid extracellular Ca2+ entry, which was inhibited by preincubation with SKF96365. Solid lines indicate the mean. Dotted lines indicate the SEM. (G) Western blot analyses showing that hMEC-1 and TIME endothelial cells treated with recMFAP5 had higher expression of p-FAK (Y861), p–PLC-γ1 (Y783), p-PKCθ (T538), p-ERK1/2 (T202/Y204), p-MLC2 (T18/S19), p-CREB (S133), c-Jun, and p–c-Jun (S73) compared with control cells. Relative normalized protein expression levels with respect to the corresponding controls are shown. Note: The blot groupings for p-CREB in hMEC-1 and TIME MECs and p-PKCθ in TIME MECs were generated from multiple gels that were run in parallel. (H) MFAP5-induced microvascular endothelial cell motility was suppressed in MECs treated with anti–αVβ3 integrin antibodies. hMEC-1 and TIME MECs were treated with 50 ng/ml recMFAP5 in the presence of an anti-α5 antibody, an anti-αvβ3 antibody, or the control IgG, and the effect on cell motility was determined by a Boyden chamber cell motility assay (mean ± SEM of 3 independent experiments; P < 0.01, by 2-tailed Student’s t test).
Next, we focused on intermediate signaling molecules that are implicated in the mediation of cell motility and calcium signaling. Compared with control cells, MECs treated with recMFAP5 had higher expression of phosphorylated FAK (p-FAK) (Y861), p–PLC-γ1 (Y783), p-PKCθ (T538), p-ERK1/2 (T202/Y204), phosphorylated myosin regulatory light chain 2 (p-MLC2) (T18/S19), phosphorylated cyclic AMP–responsive element-binding protein (p-CREB) (S133), c-Jun, and p–c-Jun (S73), which may have led to the upregulation of LPP expression and thus increased cell motility and permeability (Figure 7G).
Because our data demonstrated that MFAP5-induced microvascular endothelial cell motility was suppressed in cells that had been pretreated with an anti–αVβ3 integrin antibody (Figure 7H) and that MFAP5-upregulated p-FAK (Y861) expression was suppressed in cells that had been pretreated with BAPTA-AM (1,2-bis-[2-aminophenoxy]-ethane-N,N,N′,N′-tetraacetic acid, tetraacetoxymethyl ester) (Supplemental Figure 9A and Supplemental Figure 10A), we hypothesized that MFAP5-mediated responses in endothelial cells require the binding of MFAP5 to αVβ3 integrin, which leads to the activation of FAK. Activated FAK, in turn, activates PKCθ, which can regulate Ca2+ influx (29). Ca2+ mobilization activates ERK1/2 and leads to the phosphorylation of MLC2 and activation of CREB. The translocation of CREB to the nucleus and the binding of activated CREB to the cAMP response element of c-Jun may transcriptionally upregulate the expression of LPP, which contains multiple AP1-binding sites in its promoter sequence. The potential MFAP5-mediated signaling pathways are illustrated in Supplemental Figure 11.
To determine whether the binding of MFAP5 to αVβ3 integrin and the formation of the FAK-αVβ3 complex mediates MFAP5-induced FAK and PLC-γ1 phosphorylation, we pretreated hMEC-1 and TIME cells with an anti–αVβ3 integrin antibody (LM609), an anti-α5 antibody, or control IgG and then treated them with recMFAP5. The effect of recMFAP5 on FAK and PLC-γ1 phosphorylation was abrogated in cells pretreated with the anti–αVβ3 integrin antibody but not the anti-α5 antibody or control IgG (Supplemental Figure 9, B and C, and Supplemental Figure 10, B and C).
Next, we determined whether FAK phosphorylation mediates the MFAP5-induced phosphorylation of PKCθ in MECs. Western blot analysis of PKCθ in hMEC-1 and TIME cells treated with MFAP5 in the presence or absence of the FAK inhibitor PF573228 (Sigma-Aldrich) revealed that p-PKCθ expression was increased only in the absence of the FAK inhibitor (Supplemental Figure 9D and Supplemental Figure 10D).
