

SUMMARY
There is considerable evidence that chromosome structure plays important roles in gene control, but we have limited understanding of the proteins that contribute to structural interactions between gene promoters and their enhancer elements. Large DNA loops that encompass genes and their regulatory elements depend on CTCF-CTCF interactions, but most enhancer-promoter interactions do not employ this structural protein. Here, we show that the ubiquitously expressed transcription factor Yin Yang 1 (YY1) contributes to enhancer-promoter structural interactions in a manner analogous to DNA interactions mediated by CTCF. YY1 binds to active enhancers and promoter-proximal elements and forms dimers that facilitate the interaction of these DNA elements. Deletion of YY1 binding sites or depletion of YY1 protein disrupts enhancer-promoter looping and gene expression. We propose that YY1-mediated enhancer-promoter interactions are a general feature of mammalian gene control.
Graphical abstract
INTRODUCTION
Cell-type-specific gene expression programs in humans are generally controlled by gene regulatory elements called enhancers (Buecker and Wysocka, 2012; Bulger and Groudine, 2011; Levine et al., 2014; Ong and Corces, 2011; Ren and Yue, 2015). Transcription factors (TFs) bind these enhancer elements and regulate transcription from the promoters of nearby or distant genes through physical contacts that involve looping of DNA between enhancers and promoters (Bonev and Cavalli, 2016; Fraser et al., 2015; Heard and Bickmore, 2007; de Laat and Duboule, 2013; Pombo and Dillon, 2015; Spitz, 2016). Despite the fundamental importance of proper gene control to cell identity and development, the proteins that contribute to structural interactions between enhancers and promoters are poorly understood.
There is considerable evidence that enhancer-promoter interactions can be facilitated by transcriptional cofactors such as Mediator, structural maintenance of chromosomes (SMC) protein complexes such as cohesin, and DNA binding proteins such as CTCF. Mediator can physically bridge enhancer-bound transcription factors and the promoter-bound transcription apparatus (Allen and Taatjes, 2015; Jeronimo et al., 2016; Kagey et al., 2010; Malik and Roeder, 2010; Petrenko et al., 2016). Cohesin is loaded at active enhancers and promoters by the Mediator-associated protein NIPBL and may transiently stabilize enhancer-promoter interactions (Kagey et al., 2010; Schmidt et al., 2010). CTCF proteins bound at enhancers and promoters can interact with one another and may thus facilitate enhancer-promoter interactions (Guo et al., 2015; Splinter et al., 2006), but CTCF does not generally occupy these interacting elements (Cuddapah et al., 2009; Kim et al., 2007; Phillips-Cremins et al., 2013; Wendt et al., 2008).
Enhancer-promoter interactions generally occur within larger chromosomal loop structures formed by the interaction of CTCF proteins bound to each of the loop anchors (Gibcus and Dekker, 2013; Gorkin et al., 2014; Hnisz et al., 2016a; Merkenschlager and Nora, 2016). These loop structures, variously called topologically associating domains (TADs), loop domains, CTCF contact domains and insulated neighborhoods, tend to insulate enhancers and genes within the CTCF-CTCF loops from elements outside those loops (Dixon et al., 2012; Dowen et al., 2014; Hnisz et al., 2016b; Ji et al., 2016; Lupiáñez et al., 2015; Narendra et al., 2015; Nora et al., 2012; Phillips-Cremins et al., 2013; Rao et al., 2014; Tang et al., 2015). Constraining DNA interactions within CTCF-CTCF loop structures in this manner may facilitate proper enhancer-promoter contacts.
Evidence that CTCF-CTCF interactions play important global roles in chromosome loop structures but are only occasionally directly involved in enhancer-promoter contacts (Phillips and Corces, 2009) led us to consider the possibility that a bridging protein analogous to CTCF might generally participate in enhancer-promoter interactions. We report here that Yin Yang 1 (YY1) contributes to enhancer-promoter interactions in a manner analogous to DNA looping mediated by CTCF. YY1 and CTCF share many features: both are essential, ubiquitously expressed, zinc-coordinating proteins that bind hypo-methylated DNA sequences, form homodimers, and thus facilitate loop formation. The two proteins differ in that YY1 preferentially occupies interacting enhancers and promoters, whereas CTCF preferentially occupies sites distal from these regulatory elements that tend to form larger loops and participate in insulation. Deletion of YY1 binding sites or depletion of YY1 can disrupt enhancer-promoter contacts and normal gene expression. Thus, YY1-mediated structuring of enhancer-promoter loops is analogous to CTCF-mediated structuring of TADs, CTCF contact domains, and insulated neighborhoods. This model of YY1-mediated structuring of enhancer-promoter loops accounts for diverse functions reported previously for YY1, including contributions to both gene activation and repression and to gene dysregulation in cancer.
RESULTS
A Candidate Enhancer-Promoter Structuring Factor in Embryonic Stem Cells
We sought to identify a protein factor that might contribute to enhancer-promoter interactions in a manner analogous to that of CTCF at insulators. Such a protein would be expected to bind active enhancers and promoters, be essential for cell viability, show ubiquitous expression, and be capable of dimerization. To identify proteins that bind active enhancers and promoters, we sought candidates from chromatin immunoprecipitation with mass spectrometry (ChIP-MS), using antibodies directed toward histones with modifications characteristic of enhancer and promoter chromatin (H3K27ac and H3K4me3, respectively) (Creyghton et al., 2010), conducted previously in murine embryonic stem cells (mESCs) (Ji et al., 2015). Of 26 transcription factors that occupy both enhancers and promoters (Figure 1A), four (CTCF, YY1, NRF1, and ZBTB11) are essential based on a CRISPR cell-essentiality screen (Figure 1B) (Wang et al., 2015) and two (CTCF, YY1) are expressed in >90% of tissues examined (Figure 1C). YY1 and CTCF share additional features: like CTCF, YY1 is a zinc-finger transcription factor (Klenova et al., 1993; Shi et al., 1991), essential for embryonic and adult cell viability (Donohoe et al., 1999; Heath et al., 2008) and capable of forming homodimers (López-Perrote et al., 2014; Saldaña-Meyer et al., 2014) (Table S1). YY1, however, tends to occupy active enhancers and promoters, as well as some insulators, whereas CTCF preferentially occupies insulator elements (Figures 1D and S1A–S1C).
Figure 1. YY1 Is a Candidate Enhancer-Promoter Structuring Factor.
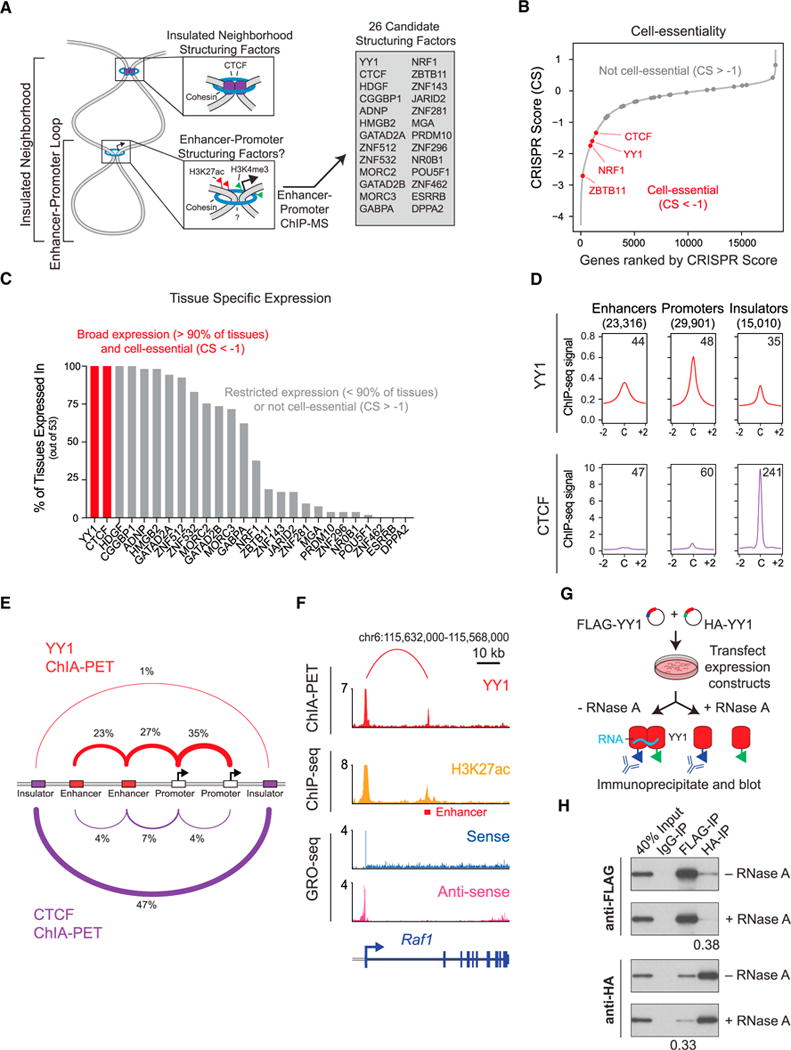
(A) Model depicting an enhancer-promoter loop contained within a larger insulated neighborhood loop. Candidate enhancer-promoter structuring transcription factors were identified by ChIP-MS of histones with modifications characteristic of enhancer and promoter chromatin. (B) CRISPR scores (CS) of all genes in KBM7 cells from Wang et al. (2015). Candidate enhancer-promoter structuring factors identified by ChIP-MS are indicated as dots, and those identified as cell essential (CS < −1) are shown in red.
(C) Histogram showing the number of tissues in which each candidate enhancer-promoter structuring factor is expressed across 53 tissues surveyed by GTEx. Candidates that are both broadly expressed (expressed in greater than 90% of tissues surveyed) and cell essential are shown in red.
(D) Metagene analysis showing the occupancy of YY1 and CTCF at enhancers, promoters, and insulator elements in mouse embryonic stem cells.
(E) Summary of the classes of high-confidence interactions identified by YY1 and CTCF ChIA-PET in mESCs.
(F) Example of a YY1-YY1 enhancer-promoter interaction at the Raf1 locus in mESCs.
(G) Model depicting co-immunoprecipitation assay to detect YY1 dimerization and evaluate dependence on RNA for YY1 dimerization.
(H) Western blot results showing co-immunoprecipitation of FLAG-tagged YY1 and HA-tagged YY1 protein from nuclear lysates prepared from transfected cells. Quantification of the remaining signal normalized to input after RNase A treatment for the co-immunoprecipitated tagged YY1 is displayed under the relevant bands.
See also Table S1 and Figure S1. See STAR Methods for detailed description of genomics analyses. Datasets used in this figure are listed in Table S4.
If YY1 contributes to enhancer-promoter interactions, then chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) (Fullwood et al., 2009) for YY1 should show that YY1 is preferentially associated with these interactions. CTCF ChIA-PET, in contrast, should show that CTCF is preferentially associated with insulator DNA interactions. We generated ChIA-PET data for YY1 and CTCF in mESCs and compared these two datasets. The results showed that the majority of YY1-associated interactions connect active regulatory elements (enhancer-enhancer, enhancer-promoter, and promoter-promoter, which we will henceforth call enhancer-promoter interactions), whereas the majority of CTCF-associated interactions connect insulator elements (Figures 1E and S1D). Some YY1-YY1 interactions involved simple enhancer-promoter contacts, as seen in the Raf1 locus (Figure 1F), and others involved more complex contacts among super-enhancer constituents and their target promoters, as seen in the Klf9 locus (Figure S1E). Super-enhancers were generally occupied by YY1 at relatively high densities and exhibited relatively high YY1-YY1 interaction frequencies (Figures S1E–S1H). For both YY1 and CTCF, there was also evidence of enhancer-insulator and promoter-insulator interactions, but these were more pronounced for CTCF (Figure S1D).
Previous studies have reported that YY1 can form dimers (López-Perrote et al., 2014). To confirm that YY1 dimerization occurs, FLAG-tagged and HA-tagged versions of YY1 protein were expressed in cells, nuclei were isolated and the tagged YY1 proteins in nuclear extracts were immunoprecipitated with either anti-FLAG or anti-HA antibodies. The results show that the FLAG-tagged and HA-tagged YY1 proteins interact (Figures 1G, 1H, S1I, and S1J), consistent with prior reports that YY1 proteins oligomerize (López-Perrote et al., 2014). Other highly expressed nuclear proteins such as OCT4 did not co-precipitate, indicating that the assay was specific (Figure S1J). We previously reported that YY1 can bind both DNA and RNA independently, and that YY1 binding of active regulatory DNA elements is enhanced by the binding of RNA species that are transcribed at these loci (Sigova et al., 2015). It is therefore possible that YY1-YY1 interactions may be enhanced by the ability of each of the YY1 proteins to bind RNA species. Indeed, when we repeated the experiment described above with nuclear extracts containing the tagged YY1 proteins, and a portion of the sample was treated with RNase A prior to immunoprecipitation with anti-tag antibodies, there was an ~60% reduction in the amount of co-immunoprecipitated YY1 partner protein (Figures 1G and 1H). These results suggest that stable YY1-YY1 interactions may be facilitated by RNA.
YY1 Generally Occupies Enhancers and Promoters in Mammalian Cells
YY1 is ubiquitously expressed in mammalian cells, so we investigated whether YY1 generally occupies enhancers and promoters in a broad spectrum of mammalian cell types. Examination of sites bound by YY1 across human cell types showed that YY1 does generally occupy enhancers and promoters genome-wide, and, as expected, enhancer occupancy tends to be cell type specific (Figures 2A, 2B, and S2A–S2F). As with mESCs, YY1 was also found at a subset of insulators in the human cells (Figures S2A–S2F). Examination of YY1 ChIP sequencing (ChIP-seq) data in multiple murine cell types confirmed that YY1 generally occupies enhancers and promoters and is present at some insulators (Figures S2G–S2J). These results indicate that YY1 generally occupies enhancer and promoter elements in mammalian cells.
Figure 2. YY1 Generally Occupies Enhancers and Promoters in Mammalian Cells.
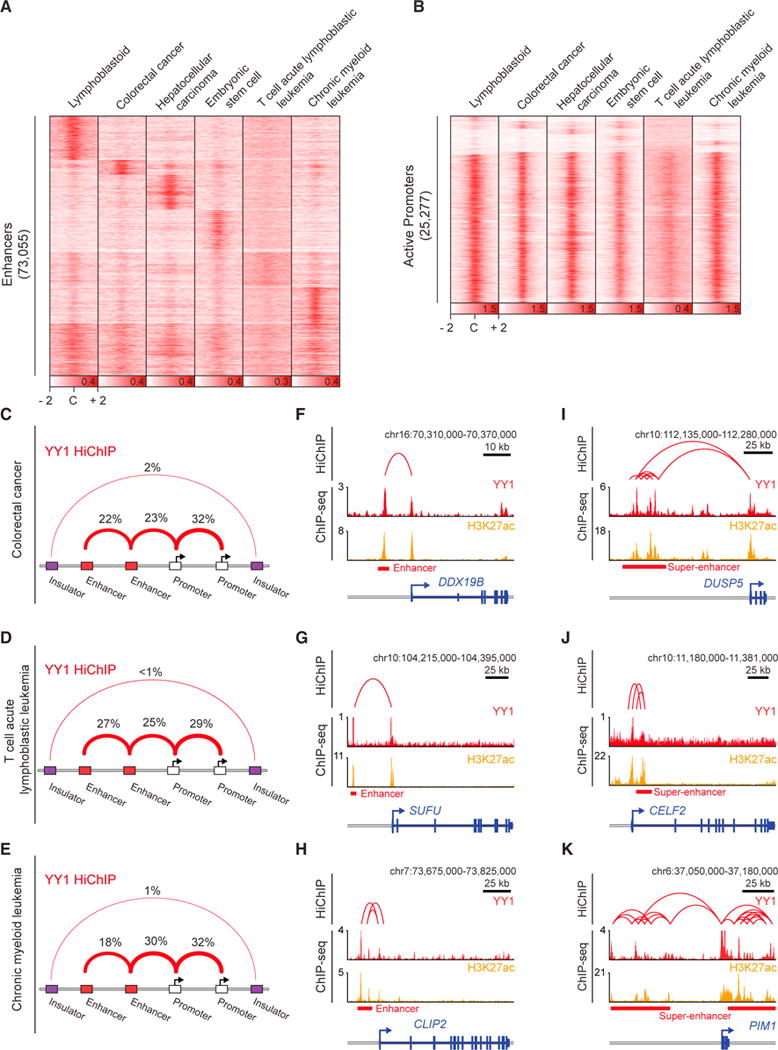
(A and B) Heatmaps displaying the YY1 occupancy at enhancers (A) and active promoters (B) in six human cell types. (C–E) Summaries of the major classes of high-confidence interactions identified with YY1 HiChIP in colorectal cancer cells (C), T cell acute lymphoblastic leukemia cells (D), and chronic myeloid leukemia cells (E).
(F–K) Examples of YY1-YY1 enhancer-promoter interactions in three human cell types: colorectal cancer (F and I), T cell acute lymphoblastic leukemia (G and J), and chronic myeloid leukemia (H and K). Displayed examples show YY1-YY1 enhancer-promoter interactions involving typical enhancers (F–H) and involving super-enhancers (I–K).
See also Figure S2. See STAR Methods for detailed description of genomics analyses. Datasets used in this figure are listed in Table S4.
To determine whether YY1 is associated with sites of enhancer-promoter interactions in human cells, we conducted YY1 HiChIP experiments (Mumbach et al., 2016) in three different cell types. These experiments revealed that YY1 is predominantly associated with enhancer-promoter interactions (Figures 2C–2K). YY1 was also associated with some insulator-enhancer and insulator-promoter interactions, suggesting that the factor may also occasionally participate in such interactions (Figures S2K–S2M). In summary, the HiChIP results indicate that YY1 generally occupies sites involved in enhancer-promoter interactions, and occasionally occupies sites of insulator interactions, in mammalian cells.
YY1 Can Enhance DNA Interactions In Vitro
CTCF proteins can form homodimers and larger oligomers and thus when bound to two different DNA sites can form a loop with the intervening DNA (Saldaña-Meyer et al., 2014). The observation that YY1 is bound to interacting enhancers and promoters, coupled with the evidence that YY1-YY1 interactions can occur in vitro and in cell extracts, is consistent with the idea that YY1-YY1 interactions can contribute to loop formation between enhancers and promoters. To obtain evidence that YY1 can have a direct effect on DNA interactions, we used an in vitro DNA circularization assay to determine whether purified YY1 can enhance the rate of DNA interaction in vitro. The rate of DNA circularization catalyzed by T4 DNA ligase has been used previously to measure persistence length and other physical properties of DNA (Shore et al., 1981). We reasoned that if YY1 bound to DNA is capable of dimerizing and thereby forming DNA loops, then incubating a linear DNA template containing YY1 binding sites with purified YY1 protein should bring the ends into proximity and increase the rate of circularization (Figures 3A and 3D). Recombinant YY1 protein was purified and shown to have DNA binding activity using a mobility shift assay (Figures S3A and S3B). This recombinant YY1 was then tested in the DNA circularization assay; the results showed that YY1 increased the rate of circularization and that this depended on the presence of YY1 motifs in the DNA (Figures 3B and 3C). The addition of an excess of a competing 200-bp DNA fragment containing the YY1 consensus binding sequence abrogated circularization of the larger DNA molecule (Figures 3D–3F). The addition of bovine serum albumin (BSA) did not increase the rate of DNA ligation (Figures 3C and 3F). These results support the idea that YY1 can directly facilitate DNA interactions.
Figure 3. YY1 Can Enhance DNA Interactions In Vitro.
