

Abstract
Acute myeloid leukemia (AML) is a disease associated with epigenetic dysregulation. 11q23 translocations involving the H3K4 methyltransferase MLL1 (KMT2A) generate oncogenic fusion proteins with deregulated transcriptional potential. The Polymerase Associated Factor complex (PAFc) is an epigenetic co-activator complex that makes direct contact with MLL fusion proteins and is involved in AML, however its functions are not well understood. Here, we explored the transcriptional targets regulated by the PAFc that facilitate leukemia by performing RNA-sequencing after conditional loss of the PAFc subunit Cdc73. We found Cdc73 promotes expression of an early hematopoietic progenitor gene program that prevents differentiation. Among the target genes, we confirmed the protein arginine methyltransferase Prmt5 is a direct target that is positively regulated by a transcriptional unit that includes the PAFc, MLL1, HOXA9 and STAT5 in leukemic cells. We observed reduced PRMT5-mediated H4R3me2s following excision of Cdc73 placing this histone modification downstream of the PAFc and revealing a novel mechanism between the PAFc and Prmt5. Knock down or pharmacologic inhibition of Prmt5 causes a G1 arrest and reduced proliferation resulting in extended leukemic disease latency in vivo. Overall, we demonstrate the PAFc regulates Prmt5 to facilitate leukemic progression and is a potential therapeutic target for AMLs.
Keywords: Leukemia, MLL, Epigenetics, PAFc, PRMT5, small molecule inhibitors
Introduction
Dysregulation of transcriptional and epigenetic mechanisms are increasingly recognized as critical events in the development of both acute myeloid (AML) and acute lymphoid leukemia (ALL). Over 70% of AML patients are estimated to harbor mutations in epigenetic regulatory proteins1. The Polymerase Associated Factor complex (PAFc) is an epigenetic co-activator protein complex that is essential for the proliferation of various forms of AML2-4, including those harboring MLL1 translocations. The PAFc is also linked to solid tumors where subunits are mutated or overexpressed in various cell types, suggesting context dependent functions5-8. The PAFc is composed of six subunits consisting of CDC73, PAF1, CTR9, LEO1, RTF1 and SKI8 in human cells9-11. Through the CDC73 subunit, the PAFc associates with the CTD of RNA Pol II facilitating transcriptional initiation, elongation, and termination12, 13. Once localized to targets, the PAFc helps recruit transcriptional and histone modifying complexes associated with transcriptional activation14-17. For example, BRE1 of the E2/E3 ubiquitin ligase complex RAD6/BRE1, which catalyzes mono-ubiquitination of histone H2BK120, directly binds to the PAF1 subunit of the PAFc18. Additionally, the Rtf1 subunit was recently described to directly interact with Rad6 to regulate H2B ubiquitylation19. Further, we and others have shown that the PAF1 and CTR9 subunits make direct contact with the H3K4 methyltransferase Mixed Lineage Leukemia 1 (MLL1)2, 4, 20.
MLL1 is involved in chromosomal translocations to one of over 70 fusion partners resulting in expression of oncogenic MLL fusion proteins21. In adult AML and ALL, around 10% of patients present with MLL-translocations, which increases to 50% for infant AMLs22. Interestingly, while the PAFc-MLL1 interaction is essential for the proliferation of several subtypes of AML (including those with MLL translocations), disruption of the PAFc-MLL1 interaction is tolerated in hematopoietic stem cells, thus identifying the PAFc as a potential therapeutic target3, 4. In addition to leukemia, various subunits of the PAFc have been implicated in a variety of solid tumors. For example, PAF1 is overexpressed in pancreatic cancer7. While, germline mutations in the CTR9 subunit have been described in Wilms tumor6, overexpressed CTR9 correlates with a poor prognosis through increased transcriptional activation in ERα+ breast cancer8. The CDC73 subunit coded by the HRPT2 gene, is commonly mutated in hyperparathyroidism-jaw tumors pointing to a tumor suppressor function5. However, CDC73 is overexpressed in liver and breast cancer23. These data point to context dependent functions for subunits of the PAFc to act as oncogenes and tumor suppressors. In leukemic cells, our data demonstrated the PAFc is necessary for MLL1 recruitment to and activation of Hoxa9 and Meis1, which are thought to act as oncogenic drivers in as much as 50% of human AML24. Despite a clear requirement for the PAFc in leukemic cells, the transcriptional programs controlled by the PAFc remain unclear.
Here, we investigated the gene expression programs controlled by the PAFc in AML cells harboring MLL fusion proteins. We discovered the PAFc promotes expression of a subset of pro-leukemic genes associated with self-renewal and a histone methyltransferase program. Within the latter program, we identified protein arginine methyltransferase 5 (Prmt5), amongst several Prmts, as a direct transcriptional target of the PAFc. PRMT5 deposits symmetric methyl marks on histone residues H4R3, H3R2, and H3R825-27 and is overexpressed in several solid cancers28, 29. As such, clinical trials are underway testing small molecules designed to inhibit PRMT5 enzymatic activity in patients with relapsed solid tumors and non-Hodgkin's lymphoma (ClinicalTrials.gov Identifier: NCT02783300). Still, conditional loss of Prmt5 in the hematopoietic system is lethal due to defective HSC cycling underscoring a need to better understand the regulation and function of Prmt5 30. Investigating the role of Prmt5 in AML, we discovered the PAFc along with STAT5, MLL1, and HOXA9, bind to the Prmt5 locus and regulate expression in leukemic cells. Our data shows chemical inhibition or genetic knockdown of Prmt5 extends AML in vivo consistent with recent work implicating Prmt5 in the progression of hematologic malignancies31-34. Our data identify the PAFc as a direct regulator of the Prmt5 locus, connecting the PAFc to the deposition of H4R3me2s. Together, these data place the PAFc atop a pro-leukemogenic gene program that includes, not only Hoxa9 and Meis1, but also Prmt5 and illustrates the potential of therapeutic targeting the PAFc and Prmt5 in AML.