Because previous studies demonstrated that PLC-γ1 phosphorylation can be stimulated not only by αVβ3 engagement alone (30) but also by the formation of a FAK-αVβ3 complex (31), we determined whether PLC-γ1 phosphorylation is FAK dependent. The MFAP5-induced phosphorylation of PLC-γ1 (Y783) was attenuated in hMEC-1 and TIME cells that had been pretreated with a FAK inhibitor (Supplemental Figure 9E and Supplemental Figure 10E), which suggests that MFAP5-induced PLC-γ1 (Y783) phosphorylation is FAK dependent. The MFAP5-induced phosphorylation of PKCθ was also attenuated in hMEC-1 and TIME cells that had been treated with a PLC inhibitor (U73122; sc-3574; Santa Cruz Biotechnology) (Supplemental Figure 9F and Supplemental Figure 10F), which suggests that PLC-γ1 phosphorylation regulates PKCθ activation. However, as described in a report of MFAP5-stimulated signaling in ovarian cancer cells (18), the upregulation of p–PLC-γ1 expression was abolished in cells treated with a PKCθ inhibitor, indicating that PKCθ phosphorylation and PLC-γ1 phosphorylation are interdependent (Supplemental Figure 9G and Supplemental Figure 10G).
To determine whether the ERK1/2 and CREB activation induced by MFAP5 via PKCθ and PLC-γ1 is Ca2+ dependent and mediated by the activation of an αVβ3 integrin/FAK/PKCθ pathway, we treated hMEC-1 and TIME cells with MFAP5 in the presence or absence of BAPTA-AM and a PKCθ pseudosubstrate inhibitor. Western blot analysis revealed that the phosphorylation of PKCθ, PLC-γ1, ERK1/2, and CREB after recMFAP5-based treatment was attenuated in BAPTA-AM–loaded cells (Supplemental Figure 9, H–K and Supplemental Figure 10, H–K). These data suggest that MFAP5-induced activation of both ERK and CREB is calcium dependent. In addition, ERK1/2 phosphorylation was abrogated in cells treated with the PKCθ pseudosubstrate inhibitor (Supplemental Figure 9L and Supplemental Figure 10L), which suggests that MFAP5-induced ERK1/2 activation depends on the αVβ3 integrin/FAK/PKCθ pathway.
Previous studies reported that MLC2 and CREB activation depends on ERK (32, 33); therefore, we determined whether calcium-dependent ERK1/2 phosphorylation mediates the activation of MLC2 and CREB. The recMFAP5-stimulated phosphorylation of MLC2 and CREB was attenuated in hMEC-1 and TIME cells treated with an ERK1/2 inhibitor (FR180204; Merck) (Supplemental Figure 9, M and N, and Supplemental Figure 10, M and N), demonstrating that MFAP5 induces MLC2 and CREB activation via ERK1/2.
Furthermore, our data demonstrated that c-Jun, which contains a cAMP response element in its promoter, upregulated expression in MFAP5-treated hMEC-1 and TIME cells. Promoter analysis revealed that the LPP promoter consists of multiple potential AP1-binding sites (Supplemental Figure 3), suggesting that the transcriptional upregulation of LPP expression is controlled by CREB-mediated c-Jun expression. To confirm this, we evaluated the effects of a CREB-binding protein–CREB (CBP-CREB) interaction inhibitor and the c-Jun inhibitor SP600125 on MFAP5-treated cells. The CBP-CREB interaction inhibitor attenuated the upregulation of both total c-Jun and p–c-Jun expression, whereas the c-Jun inhibitor SP600125 abrogated the upregulation of LPP expression (Supplemental Figure 9, O and P, and Supplemental Figure 10, O, and P). These data confirm that the MFAP5-induced increase in LPP expression is calcium dependent and is mediated by the upregulation of c-Jun expression by CREB activation.