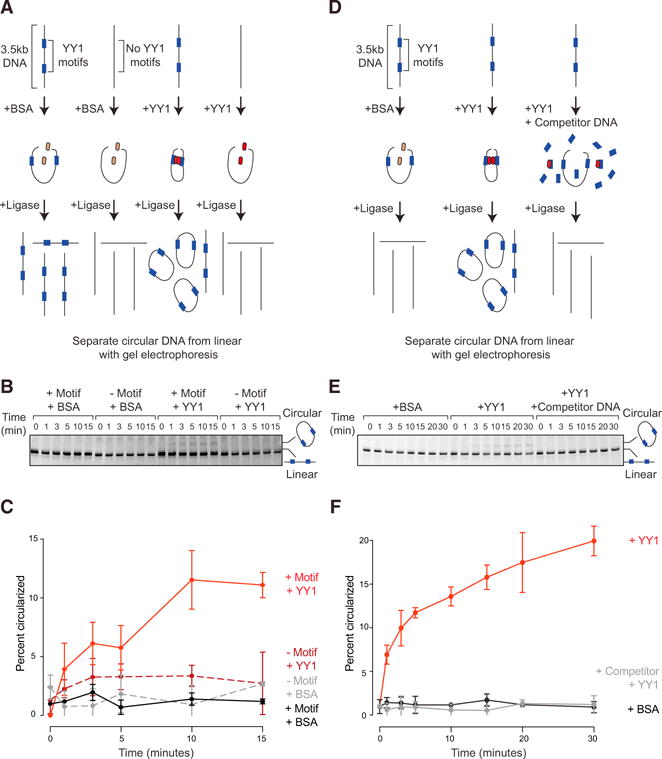
(A and D) Models depicting the in vitro DNA circularization assays used to detect the ability of YY1 to enhance DNA looping interactions with no motif control (A) or competitor DNA control (D).
(B and E) Results of the in vitro DNA circularization assay visualized by gel electrophoresis with no motif control (B) or competitor DNA control (E). The dominant lower band reflects the starting linear DNA template, while the upper band corresponds to the circularized DNA ligation product.
(C and F) Quantifications of DNA template circularization as a function of incubation time with T4 DNA ligase for no motif control (C) or competitor DNA control (F). Values correspond to the percent of DNA template that is circularized and represents the mean and SD of four experiments.
See also Figure S3.
Enhancer-Promoter Interactions Depend on YY1 in Living Cells
To test whether enhancer-promoter interactions in living cells depend on YY1 binding sites in these elements, a CRISPR/Cas9 system was used to generate a small deletion of a YY1 binding motif in the regulatory regions of two genes (Figure 4A). Deletion of the optimal DNA-binding motif for YY1 in the promoter of the Raf1 gene resulted in decreased YY1 binding at the promoter, reduced contact frequency between the enhancer and promoter, and a decrease in Raf1 mRNA levels (Figures 4B and S4A). Deletion of the optimal DNA-binding motif for YY1 in the promoter of the Etv4 gene also resulted in decreased YY1 binding and decreased enhancer-promoter contact frequency, although it did not significantly affect the levels of Etv4 mRNA (Figures 4C and S4B). These results suggest that the YY1 binding sites contribute to YY1 binding and enhancer-promoter contact frequencies at both Raf1 and Etv4, although the reduction in looping frequencies at Etv4 was not sufficient to have a significant impact on Etv4 mRNA levels. The lack of an effect on Etv4 mRNA levels may be a consequence of the residual YY1 that is bound to the Etv4 promoter region, where additional CCAT motifs are observed (Figure 4C). Indeed, when YY1 protein is depleted (see below; Figure S6E), the levels of both Raf1 and Etv4 mRNA decrease.
Figure 4. Deletion of YY1 Binding Sites Causes Loss of Enhancer-Promoter Interactions.
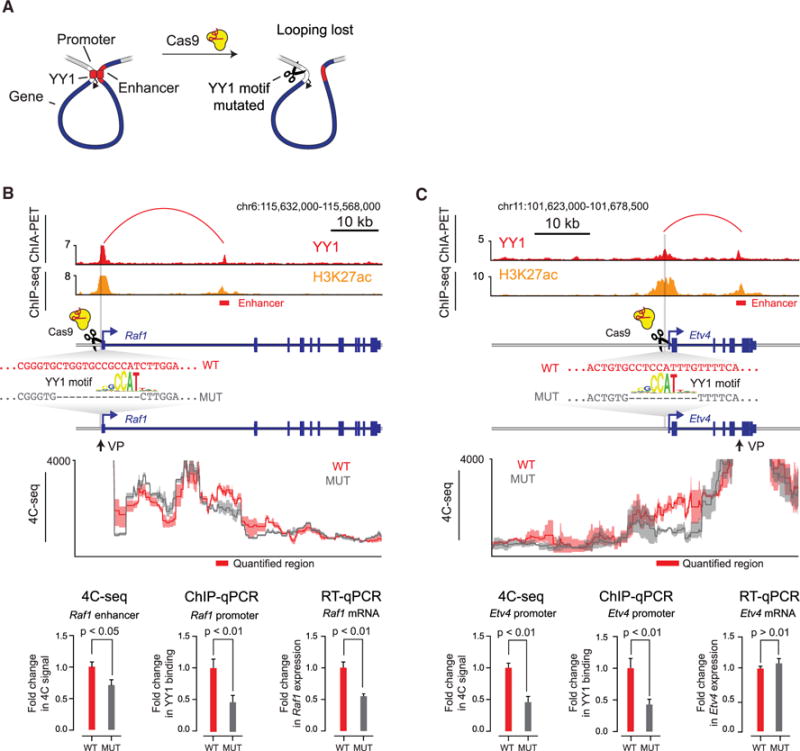
(A) Model depicting CRISPR/Cas9-mediated deletion of a YY1 binding motif in the regulatory region of a gene.
(B and C) CRISPR/Cas9-mediated deletion of YY1 binding motifs in the regulatory regions of two genes, Raf1 (B) and Etv4 (C), was performed, and the effects on YY1 occupancy, enhancer-promoter looping, and mRNA levels were measured. The positions of the targeted YY1 binding motifs, the genotype of the wild-type and mutant lines, and the 4C sequencing (4C-seq) viewpoint are indicated. The mean 4C-seq signal is represented as a line (individual replicates are shown in Figure S4), and the shaded area represents the 95% confidence interval. Three biological replicates were assayed for 4C-seq and ChIP-qPCR experiments, and six biological replicates were assayed for RT-qPCR experiments. Error bars represent the SD. All p values were determined using the Student’s t test.
See also Figure S4. See STAR Methods for detailed description of genomics analyses. Datasets used in this figure are listed in Table S4.
Previous studies have reported that YY1 is an activator of some genes and a repressor of others, but a global analysis of YY1 dependencies has not been described with a complete depletion of YY1 in mESCs (Gordon et al., 2006; Shi et al., 1997; Thomas and Seto, 1999). We used an inducible degradation system (Erb et al., 2017; Huang et al., 2017; Winter et al., 2015) to fully deplete YY1 protein levels and measured the impact on gene expression in mESCs genome-wide through RNA sequencing (RNA-seq) analysis (Figures 5A and 5B). Depletion of YY1 led to significant (adjusted p value <0.05) changes in expression of 8,234 genes, divided almost equally between genes with increased expression and genes with decreased expression (Figure 5C; Table S2; Table S3). The genes that experienced the greatest changes in expression with YY1 depletion were generally occupied by YY1 (Figure 5D).
Figure 5. Depletion of YY1 Disrupts Gene Expression.
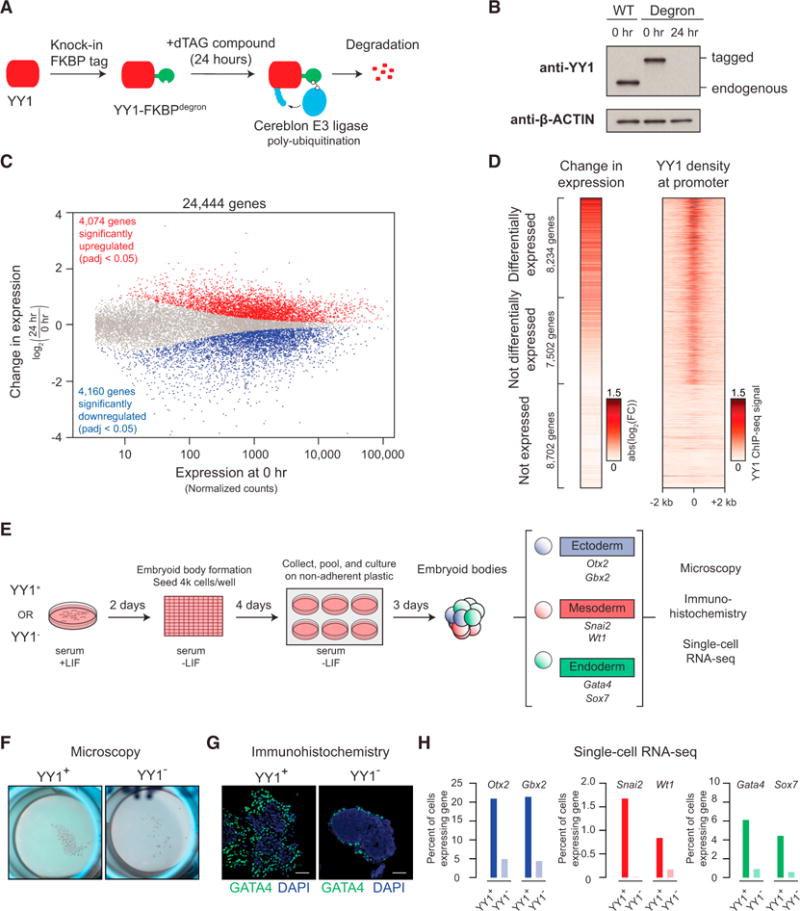
(A) Model depicting dTAG system used to rapidly deplete YY1 protein.
(B) Western blot validation of knockin of FKBP degron tag and ability to inducibly degrade YY1 protein.
(C) Change in gene expression (log2 fold change) upon degradation of YY1 for all genes plotted against the expression in untreated cells. Genes that displayed significant changes in expression (false discovery rate [FDR] adjusted p value <0.05) are colored with upregulated genes plotted in red and downregulated genes plotted in blue.
(D) Heatmaps displaying the change in expression of each gene upon degradation of YY1 and wild-type YY1 ChIP-seq signal in a ±2-kb region centered on the transcription start site (TSS) of each gene. Each row represents a single gene, and genes are ranked by their adjusted p value for change in expression upon YY1 degradation.
(E) Model depicting experimental outline to test the effect of YY1 degradation on embryonic stem cell differentiation into the three germ layers via embryoid body formation from untreated cells (YY1+) and cells treated with dTAG compound to degrade YY1 (YY1−).
(F) Microscopy images of embryoid bodies formed from YY1+ and YY1− cells.
(G) Immunohistochemistry images of embryoid bodies formed from YY1+ and YY1− cells. GATA4 is displayed in green, and DNA stained using DAPI is displayed in blue. The scale bar represents 50 μm.
(H) Quantification of single-cell RNA-seq results for embryoid bodies formed from YY1+ and YY1− cells. The percentage of cells expressing various differentiation-specific genes is displayed for YY1+ and YY1− embryoid bodies.
See also Table S2, Table S3, and Figure S5. See STAR Methods for detailed description of genomics analyses. Datasets used in this figure are listed in Table S4.
Previous studies have shown that YY1 is required for normal embryonic development (Donohoe et al., 1999). We therefore investigated whether the loss of YY1 leads to defects in embryonic stem cell (ESC) differentiation into the three germ layers (Figure 5E). mESCs and isogenic cells that were subjected to inducible degradation of YY1 were stimulated to form embryoid bodies (Figure 5F), and the cells in these bodies were subjected to immunohistochemistry staining and single-cell RNA-seq to monitor expression of differentiation-specific factors. The results showed that cells lacking YY1 showed pronounced defects in expression of the master transcription factors that drive normal differentiation (Figures 5G, 5H, and S5).
We next investigated whether changes in DNA looping occur upon global depletion of YY1 in mESCs. HiChIP for H3K27ac, a histone modification present at both enhancers and promoters, was performed before and after YY1 depletion to detect differences in enhancer-promoter interaction frequencies. Prior to YY1 depletion, the results of the HiChIP experiment showed interactions between the various elements that were similar to the earlier YY1 ChIA-PET results (Figures S6A and S6B). After YY1 depletion, the interactions between YY1-occupied enhancers and promoters decreased significantly (Figures 6A and 6B). The majority (60%) of genes connected by YY1 enhancer-promoter loops showed significant changes in gene expression (Figures 6C and S6D). Examination of the HiChIP DNA interaction profiles at specific genes confirmed these effects. For example, with YY1 depletion the Slc7a5 promoter and its enhancer showed an ~50% reduction in interaction frequency, and Slc7a5 expression levels were reduced by ~27% (Figure 6D). Similarly, after YY1 depletion the Klf9 promoter and its super-enhancer showed an ~40% reduction in interaction frequency, and Klf9 expression levels were reduced by ~50% (Figure 6E).
Figure 6. Depletion of YY1 Disrupts Enhancer-Promoter Looping.
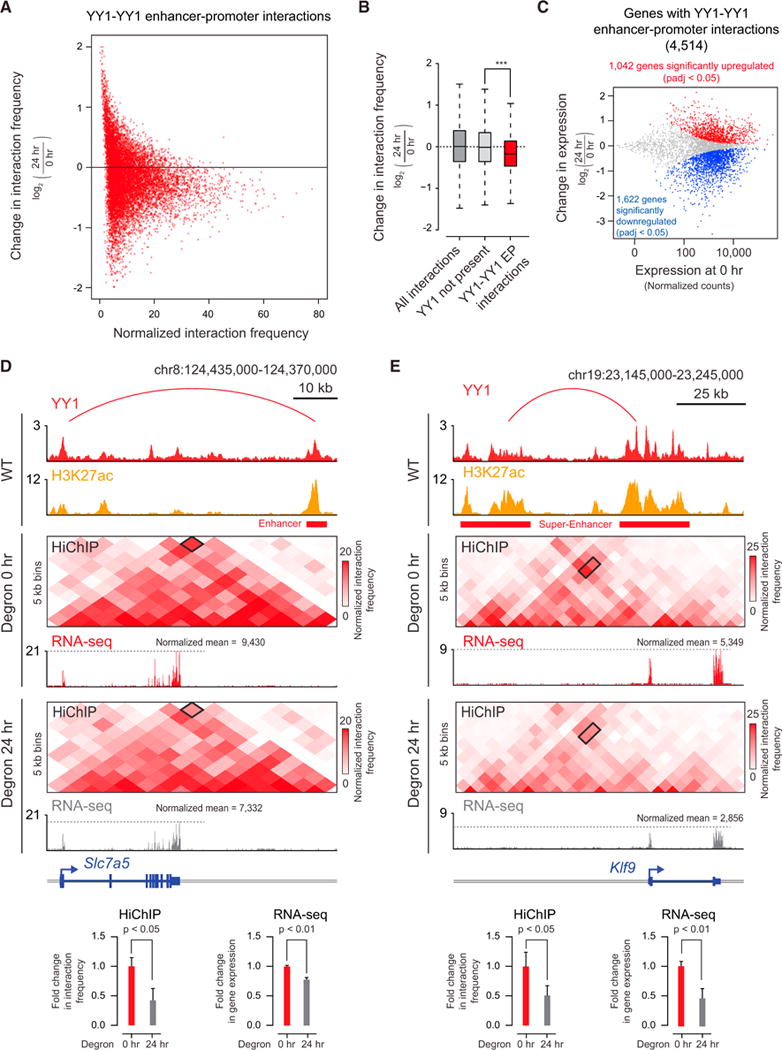
(A) Scatterplot displaying for all YY1-YY1 enhancer-promoter interactions the change in normalized interaction frequency (log2 fold change) upon degradation of YY1, as measured by H3K27ac HiChIP, and plotted against the normalized interaction frequency in untreated cells.
(B) Change in normalized interaction frequency (log2 fold change) upon degradation of YY1 for three different classes of interactions: all interactions, interactions not associated with YY1 ChIP-seq peaks, and YY1-YY1 enhancer-promoter interactions.
(C) Scatterplot displaying for each gene associated with a YY1-YY1 enhancer-promoter interaction the change in gene expression (log2 fold change) upon degradation of YY1 plotted against the expression in untreated cells. Genes that showed significant changes in expression (FDR adjusted p value <0.05) are colored with upregulated genes plotted in red and downregulated genes plotted in blue.
(D and E) Effect of YY1 degradation at the Slc7a5 locus (D) and Klf9 locus (E) on enhancer-promoter interactions and gene expression. The top of each panel shows an arc representing an enhancer-promoter interaction detected in the HiChIP data. Signal in the outlined pixels was used to quantify the change in normalized interaction frequency upon YY1 degradation. Three biological replicates were assayed per condition for H3K27ac HiChIP, and two biological replicates were assayed for RNA-seq. Error bars represent the SD. p values for HiChIP were determined using the Student’s t test. p values for RNA-seq were determined using a Wald test.
See also Figure S6. See STAR Methods for detailed description of genomics analyses. Datasets used in this figure are listed in Table S4.
Rescue of Enhancer-Promoter Interactions in Cells
The ability of an artificially tethered YY1 protein to rescue defects associated with a YY1 binding site mutation would be a strong test of the model that YY1 mediates enhancer-promoter interactions (Figure 7A). We carried out such a test with a dCas9-YY1 fusion protein targeted to a site adjacent to a YY1 binding site mutation in the promoter-proximal region of Etv4 (Figures 7B and 7C). We found that artificially tethering YY1 protein to the promoter led to increased contact frequency between the Etv4 promoter and its enhancer and caused increased transcription from the gene (Figure 7D). These results support the model that YY1 is directly involved in structuring enhancer-promoter loops.
Figure 7. Rescue of Enhancer-Promoter Interactions in Cells.
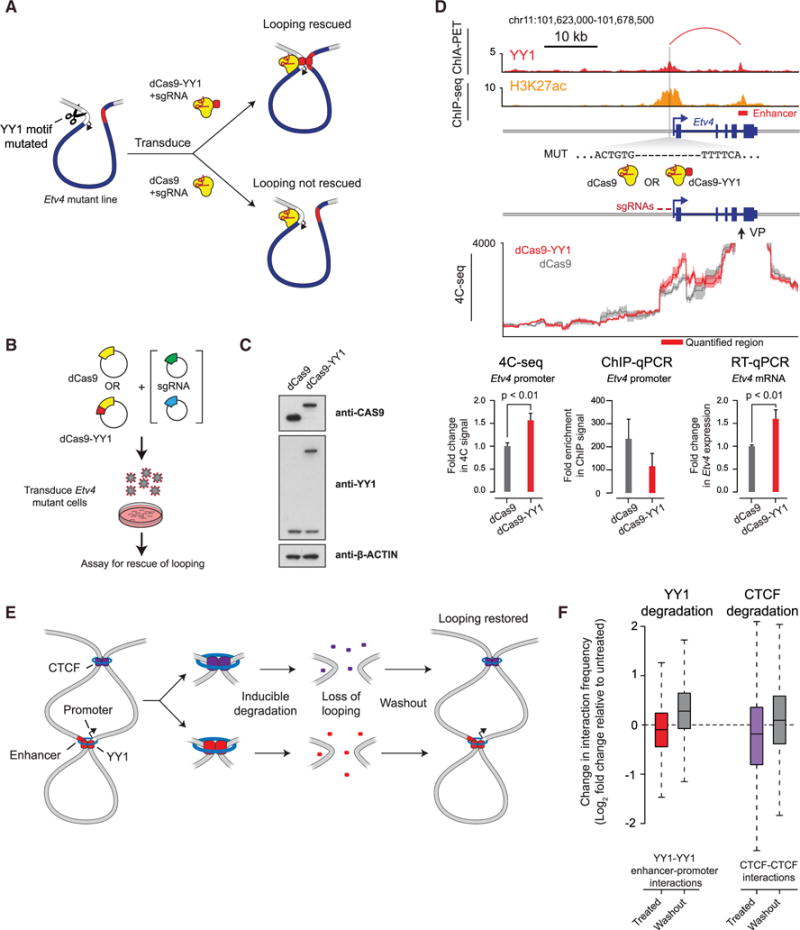
(A) Model depicting use of dCas9-YY1 to artificially tether YY1 to a site adjacent to the YY1 binding site mutation in the promoter-proximal region of Etv4 in order to determine whether artificially tethered YY1 can rescue enhancer-promoter interactions.