Results
Conditional Excision of Cdc73 Induces Differentiation and Alters the Histone Modifications of AML Cells
To evaluate the role of the PAFc in leukemic cells, we utilized a Cdc73 floxed mouse to generate MLL-AF9 AML cell lines containing a tamoxifen (4OHT) inducible CreER (MA9-Cdc73fl-CreER) and control cell lines (MA9-CreER) (Figure 1A, S1A). Our past work demonstrates that conditional excision of the PAFc subunit Cdc73, reduces the proliferation of MA9 leukemic cells and MLL fusion protein target gene expression3. We sought to explore the cellular phenotypes associated with Cdc73 excision that would elucidate its role in leukemia. Upon 4OHT treatment, MA9-Cdc73fl-CreER cells displayed morphology changes consistent with cellular myeloid differentiation including increased cytoplasmic to nuclear ratios (Figure 1B) and reduced c-Kit and increased Cd14 cell surface expression (Figure 1C). qPCR analysis of genes associated with myeloid differentiation (Mmp8, Mmp12, Id2, Ltf and Cd80) showed upregulation upon Cdc73 excision (Figure 1D)35, 36. Cell cycle analysis revealed Cdc73 excision results in a G1 phase cell cycle block (Figure 1E), while changes in apoptosis were mild (Figure S1B). We analyzed global histone modifications in MA9-Cdc73fl-CreER cells and detected a reduction of H3K4me3, H3K79me2, and an almost complete ablation of H2BK120ub following excision of Cdc73 (Figure 1F). Additionally, loss of Cdc73 reduced colony formation of MA9-Cdc73fl-CreER cells (Figure S1C). Overall, these data suggest that loss of Cdc73 partially induces differentiation of leukemic cells and alters global H3K4me3, H3K79me2 and H2B120ub.
Figure 1. Conditional Excision of Cdc73 Induces Differentiation and Alters Epigenetic Landscape in MA9 Leukemic Cells.
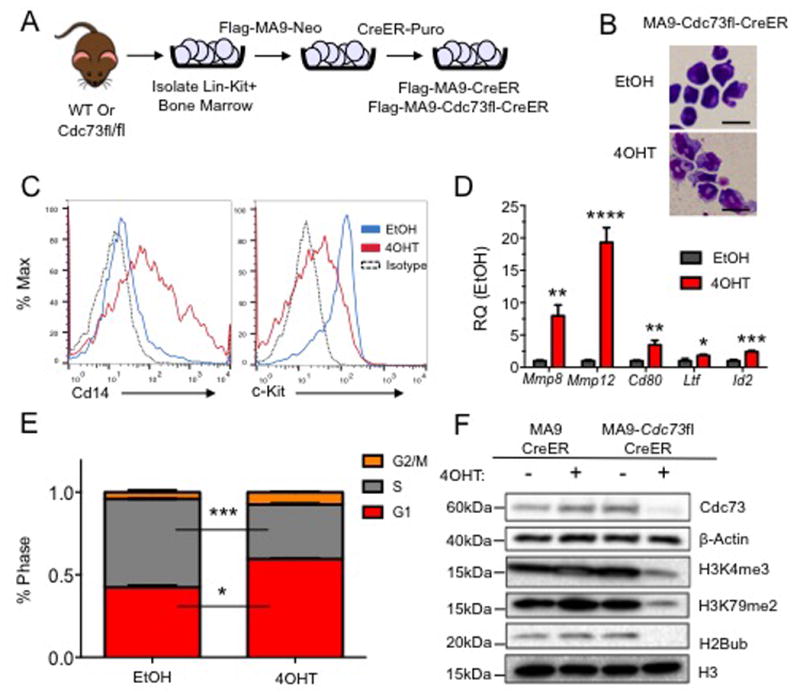
A) Scheme showing generation of Flag-tagged MA9 leukemic Cdc73 excision cells driven by inducible CreER for phenotypic analysis. B) MA9-Cdc73fl-CreER cells were treated with either EtOH or 7.5nM 4OHT for 48 hours, cytospun onto slides and stained with the Hema3 staining kit. Representative images at 100× show morphology changes. Scale bar = 20μM C) Excision of Cdc73 increases level of differentiation as measured by c-Kit, Cd14 cell surface markers on MA9 leukemic cells by flow cytometry. D) Cdc73 excision increases differentiation gene signature expression including Mmp8, Mmp12, Cd80, Ltf and Id2. All samples were scaled to β-actin and normalized to EtOH control expression. * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001, **** = p-value < 0.0001. Student t-test. E) Cdc73 excision results in a G1 cell cycle block. MA9 cells were treated as in (B) and cell cycle analysis performed using ModFit software. * = p-value < 0.05, *** = p-value < 0.001 Student t-test. F) Excision of Cdc73 alters global epigenetic landscape. Cells were treated as in (B), and ∼2×106 cells were harvested after 72 hours and subjected to histone extraction and probed with the indicated antibodies. H3 and β-Actin are used as loading controls. All experiments were performed in biological duplicate and technical duplicate for phenotypic analysis or technical triplicate for qPCR.
Prmt5 Expression is Dependent on Cdc73 in MLL-AF9 Cells
Next, we asked what gene programs were controlled by the PAFc that may contribute to blocking differentiation of leukemic cells. To that end, we performed RNA-sequencing using our MA9-Cdc73fl-CreER and MA9-CreER cell lines. Cell lines were treated with 4OHT for 48 hours and RNA harvested for sequencing (Figure 2A). To control for gene expression differences between cell lines or variation in MLL-AF9 expression, we also isolated RNA from the MA9-Cdc73fl-CreER cell line treated with EtOH (Figure 2A). A comparison of gene expression changes in MA9-Cdc73fl-CreER cells treated with EtOH or 4OHT (EtOH control) to MA9-Cdc73fl-CreER and MA9-CreER cells treated with 4OHT (Cre control) revealed a slope of 0.98 (Figure 2B) revealing remarkably similar gene expression changes. This indicates the changes in gene expression are in response to loss of Cdc73. We observed that only a subset of genes were significantly changed (fold-change > 1.5, FDR q-value <0.01) and that genes were both upregulated and downregulated upon Cdc73 excision perhaps pointing to a role for the PAFc in both activation and repression (Figure 2C)37. We report 1835 upregulated and 2151 downregulated genes at 48 hours following Cdc73 excision.
Figure 2. Prmt5 Identified as a Potential Downstream Target Gene of the PAFc.