After finding that MFAP5 can activate the FAK/ERK/CREB signaling network to upregulate LPP expression, we determined whether LPP can modulate the effect of MFAP5 on downstream signaling network activation via a positive feedback loop, as LPP can be recruited to focal adhesions in MDCK epithelial cells and interacts with α-actinin in the focal adhesion complex (13, 14, 34). Western blot analysis of FAK phosphorylation in recMFAP5-treated hMEC-1 and TIME cells transfected with LPP-targeting siRNAs or control scrambled siRNA revealed that LPP-targeting siRNA successfully abrogated MFAP5-induced FAK phosphorylation (Supplemental Figure 9Q and Supplemental Figure 10Q). In addition, knockdown of LPP expression attenuated an MFAP5-stimulated increase in focal adhesion formation (Figure 6, C and D). These findings suggest that focal adhesion targeting by LPP plays an essential role in focal adhesion complex formation and downstream signaling molecule activation, both of which mediate the effect of MFAP5 on endothelial cell motility and permeability.
Discussion
The present study demonstrates, for the first time to our knowledge, that a CAF-derived mediator elevates LPP expression in cancer-associated MECs in the tumor microenvironment and that LPP, a prognostic marker associated with poor survival rates in HGSC patients, confers paclitaxel resistance by increasing the motility and monolayer permeability of endothelial cells. Collectively, our data show, for the first time to our knowledge, that LPP can increase the motility of MECs and promote tumor progression. In addition, we believe our findings provide important information on the roles of CAFs in the modulation of tumor angiogenesis and chemoresistance.
MECs in tumor vessels are known to form abnormal monolayers, and they do not have a normal barrier function (35). These cells are disorganized and irregularly shaped. They also have loose interconnections and focal intercellular openings, which are probably responsible for increased vessel leakiness. We found that increased LPP expression facilitated the formation of focal adhesion complexes, increased cell traction force in endothelial cells, and increased leakiness in endothelial cell monolayers, suggesting that LPP plays an important role in the formation of disorganized microvessels within the tumor tissue. The increased focal adhesion, stress fiber formation, and traction force in cells enabled the establishment of contractile forces that pull apart the interendothelial cell junctions, thus increasing permeability.
In the present study, we showed that LPP expression modulates tumor vessel integrity. Blood vessel leakiness not only plays a role in angiogenesis, tumor growth, and metastasis but also affects drug delivery and drug resistance. Despite a severely defective barrier function and increase in diameter, tumor vessels do not facilitate drug delivery, because their high interstitial pressure limits the extravasation of fluid and macromolecules (36, 37). Our in vivo data show that LPP silencing significantly increased paclitaxel delivery to the tumor tissue in mice, indicating that LPP in MECs plays an important role in microvessel leakiness and paclitaxel delivery to tumor cells. Our data also suggest that targeting LPP normalizes tumor blood vessels, thereby facilitating drug delivery to tumor tissue and increasing drug efficacy.
Both cancer cells and stromal cells produce VEGF glycoproteins and proangiogenic factors, including FGFs and PDGFs. These relatively cell-type–nonspecific factors are important regulators of tumor angiogenesis, but the crucial roles of stromal-specific proangiogenic factors in tumor progression remain unclear. Antiangiogenic agents have been used to suppress uncontrolled tumor vessel formation and therefore normalize the vessel system for improved drug delivery (38–41). However, several clinical trials in cancer patients have demonstrated that agents targeting VEGF family members convey a progression-free survival advantage but rarely an overall survival advantage, possibly because other potent proangiogenic factors and their downstream effector molecules are present in the tumor microenvironment and endothelial cells, respectively, leading to insufficient suppression of tumor angiogenesis (1, 2, 42).
In the present study, we demonstrated for the first time to our knowledge that MFAP5, a novel CAF-derived proangiogenic marker (18), induced endothelial cell permeability and leakiness by upregulating LPP. We also showed that MFAP5 bound to αVβ3 integrin in MECs and thus activated a calcium-dependent FAK/ERK/MLC2/CREB signaling network to upregulate LPP. In addition, we demonstrated that LPP silencing significantly decreased MFAP5-activated FAK phosphorylation in endothelial cells, indicating that LPP in the focal adhesion complex not only facilitates the formation of stress fibers but also plays a role in activating the MFAP5 downstream signaling network. Together, these networks of CAF–endothelial cell crosstalk may decrease the effectiveness of current antiangiogenic agents that target VEGF family members.