(B) Model depicting dCas9-YY1 rescue experiments. Etv4 promoter-proximal YY1 binding motif mutant cells were transduced with lentivirus to stably express either dCas9 or dCas9-YY1, and two sgRNAs to direct their localization to the sequences adjacent to the deleted YY1 binding motif in the Etv4 promoter-proximal region. The ability to rescue enhancer-promoter looping was assayed by 4C-seq.
(C) Western blot results showing that Etv4 promoter-proximal YY1 binding motif mutant cells transduced with lentivirus to stably express either dCas9 or dCas9-YY1 successfully express dCas9 or dCas9-YY1.
(D) Artificial tethering of YY1 using dCas9-YY1 was performed at sites adjacent to the YY1 binding site mutation in the promoter-proximal region of Etv4. The effects of tethering YY1 using dCas9-YY1 on enhancer-promoter looping and expression of the Etv4 gene were measured and compared to dCas9 alone. The genotype of the Etv4 promoter-proximal YY1 binding motif mutant cells and the 4C-seq viewpoint (VP) is shown. The 4C-seq signal is displayed as the smoothed average reads per million per base pair. The mean 4C-seq signal is represented as a line, and the shaded area represents the 95% confidence interval. Three biological replicates were assayed for 4C-seq and CAS9 ChIP-qPCR experiments, and six biological replicates were assayed for RT-qPCR experiments. Error bars represent the SD. All p values were determined using the Student’s t test.
(E) Model depicting the loss of looping interactions after the inducible degradation of the structuring factors CTCF and YY1 followed by restoration of looping upon washout of degradation compounds.
(F) Change in normalized interaction frequency (log2 fold change) after YY1 and CTCF degradation (treated) and recovery (washout) relative to untreated cells. For YY1 degradation, change in normalized interaction frequency is plotted for YY1-YY1 enhancer-promoter interactions. For CTCF degradation, change in normalized interaction frequency is plotted for CTCF-CTCF interactions.
See also Figure S7. See STAR Methods for detailed description of genomics analyses. Datasets used in this figure are listed in Table S4.
To more globally test whether YY1 can rescue the loss of enhancer-promoter interactions after YY1 degradation, we subjected mESCs to YY1 degradation with the dTAG method and then washed out the dTAG compound and allowed YY1 to be restored to normal levels (Figures 7E, S7A, and S7B). Enhancer-promoter frequencies were monitored with H3K27ac HiChIP. Consistent with our previous experiment (Figure 6), the loss of YY1 caused a loss in enhancer-promoter interactions, but the recovery of YY1 levels was accompanied by a substantial increase in enhancer-promoter interactions (Figure 7F). These results were comparable to the effects observed with the rescue of CTCF-CTCF interactions in a similar experiment described recently (Figures 7F and S7C) (Nora et al., 2017), and support the model that YY1 contributes to structuring of a large fraction of enhancer-promoter loops genome-wide.
DISCUSSION
We describe here evidence that the transcription factor YY1 contributes to enhancer-promoter structural interactions. For a broad spectrum of genes, YY1 binds to active enhancers and promoters and is required for normal levels of enhancer-promoter interaction and gene transcription. YY1 is ubiquitously expressed, occupies enhancers and promoters in all cell types examined, is associated with sites of DNA looping in cells where such studies have been conducted, and is essential for embryonic and adult cell viability, so it is likely that YY1-mediated enhancer-promoter interactions are a general feature of mammalian gene control.
Evidence that CTCF-CTCF interactions play important roles in chromosome loop structures but are only occasionally involved in enhancer-promoter interactions led us to consider the possibility that a bridging protein analogous to CTCF might generally participate in enhancer-promoter interactions. CTCF and YY1 share many features: they are DNA-binding zinc-finger factors (Klenova et al., 1993; Shi et al., 1991) that selectively bind hypo-methylated DNA sequences (Bell and Felsenfeld, 2000; Yin et al., 2017), are ubiquitously expressed (Figure 1C) (Mele et al., 2015), are essential for embryonic viability (Donohoe et al., 1999; Heath et al., 2008), and are capable of dimerization (Figures 1G, 1H, S1I, and S1J) (López-Perrote et al., 2014; Saldaña-Meyer et al., 2014). The two proteins differ in several important ways. CTCF-CTCF interactions occur predominantly between sites that can act as insulators and to a lesser degree between enhancers and promoters (Figures 1E and S1A–S1D). YY1-YY1 interactions occur predominantly between enhancers and promoters and to a lesser extent between insulators (Figures 1E and S1A–S1D). At insulators, CTCF binds to a relatively large and conserved sequence motif (when compared to those bound by other transcription factors); these same sites tend to be bound in many different cell types, which may contribute to the observation that TAD boundaries tend to be preserved across cell types. At enhancers and promoters, YY1 binds to a relatively small and poorly conserved sequence motif within these regions, where RNA species are produced that can facilitate stable YY1 DNA binding (Sigova et al., 2015). The cell-type-specific activity of enhancers and promoters thus contributes to the observation that YY1-YY1 interactions tend to be cell type specific.
The model that YY1 contributes to structuring of enhancer-promoter loops can account for the many diverse functions previously reported for YY1, including activation and repression, differentiation, and cellular proliferation. For example, following its discovery in the early 1990s (Hariharan et al., 1991; Park and Atchison, 1991; Shi et al., 1991), YY1 was intensely studied and reported to act as a repressor for some genes and an activator for others; these context-specific effects have been attributed to many different mechanisms (reviewed in Gordon et al., 2006; Shi et al., 1997; Thomas and Seto, 1999). There are many similar reports of context-specific activation and repression by CTCF (reviewed in Ohlsson et al., 2001; Phillips and Corces, 2009). Although it is reasonable to assume that YY1 and CTCF can act directly as activators or repressors at some genes, the evidence that these proteins contribute to structuring of DNA loops makes it likely that the diverse active and repressive roles that have been attributed to them are often a consequence of their roles in DNA structuring. In this model, the loss of CTCF or YY1 could have positive or negative effects due to other regulators that were no longer properly positioned to produce their regulatory activities.
Previous studies have hinted at a role for YY1 in long-distance DNA interactions. CTCF, YY1, and cohesin have been implicated in the formation of DNA loops needed for V(D)J rearrangement at the immunoglobulin locus during B cell development (Degner et al., 2011; Guo et al., 2011; Liu et al., 2007). B cell-specific deletion of YY1 causes a decrease in the contraction of the immunoglobulin H (IgH) locus, thought to be mediated by DNA loops, and a block in the development of B cells (Liu et al., 2007). Knockdown of YY1 has also been shown to reduce intrachromosomal interactions between the Th2 locus control region (LCR) and the IL4 promoter (Hwang et al., 2013). As this manuscript was completed, a paper appeared reporting that YY1 is present at the base of interactions between neuronal precursor cell-specific enhancers and genes and that YY1 knockdown causes a loss of these interactions (Beagan et al., 2017). The results described here argue that YY1 is more of a general structural regulator of enhancer-promoter interactions for a large population of genes, both cell type specific and otherwise, in all cells. Thus, the tendency of YY1 to be involved in cell-type-specific loops is a reflection of the cell-type specificity of enhancers and, consequently, their interactions with genes that can be expressed in a cell-specific or a more general manner.
YY1 plays an important role in human disease, YY1 haploinsufficiency has been implicated in an intellectual disability syndrome, and YY1 overexpression occurs in many cancers. A cohort of patients with various mutations in one allele and exhibiting intellectual disability have been described as having a “YY1 syndrome,” and lymphoblastoid cell lines from these patients show reduced occupancy of regulatory regions and small changes in gene expression at a subset of genes associated with YY1 binding (Gabriele et al., 2017). These results are consistent with the model we describe for YY1 in global enhancer-promoter structuring, and with the idea that higher neurological functions are especially sensitive to such gene dysregulation. YY1 is overexpressed in a broad spectrum of tumor cells, and this overexpression has been proposed to cause unchecked cellular proliferation, tumorigenesis, metastatic potential, resistance to immune-mediated apoptotic stimuli, and resistance to chemotherapeutics (Gordon et al., 2006; Zhang et al., 2011). The mechanisms that have been reported to mediate these effects include YY1-mediated downregulation of p53 activity, interference with poly-ADP-ribose polymerase, alteration in c-Myc and NF-κB expression, regulation of death genes and gene products, differential YY1 binding in the presence of inflammatory mediators, and YY1 binding to the oncogenic c-Myc transcription factor (Gordon et al., 2006; Zhang et al., 2011). Although it is possible that YY1 carries out all these functions, its role as a general enhancer-promoter structuring factor is a more parsimonious explanation of these pleiotropic phenotypes.
Many zinc-coordinating transcription factors are capable of homo- and hetero-dimerization (Amoutzias et al., 2008; Lamb and McKnight, 1991), and, because these comprise the largest class of transcription factors in mammals (Weirauch and Hughes, 2011), we suggest that a combination of cell-type-specific and cell-ubiquitous transcription factors make a substantial and underappreciated contribution to enhancer-promoter loop structures. There are compelling studies of bacterial and bacteriophage transcription factors that contribute to looping of regulatory DNA elements through oligomerization (Adhya, 1989; Schleif, 1992), and reports of several eukaryotic factors with similar capabilities (Matthews, 1992). Nonetheless, most recent study of eukaryotic enhancer-promoter interactions has focused on cofactors that lack DNA binding capabilities and bridge enhancer-bound transcription factors and promoter-bound transcription apparatus (Allen and Taatjes, 2015; Deng et al., 2012; Jeronimo et al., 2016; Kagey et al., 2010; Malik and Roeder, 2010; Petrenko et al., 2016), with the notable exception of the proposals that some enhancer-promoter interactions are determined by the nature of transcription factors bound at the two sites (Muerdter and Stark, 2016). We predict that future studies will reveal additional transcription factors that belong in the class of DNA binding proteins whose predominant role is to contribute to chromosome structure.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| YY1 | Santa Cruz | Cat# sc-1703X |
| YY1 | Abcam | Cat# ab109237 |
| CTCF | EMD-Millipore | Cat# 07-729 |
| YY1 | Abcam | Cat# ab199998 |
| Beta-Actin | Sigma | Cat# A5441 |
| HA-HRP | Cell Signaling | Cat# 2999 |
| HA | Abcam | Cat# ab9110 |
| FLAG | Sigma | Cat# F7425 |
| FLAG-HRP | Sigma | Cat# A8592 |
| Rabbit IgG | Millipore | Cat# 12-370 |
| H3K27ac | Abcam | Cat# ab4729 |
| H3K27ac | Active Motif | Cat# 39133 |
| OCT3/4 | Santa Cruz | Cat# sc-5279 |
| Cas9 | Cell Signaling | Cat# 14697 |
| TUJI | BioLegend | Cat# 801201 |
| GFAP | Dako | Cat# Z0344 |
| GATA4 | Abcam | Cat# ab84593 |
| Bacterial and Virus Strains | ||
| BL21-CodonPlus (DE3)-RIL cells | Stratagene | Cat# 230245 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human Recombinant YY1 | This study | N/A |
| dTAG-47 compound | This study | N/A |
| Critical Commercial Assays | ||
| LightShift Chemiluminescent EMSA Kit | Thermo Scientific | Cat# 20148 |
| TruSeq DNA Sample Preparation v2 Kit | Illumina | Cat# RS-122-2001 |
| Nextera DNA Library Preparation Kit | Illumina | Cat# FC-121-1030 |
| Expand Long Template Polymerase | Roche | Cat# 11759060001 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# RS-122-2101 |
| Pierce BCA Assay | Thermo Scientific | Cat# 23225 |
| Lipofectamine 3000 Transfection Reagent | Life Technologies | Cat# L3000001 |
| Power SYBR Green Master Mix | Applied Biosystems | Cat# 4368577 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | Cat# 4304437 |
| Deposited Data | ||
| Raw and analyzed data | This study | GEO: GSE99521 |
| See Table S4 for deposited genomics datasets | N/A | N/A |
| mES ChIP-MS data | Ji et al., 2015 | http://www.pnas.org/content/suppl/2015/03/06/1502971112.DCSupplemental/pnas.1502971112.st01.xls |
| CRISPR cell essentiality screen | Wang et al., 2015 | http://science.sciencemag.org/content/sci/suppl/2015/10/14/science.aac7041.DC1/aac7041_SM_Table_S3.xlsx |
| GTEx gene expression values | Genotype-Tissue Expression Project | https://www.gtexportal.org/home/ |
| Mouse reference genome, NCBI build 37, NCBI37/mm9 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/mouse |
| Human reference genome, NCBI build 37, GRCh37/hg19 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/human |
| Super-enhancer and typical enhancer constituents | Whyte et al., 2013 | http://www.cell.com/cms/attachment/2031389007/2048455341/mmc1.zip |
| Experimental Models: Cell Lines | ||
| V6.5 Murine Embryonic Stem Cells | R. Jaenisch | N/A |
| V6.5 mESC YY1-dTAG knock-in line | This study | N/A |
| HCT116 | ATCC | Cat# CCL-247 |
| Jurkat | ATCC | Cat# TIB-152 |
| K562 | ATCC | Cat# CCL-243 |
| HEK293T | ATCC | Cat# CRL-3216 |
| Oligonucleotides | ||
| See Table S5 | N/A | N/A |
| Recombinant DNA | ||
| His6-YY1 | Shi Y. | N/A |
| Cas9-GFP | Jaenisch R. | N/A |
| pAW62.YY1.FKBP.knock-in.mCherry | This study | Deposited to Addgene Cat# 104370 |
| pAW63.YY1.FKBP.knock-in.BFP | This study | Deposited to Addgene Cat# 104371 |
| pAW91.dCas9 | This study | Deposited to Addgene Cat# 104372 |
| pAW90.dCas9-YY1 | This study | Deposited to Addgene Cat# 104373 |
| pAW12.lentiguide.GFP | This study | Deposited to Addgene Cat# 104374 |
| pAW13.lentiguide.mCherry | This study | Deposited to Addgene Cat# 104375 |
| pAW49.pUC19.YY1 | This study | Deposited to Addgene Cat# 104376 |
| pAW79.pUC19.noYY1 | This study | Deposited to Addgene Cat# 104377 |
| Software and Algorithms | ||
| Bowtie | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net |
| MACS | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/index.html |
| BEDTools | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ |
| bamToGFF | Bradner Lab | https://github.com/BradnerLab/pipeline |
| UCSC Genome Browser | Kent et al., 2002 | http://genome.ucsc.edu/cgi-bin/hgGateway |
| Kallisto | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| Deseq2 | Love et al., 2014 | https://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| PANTHER GO | Mi et al., 2013, 2017 | http://pantherdb.org |
| Origami | This study | https://github.com/younglab/origami |
| Cell Ranger | 10X Genomics | https://www.10xgenomics.com |
| Loupe Cell Browser | 10X Genomics | https://www.10xgenomics.com |
| Other | ||
| Processed insulated neighborhood calls in mouse and human stem cells. | Hnisz et al., 2016a | http://younglab/wi.mit.edu/insulatedneighborhoods.html |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard A. Young (young@wi.mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
V6.5 murine embryonic stem were a gift from R. Jaenisch of the Whitehead Institute. V6.5 are male cells derived from a C57BL/6(F) × 129/sv(M) cross. Cells were negative for mycoplasma (tested every three months).
Cell Culture Conditions
V6.5 murine embryonic stem (mES) cells were grown in serum + LIF on irradiated murine embryonic fibroblasts (MEFs) or in 2i + LIF conditions. For all experiments except for the washout experiment (Figure 7) cells were grown in serum + LIF on irradiated MEFs and then passaged twice off of MEFs before harvesting. Genome editing was done in 2i + LIF conditions. Cells were always grown on 0.2% gelatinized (Sigma, G1890) tissue culture plates. For the washout experiment (Figure 7) cells were grown on 2i + LIF.
The media used for general culturing in serum + LIF conditions is as follows: DMEM-KO (Invitrogen, 10829-018) supplemented with 15% fetal bovine serum (Hyclone, characterized SH3007103), 1,000 U/ml LIF (ESGRO, ESG1106), 100 mM nonessential amino acids (Invitrogen, 11140-050), 2 mM L-glutamine (Invitrogen, 25030-081), 100 U/mL penicillin, 100 mg/mL streptomycin (Invitrogen, 15140-122), and 8 ul/mL of 2-mercaptoethanol (Sigma, M7522).
The media used for 2i + LIF media conditions is as follows: 967.5 mL DMEM/F12 (GIBCO 11320), 5 mL N2 supplement (GIBCO 17502048), 10 mL B27 supplement (GIBCO 17504044), 0.5 mM L-glutamine (GIBCO 25030), 0.5X non-essential amino acids (GIBCO 11140), 100 U/mL Penicillin-Streptomycin (GIBCO 15140), 0.1 mM β-mercaptoethanol (Sigma), 1 uM PD0325901 (Stemgent 04-0006), 3 uM CHIR99021 (Stemgent 04-0004), and 1000 U/mL recombinant LIF (ESGRO ESG1107).
Prior to differentiation mESCs were cultured in serum + LIF media as follows: DMEM (Invitrogen, 11965-092) supplemented with 15% fetal bovine serum (Hyclone, characterized SH3007103), 100 mM nonessential amino acids (Invitrogen, 11140-050), 2 mM L-glutamine (Invitrogen, 25030-081), 100 U/mL penicillin, 100 mg/mL streptomycin (Invitrogen, 15140-122), 0.1mM β-mercaptoethanol (Sigma Aldrich) and 2×106 units of leukemia inhibitory factor (LIF).
The media used for embryoid body formation (serum - LIF) is as follows: DMEM (Invitrogen, 11965-092) supplemented with 15% fetal bovine serum (Hyclone, characterized SH3007103), 100 mM nonessential amino acids (Invitrogen, 11140-050), 2 mM L-glutamine (Invitrogen, 25030-081), 100 U/mL penicillin, 100 mg/mL streptomycin (Invitrogen, 15140-122).
HCT-116 (male) cells were purchased from ATCC (CCL-247) and cultured in DMEM, high glucose, pyruvate (GIBCO 11995-073) with 10% fetal bovine serum (Hyclone, characterized SH3007103), 100 U/mL Penicillin-Streptomycin (GIBCO 15140), 2 mM L-glutamine (Invitrogen, 25030-081). Cells were negative for mycoplasma (tested every 3 months).
Jurkat (male) cells were purchased from ATCC (TIB-152) and cultured in RPMI-1640 (GIBCO 61870-127) with 10% fetal bovine serum (Hyclone, characterized SH3007103), 100 U/mL Penicillin-Streptomycin (GIBCO 15140). Cells were negative for mycoplasma (tested every 3 months).
K562 cells (female) were purchased from ATCC (CCL-243) and cultured in in RPMI-1640 (GIBCO 61870-127) with 10% fetal bovine serum (Hyclone, characterized SH3007103), 100 U/mL Penicillin-Streptomycin (GIBCO 15140). Cells were negative for mycoplasma (tested every 3 months).