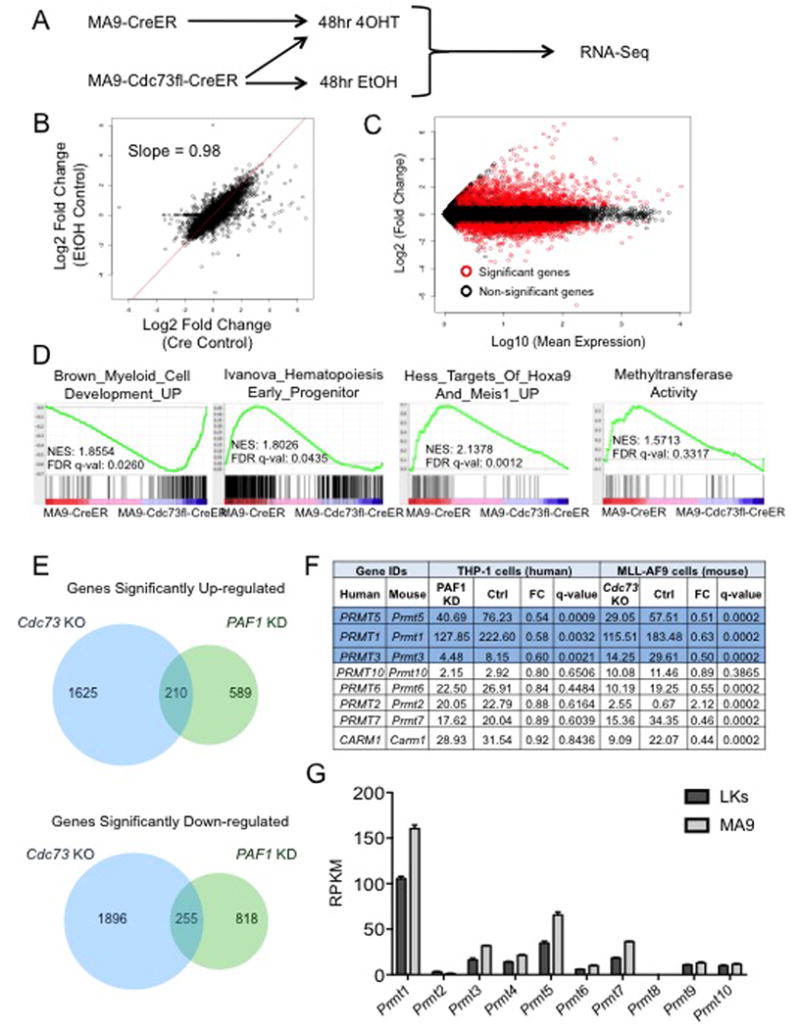
A) Scheme used for RNA-sequencing. B) Scatter-plot of changed genes between MA9-Cdc73fl-CreER 4OHT treated cells compared to either EtOH or CreER controls. Best fit displayed with slope. C) MA plot at 48 hours post 4OHT treatment showing fold change of genes in excision cells over MA-CreER control relative to the average RPKM value of the given gene across all samples. Red circles represent genes with 1.5 fold change up or down and a q-value < 0.01. D) GSEA analysis shows MA9 samples aligns with early progenitor and Hoxa9/Meis1 targets as expected while Cdc73 excision cells align with a myeloid differentiation gene set. Genes expressed in MA9 cells enrich with a methyltransferase gene program that is lost after excision. NES and q-value are displayed. E) Venn diagrams depict both overlapping and distinct genes are up and down regulated with excision of Cdc73 and knockdown of PAF1. Comparison of our Cdc73 excision dataset with a PAF1 KD data set in human THP-1 (MLL-AF9 driven) leukemic cells displayed 210 overlapping upregulated genes and 255 overlapping down-regulated genes. F) Table shows Prmt family gene expression changes following knockdown of PAF1 in THP1 cells and excision of Cdc73 in MA9 cells. Blue rows indicate genes, which displayed significant expression changes (FC > 1.5, q-value < 0.01) in both Cdc73 and PAF1 datasets. G) The Prmt family is over-expressed in MA9 leukemic cells relative to normal Lin-Kit+ bone marrow by RPKM using an FDR < 0.5. RNA-sequencing samples were submitted in biological duplicate.
To determine whether the genes altered by loss of Cdc73 are associated with specific gene programs or pathways, we performed GAGE analysis. Upon Cdc73 excision, several gene pathways associated with differentiation such as immune response were enriched in our upregulated gene set, while methyltransferase activity and transfer of methyl groups were enriched in the downregulated gene set (Figure S2A). To explore specific gene programs regulated by the PAFc in leukemic cells we also utilized Gene Set Enrichment Analysis (GSEA). Consistent with the differentiation phenotype described above, excision of Cdc73 increased expression of a gene program associated with myeloid development while control MA9-CreER cells expressed an early hematopoietic progenitor program along with a Hoxa9/Meis1 gene program characteristic of MLL-fusion driven leukemias (Figure 2D). Interestingly, 25 of 129 direct MLL-AF9 target genes and 42 of 165 direct MLL-ENL targets showed significant expression changes upon loss of Cdc73 in our MA9-Cdc73fl-CreER cells 38, 39 (Table S3, S4, S5). These data are suggestive of PAFc functions that are both cooperative and independent of MLL fusion proteins. Consistent with the GAGE analysis, expression of a methyltransferase activity program was enriched in genes that were downregulated in MA9-Cdc73fl-CreER cells following 4OHT treatment (Figure 2D). Among the genes altered in the methyltransferase activity program were several members of the protein arginine methyltransferase (Prmt) family. Thus, we investigated the fold change of Prmt genes with an RPKM > 5 following excision of Cdc73 and observed most were downregulated (Figure S2B).
We next compared our mouse Cdc73 excision expression data set with a knockdown dataset from human leukemic THP-1 cells targeting another PAFc subunit, PAF1 and discovered both unique and overlapping genes from the up and downregulated groups (Figure 2E, Table S2)40. Among the commonly downregulated genes PRMT5, PRMT1, and PRMT3 were all significantly downregulated following loss of either Cdc73 or PAF1 (q<0.05) (Figure 2F). These data implied that not only are these genes most likely regulated by the PAFc vs a CDC73 specific target, but the regulation is conserved between mouse and human. Next, we compared how Prmt expression differs between normal and leukemic cells. While most Prmt genes are upregulated in MLL-AF9 cells compared to normal lin-c-kit+ mouse bone marrow cells, Prmt5 showed one of the highest percentage increases in expression (Figure 2G). Taken together, our analysis directed us to investigate the role of several Prmt's in MLL-fusion leukemia.
Prmt5 is Necessary for Leukemia Cell Growth In Vitro and In Vivo
To investigate the role of Prmt genes in leukemia, we narrowed our evaluation to Prmt1, Prmt4, and Prmt5 based on several criteria: 1) the identification of Prmt1 and Prmt5 in both PAF1 and Cdc73 expression analyses (Figure 2F), 2) genetic deletion of Prmt1, Prmt4, and Prmt5 shows embryonic lethality in mice suggesting non-overlapping function and 3) reported roles of all three in various cancers including leukemia31-34, 41-43. We confirmed our RNA-seq with qPCR, which showed downregulation of Prmt1, Prmt4, and Prmt5 following loss of Cdc73 (Figure S2C). We generated constitutively active pSM2C-shRNA vectors targeting these Prmts and validated them in MA9 cells by qPCR (Figure S3B, top panel). Knockdown of Prmt1, Prmt4, and Prmt5 results in a significant decrease in cell number of MLL-AF9 transformed cells (Figure S3C). However, a significant decrease of colony formation was only observed following knockdown of Prmt4 and Prmt5 (Figure S3D). Interestingly, only overexpression of Prmt5 significantly increased proliferation and colony formation in MA9 cells (Figure S3E, S3F)44. Since knockdown and overexpression of Prmt5 showed significant phenotypic effects and we identified this gene in both PAF1 and Cdc73 expression data sets, we focused on the role of Prmt5 in leukemic cells and its regulation by the PAFc.