Our studies focused on the use of NOFs and CAFs derived from the ovarian site, since the ovary is the preferred site for ovarian cancer development. Cancer cells, derived either from the tubal epithelium or from the ovarian surface epithelium, interact with local ovarian fibroblasts or fibroblasts recruited to the ovarian site during tumor development. As the disease progresses, tumor spreads to the omentum, which is the preferred metastatic disease site for ovarian cancer cells (43). Further study of the interaction between omental CAFs and metastatic ovarian cancer cells could provide additional insights into the roles of CAFs in ovarian cancer progression.
In conclusion, we characterized the roles of LPP in ovarian cancer angiogenesis and delineated the underlying mechanism by which CAF-derived MFAP5 modulates LPP expression in endothelial cells. In addition, our data highlight the importance of the activation of CAF–endothelial cell crosstalk signaling in modulating chemoresistance in patients with ovarian cancer. More important, we demonstrated the feasibility and improved the efficacy of using LPP-targeting siRNA in combination with cytotoxic drugs as a treatment for ovarian cancer. Our findings support the idea that therapies targeting both CAFs and endothelial cells in the ovarian tumor microenvironment may synergize with other cancer cell–targeting regimens to increase treatment efficacy.
Methods
Cell lines and culture conditions.
hMEC-1 cells were cultured in MCDB131 medium supplemented with 10% FBS, 10 mM L-glutamine, 10 ng/ml EGF, and 1 μg/ml hydrocortisone. TIME cells were cultured in endothelial cell growth medium-2 (Lonza). Both endothelial cell lines were obtained from the ATCC. The ovarian adenocarcinoma cell lines A224 (gift of Michael Birrer’s laboratory, University of Alabama, Tuscaloosa, Alabama, USA) and OVCA432 (gift of Robert Bast’s laboratory, The University of Texas MD Anderson Cancer Center) were maintained in RPMI 1640 medium supplemented with 10% FBS and 2 mM glutamine. Human fibroblast cultures were maintained in 1:1 MCDB105/199 medium supplemented with 10% FBS and 1 ng/ml EGF.
In vivo silencing of endothelial Lpp.
To evaluate the effects of endothelial LPP expression on ovarian tumor progression in vivo, we i.p. injected 2 × 106 luciferase-labeled OVCA432 cells into 6-week-old female nude mice (Envigo). OVCA432 ovarian tumor–bearing mice were given twice-weekly tail-vein injections of chitosan nanoparticles with 5 μg control scrambled siRNA, murine Lpp–targeting siRNA 1, or murine Lpp–targeting siRNA 2 and weekly i.p. injections of either sterile PBS or paclitaxel (3.5 mg/kg) for 6 weeks. For each experiment group, half the animals were given 100 μl of 10 mg/ml FITC-dextran (Sigma-Aldrich) via the tail vein before evaluation of intratumoral blood vessel leakiness; the remaining animals received Oregon Green 488–conjugated paclitaxel (1 mg/kg; Life Technologies, Thermo Fisher Scientific) via i.v. injection 1 hour before they were evaluated for paclitaxel biodistribution. All mice in all treatment groups were euthanized at the experimental endpoint. Intraperitoneal tumor nodules were harvested, weighed, and fixed for histological analysis. In addition to formalin tissue sections, 6-μm frozen tissue sections were prepared from tumors harvested using a CM1850 cryostat (Leica Microsystems) to evaluate the effect of LPP silencing on intratumoral microvessels and the bioavailability of paclitaxel by fluorescence microscopy.
In vivo silencing of stromal Mfap5.
To determine the roles of MFAP5 in regulating endothelial LPP expression and modulating tumor progression and angiogenesis in vivo, we injected 2 × 106 A224 ovarian cancer cells i.p. into 6-week-old female nude mice (Envigo). Two weeks after tumor cell injection, ovarian cancer–bearing mice were injected twice weekly via the tail vein with chitosan nanoparticles incorporated with 1 of 2 different murine Mfap5–targeting siRNAs or control scrambled siRNA for a total of 6 weeks. Tumor progression was monitored using an IVIS 200 Bioluminescence and Fluorescence Imaging System (Caliper Life Sciences) throughout the course of the experiment. By week 6, all animals were euthanized, and tumor tissues were resected and weighted. Immunolocalization of murine Mfap5, CD34, and Lpp on paraffin-embedded sections of ovarian tumors from mice was performed.