HEK293T cells were purchased from ATCC (ATCC CRL-3216) and cultured in DMEM, high glucose, pyruvate (GIBCO 11995-073) with 10% fetal bovine serum (Hyclone, characterized SH3007103), 100 U/mL Penicillin-Streptomycin (GIBCO 15140), 2 mM L-glutamine (Invitrogen, 25030-081). Cells were negative for mycoplasma (tested every 3 months).
METHOD DETAILS
Experimental Design
All experiments were replicated. For the specific number of replicates done see either the figure legends or the specific section below. No aspect of the study was done blinded. Sample size was not predetermined and no outliers were excluded.
Recombinant YY1 purification and characterization
YY1 purification
YY1 protein was purified using methods established by the Lee Lab (Jeon and Lee, 2011) and previously described in (Sigova et al., 2015). A plasmid containing N-terminal His6-tagged human YY1 coding sequence (a gift from Dr. Yang Shi) was transformed into BL21-CodonPlus (DE3)-RIL cells (Stratagene, 230245). A fresh bacterial colony was inoculated into LB media containing ampicillin and chloramphenicol and grown overnight at 37°C. These bacteria were diluted 1:10 in 500 mL pre-warmed LB with ampicillin and chloramphenicol and grown for 1.5 hours at 37°C. After induction of YY1 expression with 1mM IPTG, cells were grown for another 5 hours, collected, and stored frozen at −80°C until ready to use.
Pellets from 500mL cells were resuspended in 15mL of Buffer A (6M GuHCl, 25mM Tris, 100mM NaCl, pH8.0) containing 10mM imidazole, 5mM β-mercaptoethanol, cOmplete protease inhibitors (Roche, 11873580001) and sonicated (ten cycles of 15 s on, 60 s off). The lysate was cleared by centrifugation at 12,000 g for 30 minutes at 4°C and added to 1mL of Ni-NTA agarose (Invitrogen, R901-15) pre-equilibrated with 10X volumes of Buffer A. Tubes containing this agarose lysate slurry were rotated at room temperature for 1 hour. The slurry was poured into a column, and the packed agarose washed with 15 volumes of Buffer A containing 10mM imidazole. Protein was eluted with 4 × 2 mL Buffer A containing 500mM imidazole.
Fractions were run out by SDS-PAGE gel electrophoresis and stained with Coomassie Brilliant Blue (data not shown). Fractions containing protein of the correct size and high purity were combined and diluted 1:1 with elution buffer. DTT was added to a final concentration of 100mM and incubated at 60°C for 30 minutes. The protein was refolded by dialysis against 2 changes of 1 Liter of 25mM Tris-HCl pH 8.5, 100mM NaCl, 0.1mM ZnCl2, and 10mM DTT at 4°C followed by 1 change of the same dialysis buffer with 10% glycerol. Protein was stored in aliquots at −80°C.
YY1 characterization
The purity of the recombinant YY1 was assessed by SDS-PAGE gel electrophoresis followed by Coomassie Brilliant Blue staining and western blotting (Figure S3A). The activity of the recombinant protein was assessed by EMSA (Figure S3B).
EMSA was performed using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific #20148) following the manufacturer’s recommendations. Briefly, recombinant protein was incubated with a biotinylated probe in the presence or absence of a cold competitor. Reactions were separated using a native gel and transferred to a membrane. Labeled DNA was detected using chemiluminescence.
To generate the biotin labeled probe, 30-nucleotide-long 5′ biotinylated single stranded oligonucleotides (IDT) were annealed in 10 mM Tris pH 7.5, 50 mM NaCl, and 1mM EDTA at a 50 uM concentration. The same protocol was used to generate the cold competitor. The probe was serially diluted to a concentration of 10 fmol/μL and cold competitor to a concentration of 2 pmol/μL. 2 μL of diluted probe and cold competitor were used for each binding reaction for a final amount of 20 fmol labeled probe and 4 pmol cold competitor (200 fold excess) in each reaction.
Binding reactions were set-up in a 20 μL volume containing 1× Binding Buffer (10 mM Tris, 50 mM KCl, 1 mM DTT; pH 7.5), 2.5% Glycerol, 5 mM MgCl2, 50 ng/μL Poly dI dC, 0.05% NP-40 0.1 mM ZnCl2, 10 mM HEPES, and 2 μg of recombinant YY1 protein. Binding reactions were pre-incubated for 20 mins at room temperature with or without the cold competitor. Labeled probe was then added to binding reactions and incubated for 80 minutes at room temperature. After the 80 min incubation 5× Loading Buffer (Thermo Scientific #20148) was added to the reaction and run on a 4%–12% TBE gel using 0.5× TBE at 40 mA for 2.5 hr at 4°C. The TBE gel was pre-run for 1 hr at 4°C. DNA was then electrophoretically transferred to a Biodyne B Nylon Membrane (pre-soaked in cold 0.5× TBE for 10 mins) at 380 mA for 30 mins at 4°C. The DNA was then crosslinked to the membrane by placing the membrane on a Dark Reader Transilluminator for 15 mins. The membrane was allowed to air dry at room temperature overnight and chemiluminescence detected the following day.
Detection of biotin-labeled DNA was done as follows. The membrane was blocked for 20 mins using Blocking Buffer (Thermo Scientific #20148). The membrane was then incubated in conjugate/blocking buffer (Thermo Scientific #20148) for 15 mins. The membrane was then washed four times with 1× Wash Buffer (Thermo Scientific #20148) for 5 mins. The membrane was then incubated in Substrate Equilibration Buffer (Thermo Scientific #20148) for 5 mins and then incubated in Substrate Working Solution (Thermo Scientific #20148) for 5 mins. The membrane was then imaged using a CCD camera using a 120 s exposure. All of these steps were performed at room temperature.
Genome Editing
The CRISPR/Cas9 system was used to genetically engineer ESC lines. Target-specific oligonucleotides were cloned into a plasmid carrying a codon-optimized version of Cas9 with GFP (gift from R. Jaenisch). The oligos used for the cloning are included in Table S3. The sequences of the DNA targeted (the protospacer adjacent motif is underlined) are listed below:
| Locus | Targeted DNA |
|---|---|
| Raf1_promoter | 5′-ACTCCCGCCATCCAAGATGGCGG-3′ |
| Etv4_promoter | 5′-GAGCTACTTGAAAACAAATGGAGG-3′ |
| YY1_stop_codon | 5′-GTCTTCTCTCTTCTTTTCACTGG-3′ |
For the motif deletions, five hundred thousand mES cells were transfected with 2.5 μg plasmid and sorted 48 hours later for the presence of GFP. Thirty thousand GFP-positive sorted cells were plated in a six-well plate in a 1:2 serial dilution (first well 15,000 cells, second well 7,500 cells, etc.). The cells were grown for approximately one week in 2i + LIF. Individual colonies were picked using a stereoscope into a 96-well plate. Cells were expanded and genotyped by PCR and Sanger sequencing. Clones with deletions spanning the motif were further expanded and used for experiments.
For the generation of the endogenously tagged lines, five hundred thousand mES cells were transfected with 2.5 μg Cas9 plasmid and 1.25 μg non-linearized repair plasmid 1 (pAW62.YY1.FKBP.knock-in.mCherry) and 1.25 μg non-linearized repair plasmid 2 (pAW63.YY1.FKBP.knock-in.BFP). Cells were sorted after 48 hours for the presence of GFP. Cells were expanded for five days and then sorted again for double positive mCherry and BFP cells. Thirty thousand mCherry+/BFP+ sorted cells were plated in a six-well plate in a 1:2 serial dilution (first well 15,000 cells, second well 7,500 cells, etc). The cells were grown for approximately one week in 2i medium and then individual colonies were picked using a stereoscope into a 96-well plate. Cells were expanded and genotyped by PCR (YY1_gPCR_3F/3R, Table S3). Clones with a homozygous knock-in tag were further expanded and used for experiments.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed as described in (Lee et al., 2006) with a few adaptations. mES cells were depleted of MEFs by splitting twice onto newly gelatinized plates without MEFs. Approximately 50 million mES cells were crosslinked for 15 minutes at room temperature by the addition of one-tenth volume of fresh 11% formaldehyde solution (11% formaldehyde, 50 mM HEPES pH 7.3, 100 mM NaCl, 1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0) to the growth media followed by 5 min quenching with 125 mM glycine. Cells were rinsed twice with 1X PBS and harvested using a silicon scraper and flash frozen in liquid nitrogen. Jurkat cells were crosslinked for 10 minutes in media at a concentration of 1 million cells /mL. Frozen crosslinked cells were stored at −80°C.
100μl of Protein G Dynabeads (Life Technologies #10009D) were washed 3X for 5 minutes with 0.5% BSA (w/v) in PBS. Magnetic beads were bound with 10 μg of anti-YY1 antibody (Santa Cruz, sc-281X) overnight at 4°C, and then washed 3X with 0.5% BSA (w/v) in PBS.
Cells were prepared for ChIP as follows. All buffers contained freshly prepared 1 3 cOmplete protease inhibitors (Roche, 11873580001). Frozen crosslinked cells were thawed on ice and then resuspended in lysis buffer I (50 mM HEPES-KOH, pH 7.5, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 1 3 protease inhibitors) and rotated for 10 minutes at 4°C, then spun at 1350 rcf. for 5 minutes at 4°C. The pellet was resuspended in lysis buffer II (10 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1 3 protease inhibitors) and rotated for 10 minutes at 4°C and spun at 1350 rcf. for 5 minutes at 4°C. The pellet was resuspended in sonication buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA pH 8.0, 0.1% SDS, and 1% Triton X-100, 1 3 protease inhibitors) and then sonicated on a Misonix 3000 sonicator for 10 cycles at 30 s each on ice (18-21 W) with 60 s on ice between cycles. Sonicated lysates were cleared once by centrifugation at 16,000 rcf. for 10 minutes at 4°C. 50 μL was reserved for input, and then the remainder was incubated overnight at 4°C with magnetic beads bound with antibody to enrich for DNA fragments bound by the indicated factor.
Beads were washed twice with each of the following buffers: wash buffer A (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 0.1% Na-Deoxycholate, 1% Triton X-100, 0.1% SDS), wash buffer B (50 mM HEPES-KOH pH 7.9, 500 mM NaCl, 1 mM EDTA pH 8.0, 0.1% Na-Deoxycholate, 1% Triton X-100, 0.1% SDS), wash buffer C (20 mM Tris-HCl pH8.0, 250 mM LiCl, 1 mM EDTA pH 8.0, 0.5% Na-Deoxycholate, 0.5% IGEPAL C-630, 0.1% SDS), wash buffer D (TE with 0.2% Triton X-100), and TE buffer. DNA was eluted off the beads by incubation at 65°C for 1 hour with intermittent vortexing in 200 μL elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS). Cross-links were reversed overnight at 65°C. To purify eluted DNA, 200 μL TE was added and then RNA was degraded by the addition of 2.5 μL of 33 mg/mL RNase A (Sigma, R4642) and incubation at 37°C for 2 hours. Protein was degraded by the addition of 10 μL of 20 mg/mL proteinase K (Invitrogen, 25530049) and incubation at 55°C for 2 hours. A phenol:chloroform:isoamyl alcohol extraction was performed followed by an ethanol precipitation. The DNA was then resuspended in 50 μL TE and used for either qPCR or sequencing.
For ChIP-qPCR experiments, qPCR was performed using Power SYBR Green mix (Life Technologies #4367659) on either a QuantStudio 5 or a QuantStudio 6 System (Life Technologies). Values displayed in the figures were normalized to the input, a negative control region, and wild-type values according to the following formulas:
qPCRs were performed in technical triplicate, and ChIPs were performed in biological triplicate. Values were comparable across replicates. The average WT norm values and standard deviation are displayed (Figures 4A and 4B). The primers used are listed in Table S3.
For ChIP-seq experiments, purified ChIP DNA was used to prepare Illumina multiplexed sequencing libraries. Libraries for Illumina sequencing were prepared following the Illumina TruSeq DNA Sample Preparation v2 kit. Amplified libraries were size-selected using a 2% gel cassette in the Pippin Prep system from Sage Science set to capture fragments between 200 and 400 bp. Libraries were quantified by qPCR using the KAPA Biosystems Illumina Library Quantification kit according to kit protocols. Libraries were sequenced on the Illumina HiSeq 2500 for 40 bases in single read mode.
ChIA-PET
ChIA-PET was performed using a modified version (Tang et al., 2015) of a previously described protocol (Fullwood et al., 2009). mES cells (~500 million cells, grown to 80% confluency) were crosslinked with 1% formaldehyde at room temperature for 15 min and then neutralized with 125mM glycine. Crosslinked cells were washed three times with ice-cold PBS, snap-frozen in liquid nitrogen, and stored at −80°C before further processing. Nuclei were isolated as previously described above, and chromatin was fragmented using a Misonix 3000 sonicator. Either CTCF or YY1 antibodies were used to enrich protein-bound chromatin fragments exactly as described in the ChIP-seq section. A portion of ChIP DNA was eluted from antibody-coated beads for concentration quantification and for enrichment analysis using qPCR. For ChIA-PET library construction ChIP DNA fragments were end-repaired using T4 DNA polymerase (NEB # M0203) followed by A-tailing with Klenow (NEB M0212). Bridge linker oligos (Table S5) were annealed to generate a double stranded bridge linker with T-overhangs. 800 ng of bridge linker was added and the proximity ligation was performed overnight at 16°C in 1.5 mL volume. Unligated DNA was then digested with exonuclease and lambda nuclease (NEB M0262S, M0293S). DNA was eluted off the beads by incubation at 65°C for 1 hour with intermittent vortexing in 200 μL elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS). Cross-links were reversed overnight at 65°C. To purify eluted DNA, 200 μL TE was added and then RNA was degraded by the addition of 2.5 μL of 33 mg/mL RNase A (Sigma, R4642) and incubation at 37°C for 2 hours. Protein was degraded by the addition of 10 μL of 20 mg/mL proteinase K (Invitrogen, 25530049) and incubation at 55°C for 2 hours.
A phenol:chloroform:isoamyl alcohol extraction was performed followed by an ethanol precipitation. Precipitated DNA was resuspended in Nextera DNA resuspension buffer (Illumina FC-121-1030). The DNA was then tagmented with the Nextera Tagmentation kit (Illumina FC-121-1030). 5 μL of transposon was used per 50 ng of DNA. The tagmented library was purified with a Zymo DNA Clean & Concentrator (Zymo D4003) and bound to streptavidin beads (Life Technologies #11205D) to enrich for ligation junctions (containing the biotinylated bridge linker). 12 cycles of the polymerase chain reaction were performed to amplify the library using standard Nextera primers (Illumina FC-121-1030). The amplified library was size-selected (350-500 bp) and sequenced using paired-end sequencing on an Illumina Hi-Seq 2500 platform.
HiChIP
HiChIP was performed as described in (Mumbach et al., 2016) with a few modifications. Ten million cells were cross-linked for 10 min at room temperature with 1% formaldehyde in growth media and quenched in 0.125 M glycine. After washing twice with ice-cold PBS, the supernatant was aspirated and the cell pellet was flash frozen in liquid nitrogen and stored at −80°C.
Cross-linked cell pellets were thawed on ice, resuspended in 800 μL of ice-cold Hi-C lysis buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, and 0.2% IGEPAL CA-630 with 1× cOmplete protease inhibitor (Roche, 11697498001)), and incubated at 4°C for 30 minutes with rotation. Nuclei were pelleted by centrifugation at 2500 rcf. for 5 min at 4°C and washed once with 500 μL of ice-cold Hi-C lysis buffer. After removing supernatant, nuclei were resuspended in 100 μL of 0.5% SDS and incubated at 62°C for 10 minutes. SDS was quenched by adding 335 μL of 1.5% Triton X-100 and incubating for 15 minutes at 37°C. After the addition of 50 μL of 10X NEB Buffer 2 (NEB, B7002) and 375 U of MboI restriction enzyme (NEB, R0147), chromatin was digested at 37°C for 2 hours with rotation. Following digestion, MboI enzyme was heat inactivated by incubating the nuclei at 62°C for 20 min.
To fill in the restriction fragment overhangs and mark the DNA ends with biotin, 52 μL of fill-in master mix, containing 37.5 μL of 0.4 mM biotin-dATP (Invitrogen, 19524016), 1.5 μL of 10 mM dCTP (Invitrogen, 18253013), 1.5 μL of 10 mM dGTP (Invitrogen, 18254011), 1.5 μL of 10 mM dTTP (Invitrogen, 18255018), and 10 μL of 5 U/μL DNA Polymerase I, Large (Klenow) Fragment (NEB, M0210), was added and the tubes were incubated at 37°C for 1 hour with rotation. Proximity ligation was performed by addition of 947 μL of ligation master mix, containing 150 μL of 10X NEB T4 DNA ligase buffer (NEB, B0202), 125 μL of 10% Triton X-100, 7.5 μL of 20 mg/mL BSA (NEB, B9000), 10 μL of 400 U/μL T4 DNA ligase (NEB, M0202), and 655.5 μL of water, and incubation at room temperature for 4 hours with rotation.
After proximity ligation, nuclei were pelleted by centrifugation at 2500 rcf. for 5 minutes and resuspended in 1 mL of ChIP sonication buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1 mM EGTA pH 8.0, 1% Triton X-100, 0.1% sodium deoxycholate, and 0.1% SDS with protease inhibitor). Nuclei were sonicated using a Covaris S220 for 6 minutes with the following settings: fill level 8, duty cycle 5, peak incidence power 140, cycles per burst 200. Sonicated chromatin was clarified by centrifugation at 16,100 rcf. for 15 min at 4°C and supernatant was transferred to a tube. 60 μL of protein G magnetic beads were washed three times with sonication buffer, resuspended in 50 μL of sonication buffer. Washed beads were then added to the sonicated chromatin and incubated for 1 hour at 4°C with rotation. Beads were then separated on a magnetic stand and the supernatant was transferred to a new tube. 7.5 μg of H3K27ac antibody (Abcam, ab4729) or 7.5 μg of YY1 antibody (Abcam, ab109237) was added to the tube and the tube was incubated overnight at 4°C with rotation. For YY1 six reactions were carried out and pooled prior to tagmentation. The next day, 60 μL of protein G magnetic beads were washed three time in 0.5% BSA in PBS and washed once with sonication buffer before being resuspended in 100 μL of sonication buffer and added to each sample tube. Samples were incubated for 2 hours at 4°C with rotation. Beads were then separated on a magnetic stand and washed three times with 1 mL of high salt sonication buffer (50 mM HEPES-KOH pH 7.5, 500 mM NaCl, 1 mM EDTA pH 8.0, 1 mM EGTA pH 8.0, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) followed by three times with 1 mL of LiCl wash buffer (20 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 250 mM LiCl, 0.5% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS) and once with 1 mL of TE with salt (10 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 50 mM NaCl). Beads were then resuspended in 200 μL of elution buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA pH 8.0, 1% SDS) and incubated at 65°C for 15 minutes to elute. To purify eluted DNA, RNA was degraded by the addition of 2.5 μL of 33 mg/mL RNase A (Sigma, R4642) and incubation at 37°C for 2 hours. Protein was degraded by the addition of 10 μL of 20 mg/mL proteinase K (Invitrogen, 25530049) and incubation at 55°C for 45 minutes. Samples were then incubated at 65°C for 5 hours to reverse crosslinks. DNA was then purified using Zymo DNA Clean and Concentrate 5 columns (Zymo, D4013) according to manufacturer’s protocol and eluted in 14 μL water. The amount of eluted DNA was quantified by Qubit dsDNA HS kit (Invitrogen, Q32854).