We generated an inducible TetON Prmt5 knockdown system to more precisely assess the function of Prmt5 in leukemic cells. We observed reduced mRNA and protein following shRNA induction (Figure S3A, S3B lower panel). We performed competition assays using MA9-TetON-shPrmt5 or control MA9-TetON-shRenilla cell lines cocultured with parental MA9-TetOn cells. Cells with Prmt5 knockdown were outcompeted by parental MA9-TetON cells while those expressing the shRenilla construct remained stable through the experiment (Figure 3A-B). This illustrates a competitive growth disadvantage in leukemic cells with reduced Prmt5 expression. Similarly, growth assays show knockdown of Prmt5 causes a proliferative defect in MA9 cells (Figure 3C). Interestingly, c-Kit and Cd14 cell surface expression only mildly change after Prmt5 knockdown suggesting the differentiation of leukemic cells observed following excision of Cdc73, (Figure 1) is not facilitated by Prmt5 (Figure 3D). However, reduction of Prmt5 does lead to G1 cell cycle arrest similarly to Cdc73 excision suggesting Prmt5 contributes to cell cycle progression in leukemic cells downstream of the PAFc (Figure 3E).
Figure 3. Modulation of Prmt5 Disrupts Leukemic Progression in MA9 Cells.
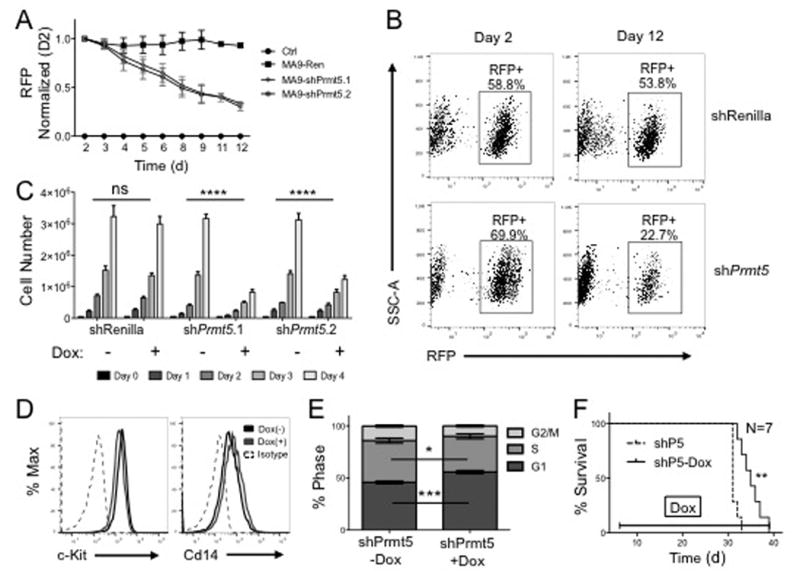
A) Loss of Prmt5 reduces cell fitness. Cells were plated in duplicate in a 3:1 ratio of parent MA9-TetON cells to MA9-TetON shPrmt5 knockdown cells and treated with vehicle or Dox. RFP signal was read every two days by flow cytometry. B) Gating of RFP readout is shown at Day 2 and Day 12. C) Prmt5 knockdown reduces cell proliferation. Cells were treated with 1μg/mL Dox once per day and grown for 4 days. shRenilla is used as a control. **** = p-value < 0.0001 by Two-way ANOVA. D) Prmt5 knockdown has minimal effect on differentiation. Cells were treated as in (C) and harvested at 48 hours, incubated with c-Kit or Cd14 antibody and analyzed by flow cytometry. E) Loss of Prmt5 leads to G1 cell cycle arrest. Cells were treated as in (C) and stained with DAPI and analyzed by flow cytometry. * = p-value < 0.05, *** = p-value < 0.001 by two-tailed student's t-test. F) Prmt5 knockdown extends disease latency in Hoxa9/Meis1 leukemic model. Survival curve of a secondary transplant of TetON-MA9-shPrmt5 cells transplanted into sub-lethally irradiated recipients treated with vehicle or Dox beginning on day 4. N=7, ** = p-value < 0.01 by Wilcoxon test. All experiments were performed in biological duplicate and technical duplicate.
We next used our inducible knockdown system to test how loss of Prmt5 affects AML disease latency in vivo. To examine the role of Prmt5 in a broader group of AML, we knocked down expression in leukemic cells driven by overexpression of Hoxa9 and Meis1, which are direct MLL-fusion targets overexpressed in ∼50% of AML24. Here, we transduced primary TetON Hoxa9/Meis1 leukemic cells with TRMPV-shPrmt5 vectors and injected them into syngeneic recipient mice followed by knock down of Prmt5 with doxycycline (Dox). We verified that cells showed activated shRNA and knocked down Prmt5 prior to injection (Figure S4A). Dox mediated Prmt5 knockdown significantly extended disease latency by 4.5 days compared to our non-Dox treated group (Figure 3F). Leukemic cell infiltration in the liver, spleen, and bone marrow was comparable between both groups as was the spleen weight at the moribund state (Figure S4B-D). Interestingly, we observe similar levels of Prmt5 expression suggesting a selective pressure to maintain Prmt5 expression (Figure S4E). We repeated this assay using MA9-TetON cells and see no statistical latency extension between our treatment groups (Figure S5A, B). We observe a significant reduction in spleen weight in the Dox treated group at the moribund state, but comparable levels of leukemic cells in the BM, tissue infiltration, and Prmt5 expression (Figure S5C-F). The long disease latency in the MLL-AF9 model likely provides additional time to select cells that escape Prmt5 knock down. These data indicate an important role for Prmt5 in promoting cell cycle progression and leukemogenesis in AML displaying overexpression of the Hoxa9/Meis1 gene program such as MLL fusion leukemias.