In vivo implantation of Matrigel plugs.
To determine the extent to which MFAP5 protein promotes endothelial LPP expression, tumor progression, and angiogenesis in vivo, mice were implanted i.p. with Matrigel plugs reconstituted in recMFAP5 or control buffer. Five days after implantation, the Matrigel plugs were resected, and a phenotypic analysis of CD31-positive endothelial cells was performed using the angiogenesis module of MetaMorph Imaging Analysis software (Molecular Devices). To determine whether recMfap5 directly upregulates endothelial Lpp in vivo, we performed transcriptome profiling on total RNA samples isolated from mouse endothelial cells that invaded into the Matrigel plugs. Total RNA (100 ng) from each group of Matrigel plugs was used to generate biotin-labeled RNA with a MessageAmp Premier RNA Amplification Kit (Life Technologies, Thermo Fisher Scientific) according to the manufacturer’s protocol. Biotin-labeled RNA samples from mouse endothelial cells were then subjected to whole-genome transcriptome profiling using a GeneChip Mouse Genome 430 2.0 Array (Affymetrix). qRT-PCR and immunostaining were performed to further validate the upregulation of LPP expression by MFAP5.
Accession numbers.
Data files from the transcriptome profiling analysis were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE70344 and GSE106519).
Statistics.
SPSS 19 (IBM Corporation) and GraphPad Prism 5.0 (GraphPad Software) were used to perform statistical analyses. All in vitro experiments were repeated independently in triplicate, and a 2-tailed Student’s t test was used to determine differences in sample means. The Mann-Whitney U test was used in animal studies. For transcriptome analyses, Genespring GX Bioinformatics Suite, version 14.9 (Agilent Technologies) was used. A P value of less than 0.05 was considered statistically significant, and a moderated t test and Benjamini-Hochberg multiple testing correction were used as appropriate.
Study approval.
Patients’ tissue samples were collected from the Ovarian Cancer Repository under protocols approved by the IRB of The University of Texas MD Anderson Cancer Center, and all animal experiments were approved by the IACUC of The University of Texas MD Anderson Cancer Center.
Additional information is provided in the Supplemental Methods.
Author contributions
CSL, TLY, MJB, and SCM conceptualized the study. CSL, TLY, KPY, SYH, LSM, AKS, and GLB designed the experiments. CSL, TLY, KKW, JS, and STCW performed formal data analysis. CSL, TLY, KPY, and SYH conducted the experiments. CSL, TLY, KPY, JS, STCW, MJB, and SCM wrote the manuscript. MJB and SCM supervised the study.
Supplementary Material
Acknowledgments
This study was supported in part by NIH grants (R01 CA133057, R01 CA142832, RC4 CA156551, U54 CA151668, U01 188388, CA177909, and UH2 TR000943); a University of Texas MD Anderson Cancer Center Ovarian Cancer Specialized Programs of Research Excellence (SPORE) grant (P50 CA083639), a Uterine Cancer SPORE grant (P50 CA098258), and an NIH Cancer Center Support grant (P30 CA016672); the U.S. Department of Health and Human Services; the Gilder Foundation; Cancer Prevention & Research Institute of Texas grants (RP100094 and RP110595); US Department of Defense grants (W81XWH-16-2-0038 and W81XWH-17-1-0126); the Mary K. Chapman Foundation; and the Ovarian Cancer Research Fund. Editorial support was provided by the Department of Scientific Publications at The University of Texas MD Anderson Cancer Center.
Version 1. 12/18/2017
Electronic publication
Version 2. 02/01/2018
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: J Clin Invest. 2018;128(2):589–606.https://doi.org/10.1172/JCI95200.
Contributor Information
Tsz-Lun Yeung, Email: TYeung@mdanderson.org.
Kay-Pong Yip, Email: dyip@health.usf.edu.
Kwong-Kwok Wong, Email: kkwong@mdanderson.org.
Gabriel Lopez-Berestein, Email: glopez@mdanderson.org.
Jianting Sheng, Email: jsheng@houstonmethodist.org.