Tagmentation of ChIP DNA was performed using the Illumina Nextera DNA Library Prep Kit (Illumina, FC-121-1030). First, 5 μL of streptavidin C1 magnetic beads (Invitrogen, 65001) was washed with 1 mL of tween wash buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA pH 8.0, 1 M NaCl, 0.05% Tween-20) and resuspended in 10 μL of 2X biotin binding buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8.0, 2 M NaCl). 54.19 ng purified DNA was added in a total volume of 10 μL of water to the beads and incubated at room temperature for 15 minutes with agitation every 5 minutes. After capture, beads were separated with a magnet and the supernatant was discarded. Beads were then washed twice with 500 μL of tween wash buffer, incubating at 55°C for 2 minutes with shaking for each wash. Beads were resuspended in 25 μL of Nextera Tagment DNA buffer. To tagment the captured DNA, 3.5 μL of Nextera Tagment DNA Enzyme 1 was added with 21.5 μL of Nextera Resuspension Buffer and samples were incubated at 55°C for 10 minutes with shaking. Beads were separated on a magnet and supernatant was discarded. Beads were washed with 500 μL of 50 mM EDTA at 50°C for 30 minutes, then washed three times with 500 μL of tween wash buffer at 55°C for 2 minutes each, and finally washed once with 500 μL of 10 mM Tris-HCl pH 7.5 for 1 minute at room temperature. Beads were separated on a magnet and supernatant was discarded.
To generate the sequencing library, PCR amplification of the tagmented DNA was performed while the DNA is still bound to the beads. Beads were resuspended in 15 μL of Nextera PCR Master Mix, 5 μL of Nextera PCR Primer Cocktail, 5 μL of Nextera Index Primer 1, 5 μL of Nextera Index Primer 2, and 20 μL of water. DNA was amplified with 8 cycles of PCR. After PCR, beads were separated on a magnet and the supernatant containing the PCR amplified library was transferred to a new tube, purified using the Zymo DNA Clean and Concentrate-5 (Zymo D4003T) kit according to manufacturer’s protocol, and eluted in 14 μL water. Purified HiChIP libraries were size selected to 300-700 bp using a Sage Science Pippin Prep instrument according to manufacturer’s protocol and subject to paired-end sequencing on an Illumina HiSeq 2500. H3K27ac libraries were initially sequenced with 100×100 bp paired-end sequencing. A second round of sequencing was done on the same libraries with 50×50 bp paired-end sequencing.
4C-seq
A modified version of 4C-seq (van de Werken et al., 2012a, 2012b) was developed. The major change was the proximity ligation is performed in intact nuclei (in situ). This change was incorporated because previous work has noted that in situ ligation dramatically decreases the rate of chimeric ligations and background interactions (Nagano et al., 2015; Rao et al., 2014).
Approximately 5 million mES cells were trypsinized and then resuspended in 5 mL 10% FBS/PBS. 5 mL of 4% formaldehyde in 10% FBS/PBS was added and cells were crosslinked for 10 minutes. Glycine was added to a final concentration of 0.125 M and cells were centrifuged at 300 rcf. for 5 minutes. Cells were washed twice with PBS, transferred to a 1.5 mL Eppendorf tube, snap frozen and stored at −80.
Pellets were gently resuspended in Hi-C lysis buffer (10 mM Tris-HCl pH 8, 10 mM NaCl, 0.2% Igepal) with 1× cOmplete protease inhibitors (Roche 11697498001). Cells were incubated on ice for 30 minutes then washed once with 500 μL of ice-cold Hi-C lysis buffer with no protease inhibitors. Pellets were resuspended in 50 μL of 0.5% SDS and incubated at 62°C for 7 minutes. 145 μL of H2O and 25 μL of 10% Triton X-100 were added and tubes incubated at 37°C for 15 minutes. 25 μL of the appropriate 10X New England Biolabs restriction enzyme buffer and 200 units of enzyme were added and the chromatin was incubated at 37°C degrees in a thermomixer at 500 RPM for four hours, 200 more units of enzyme was added and the reaction was incubated overnight at 37°C degrees in a thermomixer at 500 RPM, then 200 more units were added and the reaction was incubated another four hours at 37°C degrees in a thermomixer at 500 RPM. DpnII (NEB R0543) was used as the primary cutter for both Raf1 and Etv4. Restriction enzyme was inactivated by heating to 62°C for 20 minutes while shaking at 500 rpm. Proximity ligation was performed in a total of 1200 μL with 2000 units of T4 DNA ligase (NEB M020) for six hours at room temperature. After ligation samples were spun down for 5 minutes at 2500 rcf. and resuspended in 300 μL 10 mM Tris-HCl, 1% SDS and 0.5 mM NaCl with 1000 units of Proteinase K. Cross-links were reversed by incubation overnight at 65°C.
Samples were then phenol-chloroform extracted and ethanol precipitated and the second digestion was performed overnight in 450 μL with 50 units of restriction enzyme. BfaI (NEB R0568S) was used for Etv4 and CviQI (NEB R0639S) was used for Raf1. Samples were phenol-chloroform extracted and ethanol precipitated and the second ligation was performed in 14 mL total with 6700 units of T4 DNA ligase (NEB M020) at 16°C overnight. Samples were ethanol precipitated, resuspended in 500 μL QIAGEN EB buffer, and purified with a QIAGEN PCR purification kit.
PCR amplification was performed with 16 50 μL PCR reactions using Roche Expand Long Template polymerase (Roche 11759060001). Reaction conditions are as follows: 11.2 μL Roche Expand Long Template Polymerase, 80 μL of 10 × Roche Buffer 1, 16 μL of 10 mM dNTPs (Promega PAU1515), 112 μL of 10 uM forward primer, 112 μL of 10 uM reverse primer (Table S5), 200 ng template, and milli-q water until 800 μL total. Reactions were mixed and then distributed into 16 50 μL reactions for amplification. Cycling conditions were a “Touchdown PCR” based on reports that this decreases non-specific amplification of 4C libraries (Ghavi-Helm et al., 2014). The conditions are: 2′ 94°C, 10” 94°C, 1’ 63°C, 3′ 68°C, repeat steps 2-4 but decrease annealing temperature by one degree, until 53°C is reached at which point the reaction is cycled an additional 15 times at 53°C, after 25 total cycles are performed the reaction is held for 5′ at 68°C and then 4°C. Libraries were cleaned-up using a Roche PCR purification kit (Roche 11732676001) using 4 columns per library. Reactions were then further purified with Ampure XP beads (Agencourt A63882) with a 1:1 ratio of bead solution to library following the manufactures instructions. Samples were then quantified with Qubit and the KAPA Biosystems Illumina Library Quantification kit according to kit protocols. Libraries were sequenced on the Illumina HiSeq 2500 for 40 bases in single read mode.
RNA-isolation, qRT-PCR and sequencing
RNA was isolated using the RNeasy Plus Mini Kit (QIAGEN, 74136) according to manufacturer’s instructions.
For RT-qPCR assays, reverse transcription was performed using SuperScript III Reverse Transcriptase (Invitrogen, 18080093) with oligo-dT primers (Promega, C1101) according to manufacturers’ instructions. Quantitative real-time PCR was performed on Applied Biosystems 7000, QuantStudio 5, and QuantStudio 6 instruments using TaqMan probes for Raf1 (Applied Biosystems, Mm00466513_m1) and Etv4 (Applied Biosystems, Mm00476696_m1) in conjunction with TaqMan Universal PCR Master Mix (Applied Biosystems, 4304437) according to manufacturer’s instructions.
For RNA-seq experiments, stranded polyA selected libraries were prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina, RS-122-2101) according to manufacturer’s standard protocol. Libraries were subject to 40 bp single end sequencing on an Illumina HiSeq 2500 instrument.
YY1 degradation
A clonal homozygous knock-in line expressing FKBP tagged YY1 was used for the degradation experiments. Cells were grown two passages off MEFs and then treated with dTAG-47 at a concentration of 500 nM for 24 hours.
dTAG-47 Washout Experiments
The homozygous knock-in line expressing FKBP tagged YY1 was cultured on 2i + LIF media. Cells were treated with dTAG-47 at a concentration of 500 nM for 24 hours. After 24 hours of drug treatment, cells were washed three times with PBS and passaged onto a new plate. Cells were then fed daily and passaged onto a new plate every 48 hours until YY1 protein levels were restored (5 days after drug withdrawal). Cells were then harvested for protein or RNA extraction or cross-linked for ChIP or HiChIP.
dTAG-47 synthesis
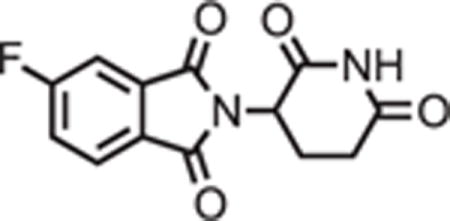
2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione
4-fluorophthalic anhydride (3.32 g, 20 mmol, 1 eq) and 3-aminopiperidine-2,6-dione hydrochloride salt (3.620 g, 22 mmol, 1.1 were dissolved in AcOH (50 mL) followed by potassium acetate (6.08 g, 62 mmol, 3.1 eq). The mixture was fitted with an air condenser and heated to 90°C. After 16 hours, the mixture was diluted with 200 mL water and cooled over ice. The slurry was then centrifuged (4000 rpm, 20 minutes, 4°C) and decanted. The remaining solid was then resuspended in water, centrifuged and decanted again. The solid was then dissolved in MeOH and filtered through a silica plug (that had been pre-wetted with MeOH), washed with 50% DCM and concentrated under reduced pressure to yield the desired product as a gray solid (2.1883 g, 7.92 mmol, 40%).
1H NMR (500 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.01 (dd, J = 8.3, 4.5 Hz, 1H), 7.85 (dd, J = 7.4, 2.2 Hz, 1H), 7.72 (ddd, J = 9.4, 8.4, 2.3 Hz, 1H), 5.16 (dd, J = 12.9, 5.4 Hz, 1H), 2.89 (ddd, J = 17.2, 13.9, 5.5 Hz, 1H), 2.65 – 2.51 (m, 2H), 2.07 (dtd, J = 12.9, 2.2 Hz, 1H).
LCMS 277.22 (M+H).
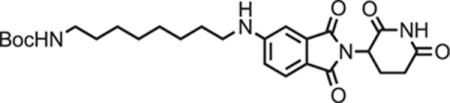
tert-butyl (8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)octyl)carbamate
2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (294 mg, 1.06 mmol, 1 eq) and tert-butyl (8-aminooctyl)carbamate (286 mg, 1.17 mmol, 1.1 eq) were dissolved in NMP (5.3 mL, 0.2M). DIPEA (369 μL, 2.12 mmol, 2 eq) was added and the mixture was heated to 90°C. After 19 hours, the mixture was diluted with ethyl acetate and washed with water and three times with brine. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. Purification by column chromatography (ISCO, 12 g column, 0%–10% MeOH/DCM, 30 minute gradient) gave the desired product as a brown solid (0.28 g, 0.668 mmol, 63%).
1H NMR (500 MHz, Chloroform-d) δ 8.12 (s, 1H), 7.62 (d, J = 8.3 Hz, 1H), 7.02 (s, 1H), 6.81 (d, J = 7.2 Hz, 1H), 4.93 (dd, J = 12.3, 5.3 Hz, 1H), 4.51 (s, 1H), 3.21 (t, J = 7.2 Hz, 2H), 3.09 (d, J = 6.4 Hz, 2H), 2.90 (dd, J = 18.3, 15.3 Hz, 1H), 2.82 – 2.68 (m, 2H), 2.16 – 2.08 (m, 1H), 1.66 (p, J = 7.2 Hz, 2H), 1.37 (d, J = 62.3 Hz, 20H).
LCMS 501.41 (M+H).
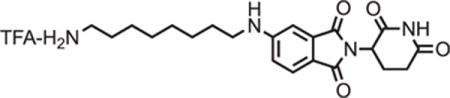
5-((8-aminooctyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione trifluoroacetate
tert-butyl (8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)octyl)carbamate (334.5 g, 0.668 mmol, 1 eq) was dissolved in TFA (6.7 mL) and heated to 50°C. After 1 hour, the mixture was cooled to room temperature, diluted with DCM and concentrated under reduced pressure. The crude material was triturated with diethyl ether and dried under vacuum to give a dark yellow foam (253.1 mg, 0.492 mmol, 74%).
1H NMR (500 MHz, Methanol-d4) δ 7.56 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 2.1 Hz, 1H), 6.83 (dd, J = 8.4, 2.2 Hz, 1H), 5.04 (dd, J = 12.6, 5.5 Hz, 1H), 3.22 (t, J = 7.1 Hz, 2H), 2.94 – 2.88 (m, 2H), 2.85 – 2.68 (m, 3H), 2.09 (ddd, J = 10.4, 5.4, 3.0 Hz, 1H), 1.70 – 1.61 (m, 4H), 1.43 (d, J = 19.0 Hz, 8H).
LCMS 401.36 (M+H).
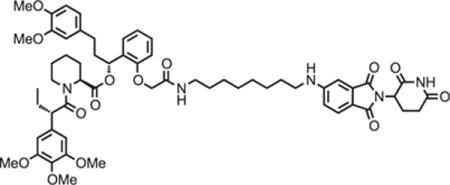
(2S)-(1R)-3-(3,4-dimethoxyphenyl)-1-(2-(2-((8-((2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindolin-5-yl)amino)octyl)amino)-2-oxoethoxy)phenyl)propyl 1-((S)-2-(3,4,5-trimethoxyphenyl)butanoyl)piperidine-2-carboxylate (dTAG47)
5-((8-aminooctyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione trifluoroacetate salt (10.3 mg, 0.020 mmol, 1 eq) was added to 2-(2-((R)-3-(3,4-dimethoxyphenyl)-1-(((S)-1-((S)-2-(3,4,5-trimethoxyphenyl)butanoyl)piperidine-2-carbonyl)oxy)propyl)phenoxy)acetic acid (13.9 mg, 0.020 mmol, 1 eq) as a 0.1 M solution in DMF (200 μl) at room temperature. DIPEA (10.5 μl, 0.060 mmol, 3 eq) and HATU (7.6 mg, 0.020 mmol, 1 eq) were then added. After 29.5 hours, the mixture was diluted with EtOAc, and washed with 10% citric acid (aq), brine, saturated sodium bicarbonate, water and brine. The organic layer was dried over sodium sulfate, filtered and condensed. Purification by column chromatography (ISCO, 4 g silica column, 0%–10% MeOH/DCM, 25 minute gradient) gave the desired product as a yellow solid (14.1 mg, 0.0131 mmol, 65%).
1H NMR (500 MHz, Methanol-d4) δ 7.55 (d, J = 8.4 Hz, 1H), 7.26 – 7.20 (m, 1H), 6.99 – 6.93 (m, 1H), 6.89 (t, J = 7.7 Hz, 2H), 6.82 (dd, J = 8.4, 2.3 Hz, 2H), 6.77 (d, J = 7.5 Hz, 1H), 6.74 (d, J = 1.9 Hz, 1H), 6.63 (d, J = 9.6 Hz, 2H), 6.12 (dd, J = 8.1, 6.0 Hz, 1H), 5.40 (d, J = 4.3 Hz, 1H), 5.03 (dd, J = 13.1, 5.5 Hz, 1H), 4.57 (d, J = 14.9 Hz, 1H), 4.46 – 4.39 (m, 1H), 4.11 (d, J = 13.6 Hz, 1H), 3.86 (t, J = 7.3 Hz, 1H), 3.80 – 3.76 (m, 7H), 3.71 – 3.65 (m, 8H), 3.14 (ddt, J = 17.2, 13.3, 7.1 Hz, 4H), 2.90 – 2.80 (m, 1H), 2.77 – 2.40 (m, 6H), 2.24 (d, J = 13.8 Hz, 1H), 2.12 – 1.97 (m, 3H), 1.92 (dq, J = 14.0, 7.8 Hz, 1H), 1.67 (ddt, J = 54.1, 14.7, 7.1 Hz, 5H), 1.50 (dd, J = 46.1, 14.1 Hz, 3H), 1.38 (dt, J = 14.5, 7.1 Hz, 4H), 1.28 – 1.17 (m, 6H), 0.87 (t, J = 7.3 Hz, 3H).
13C NMR (126 MHz, MeOD) δ 174.78, 174.69, 172.53, 171.71, 170.50, 169.66, 169.31, 156.22, 155.41, 154.62, 150.36, 148.83, 138.05, 136.90, 136.00, 134.93, 130.54, 128.40, 126.21, 123.14, 121.82, 117.94, 116.62, 113.58, 113.05, 112.73, 106.59, 70.69, 68.05, 61.06, 56.59, 56.51, 56.45, 53.42, 50.99, 50.31, 45.01, 44.09, 40.07, 37.44, 32.22, 32.17, 30.38, 30.32, 30.18, 29.84, 29.32, 28.05, 27.80, 27.58, 26.38, 23.87, 21.95, 12.57.
LCMS: 1077.35 (M+H).
In vitro DNA circularization assay
First, two plasmids (pAW49, pAW79) were generated. pAW49 contains YY1 binding sites separated by ~3.5 kb of intervening DNA. pAW79 is identical except it contains filler DNA instead of the YY1 motifs. The intervening DNA was chosen based on looking at YY1 ChIP-seq and motif distribution in mES cells to identify regions that lacked YY1 occupancy and YY1 binding motifs. The YY1 binding motifs were chosen based on successful EMSAs (Sigova et al., 2015). Approximately 200 bp of sequence was added between the binding motifs and the termini in order to provide flexibility for the termini to ligate. The plasmid was built using Gibson assembly.
Next, a PCR was run using plasmid as a template to generate a linear piece of DNA (Table S5). This PCR product was PCR purified (QIAGEN 28104) and then digested with BamHI (NEB R3136) and PCR purified. The BamHI digested template was used in the ligation assay.
The ligation assay was carried out as follows. Reactions were prepared on ice in 66 μL with the following components:
BSA control: 0.25 nM DNA, 1× T4 DNA ligase buffer (NEB B0202S), H2O 0.12 μg/μL of BSA.
YY1: 0.25 nM DNA, 1× T4 DNA ligase buffer (NEB B0202S), H2O 0.12 μg/μL of YY1.
YY1 + competitor: 0.25 nM DNA, 1× T4 DNA ligase buffer (NEB B0202S), H2O 0.12 μg/μL of YY1, 100 nM competitor DNA (Table S5)
Assuming an extinction coefficient for YY1 of 19940 M−1 cm−1 and 75% purity, that gives an approximate YY1 molar concentration of ~3 uM.
Reactions were incubated at 20°C for 20 minutes to allow binding of YY1 to the DNA. For each time point 6 μL of the reaction was withdrawn and quenched in a total volume of 9 μL with a final concentration of 30 mM EDTA, 1× NEB loading dye (NEB, B7024S), 1 μg/μL of proteinase K, and heated at 65°C for 5 minutes. Time point 0 was taken and then 600 units of T4 DNA ligase (NEB M0202) was added and the reaction was carried out at 20°C. Indicated time points were taken and then samples were run on a 4%–20% TBE gradient gel for three hours at 120 V. The gel was stained with SYBR Gold (Life Technologies S11494) and imaged with a CCD camera.
Quantification was done using Image Lab version 5.2.1 (Bio-Rad Laboratories). First, band density of the starting product and ligation product were measured. Then the percent circularized was calculated: (ligation product)/(ligation product + starting band)*100. In Figure 3 to facilitate visualization overexposed gels are shown. For the quantification exposures were used that did not have any overexposed pixels.