MLL-AF9 Cells are Dependent on PRMT5 Enzymatic Activity
We next asked whether the enzymatic activity of Prmt5 was required. We transduced our MA9-TetOn-shPrmt5 cells with either wild-type PRMT5 (WT-P5) or a catalytic dead PRMT5 mutant (CD-P5) as described27, and subjected them to growth assays. While the growth defect in MA9-TetON-shPrmt5 cells after Prmt5 knockdown is rescued by WT-P5 expression, the CD-P5 mutant did not suggesting the enzymatic activity of Prmt5 is necessary for MLL-AF9 leukemic cell growth (Figure 4A). Thus, we investigated histone modifications in MLL-AF9 cells following knockdown of Prmt5. Western blot analysis revealed global reduction of pan symmetric di-methylation of arginine (SDMA) and H4R3me2s following knockdown of Prmt5 with little change on H3K4me3, H3K79me2, and H2Bub (Figure 4B). We similarly detected reduced H4R3me2s and pan SDMA after Cdc73 excision consistent with the PAFc regulating Prmt5 (Figure 4C). This effect was specific since H4R3 asymmetric methylation (H4R3me2a) catalyzed by Prmt1 and Prmt6 remained unchanged (Figure S6B). Additionally, knockdown of the PAF1 subunit in HeLa cells also reduces PRMT5 protein level consistent with the PAFc regulating PRMT5 expression (Figure S6D). To test whether Prmt5 overexpression can rescue proliferation following loss of Cdc73, we stably transduced WT-Prmt5 and CD-Prmt5 into our MA9-Cdc73fl-CreER cells. Ectopic expression of neither WT-Prmt5 nor CD-Prmt5 rescues the growth of cells lacking Cdc73 consistent with a multitude of important PAFc target genes (Figure S6A). Because Prmt5 knockdown did not completely arrest MA9 cell growth, we asked whether this might be due to escapees. To this end, we maintained MA9-TetON-shPrmt5 cells in Dox for 9 days and monitored Prmt5 expression by western blot. Prmt5 protein expression does recover after initial knockdown suggesting a selective pressure for maintaining Prmt5 expression in leukemic cells (Figure 4D and S6C). These data imply that MLL-AF9 leukemic cells require the PRMT5 protein, and its enzymatic activity to maintain and promote oncogenesis.
Figure 4. The catalytic activity of Prmt5 is Required for MA9 leukemic cells.
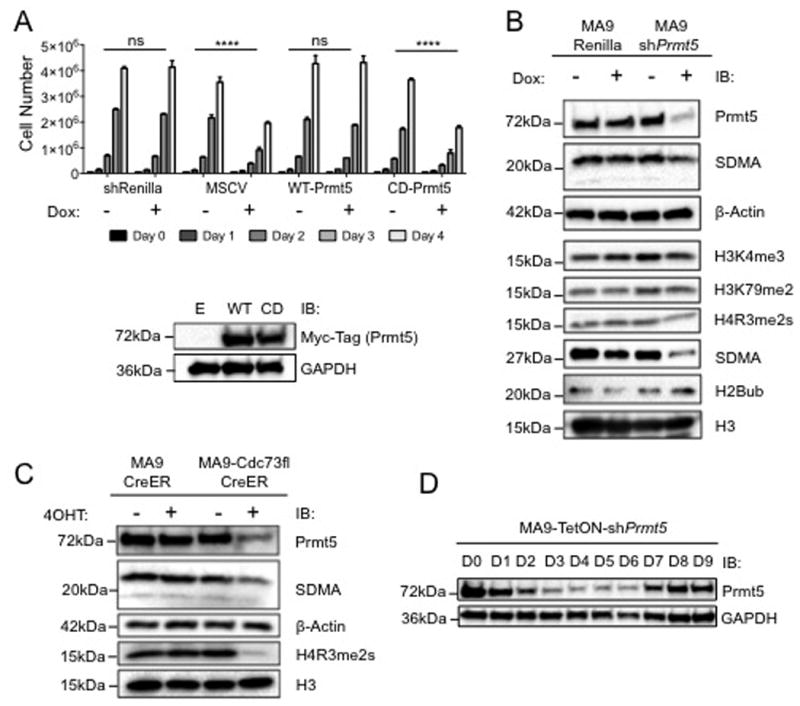
A) Expression of catalytic dead Prmt5 mutant cannot rescue growth of MA9 cells. WT-Prmt5 or CD-Prmt5 were transduced into MA9-TetON-shPrmt5 cells to generate stable lines. Cells were treated with either PBS or Dox every day for 4 days. **** = p-value < 0.0001 by Two-way ANOVA. Performed in biological triplicate and technical triplicate. Protein level of expression vectors is shown below. B) Knockdown of Prmt5 specifically alters its histone modification targets. MA9-TetON cells containing either shRenilla or shPrmt5 were treated with 1μg/mL Doxycycline every day for 3 days followed by histone extraction or whole cell extraction and western blots. Data indicates global reduction of SDMA and H4R3me2s without changes to H3K4me3, H3K79me2 and H2Bub. Prmt5 antibodies were used as confirmation of knockdown with whole cell lysate. H3 and β-Actin are used as loading controls. C) Cdc73 excision in MA9 cells reduces PRMT5 histone modification targets. Cells were treated as in (B) with the exception of 7.5nM EtOH or 4OHT instead of doxycycline. Reduced H4R3me2s and SDMA is observed. H3 and β-Actin are used as loading controls. D) MA9 cells show strong selective pressure to maintain PRMT5. Cells were maintained in the presence of 1μg/mL Dox and passaged as needed. 1×106 cells were harvested per day and PRMT5 level in the lysate was measured with Prmt5 antibody and GAPDH as a loading control. Westerns performed in biological duplicate.
The PAFc is a Direct Transcriptional Regulator of the Prmt5 Locus
In yeast, the PAFc regulates about 20% of genes45. Similarly, in human cells PAF1 knockdown only modestly changed gene expression suggesting the PAFc governs a specific program40. To investigate whether the PAFc directly regulates Prmt5, we analyzed ChIP-sequencing data targeting the PAFc generated from the human MLL-AF9 driven AML cell line, THP-140. We observed a distinct binding peak of the PAFc, represented by CDC73 and LEO1 that overlaps with RNA polymerase II at the transcriptional start site of the PRMT5 gene (Figure 5A). To confirm this enrichment in mouse cells, we performed ChIP-qPCR experiments on the Prmt5 locus in MA9-Cdc73fl-CreER leukemic cells with and without Cdc73 excision (Figure 5B). Cdc73, Leo1, Mll1c, and MLL-AF9 were enriched at the Prmt5 locus. Cdc73 excision reduces enrichment of all four proteins suggesting Cdc73 is necessary for the localization of these proteins to Prmt5 (Figure 5C), similar to the known MLL and PAFc target gene Meis1 (Figure S7A and S7B). Loss of Cdc73 also leads to a decrease in H3K4me3 consistent with reduced transcriptional activity (Figure 5C). Consistent with the PAFc regulating Prmt5 expression, a Prmt5-Luciferase reporter was activated in a dose dependent manner to increasing levels of PAFc in the presence of MLL-AF9 (Figure S7C). Recent work has shown STAT5 is a direct regulator of PRMT532and overlaps with HOXA9 binding sites46. Thus, we analyzed HOXA9 and STAT5 binding at the Prmt5 locus. To test this, we generated 4OHT inducible Cdc73 floxed leukemic cell lines transformed with Myc-HOXA9 and HA-MEIS1. We observed Stat5 binding at the Prmt5 transcriptional start site that was strongly dependent on Cdc73 (Figure 5D). Ectopically expressed Myc-HOXA9 also enriches at the Prmt5 locus, but was less responsive to Cdc73 excision (Figure 5D). These data support that Prmt5 is directly regulated by the PAFc, which promotes expression in conjunction with other oncogenic factors such as STAT5 and HOXA9 in leukemic cells.