References
- 1.Choi HJ, Armaiz Pena GN, Pradeep S, Cho MS, Coleman RL, Sood AK. Anti-vascular therapies in ovarian cancer: moving beyond anti-VEGF approaches. Cancer Metastasis Rev. 2015;34(1):19–40. doi: 10.1007/s10555-014-9538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burger RA. Overview of anti-angiogenic agents in development for ovarian cancer. Gynecol Oncol. 2011;121(1):230–238. doi: 10.1016/j.ygyno.2010.11.035. [DOI] [PubMed] [Google Scholar]
- 3.Perren TJ, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. doi: 10.1056/NEJMoa1103799. [DOI] [PubMed] [Google Scholar]
- 4.Aghajanian C, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. doi: 10.1200/JCO.2012.42.0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pujade-Lauraine E, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32(13):1302–1308. doi: 10.1200/JCO.2013.51.4489. [DOI] [PubMed] [Google Scholar]
- 6.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5(15):1597–1601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 7.Yeung TL, Leung CS, Li F, Wong SS, Mok SC. Targeting stromal-cancer cell crosstalk networks in ovarian cancer treatment. Biomolecules. 2016;6(1):3. doi: 10.3390/biom6010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeung TL, et al. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013;73(16):5016–5028. doi: 10.1158/0008-5472.CAN-13-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 10.Kitadai Y. Cancer-stromal cell interaction and tumor angiogenesis in gastric cancer. Cancer Microenviron. 2010;3(1):109–116. doi: 10.1007/s12307-009-0032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Au Yeung CL, et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. 2016;7:11150. doi: 10.1038/ncomms11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petit MM, Mols R, Schoenmakers EF, Mandahl N, Van de Ven WJ. LPP, the preferred fusion partner gene of HMGIC in lipomas, is a novel member of the LIM protein gene family. Genomics. 1996;36(1):118–129. doi: 10.1006/geno.1996.0432. [DOI] [PubMed] [Google Scholar]
- 13.Petit MM, et al. LPP, an actin cytoskeleton protein related to zyxin, harbors a nuclear export signal and transcriptional activation capacity. Mol Biol Cell. 2000;11(1):117–129. doi: 10.1091/mbc.11.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Itallie CM, et al. Biotin ligase tagging identifies proteins proximal to E-cadherin, including lipoma preferred partner, a regulator of epithelial cell-cell and cell-substrate adhesion. J Cell Sci. 2014;127(Pt 4):885–895. doi: 10.1242/jcs.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H, Chen X, Bollag WB, Bollag RJ, Sheehan DJ, Chew CS. Lasp1 gene disruption is linked to enhanced cell migration and tumor formation. Physiol Genomics. 2009;38(3):372–385. doi: 10.1152/physiolgenomics.00048.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thibault B, Castells M, Delord JP, Couderc B. Ovarian cancer microenvironment: implications for cancer dissemination and chemoresistance acquisition. Cancer Metastasis Rev. 2014;33(1):17–39. doi: 10.1007/s10555-013-9456-2. [DOI] [PubMed] [Google Scholar]
- 17.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307(5706):58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 18.Leung CS, et al. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat Commun. 2014;5:5092. doi: 10.1038/ncomms6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingber DE, Folkman J. Mechanochemical switching between growth and differentiation during fibroblast growth factor-stimulated angiogenesis in vitro: role of extracellular matrix. J Cell Biol. 1989;109(1):317–330. doi: 10.1083/jcb.109.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumoto T, Yung YC, Fischbach C, Kong HJ, Nakaoka R, Mooney DJ. Mechanical strain regulates endothelial cell patterning in vitro. Tissue Eng. 2007;13(1):207–217. doi: 10.1089/ten.2006.0058. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh K, Thodeti CK, Dudley AC, Mammoto A, Klagsbrun M, Ingber DE. Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc Natl Acad Sci U S A. 2008;105(32):11305–11310. doi: 10.1073/pnas.0800835105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scherr DS. Commentary on “tissue-specific mutagenesis by N-butyl-N-(4-hydroxybutyl) nitrosamine as the basis for urothelial cell carcinogenesis.” He Z, Kosinska W, Zhao ZL, Wu XR, Guttenplan JB, Department of Basic Science, New York University Dental College, NY, USA.: Mutat Res 2012;742(1-2):92-5 [Epub 2011 Dec 4] Urol Oncol. 