Co-immunoprecipitation
V6.5 mESCs were transfected with pcDNA3_FLAG_YY1 and pcDNA3_FLAG_HA using Lipofectamine 3000 (Life Technologies #L3000001) according to the manufacturer’s instructions. Briefly, cells were split and 8 million cells were plated onto a gelatinized 15 cm plate. 7.5 μg of each plasmid was mixed with 30 μL P3000 reagent and 75 μL Lipofectamine 3000 reagent (Life Technologies #L3000001) in 1250 μL of DMEM (Life technologies #11995-073). After ~12-16 hours media was changed.
Cells were harvested 48 hours after transfection by washing twice with ice-cold PBS and collected by scraping in ice-cold PBS. Harvested cells were centrifuged at 1,000 rcf. for 3 minutes to pellet cells. Supernatant was discarded and cell pellets were flash frozen and stored at −80°C until ready to prepare nuclear extract. For each 15 cm plate of cells, frozen cell pellets were resuspended in 5 mL of ice-cold hypotonic lysis buffer (20 mM HEPES-KOH pH 7.5, 20% glycerol, 10 mM NaCl, 0.1% Triton X-100, 1.5 mM MgCl2, 0.5 mM DTT and protease inhibitor (Roche, 11697498001)) and incubated on ice for 10 minutes to extract nuclei. Nuclei were pelleted by centrifugation at 14,000 rcf. for 10 minutes at 4°C. Supernatant was discarded and nuclei were resuspended in 0.5 mL of ice-cold nuclear extraction buffer (20 mM HEPES-KOH pH 7.5, 20% glycerol, 250 mM NaCl, 0.1% Triton X-100, 1.5 mM MgCl2 and protease inhibitor) and incubated for 1 hour at 4°C with rotation. Lysates were clarified by centrifugation at 14,000 rcf. for 10 minutes at 4°C. Nuclear extract, supernatant, was transferred to a new tube and diluted with 1 mL of ice-cold dilution buffer (20 mM HEPES-KOH pH 7.5, 10% glycerol, 100 mM NaCl, 0.1% Triton X-100, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT and protease inhibitor). Protein concentration of extracts was quantified by BCA assay (Thermo Scientific, 23225) and protein concentration was adjusted to 400 μg/mL by addition of appropriate volume of 1:2 nuclear extraction buffer:dilution buffer. For RNase A-treated nuclear extract experiments, 250 μL of nuclear extract (100 μg) was treated by addition of 7.5 μL of 33 mg/mL RNase A (Sigma, R4642) or 18.75 μL of 20 U/μL SUPERase In RNase Inhibitor (Invitrogen, AM2696) followed by incubation at 37°C for 10 minutes. For all experiments, an aliquot of extract was saved and stored at 80°C for use as an input sample after immunoprecipitation.
To prepare beads for immunoprecipitation of FLAG-tagged and HA-tagged YY1 from nuclear extract, 50 μL of protein G magnetic beads per immunoprecipitation was washed three times with 1 mL of blocking buffer (0.5% BSA in PBS), rotating for 5 minutes at 4°C for each wash. After separation on a magnet, beads were resuspended in 250 μL of blocking buffer. After addition of 5 μg of anti-FLAG (Sigma, F7425)), anti-HA (Abcam, ab9110), or normal IgG (Millipore, 12-370) antibody, beads were allowed to incubate for at least 1 hour at 4°C with rotation to bind antibody. After incubation, beads were washed three times with 1 mL of blocking buffer, rotating for 5 minutes at 4°C for each wash.
Washed beads were separated on a magnet and the supernatant was discarded before resuspending in 250 μL of nuclear extract (100 μg). Beads were allowed to incubate with extract overnight at 4°C with rotation. The following morning, beads were washed five times with 1 mL of ice-cold wash buffer, rotating for 5 minutes at 4°C for each wash. Washed beads were resuspended in 100 μL of 1X XT sample buffer (Biorad, 1610791) with 100 mM DTT and incubated at 95°C for 10 min. Beads were separated on a magnet and supernatant containing immunoprecipitated material was transferred to a new tube.
To assay immunoprecipitation results by western blot, 10 μL of each samples was run on a 4%–20% Bis-Tris gel (Bio-rad, 3450124) using XT MOPS running buffer (Bio-rad, 1610788) at 80 V for 20 minutes, followed by 150 V until dye front reached the end of the gel. Protein was then wet transferred to a 0.45 μm PVDF membrane (Millipore, IPVH00010) in ice-cold transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) at 250 mA for 2 hours at 4°C. After transfer the membrane was blocked with 5% non-fat milk in TBS for 1 hour at room temperature, shaking. Membrane was then incubated with 1:50,000 anti-FLAG-HRP (Sigma, A8592), 1:25:000 anti-HA-HRP (Cell Signaling, 2999), or anti-OCT3/4 (C-10, Santa Cruz sc-5279) 1:2000 antibody diluted in 5% nonfat milk in TBST and incubated overnight at 4°C, with shaking. In the morning, the membrane was washed three times with TBST for 5 min at room temperature shaking for each wash. Membranes were developed with ECL substrate (Thermo Scientific, 34080) and imaged using a CCD camera or exposed using film.
Embryoid Body Formation
Prior to differentiation, YY1-FKBP tagged knock-in mESCs were cultured in serum + LIF on irradiated MEFs. Starting 48 hours prior to the differentiation and continuing throughout the entire experiment the YY1− condition were exposed to 500 nM dTAG-47. 4,000 cells (either YY1− or YY1+) were then plated into each well of a 96-well plate (Nunclon Sphera, ThermoFisher) in Embryoid Body formation media (serum - LIF). Three plates were generated for each condition. The EBs were cultured in 96-well plates for 4 days and then pooled and cultured in ultra-low attachment culture plates (Costar, Corning). After three days, cells were harvested for single-cell RNA-seq (day 7 of differentiation). Cells were harvested for single-cell RNA-seq by dissociation with Accutase for 30 minutes at 37°C. The cells were then resuspended in PBS with 0.04% BSA and then prepared for sequencing (see section on single-cell RNA-seq). Immunohistochemistry was performed after four days (day 8 of differentiation).
Immunohistochemistry
Cells were fixed in 4% paraformaldehyde in PBS and embedded in paraffin. Cells were sectioned and stained according to standard protocols using TUJI (Biolegend 801201, 1:1000), GFAP (Dako Z0344, 1:200), and Gata-4 (Abcam ab84593 1:100) primary antibodies and appropriate Alexa Fluor dye conjugated secondary antibodies (1:1000, ThermoFisher) and DAPI. Slides were mounted with Fluoro-mount G (Electron Microscopy Science) and imaged using a Zeiss LSM 710 laser scanning confocal microscope. In all images scale bars are 50 μm.
Single-cell RNA-seq library preparation
Single-cell RNA-seq libraries were prepared using the Chromium Controller (10X Genomics). Briefly, single cells in 0.04% BSA in PBS were separated into droplets and then reverse transcription and library construction was performed according to the 10X Chromium Single Cell 3′ Reagent Kit User Guide and sequenced on an Illumina Hi-seq 2500.
dCas9-YY1 tethering
First two lentiviral constructs were generated by modifying lenti dCAS-VP64_Blast (lenti dCAS-VP64_Blast was a gift from Feng Zhang (Addgene plasmid # 61425) (Konermann et al., 2015)). The VP64 was removed to generate dCas9 alone (pAW91) or the human YY1 cDNA was inserted to the C terminus to generate dCas9-YY1 (pAW90).
For virus production, HEK293T cells grown to 50%–75% confluency on a 15 cm dish and then transfected with 15 μg of pAW90 or pAW91, 11.25 μg psPAX (Addgene 12260), and 3.75 μg pMD2.G (Addgene 12259). psPAX and pMD2.G were kind gifts of Didier Trono. After 12 hours, media was replaced. Viral supernatant was collected 24 hours after media replacement (36 hr post transfection) and fresh media was added. Viral supernatant was collected again 48 hours after the media replacement (60 hours post transfection). Viral supernatant was cleared of cells by either centrifugation at 500 × g for 10 minutes. The virus was concentrated with Lenti-X concentrator (Clontech 631231) per manufacturers’ instructions. Concentrated virus was resuspended in mES media (serum + LIF) and added to 5 million cells in the presence of polybrene (Millipore TR-1003) at 8 μg/mL. After 24 hours, viral media was removed and fresh media containing Blasticidin (Invitrogen ant-bl-1) at 10 μg/mL. Cells were selected until all cells on non-transduced plates died.
Two additional lentiviral constructs were generated (pAW12.lentiguide-GFP, pAW13.lentiguide-mCherry) by modifying lentiGuide-puro (lentiGuide-Puro was a gift from Feng Zhang (Addgene plasmid # 52963) (Sanjana et al., 2014)) to remove the puromycin and replace it either GFP or mCherry. The tethering guide RNAs (Table S5, etv4_p_sgT1_F&R, etv4_p_sgT2_F&R) were then cloned into pAW12 and pAW13. Virus was generated as described above and mES cells were transduced. Double positive cells were identified and collected by flow cytometry and expanded. These expanded cell lines were analyzed by 4C-seq, ChIP-qPCR (anti-Cas9, CST 14697), and RT-qPCR exactly as described elsewhere in the methods.
QUANTIFICATION AND STATISTICAL ANALYSIS
ChIP-MS data analysis
Previously published ChIP-ms data was downloaded (Ji et al., 2015). For each mark, the log2 ratio of the immunoprecipitation over the input and over IgG was calculated. Then a high confidence set of proteins was identified by filtering out all proteins that had a log2 fold change less than or equal to one in either the input or IgG control. Then we filtered for transcription factors using the annotation provided in the original table to end up with the 26 candidates displayed in Figure 1.
Tissue specific expression analysis
In order to identify candidate structuring factors that are broadly expressed across many tissues, tissue specific expression data from RNA-seq was downloaded from the Genotype-Tissue Expression (GTEx) Project (release V6p). Genes were considered to be expressed in a particular tissues if the median reads per million per kilobase for that tissue was greater than 5 (RPKM > 5). Broadly expressed genes were identified as genes that were expressed in greater than 90% of the 53 tissues surveyed by GTEx.
Definition of regulatory regions
Throughout the manuscript multiple analyses rely on overlaps with different regulatory regions, namely enhancers, promoters, and insulators. Here we explain how these regulatory regions were defined.
Promoters
Promoters were defined as ± 2 kilobases from the transcription start site.
Active Promoters
Active promoters were defined as ± 2 kilobases from the transcription start site that overlapped with a H3K27ac peak.
Enhancers
Enhancers were defined as H3K27ac peaks that did not overlap with a promoter.
Insulators
Insulators were defined by downloading the called insulated neighborhoods from (Hnisz et al., 2016a) (available at: http://younglab. wi.mit.edu/insulatedneighborhoods.htm). Each row represents an insulated neighborhood (defined as a SMC1 cohesin ChIA-PET interaction with both anchors overlapping a CTCF peak). The file contains six columns, columns 1-3 contain the coordinates for the left interaction anchors of the insulated neighborhoods, and columns 4-6 contain the coordinates for the right interaction anchors of the insulated neighborhoods. Columns 1-3 and 4-6 were concatenated and then filtered to identify the unique anchors. The unique loop anchors regions correspond to SMC1 ChIA-PET peaks. Insulators elements were identified as the subset of CTCF ChIP-seq peaks that overlapped the unique anchors.
Super-enhancers
Oct4/Sox2/Nanog/Med1 super-enhancers and constituents were downloaded from (Whyte et al., 2013)
Typical-enhancer constituents
Oct4/Sox2/Nanog/Med1 typical-enhancer constituents were downloaded from (Whyte et al., 2013)
ChIP-seq data analysis
Alignment
Reads from ChIP-seq experiments were aligned to the mm9 revision of the mouse reference genome using only annotated chromosomes 1-19, chrX, chrY, and chrM or to the hg19 revision of the human genome using only annotated chromosomes 1-22, chrX, chrY, and chrM. Alignment was performed using bowtie (Langmead et al., 2009) with parameters –best –k 1 –m 1 –sam and –l set to read length (mouse) or –best –k 2 –m 2 –sam and –I set to read length (human).
Read pileup for display
Wiggle files representing counts of ChIP-Seq reads across the reference genome were created using MACS (Zhang et al., 2008) with parameters –w –S –space = 50 –nomodel –shiftsize = 200. Resulting wiggle files were normalized for sequencing depth by dividing the read counts in each bin by the millions of mapped reads in each sample and were visualized in the UCSC genome browser (Kent et al., 2002).
Gene list and promoter list
For mouse data analysis 36,796 RefSeq transcripts were downloaded in the GTF format from the UCSC genome browser on February 1, 2017. For human data analysis, 39,967 RefSeq transcripts were downloaded on December 7th, 2016 in the GTF format from the UCSC genome browser on February 1, 2017. For each transcript, a promoter was created that is a 4,000 bp window centered on the transcription start site. Promoters with the same start coordinate, end coordinate, and gene name were collapsed into one representative promoter.
Peak calling
Regions with an exceptionally high coverage of ChIP-Seq reads (i.e., peaks) were identified using MACS with parameters –keep-dup = auto –p1e-9 and with corresponding input control.
Heatmaps and Metagenes
Profiles of ChIP-seq and GRO-seq signal at individual regions of interest were created by quantifying the signal in reads per million per base pair (rpm/bp) in bins that equally divide each region of interest using bamToGFF (https://github.com/BradnerLab/pipeline) with parameters –m 200 –r –d. Reads used for quantification were removed of presumed PCR duplicate reads using samtools v0.1.19-44428cd rmdup (Li et al., 2009). Promoters with the same gene id, chromosome, start, and end coordinates were collapsed into one instance.
Heatmaps of ChIP-seq profiles were used to display ChIP-seq signal at enhancer and active promoters. Each row of a heatmap represents an individual region of interest with the ChIP-seq signal profile at that region displayed in rpm/bp in a ± 2kb region centered on the region of interest. For each heatmap the number of regions of interest are displayed in parentheses in the figure panel. For murine ES cell heatmaps, ChIP-seq signal was quantified in 200 bins per region of interest. For human tissues and non-ES cell murine tissues, heatmaps were generated by quantifying ChIP-seq signal in 50 bins per region of interest.
Metagene plots were used to display the average ChIP-seq signal across related regions of interest. Metagene plots were generated for enhancer, promoter, and insulator elements, separately. The average profile (metagene) was calculated by calculating the mean ChIP-seq or GRO-seq signal profiles across the related regions of interest. For each metagene plot, the average profile is displayed in rpm/bp in a ± 2kb region centered on the regions of interest. The number of enhancers, promoters, and insulators surveyed are noted in parentheses. To facilitate comparisons of the ChIP-seq signal from a single factor between different sets of regions, the total ChIP-seq signal for each metagene analysis was quantified and is displayed in the top right corner of each metagene plot. We note that different antibodies have different immunoprecipitation efficiencies resulting in different signal intensities. Therefore, we believe that quantitative comparisons should be made across different sites in the same ChIP rather than across different ChIPs at the same site. In Figure S1F super-enhancer and typical enhancer constituent metagenes were floored by subtracting the smallest mean value across all bins in the typical enhancer constituent from each of the bins displayed.
RNA-seq data analysis
RNA-seq Analysis
RNA-seq data was aligned and quantified using kallisto (version 0.43.0) (Bray et al., 2016) with the following parameters: -b 100–single -l 180 -s 20 using the mm9 RefSeq transcriptome (downloaded on February 1, 2017). The output files represent the estimated transcript counts.
Differential gene expression analysis was performed using deseq2 (version 1.14.1) (Love et al., 2014). Analysis was performed on the gene level. To calculate the gene-level read counts, the estimated transcript counts were summed across all the isoforms of the gene. This was then input into deseq2 and adjusted p values were calculated using the default settings. Log2 fold changes and adjusted p values are included in Table S2. An FDR value of 0.05 was used as a cut off for significant differential expression. For Figure 5C, the values on the y axis are the deseq2-calculated log2 fold change values. The values on the × axis are the deseq2 calculated baseMean values.
For Figure 5D, the absolute value of the deseq2 calculated log2 fold change is plotted on the left side. On the right side the YY1 density at the promoter is plotted. Because the analysis is done on the gene level, the YY1 promoter signal for genes with multiple isoforms was averaged.
For the GO analysis the list of differentially expressed genes (Table S3) was input into the PANTHER GO analysis web tool (http://pantherdb.org/, Version 11.1) (Mi et al., 2013, 2017) and a statistical overrepresentation test was performed using the default settings.
RNA-seq Display
For displaying RNA-seq tracks, the RNA-seq data was mapped with Tophat to the mm9 RefSeq transcriptome (downloaded on February 1, 2017) using the following parameters: -n 10 tophat -p 10 –no-novel-juncs –o. Wiggle files representing counts of RNA-Seq reads across the reference genome were created using MACS (Zhang et al., 2008) with parameters –w –S –space = 50 –nomodel –shiftsize = 200. Resulting wiggle files were normalized for sequencing depth by dividing the read counts in each bin by the millions of mapped reads in each sample and were visualized in the UCSC genome browser (Kent et al., 2002).
Single-cell RNA-seq Analysis
Sequencing data was demultiplexed using the 10X Genomics Cell Ranger software (version 2.0.0) and aligned to the mm10 transcriptome. Unique molecular identifiers were collapsed into a gene-barcode matrix representing the counts of molecules per cell as determined and filtered by Cell Ranger using default parameters. Normalized expression values were generated using Cell Ranger using the default parameters. For Figure 5H the number of cells with a > 1 normalized expression value for the specified transcript were counted. For Figure S5C the cells were arranged by principal component analysis using the default Cell Ranger parameters. In Figure S5D cells were split into the two panels based on what condition they came from. The arrangement is the same as in Figure S5C. Individual cells are then colored by normalized expression level.
4C-seq data analysis
4C-seq Analysis
The 4C-seq samples were processed using fourfold (https://github.com/younglab/fourfold). Samples were first processed by removing their associated read primer sequences (Table S5) from the 5′ end of each FASTQ read. To improve mapping efficiency of the trimmed reads by making the read longer, the restriction enzyme digest site was kept on the trimmed read. After trimming the reads, the reads were mapped using bowtie with options –k 1 –m 1 against the mm9 genome assembly. All unmapped or repetitively mapping reads were discarded from further analysis. The mm9 genome was then “digested” in silico according to the restriction enzyme pair used for that sample to identify all the fragments that could be generated by a 4C experiment given a restriction enzyme pair. All mapped reads were assigned to their corresponding fragment based on where they mapped to the genome. The digestion of a sample in a 4C experiment creates a series of “blind” and “non-blind” fragments as described by the Tanay and De Laat labs (van de Werken et al., 2012a). In brief, “blind” fragments lack a secondary restriction enzyme site whereas “non-blind” fragments contain a secondary restriction enzyme site. Because of this we expect to only observe reads derived from non-blind fragments. We therefore only used reads derived from non-blind fragments.
Experiments were conducted in biological triplicate and the mutant and WT samples were quantile normalized with each other.
If no reads were detected at a non-blind fragment for a given sample when reads were detected in at least one other sample, we assigned a “0” to that non-blind fragment for the sample(s) missing reads.
4C-seq Display
To display 4C-seq genomic coverage tracks, we first smoothed the normalized 4C-seq signal using a 5kb running mean at 50bp steps across the genome for each sample. Individual replicates are displayed in Figure S4. Next, biological replicates of the same condition were combined and the mean and 95% confidence interval of the 4C-seq signal for each bin across the genome was calculated. In Figure 4 and Figure 7, the 4C-seq signal tracks display the mean 4C-seq signal along the genome as a line and the 95% confidence interval as the shaded area around the line. For each 4C-seq signal track, the viewpoint used in the 4C-seq experiment is indicated as an arrow labeled VP.