Figure 5. PAFc is a Transcriptional Regulator of Prmt5 Locus.
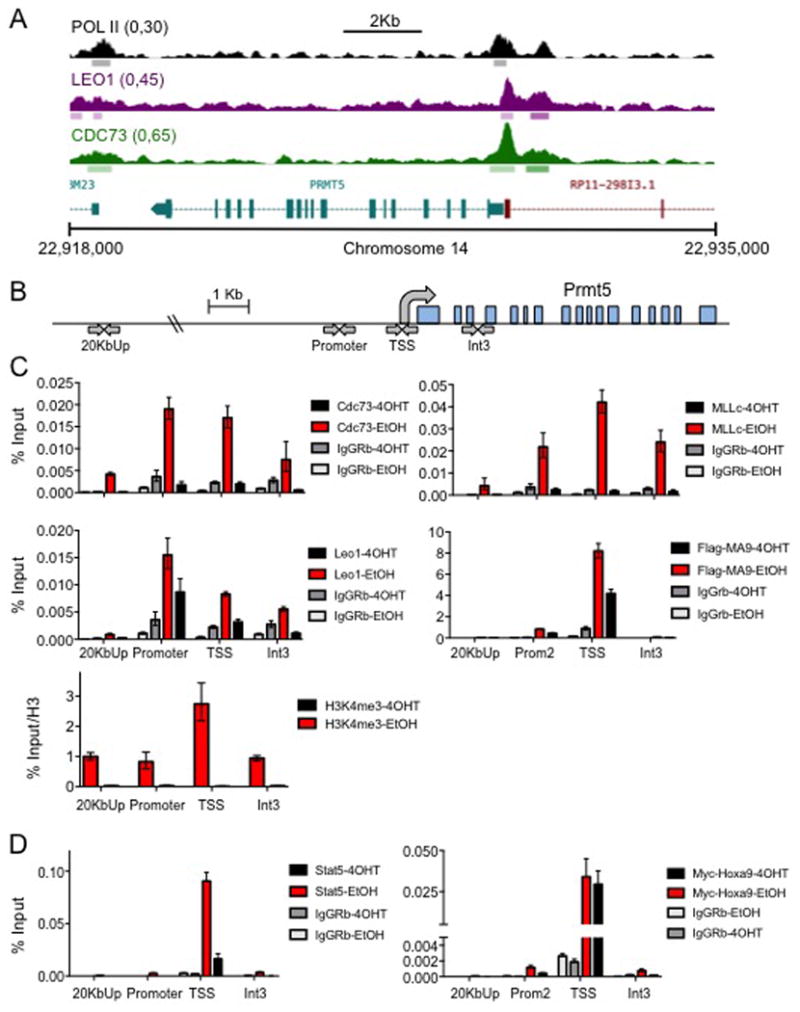
A) ChIP-sequencing tracks at the PRMT5 locus in human THP-1 leukemic cells40. Tracks for RNAPol II, CDC73, and LEO1 are shown. B) Scheme showing the locations on the Prmt5 locus that were tested by ChIP-qPCR. C) PAFc and MLL-fusion localize to the Prmt5 locus. MA9-Cdc73fl-CreER cells were treated with either 7.5nM EtOH or 4OHT and grown for 48 hours. ∼30×106 cells were harvested and chromatin immunoprecipitations were performed at the promoter, transcriptional start site (TSS), and Intron 3 of the Prmt5 locus for CDC73, LEO1, Flag tagged MLL-AF9 fusion, MLLc, and H3K4me3. A site 20kb upstream of Prmt5 Locus was used as a negative control. All values are normalized to input or H3 and IgG was used as a non-specific binding control. D) Stat5 and Hoxa9 localize to Prmt5 locus. Hoxa9/Meis1-Cdc73fl-CreERT2 cells were treated as described in (C). Stat5-Y694 and Myc-Tag-71D10 antibodies were used to detect Stat5 and Hoxa9 respectively. ChIP samples performed in biological duplicate and technical triplicate for qPCR.
Chemical inhibition of Prmt5 Reduces Leukemic Cell Growth and Prolongs Disease Latency In Vivo
PRMT5 inhibitors are currently in clinical trials for the treatment of solid tumors (ClinicalTrials.gov Identifier: NCT02783300). To understand how chemical inhibition of Prmt5 activity affects leukemic cell growth in vitro and in vivo, we treated several human leukemic cell lines with the PRMT5 inhibitor EPZ015666 and measured proliferation31. U937, NB4, and MonoMac6 cells all display a dose dependent decrease in proliferation and H4R3me2s demonstrating multiple leukemic subtypes are affected by PRMT5 inhibition (Figure 6A). Notably, NB4 cells, which display low MTAP expression, also show the greatest sensitivity to PRMT5 inhibition (Figure S8A). This is remarkably consistent with recent reports revealing cancer cell dependence on PRMT5 with the loss of the MTAP enzyme 47, 48. Similarly, mouse MLL-AF9 leukemic cells showed a dose dependent decrease in proliferation and H4R3me2s following Prmt5 inhibition (Figure S8B-C). We investigated how AML responds to Prmt5 inhibition using the EPZ015666 compound in an MLL-AF9 mouse model. Primary MLL-AF9 leukemic cells were injected into syngeneic recipients and dosed with EPZ015666 for 14 days. The median survival of mice receiving MLL-AF9 cells was extended by 4.5 days with EPZ015666 treatment (Figure 6B). While all moribund mice succumbed to leukemia (Figure 6B), reduced spleen weight and leukemic infiltration in EPZ015666 treated mice points to a less progressed disease compared to vehicle treated mice (Figure 6C and S9A-B). Potentially, these mice are more susceptible to leukemic burden due to drug toxicity, consistent with control mice tolerating the dosing schedule, but exhibiting minor weight loss (Figure S9C). We see no difference in H4R3me2s methylation at the moribund state between both treatment groups that likely reflects a strong selective pressure (Figure S9D). These results imply Prmt5 may play an important role in multiple subtypes of AML where chemical inhibition of PRMT5 may be an effective treatment.
Figure 6. Chemical Inhibition of PRMT5 Reduces Leukemic Growth and Extends Disease Latency.