2014;32(2):214. doi: 10.1016/j.urolonc.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 23.Califano JP, Reinhart-King CA. Exogenous and endogenous force regulation of endothelial cell behavior. J Biomech. 2010;43(1):79–86. doi: 10.1016/j.jbiomech.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 24.Krishnan R, et al. Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am J Physiol, Cell Physiol. 2011;300(1):C146–C154. doi: 10.1152/ajpcell.00195.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerr JS, Mousa SA, Slee AM. Alpha(v)beta(3) integrin in angiogenesis and restenosis. Drug News Perspect. 2001;14(3):143–150. [PubMed] [Google Scholar]
- 26.Eddy RJ, Pierini LM, Matsumura F, Maxfield FR. Ca2+-dependent myosin II activation is required for uropod retraction during neutrophil migration. J Cell Sci. 2000;113(Pt 7):1287–1298. doi: 10.1242/jcs.113.7.1287. [DOI] [PubMed] [Google Scholar]
- 27.Lawson MA, Maxfield FR. Ca(2+)- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377(6544):75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- 28.Coppolino MG, Woodside MJ, Demaurex N, Grinstein S, St-Arnaud R, Dedhar S. Calreticulin is essential for integrin-mediated calcium signalling and cell adhesion. Nature. 1997;386(6627):843–847. doi: 10.1038/386843a0. [DOI] [PubMed] [Google Scholar]
- 29.Manicassamy S, Sadim M, Ye RD, Sun Z. Differential roles of PKC-theta in the regulation of intracellular calcium concentration in primary T cells. J Mol Biol. 2006;355(3):347–359. doi: 10.1016/j.jmb.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 30.Nakamura I, Lipfert L, Rodan GA, Le TD. Convergence of alpha(v)beta(3) integrin- and macrophage colony stimulating factor-mediated signals on phospholipase Cgamma in prefusion osteoclasts. J Cell Biol. 2001;152(2):361–373. doi: 10.1083/jcb.152.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masson-Gadais B, Houle F, Laferrière J, Huot J. Integrin alphavbeta3, requirement for VEGFR2-mediated activation of SAPK2/p38 and for Hsp90-dependent phosphorylation of focal adhesion kinase in endothelial cells activated by VEGF. Cell Stress Chaperones. 2003;8(1):37–52. doi: 10.1379/1466-1268(2003)8<37:IVRFVA>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Curtis J, Finkbeiner S. Sending signals from the synapse to the nucleus: possible roles for CaMK, Ras/ERK, and SAPK pathways in the regulation of synaptic plasticity and neuronal growth. J Neurosci Res. 1999;58(1):88–95. doi: 10.1002/(SICI)1097-4547(19991001)58:1<88::AID-JNR9>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 33.Mavria G, et al. ERK-MAPK signaling opposes Rho-kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell. 2006;9(1):33–44. doi: 10.1016/j.ccr.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 34.Hansen MD, Beckerle MC. Alpha-Actinin links LPP, but not zyxin, to cadherin-based junctions. Biochem Biophys Res Commun. 2008;371(1):144–148. doi: 10.1016/j.bbrc.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hashizume H, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. doi: 10.1016/S0002-9440(10)65006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McDonald DM, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res. 2002;62(18):5381–5385. [PubMed] [Google Scholar]
- 37.Trédan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–1454. doi: 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- 38.Goel S, Wong AH, Jain RK. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb Perspect Med. 2012;2(3):a006486. doi: 10.1101/cshperspect.a006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goel S, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain RK. Taming vessels to treat cancer. Sci Am. 2008;298(1):56–63. doi: 10.1038/scientificamerican0108-56. [DOI] [PubMed] [Google Scholar]
- 41.Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427(6976):695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 42.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeung TL, Leung CS, Yip KP, Au Yeung CL, Wong ST, Mok SC. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am J Physiol, Cell Physiol. 2015;309(7):C444–C456. doi: 10.1152/ajpcell.00188.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.