To quantify the change in 4C-seq signal in a specific region of interest, the normalized 4C-seq signal (non-smoothed) was counted for each sample and the mean and standard deviation of the quantified signal was calculated for biological replicates of the same condition. The mean and standard deviation of the quantified signal was normalized to the appropriate control condition (either WT or dCas9) before plotting. Below each 4C-seq signal track, the quantified region is indicated as a red bar labeled “Quantified region.” The coordinates of the quantified region for Raf1 are chr6:115598005-115604631, and for Etv4 are chr11:101644625-101648624.
ChIA-PET data analysis
ChIA-PET Read Processing
For each ChIA-PET dataset, raw reads were processed in order to identify a set of putative interactions that connect interaction anchors for further statistical modeling and analysis. First, paired-end tags (PETs), each containing two paired reads, were analyzed for the presence of the bridge-linker sequence and trimmed to facilitate read mapping. PETs containing at least one instance of the bridge-linker sequence in either of the two reads were kept for further processing and reads containing the bridge-linker sequence were trimmed immediately before the linker sequence using cutadapt with options “-n 3 -O 3 -m 15 -a forward=ACGCGATATCT TATCTGACT -a reverse=AGTCAGATAAGATATCGCGT” (http://cutadapt.readthedocs.io/en/stable/). PETs that did not contain an instance of the bridge-linker sequence were not processed further. Trimmed reads were mapped individually to the mm9 mouse reference genome using Bowtie with options “-n 1 -m 1 -p 6” (Langmead et al., 2009). After alignment, paired reads were re-linked with an in-house script using read identifiers. To avoid potential artifacts arising from PCR bias, redundant PETs with identical genomic mapping coordinates and strand information were collapsed into a single PET. Potential interaction anchors were determined by identifying regions of local enrichment in the individually mapped reads using MACS with options “-g mm -p 1e-9– nolambda–nomodel–shiftsize=100” (Zhang et al., 2008). PETs with two mapped reads that each overlapped a different potential interaction anchor by at least 1 bp were used to identify putative interactions between the overlapped interaction anchors. Each putative interaction represents a connection between two interaction anchors and is supported by the number of PETs (PET count) that connect the two interaction anchors.
ChIA-PET Statistical Analysis Overview
In processing our chromatin interaction data, we sought to identify the putative interactions that represent structured chromatin contacts, defined as chromatin contacts that are structured by forces other than the fiber dynamics resulting from the linear genomic distance between the two contacting regions. In contrast, we sought to filter out putative interactions that likely result from PETs arising from non-structured chromatin contacts, defined as contacts resulting from the close linear genomic proximity of the two contacting regions, or from technical artifacts of the ChIA-PET protocol. We expect that putative interactions that represent structured chromatin contacts should be detected with greater frequency, or PET count, than expected given the linear genomic distance between the two contacting regions, allowing us to distinguish between these two classes of interactions.
To this end, we developed Origami, a statistical method to identify high-confidence interactions that are likely to represent structured chromatin contacts. Conceptually, Origami uses a semi-Bayesian two-component mixture model to estimate the probability that a putative interaction corresponds to one of two groups: structured chromatin contacts, or non-structured chromatin contacts and technical artifacts. Origami estimates this as a probability score for each putative interaction by modeling the relationship between PET count, linear genomic distance between interaction anchors, and read depth at the interaction anchors. High-confidence interactions are then identified as the subset of putative interactions that are likely to represent structured chromatin contacts, by requiring high-confidence interactions to have a probability score > 0.9.
All the methods below were developed within the origami software that is available at https://github.com/younglab/origami. The version used was version 1.1 (tagged on GitHub repository as v1.1). The software below was run with the following parameters:–iterations = 10000–burn-in = 100–prune = 0–min-dist = 4000–peak-count-filter = 5.
Origami Statistical Model
We developed Origami, a method to analyze ChIA-PET data, in order to identify putative interactions that likely represent structured chromatin contacts, and to filter out putative interactions that likely represent non-structured chromatin contacts that occur as a result of the close linear genomic proximity of contacting regions and interactions that represent technical artifacts of the ChIA-PET protocol. This includes modeling of the relationship between the number of PETs observed to support each interaction (Ii), linear genomic distance between interaction anchors (di), and the sequencing depth at the interaction anchors, to estimate the probability that each putative interaction (i) represents a structured chromatin contact given the observed PET count (Ii).
We initially assume that putative interactions classify into one of two groups, j∈ {0; 1}, such that each putative interaction, i∈ {1..N}, has a latent group identity Zi that corresponds to a value of j. Group 1 is designated as the set of putative interactions resulting from structured chromatin contacts that we expect to detect with greater frequencies than expected given the linear genomic distance between the contacting regions. Group 0 is designated as the set of putative interactions resulting from non-structured chromatin contacts due to close linear genomic proximity of the contacting regions, or from technical artifacts of the ChIA-PET protocol.
We developed a semi-Bayesian two-component mixture model to estimate the probability that each putative interaction represents a structured chromatin contact. For each group, we modeled the likelihood to observe the PET count (Ii) under that group as a Poisson process with two underlying factors. These factors are the number of PETs observed as a result of being part of the group (Gij), and the number of PETs observed as a result of the linear genomic distance between the anchors given the group (Dij). We modeled the number of PETs observed as a result of being part of the group (Gij) as a Poisson process with mean, λj. We modeled the number of PETs observed as a result of the linear genomic distance between the anchors given the group (Dij) as a Poisson process with mean, μij. Since these two factors are thought to be independent (Phanstiel et al., 2015), the total Poisson process is the summation of these two underlying factors.
We modeled the data variables under the following distributions:
We modeled our parameters with the following prior distributions:
Since wi1 is a binomial probability, wi0 = 1 wi1.
From these priors and likelihood distributions, the posterior distributions of these parameters are as follows:
Aside from Dij and μij, we estimated the parameters using the iterative process Markov Chain Monte Carlo (MCMC) with Gibbs Sampling with the appropriate posterior to sample from (Gelman et al., 2004).
To estimate μij, we modeled the function between Dij and the linear genomic distance (di) on the log10 scale using a smoothed cubic spline (via smooth.spline in R), taking μij to be the expected number of PETs to be observed due to distance (Dij) given the linear genomic distance (di), for each putative interaction (i).
The constants αi and βi were set to be as minimally informative as possible. The constant αi was set equal to the number of putative interactions sharing one anchor with i that have PET counts less than Ii. The constant βi was set equal to the number of putative interactions sharing one anchor with i that have PET counts greater than Ii plus the ratio of the depth score (si) to the median depth score with all values < 1 floored to 0. The depth score (si) for each putative interaction is defined as the product of the number of reads that map to its interaction anchors.
Origami Implementation
We implemented the model described above by Markov Chain Monte Carlo simulation. By iteratively estimating the group identity (Zi) of each putative interaction, we sought to explore the probability space for Zi and determined a probability score (pi) for each putative interaction that reflects the probability that the interaction results from a structured chromatin contact (belongs to group 1). The steps in our implementation are as follows.
For each putative interaction, we recorded the number of PETs observed that support the interaction (Ii), the linear genomic distance of the interaction between the outermost basepairs of the putative interaction’s two anchors (di), and a depth score (si), which is defined as the product of the number of the reads in the dataset that map to each anchor of the putative interaction.
To seed the parameters of the model for the first iteration, the following was performed. The mixing weights (wij) were set to be equal at 0.5 for each interaction. The group process means (λj) were assigned values of 5 and 1 for group 1 and 0, respectively. The distance process mean (μij) was initially set to 0 for all interactions.
Additionally values of αi and βi were computed for each interaction, but not used in the first iteration. In all subsequent iterations, αi and βi, are used in updating the values of the mixing weights (wij). The parameter αi was set equal to the number of putative interactions sharing one anchor with i that have PET counts less than Ii. The parameter βi was set equal to the number of putative interactions sharing one anchor with i that have PET counts greater than Ii plus the ratio of the depth score (si) over the median depth score for all putative interactions, where when this ratio is less than 1 it is floored to 0.
- For each putative interaction, we estimated the likelihood (Iij) that the putative interaction is observed with PET count (Ii), given that the putative interaction belongs to group 1 and group 0, as follows:
where dPoisson is the density function of the Poisson distribution for the mean λj+μij and evaluated on Ii. - We calculated the relative weighted likelihood (ri) of each putative interaction belonging to group 1. To do this we multiplied each of the two likelihoods calculated for each putative interaction by their respective mixing weights (wij) and evaluated as follows.
- We update the group identity (Zi) of each interaction by drawing from the binomial distribution with a probability of ri as follows.
where rBinomial means we randomly draw 1 or 0 with the probability of ri for drawing 1. - We update the mixing weights (wij) using our newly updated group identies (Zi), by drawing from the Beta distribution in the following way:
where rBeta means we randomly draw from the beta distribution with the above parameters. Since wi1 is a binomial probability, wi0 = 1 − wi1. - In order to estimate the PET counts for Gij and Dij, we randomly sampled the number of PETs for Gij and Dij by taking advantage of the fact that when two Poisson variables are known to sum to a given count, then the distribution of either variable follows a binomial distribution with probability λj / =(λj + μij). Accordingly, we estimated the PET counts for Gij and Dij in the following way:
where rBinomial means we randomly draw up to Ii PETs with the probabilty λj / =(λj + μij) of drawing each PET. - We update the group process mean (λj) using the following identity, requiring that λ1 > λ0 in order to maintain identifiability of the two groups (although during our runs this constraint was not necessary).
where rGamma means we randomly draw from the Gamma distribution with the above parameters. To update the distance process means (μij), we calculated the function between Dij and the log10(di+1), using a smoothed cubic spline (via smooth.spline in R). To simplify estimation of μij, we chose to take the maximum likelihood estimate of this process.
We iterated steps 4-10 in the following way. We performed an initial 1,000 iterations as a burn-in, which were discarded. Then we performed 10,000 iterations.
- We estimated the probability that each putative interaction belongs to group 1 by calculating a probability score (pi) for each putative interaction that equals the mean value of Zi across the 10,000 iterations. High-confidence interactions were identified as putative interactions with pi > 0.9.
HiChIP data analysis
HiChIP Processing
The HiChIP samples were processed by first identifying reads with a restriction fragment junction (i.e., a site where ligation occurred). Reads containing the restriction fragment junction were trimmed such that the information 5′ to the junction was kept. Reads without restriction fragment junctions were left untrimmed. Reads were then mapped using bowtie with options −k 1 −m 1 against the mm9 genome assembly. All unmapped or repetitively mapping reads were discarded from further analysis. Reads were joined back together in pairs by their read identifier. The genome was binned and for every pair of bins the number of PETs joining them was calculated. These data were then used as input into the Origami pipeline described above to identify significant bin to bin interaction pairs.
HiChIP Analysis
Quantitative analysis of HiChIP and Hi-C data (Figures 6 and 7) was done as follows. High-confidence interactions were identified by Origami. A union of high-confidence interactions was then created for each experiment.
| Experiment | Figure | Condition | Replicate |
|---|---|---|---|
| Degron | 6, S6 | noDrug | 1 |
| Degron | 6, S6 | noDrug | 2 |
| Degron | 6, S6 | noDrug | 3 |
| Degron | 6, S6 | yesDrug | 1 |
| Degron | 6, S6 | yesDrug | 2 |
| Degron | 6, S6 | yesDrug | 3 |
| Washout | 7 | Untreated (UT) | 1 |
| Washout | 7 | Untreated (UT) | 2 |
| Washout | 7 | Untreated (UT) | 3 |
| Washout | 7 | Treated (TR) | 1 |
| Washout | 7 | Treated (TR) | 2 |
| Washout | 7 | Washout (WO) | 1 |
| Washout | 7 | Washout (WO) | 2 |
| Washout | 7 | Washout (WO) | 3 |
| CTCF Washout | 7 | Untreated (UT) | 1 |
| CTCF Washout | 7 | Untreated (UT) | 2 |
| CTCF Washout | 7 | Treated (TR) | 1 |
| CTCF Washout | 7 | Treated (TR) | 2 |
| CTCF Washout | 7 | Washout (WO) | 1 |
| CTCF Washout | 7 | Washout (WO) | 2 |
For example, the degron high-confidence set would consist of the union of the 6 degron samples listed above. The PET counts were then normalized to each other using deseq2 (Love et al., 2014). The mean of each group was then calculated and then the fold change was then calculated by taking the ratio of the perturbed condition to the non-perturbed condition (i.e., yesDrug to noDrug or TR/UT;WO/UT) with a pseudocount of 0.5 added to both. This complete set of significant interactions is what is displayed in Figure 6B as “All Interactions.”
For subset analysis the anchor of each interaction was classified by overlapping with known genomic features as defined earlier. This resulted in a binary score for whether an anchor overlapped with an enhancer, promoter, insulator, YY1, or CTCF. The interactions were then subset to identify the following groups:
YY1 not present (Figure 6): no YY1 at either end of the interaction.
YY1 enhancer-promoter interactions (Figures 6 and 7): YY1 at both ends AND an enhancer or promoter at both ends.
CTCF-CTCF interaction: CTCF at both ends.
The log2 fold change for these groups is plotted in Figures 6B and 7F.
The analysis in Figure 6C was done by identifying the gene at the end of YY1 enhancer-promoter loops. This was done by intersecting promoters (as defined above) with the significant loop anchors. Genes with multiple promoters were collapsed after the intersection to generate a list of genes at the end of YY1 enhancer-promoter loops. The deseq2 calculated log2 fold change for these genes is then plotted in Figure 6C. Genes are colored based on the deseq2 calculated adjusted p value (as in Figure 5).
HiChIP Display
HiChIP interaction matrices displayed in Figures 6D and 6E. For these interaction matrices, all putative interactions are displayed and the intensity of each pixel represents the mean of the deseq2 normalized interaction frequency of all biological replicates of that condition. In Figures 6D and 6E the outlined pixel, which reflects the frequency of interaction between sites at the base of the diagonals, was used to quantify the change in normalized interaction frequency upon YY1 degradation.
In Figure 2, high-confidence HiChIP interactions are displayed as arcs. For display, the interactions displayed were filtered to remove bin to adjacent bin contacts and non-enhancer-promoter interactions. Arcs were centered on the relevant genomic feature within the bin (for example a ChIP-seq peak summit or transcription start site).
Interaction classification
High-confidence ChIA-PET and HiChIP interactions were classified based on the presence of enhancer, promoter, and insulator elements at the anchors of each interaction as defined above. In the case where an interaction anchor overlapped both an enhancer and an insulator or a promoter and an insulator a hierarchy where anchors were considered first as promoters, then enhancers, then insulators. For example, if there is an interaction where the left anchor is insulator/promoter and the right anchor is enhancer/insulator it would be counted as an enhancer-promoter interaction and not an insulator-insulator interaction.
To display summaries of the classes of high-confidence interactions, each class of interactions is displayed as an arc between the relevant enhancer, promoter, and insulator elements. The thickness of the arcs approximately reflects the percentage of interactions of that class relative to the total number of interactions that were classified. In the main figures, enhancer-enhancer, enhancer-promoter, promoter-promoter, and insulator-insulator interaction classes are displayed. Extended summaries that additionally include enhancer-insulator and promoter-insulator interactions are displayed in the supplemental figures.
Figure Display
In certain figure panels displaying genome tracks, enhancer elements are indicated as red boxes labeled “Enhancer.” These regions represent the authors’ interpretation of the ChIP-seq data and are distinct from the algorithmically defined enhancers used in the quantitative genome-wide analysis.
Statistical Analysis
In order to use the unpaired t test we made two assumptions.
Populations are distributed according to a Gaussian distribution. For most experiments three replicates were used, and so sample sizes were too small to reliably calculate departure from normality (i.e., with a D’Agostino test).
The two populations have the same variance. A test for variance was not carried out.
Full p values are listed here*:
| Figure | Sub panel | Test | Biological Replicates | P value |
|---|---|---|---|---|
| 4B | 4C-seq | Student’s T-Test | 3 | 0.011 |
| 4B | ChIP-qPCR | Student’s T-Test | 3 | 0.0066 |
| 4B | RT-qPCR | Student’s T-Test | 6 | < 0.0001 |
| 4C | 4C-seq | Student’s T-Test | 3 | 0.0013 |
| 4C | ChIP-qPCR | Student’s T-Test | 3 | 0.0048 |
| 4C | RT-qPCR | Student’s T-Test | 6 | 0.0394 |
| 6B | Welch | Two Sample T-Test | 3 | < 2.2e-16 |
| 6D | HiChIP | Student’s T-Test | 3 | 0.0162 |
| 6D | RNA-seq | Wald | 2 | 7.22E-13 |
| 6E | HiChIP | Student’s T-Test | 3 | 0.0446 |
| 6E | RNA-seq | Wald | 2 | 1.25E-58 |
| 7D | 4c-seq | Student’s T-Test | 3 | 0.004717003 |
| 7D | RT-qPCR | Student’s T-Test | 6 | < 0.0001 |
| S6D | Raf1 Wald | 2 | 1.63E-53 | |
| S6D | Etv4 Wald | 2 | 2.88E-34 |
note that the Student’s t test was conducted using GraphPad Prism which sets a lower limit at 0.0001, the Welch Two Sample t test was conducted using R which sets a lower limit at 2.2e-16, Wald test was conducted using deseq2 in R which does not have a lower limit on the p value.
DATA AND SOFTWARE AVAILABILITY
All datasets used are summarized in Table S4. Oligos used can be found in Table S5.
Origami: https://github.com/younglab/origami using version v1.1-alpha-2. Fourfold: https://github.com/younglab/fourfold usingv0.1
The accession number for the data reported in this paper is GEO: GSE99521.
Supplementary Material
Highlights.
YY1 generally occupies active enhancers and promoters across cell types
YY1 can form dimers and promote DNA interactions
Perturbation of YY1 binding disrupts enhancer-promoter looping and gene expression
YY1’s structural role accounts for diverse functions reported previously
Acknowledgments
We thank the Whitehead Institute Genome Technology Core, FACS facility, and Johanna Goldmann for their assistance. This work is supported by NIH grants HG002668/GM123511 (R.A.Y.), R37HD045022/R01-NS088538/R01-MH104610 (R.J.), Ludwig Graduate Fellowship funds (A.S.W.), NSF GRFP (A.V.Z.), American Cancer Society New England Division Postdoctoral Fellowship PF-16-146-01-DMC (D.S.D.), Margaret and Herman Sokol Postdoctoral Award (D.H.), ACS Postdoctoral Fellowship PF-17-010-01-CDD (B.N.), Merck Fellow of the Damon Runyon Cancer Research Foundation DRG-2196-14 (D.L.B.), Hope Funds for Cancer Research Grillo-Marxuach Family Fellowship (B.J.A.), Emerald Foundation (M.A.C.), and Cancer Research Institute Irvington Fellowship (Y.E.G.). The Whitehead Institute filed a patent application based on this study. R.A.Y., J.E.B., and N.S.G. are the founders of Syros Pharmaceuticals. B.J.A. is a shareholder in Syros. R.A.Y. is a founder of Syros Pharmaceuticals, Marauder Therapeutics, and Omega Therapeutics. J.E.B. is a Scientific Founder of SHAPE Pharmaceuticals, Acetylon Pharmaceuticals, Tensha Therapeutics (now Roche), and C4 Therapeutics. J.E.B. is now an executive and shareholder in Novartis AG. N.S.G. is a founder and member of the Scientific Advisory Board of C4 and Petra Therapeutics. R.J. is a cofounder of Fate Therapeutics, Fulcrum Therapeutics, and Omega Therapeutics.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and five tables and can be found with this article online at https://doi.org/10.1016/j.cell.2017.11.008.