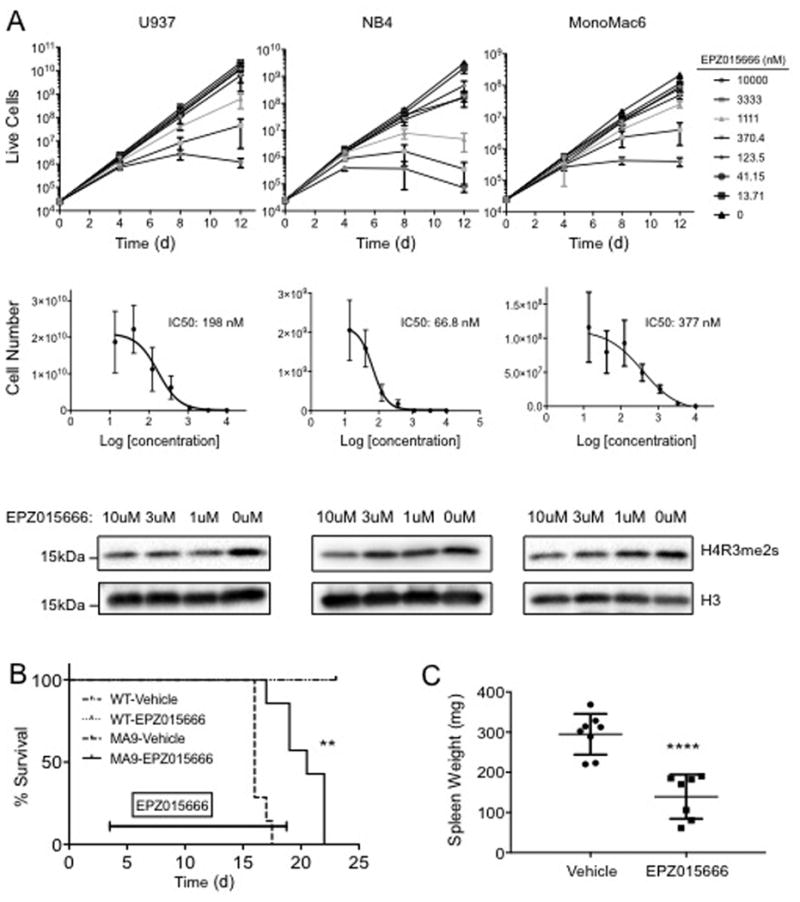
A) Human leukemic cell lines show reduced proliferation upon treatment with PRMT5 inhibitor EPZ015666. U937 (CALM-AF10), NB4 (PML-RARα), and MonoMac6 (MLL-AF9) cell lines were dosed with EPZ015666 at indicated concentrations. Cells numbers were normalized, reseeded, and redosed every 4 days over a 12-day time course. Cell number represents total cells accumulated drawn from the rate of growth between each reseeding. IC50 values were calculated at Day 12 and H4R3me2s is shown at Day 4 for the 10, 3, 1, and 0μM doses. Performed in biological triplicate and technical triplicate. B) Chemical inhibition of PRMT5 in vivo extends disease latency in a MLL-AF9 mouse model. Sublethally irradiated mice were intravenously injected with 100K primary MLL-AF9 cells (N=7 per dose group) or WT BM cells (N=4 per dose group). At Day 4 mice were subjected to vehicle or EPZ015666 treatment (200mg/kg) BID for 14 days by oral gavage (200μL). ** = p-value < 0.01 by Wilcoxon test. C) Mice dosed with Prmt5 Inhibitor have reduced spleen weight at moribund stage. Spleens were harvested from leukemic mice photographed and weighed. **** = p-value< 0.0001 by student's t-test.
Discussion
We examined the downstream gene program controlled by Cdc73 in leukemic cells and discovered that Prmt5 is a direct transcriptional target of the PAFc. Similar to yeast studies, we observed that conditional excision of Cdc73, as a surrogate for the PAFc, transcriptionally altered only a subset of genes suggesting specific gene programs under control of the PAFc45. Indeed, our data points to the PAFc maintaining gene programs that promote self-renewal and proliferation (Figure 2). Many genes from our data set were also upregulated, potentially reflecting indirect transcriptional changes associated with differentiation following loss of Cdc73 or a role for the PAFc in repressing transcription as has been reported37. Importantly, we can assign several Prmt genes to the list of PAFc targets. Past studies describe the importance of the PAFc or its individual components, as well as PRMT proteins in a variety of cancers including MLL-rearranged leukemias2, 4, 44, 49. Thus, a thorough understanding of the transcriptional mechanisms regulating PRMT genes may be used to develop novel therapies.
PRMT5 is a protein arginine methyltranferase necessary to deactivate gene transcription of targets and has a growing list of non-histone target proteins25, 27, 50-52. Studies exploring the overexpression of PRMT5 in cancer have clearly established a pro-oncogenic role which may be relevant in a diverse list of cancer types28, 33, 53, 54. Our work describes the transcriptional regulation of Prmt5 by the PAFc (Figure 5). The identification of selective cellular phenotypes associated with loss of Prmt5 (Figure 3), suggests several functional pathways lie downstream of the PAFc in leukemia. For example, while differentiation and cell cycle arrest were prevalent upon Cdc73 excision, only cell cycle arrest occurred with direct Prmt5 knockdown (Figure 1 and 3). This suggests that upregulation of known PAFc targets Hoxa9 and Meis13, 4 are predominantly responsible for blocking myeloid differentiation, while PAFc mediated regulation of Prmt5 contributes to cell cycle progression. In addition to the PAFc, we detected MLL-AF9, MLL1, STAT5, and HOXA9 at the Prmt5 locus (Figure 5). Loss of Cdc73 does not affect ectopically expressed HOXA9 enrichment at the Prmt5 locus. Thus, Hoxa9 may be a pioneering transcription factor that associates with targets independent of the PAFc. Additionally, these data are consistent with STAT protein dependence on CTR9 for localization to IL-6 responsive genes55.