AUTHOR CONTRIBUTIONS
Conceptualization, A.S.W., C.H.L., A.A.S., and R.A.Y.; Methodology, A.S.W. and C.H.L.; Software, A.S.W., C.H.L., D.S.D., and B.J.A.; Formal Analysis, A.S.W., B.J.A., and D.S.D., Investigation, A.S.W., C.H.L., A.V.Z., A.A.S., N.M.H., M.A.C., and D.H.; Resources, N.M.H., B.N., D.L.B., R.J., J.E.B., N.S.G., and Y.E.G.; Writing – Original Draft, A.S.W., C.H.L., and R.A.Y.; Writing – Review & Editing, all authors; Visualization, A.S.W.; Supervision, R.A.Y. and A.S.W.; Funding Acquisition, R.A.Y.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Arnold et al. (1996); Baniahmad et al. (1990); Castellano et al. (2009); ENCODE Project Consortium (2012); Filippova et al. (1998); Filippova et al. (2002); Gertz et al. (2013); Heidari et al. (2014); Kleiman et al. (2016); Köhne et al., 1993); Lin et al. (2010); Lobanenkov et al. (1990); Mendenhall et al. (2010); Moore et al. (2012); Natesan and Gilman (1993); Nora et al. (2016); Pope et al. (2014); Seto et al. (1991); Stadler et al. (2011); Wang et al. (2012); Wu et al. (2007); Xu et al. (2007); Yusufzai et al. (2004).
References
- Adhya S. Multipartite genetic control elements: Communication by DNA loop. Annu Rev Genet. 1989;23:227–250. doi: 10.1146/annurev.ge.23.120189.001303. [DOI] [PubMed] [Google Scholar]
- Allen BL, Taatjes DJ. The Mediator complex: A central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoutzias GD, Robertson DL, Van de Peer Y, Oliver SG. Choose your partners: Dimerization in eukaryotic transcription factors. Trends Biochem Sci. 2008;33:220–229. doi: 10.1016/j.tibs.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Arnold R, Burcin M, Kaiser B, Muller M, Renkawitz R. DNA bending by the silencer protein NeP1 is modulated by TR and RXR. Nucleic Acids Res. 1996;24:2640–2647. doi: 10.1093/nar/24.14.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baniahmad A, Steiner C, Köhne AC, Renkawitz R. Modular structure of a chicken lysozyme silencer: involvement of an unusual thyroid hormone receptor binding site. Cell. 1990;61:505–514. doi: 10.1016/0092-8674(90)90532-j. [DOI] [PubMed] [Google Scholar]
- Beagan JA, Duong MT, Titus KR, Zhou L, Cao Z, Ma J, Lachanski CV, Gillis DR, Phillips-Cremins JE. YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 2017;27:1139–1152. doi: 10.1101/gr.215160.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Bonev B, Cavalli G. Organization and function of the 3D genome. Nat Rev Genet. 2016;17:772–772. doi: 10.1038/nrg.2016.147. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Buecker C, Wysocka J. Enhancers as information integration hubs in development: Lessons from genomics. Trends Genet. 2012;28:276–284. doi: 10.1016/j.tig.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano G, Torrisi E, Ligresti G, Malaponte G, Militello L, Russo AE, McCubrey JA, Canevari S, Libra M. The involvement of the transcription factor Yin Yang 1 in cancer development and progression. Cell Cycle. 2009;8:1367–1372. doi: 10.4161/cc.8.9.8314. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddapah S, Jothi R, Schones DE, Roh TY, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502:499–506. doi: 10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- Degner SC, Verma-Gaur J, Wong TP, Bossen C, Iverson GM, Torkamani A, Vettermann C, Lin YC, Ju Z, Schulz D, et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc Natl Acad Sci USA. 2011;108:9566–9571. doi: 10.1073/pnas.1019391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012;149:1233–1244. doi: 10.1016/j.cell.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe ME, Zhang X, McGinnis L, Biggers J, Li E, Shi Y. Targeted disruption of mouse Yin Yang 1 transcription factor results in peri-implantation lethality. Mol Cell Biol. 1999;19:7237–7244. doi: 10.1128/mcb.19.10.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen JM, Fan ZP, Hnisz D, Ren G, Abraham BJ, Zhang LN, Weintraub AS, Schujiers J, Lee TI, Zhao K, Young RA. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature. 2017;543:270–274. doi: 10.1038/nature21688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova G, Lindblom L, Meincke L, Klenova E, Neiman P, Collins S, Doggett N, Lobanenkov V. A widely expressed transcription factor with multiple DNA sequence specificity, CTCF, is localized at chromosome segment 16q22.1 within one of the smallest regions of overlap for common deletions in breast and prostate cancers. Genes, Chromosom. Cancer. 1998 [PubMed] [Google Scholar]
- Filippova GN, Qi CF, Ulmer JE, Moore JM, Ward MD, Hu YJ, Loukinov DI, Pugacheva EM, Klenova EM, Grundy PE, et al. Tumor-associated zinc finger mutations in the CTCF transcription factor selectively alter tts DNA-binding specificity. Cancer Res. 2002;62:48–52. [PubMed] [Google Scholar]
- Fraser J, Williamson I, Bickmore WA, Dostie J. An overview of genome organization and how we got there: From FISH to Hi-C. Microbiol Mol Biol Rev. 2015;79:347–372. doi: 10.1128/MMBR.00006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH, et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 2009;462:58–64. doi: 10.1038/nature08497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriele M, Vulto-van Silfhout AT, Germain PL, Vitriolo A, Kumar R, Douglas E, Haan E, Kosaki K, Takenouchi T, Rauch A, et al. YY1 Haploinsufficiency Causes an Intellectual Disability Syndrome Featuring Transcriptional and Chromatin Dysfunction. Am J Hum Genet. 2017;100:907–925. doi: 10.1016/j.ajhg.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman A, Carlin JB, Stern HS, Rubin DB. Bayesian Data Analysis 2004 [Google Scholar]
- Gertz J, Savic D, Varley KE, Partridge EC, Safi A, Jain P, Cooper GM, Reddy TE, Crawford GE, Myers RM. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol Cell. 2013;52:25–36. doi: 10.1016/j.molcel.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, Furlong EEM. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014;512:96–100. doi: 10.1038/nature13417. [DOI] [PubMed] [Google Scholar]
- Gibcus JH, Dekker J. The hierarchy of the 3D genome. Mol Cell. 2013;49:773–782. doi: 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Akopyan G, Garban H, Bonavida B. Transcription factor YY1: Structure, function, and therapeutic implications in cancer biology. Oncogene. 2006;25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- Gorkin DU, Leung D, Ren B. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell. 2014;14:762–775. doi: 10.1016/j.stem.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Yoon HS, Franklin A, Jain S, Ebert A, Cheng HL, Hansen E, Despo O, Bossen C, Vettermann C, et al. CTCF-binding elements mediate control of V(D)J recombination. Nature. 2011;477:424–430. doi: 10.1038/nature10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, et al. CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell. 2015;162:900–910. doi: 10.1016/j.cell.2015.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan N, Kelley DE, Perry RP. Delta, a transcription factor that binds to downstream elements in several polymerase II promoters, is a functionally versatile zinc finger protein. Proc Natl Acad Sci USA. 1991;88:9799–9803. doi: 10.1073/pnas.88.21.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Bickmore W. The ins and outs of gene regulation and chromosome territory organisation. Curr Opin Cell Biol. 2007;19:311–316. doi: 10.1016/j.ceb.2007.04.016. [DOI] [PubMed] [Google Scholar]
- Heath H, Ribeiro de Almeida C, Sleutels F, Dingjan G, van de Nobelen S, Jonkers I, Ling KW, Gribnau J, Renkawitz R, Grosveld F, et al. CTCF regulates cell cycle progression of alphabeta T cells in the thymus. EMBO J. 2008;27:2839–2850. doi: 10.1038/emboj.2008.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidari N, Phanstiel DH, He C, Grubert F, Jahanbani F, Kasowski M, Zhang MQ, Snyder MP. Genome-wide map of regulatory interactions in the human genome. Genome Res. 2014;24:1905–1917. doi: 10.1101/gr.176586.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Day DS, Young RA. Insulated neighborhoods: Structural and functional units of mammalian gene control. Cell. 2016a;167:1188–1200. doi: 10.1016/j.cell.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016b;351:1454–1458. doi: 10.1126/science.aad9024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Seo H, Zhang T, Wang Y, Jiang B, Li Q, Buckley DL, Nabet B, Roberts JM, Paulk J, et al. MELK is not necessary for the proliferation of basal-like breast cancer cells. eLife. 2017 doi: 10.7554/eLife.26693. Published online September 19, 2017. https://doi.org/10.7554/eLife.26693. [DOI] [PMC free article] [PubMed]
- Hwang SS, Kim YU, Lee S, Jang SW, Kim MK, Koh BH, Lee W, Kim J, Souabni A, Busslinger M, Lee GR. Transcription factor YY1 is essential for regulation of the Th2 cytokine locus and for Th2 cell differentiation. Proc Natl Acad Sci USA. 2013;110:276–281. doi: 10.1073/pnas.1214682110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011;146:119–133. doi: 10.1016/j.cell.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeronimo C, Langelier MF, Bataille AR, Pascal JM, Pugh BF, Robert F. Tail and kinase modules differently regulate core mediator recruitment and function in vivo. Mol Cell. 2016;64:455–466. doi: 10.1016/j.molcel.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Dadon DB, Abraham BJ, Lee TI, Jaenisch R, Bradner JE, Young RA. Chromatin proteomic profiling reveals novel proteins associated with histone-marked genomic regions. Proc Natl Acad Sci USA. 2015;112:3841–3846. doi: 10.1073/pnas.1502971112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, Dadon DB, Powell BE, Fan ZP, Borges-Rivera D, Shachar S, Weintraub AS, Hnisz D, Pegoraro G, Lee TI, et al. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18:262–275. doi: 10.1016/j.stem.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RDD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman E, Jia H, Loguercio S, Su AI, Feeney AJ. YY1 plays an essential role at all stages of B-cell differentiation. Proc Natl Acad Sci USA. 2016;113:E3911–E3920. doi: 10.1073/pnas.1606297113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol. 1993;13:7612–7624. doi: 10.1128/mcb.13.12.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhne A, Baniahmad A, Renkawitz R. NeP1. A Ubiquitous Transcription Factor Synergizes with v-ERBA in Transcriptional Silencing. J Mol Biol. 1993;232:747–755. doi: 10.1006/jmbi.1993.1428. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb P, McKnight SL. Diversity and specificity in transcriptional regulation: The benefits of heterotypic dimerization. Trends Biochem Sci. 1991;16:417–422. doi: 10.1016/0968-0004(91)90167-t. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, Young RA. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc. 2006;1:729–748. doi: 10.1038/nprot.2006.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Cattoglio C, Tjian R. Looping back to leap forward: Transcription enters a new era. Cell. 2014;157:13–25. doi: 10.1016/j.cell.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, Mansson R, Sigvardsson M, Hagman J, Espinoza CA, Dutkowski J, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Schmidt-supprian M, Shi Y, Hobeika E, Barteneva N, Jumaa H, Pelanda R, Reth M, Skok J, Rajewsky K, et al. Yin Yang 1 is a critical regulator of B-cell development. Genes Dev. 2007;21:1179–1189. doi: 10.1101/gad.1529307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanenkov VV, Nicolas RH, Adler VV, Paterson H, Klenova EM, Polotskaja AV, Goodwin GH. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene. 1990;5:1743–1753. [PubMed] [Google Scholar]
- López-Perrote A, Alatwi HE, Torreira E, Ismail A, Ayora S, Downs JA, Llorca O. Structure of Yin Yang 1 oligomers that cooperate with RuvBL1-RuvBL2 ATPases. J Biol Chem. 2014;289:22614–22629. doi: 10.1074/jbc.M114.567040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161:1012–1025. doi: 10.1016/j.cell.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet. 2010;11:761–772. doi: 10.1038/nrg2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews KS. DNA looping. Microbiol Rev. 1992;56:123–136. doi: 10.1128/mr.56.1.123-136.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al. The human transcriptome across tissues and individuals. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Koche RP, Truong T, Zhou VW, Issac B, Chi AS, Ku M, Bernstein BE. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010;6:e1001244. doi: 10.1371/journal.pgen.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, Thomas PD. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D189. doi: 10.1093/nar/gkw1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkenschlager M, Nora EP. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu Rev Genomics Hum Genet. 2016;17:17–43. doi: 10.1146/annurev-genom-083115-022339. [DOI] [PubMed] [Google Scholar]
- Moore JM, Rabaia NA, Smith LE, Fagerlie S, Gurley K, Loukinov D, Disteche CM, Collins SJ, Kemp CJ, Lobanenkov VV, Filippova GN. Loss of maternal CTCF is associated with peri-implantation lethality of Ctcf null embryos. PLoS ONE. 2012;7:e34915. doi: 10.1371/journal.pone.0034915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muerdter F, Stark A. Gene regulation: Activation through space. Curr Biol. 2016;26:R895–R898. doi: 10.1016/j.cub.2016.08.031. [DOI] [PubMed] [Google Scholar]
- Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat Methods. 2016;13:919–922. doi: 10.1038/nmeth.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagano T, Várnai C, Schoenfelder S, Javierre BM, Wingett SW, Fraser P. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol. 2015;16:175. doi: 10.1186/s13059-015-0753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra V, Rocha PP, An D, Raviram R, Skok JA, Mazzoni EO, Reinberg D. CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science. 2015;347:1017–1022. doi: 10.1126/science.1262088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natesan S, Gilman MZ. DNA bending and orientation-dependent function of YY1 in the c-fos promoter. Genes Dev. 1993;7(12B):2497–2509. doi: 10.1101/gad.7.12b.2497. [DOI] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted degradation of CTCF decouples local insulation of chromosome domains from higher-order genomic compartmentalization. Bioriv. 2016 doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169:930–944.e22. doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson R, Renkawitz R, Lobanenkov V. CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet. 2001;17:520–527. doi: 10.1016/s0168-9525(01)02366-6. [DOI] [PubMed] [Google Scholar]
- Ong CT, Corces VG. Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Atchison ML. Isolation of a candidate repressor/activator, NF-E1 (YY-1, delta), that binds to the immunoglobulin kappa 3′ enhancer and the immunoglobulin heavy-chain mu E1 site. Proc Natl Acad Sci USA. 1991;88:9804–9808. doi: 10.1073/pnas.88.21.9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko N, Jin Y, Wong KH, Struhl K. Mediator undergoes a compositional change during transcriptional activation. Mol Cell. 2016;64:443–454. doi: 10.1016/j.molcel.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Heidari N, Snyder MP. Mango: a bias correcting ChIA-PET analysis pipeline. Bioinformatics. 2015;31:3092–3098. doi: 10.1093/bioinformatics/btv336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JE, Corces VG. CTCF: Master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong CT, Hookway TA, Guo C, Sun Y, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo A, Dillon N. Three-dimensional genome architecture: Players and mechanisms. Nat Rev Mol Cell Biol. 2015;16:245–257. doi: 10.1038/nrm3965. [DOI] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Yue F. Transcriptional enhancers: Bridging the genome and phenome. Cold Spring Harb Symp Quant Biol. 2015;80:17–26. doi: 10.1101/sqb.2015.80.027219. [DOI] [PubMed] [Google Scholar]
- Saldaña-Meyer R, González-Buendía E, Guerrero G, Narendra V, Bonasio R, Recillas-Targa F, Reinberg D. CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 2014;28:723–734. doi: 10.1101/gad.236869.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleif R. DNA looping. Annu Rev Biochem. 1992;61:199–223. doi: 10.1146/annurev.bi.61.070192.001215. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Schwalie PC, Ross-Innes CS, Hurtado A, Brown GD, Carroll JS, Flicek P, Odom DT. A CTCF-independent role for cohesin in tissue-specific transcription. Genome Res. 2010;20:578–588. doi: 10.1101/gr.100479.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E, Shi Y, Shenk T. YY1 is an initiator sequence-binding protein that directs and activates transcription in vitro. Nature. 1991;354:241–245. doi: 10.1038/354241a0. [DOI] [PubMed] [Google Scholar]
- Shi Y, Seto E, Chang LS, Shenk T. Transcriptional repression by YY1, a human GLI-Krüppel-related protein, and relief of repression by adenovirus E1A protein. Cell. 1991;67:377–388. doi: 10.1016/0092-8674(91)90189-6. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lee JS, Galvin KM. Everything you have ever wanted to know about Yin Yang 1…. Biochim Biophys Acta. 1997;1332:F49–F66. doi: 10.1016/s0304-419x(96)00044-3. [DOI] [PubMed] [Google Scholar]
- Shore D, Langowski J, Baldwin RL. DNA flexibility studied by covalent closure of short fragments into circles. Proc Natl Acad Sci USA. 1981;78:4833–4837. doi: 10.1073/pnas.78.8.4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigova AA, Abraham BJ, Ji X, Molinie B, Hannett NM, Guo YE, Jangi M, Giallourakis CC, Sharp PA, Young RA. Transcription factor trapping by RNA in gene regulatory elements. Science. 2015;350:978–981. doi: 10.1126/science.aad3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F. Gene regulation at a distance: From remote enhancers to 3D regulatory ensembles. Semin Cell Dev Biol. 2016;57:57–67. doi: 10.1016/j.semcdb.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Splinter E, Heath H, Kooren J, Palstra RJ, Klous P, Grosveld F, Galjart N, de Laat W. CTCF mediates long-range chromatin looping and local histone modification in the beta-globin locus. Genes Dev. 2006;20:2349–2354. doi: 10.1101/gad.399506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015;163:1611–1627. doi: 10.1016/j.cell.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Seto E. Unlocking the mechanisms of transcription factor YY1: Are chromatin modifying enzymes the key? Gene. 1999;236:197–208. doi: 10.1016/s0378-1119(99)00261-9. [DOI] [PubMed] [Google Scholar]
- van de Werken HJG, Landan G, Holwerda SJB, Hoichman M, Klous P, Chachik R, Splinter E, Valdes-Quezada C, Öz Y, Bouwman BAM, et al. Robust 4C-seq data analysis to screen for regulatory DNA interactions. Nat Methods. 2012a;9:969–972. doi: 10.1038/nmeth.2173. [DOI] [PubMed] [Google Scholar]
- van de Werken HJG, de Vree PJP, Splinter E, Holwerda SJB, Klous P, de Wit E, de Laat W. 4C technology: protocols and data analysis. Methods Enzymol. 2012b;513:89–112. doi: 10.1016/B978-0-12-391938-0.00004-5. [DOI] [PubMed] [Google Scholar]
- Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, Lee K, Canfield T, Weaver M, Sandstrom R, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22:1680–1688. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Hughes TR. A catalogue of eukaryotic transcription factor types, their evolutionary origin, and species distribution. A Handbook of Transcription Factors (Subcellular Biochemistry) 2011:25–73. doi: 10.1007/978-90-481-9069-0_3. [DOI] [PubMed] [Google Scholar]
- Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Shi Y, Mulligan P, Gay F, Landry J, Liu H, Lu J, Qi HH, Wang W, Nickoloff JA, et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol. 2007;14:1165–1172. doi: 10.1038/nsmb1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Donohoe ME, Silva SS, Lee JT. Evidence that homologous X-chromosome pairing requires transcription and Ctcf protein. Nat Genet. 2007;39:1390–1396. doi: 10.1038/ng.2007.5. [DOI] [PubMed] [Google Scholar]
- Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356 doi: 10.1126/science.aaj2239. Published online May 5, 2017. https://doi.org/10.1126/science.aaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai TM, Tagami H, Nakatani Y, Felsenfeld G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol Cell. 2004;13:291–298. doi: 10.1016/s1097-2765(04)00029-2. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Stovall DB, Inoue K, Sui G. The oncogenic role of Yin Yang 1. Crit Rev Oncog. 2011;16:163–197. doi: 10.1615/critrevoncog.v16.i3-4.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.