It is noteworthy that several Prmt genes were deregulated after Cdc73 excision, although only Prmt5, Prmt1, and Prmt3 were commonly misregulated in mouse and human cells following disruption of the PAFc (Figure 3). Thus, the PAFc may help regulate multiple PRMT genes with oncogenic functions, which will require further investigation. Still, our data and others point to PRMT5 (amongst several PRMT proteins) as being critically important to facilitate leukemia56. For example, PRMT1, PRMT4, and PRMT5 have all been reported to promote leukemia through a variety of mechanisms31-34, 41-43, 57, 58,59. However, some of these are also essential during development56. For example, loss of Prmt5 is embryonic lethal and conditional knockouts demonstrate it is essential for the maintenance of adult hematopoietic stem cells30, 51. Further, loss of Prmt1 or Prmt4 is lethal during embryogenesis or shortly after birth60, 61. Thus, targeting the transcriptional and epigenetic mechanisms regulating several PRMT genes, such as the PAFc, may allow for a combinatorial downregulation of Prmt genes without fully abolishing expression, which may be therapeutically advantageous. This is highlighted by our results that chemical inhibition of PRMT5 extends disease latency, but does not cure the leukemic disease (Figure 6). A combinatorial approach targeting the PAFc may facilitate altered expression of target PRMT genes that avoids tissue toxicity while exploiting the reliance of tumor cells on high PRMT expression. Further, targeting of the PAFc would also downregulate Hoxa9 and Meis1 that serve as oncogenic drivers in as much as 50% of AML24. Our previous work established the PAFc-MLL interaction as an attractive therapeutic target. Our detection of MLL and MLL-fusion proteins at the Prmt5 locus suggests that targeting of the PAFc-MLL interaction may alter Prmt5 expression in addition to Hoxa9 and Meis1. Overall, the work presented here suggests Prmt5 is one of many potential PAFc targets contributing to the progression and maintenance of MLL driven leukemia and supports the therapeutic potential of the PAFc in leukemias and other cancer types.
Materials and Methods
Mice
Female C57BL/6 mice at 8-10 weeks old were purchased from Taconic Farms (Hudson, NY). B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/J mice (TetOn mice) were purchased from Jackson laboratory (Bar Harbor, ME). All animal studies were approved by the University of Michigan Committee on Use and Care of Animals and the Unit for Laboratory Medicine.
Colony assays, cell lines and PRMT5 inhibition assay
Colony assays and cell line generation were performed as previously reported3, 62. MA9-Cdc73fl-CreER and MA9-CreER cells were treated with 7.5nM 4OHT for 48 or 72 hours to induce excision of Cdc73. For PRMT5 chemical inhibition, 10K cells were subjected to various doses of EPZ015666, counted every 4 days (3 days for MA9 cells), retreated and reseeded at original density for a total of 12 days as described 31. For shRNA knockdown, MA9 cells were re-transduced with pSM2C-shPrmt5 or control vectors and selected in puromycin (100ng/mL), plated and counted after 1 week. Prmt overexpression assay cells were re-plated for 3 rounds. Transforming TetOn mouse bone marrow with the MLL-AF9 oncogene generated stable TetON cell lines as described 62. Knockdown was achieved by transducing with the pTRMPV vector containing shRNA directed towards Prmt1, Prmt4, Prmt5 and Renilla control (Table S1) and Doxycycline treatment (1μg/ml). shRNA constructs were designed as described63. Differentiation, cell cycle, and apoptosis assays were performed as described 62 using c-Kit, Cd14, Cd11b and isotype control antibodies (BD Pharmigen, San Jose, CA & Biolegend, San Diego, CA) or Annexin V-DAPI (BD Pharmigen, San Jose, CA) co-stain and read on an LSRII Flow Cytometer (BD Biosciences, San Jose, CA). Data was analyzed using FlowJo X and ModFit Software. siRNA knockdown of Paf1 in MA9-Cdc73fl-CreER cells was performed as previously described4.
RNA Sequencing and ChIP sequencing
MA9-Cdc73fl-CreER and MA9-CreER cells were treated with 7.5nM 4OHT and harvested at 48 hours. RNA-seq libraries were prepared according to the manufacturer's instructions (Illumina, Eindhoven, The Netherlands) by the University of Michigan Sequencing Core. All data available at the Gene Expression Omnibus: GSE90136. Previously published RNA-seq and ChIP-Seq reads were downloaded from project number SRP048744 in GEO and converted to fastq format using the SRA toolkit40.
In Vivo Disease Latency
Primary Hoxa9/Meis1 transformed TetOn leukemic cells were transduced with either TRMPV-shRenilla or shPrmt5 packaged retrovirus, selected in 200 μg/mL hygromycin and injected (250K cells) into irradiated (600 rads) syngeneic recipients. Dox food (Harlan Labs 625mg/kg) and water (Sigma 2mg/mL) were used to induce knockdown. For Prmt5 chemical inhibition in vivo, mice received EPZ015666 by oral gavage at 200 mg/kg BID for 14 days31, 4 days post MA9 cell injection. Mice were sacrificed and analyzed at the moribund stage as described 62. Mice were block randomized at N=7 after power analysis and investigator blinding was not used. No mice were excluded due to failure of cell engraftment following irradiation.
ChIP-qPCR
ChIP-qPCR was performed as described64, using αCDC73, αLEO1 (Bethyl, Montgomery, TX), αMLLc (gift from Dr. Yali Dou), αFlag (Sigma), αH3K4me3, αH3 (Abcam, Cambridge, UK), IgG (Santa Cruz, Dallas, TX), Stat5-Y694 and Myc-Tag-71D10 (Cell Signaling, Danvers, MA). qPCR primer sequences are listed (Table S1).
Western Blots
Western blots were performed with whole cell lysates or acid extracted histones and probed with the following antibodies: αCDC73, αPAF1 (Bethyl, Montgomery, TX), αPRMT5, αSYM10, αGAPDH, αH2BUb (Millipore, Billerica, MA), αH3, αH3K4me3, αH3K79me2, αH4R3me2s (Abcam,Cambridge, UK), αR-Me2s (Cell Signaling, Danvers, MA), α-βActin (Sigma). Bands were detected using SuperSignal West Pico Chemiluminescent substrate (Thermo-Fisher Scientific, Waltham, MA) and imaged on a ChemiDocXRS (Bio-Rad, Hercules, CA).
Statistical Validation & Human Cell Lines
Statistics, sample sizes, replicates, and p-values are presented in figure legends or main text. Variance was factored into statistical tests where applicable. U937 sourced from ATCC while NB4 and MM6 were sourced from DSMZ. All were STR validated, but not tested for mycoplasma.
Supplementary Material
Acknowledgments
We thank Dr. Dong-Er Zhang (UCSD), Dr. Wei Xu (University of Wisconsin), Dr. Jean-Francois Rual and Dr. Tao Xu (University of Michigan) for providing the Prmt1, Prmt4, and Prmt5 constructs respectively, Dr. Yali Dou (University of Michigan) for the MLLc antibody. This work was supported by NIH grants R00 CA158136 (A.G.M.), an American Society of Hematology Scholar Award (A.G.M.), a Leukemia Research Foundation award (A.G.M), an American Cancer Society Scholar Award RSG-15-046 (A.G.M.) and Children's Leukemia Research Association (A.G.M.).
This work was supported by National Institute of Health grants R00 CA158136 (A.G.M.), an American Society of Hematology Scholar Award (A.G.M.), a Leukemia Research Foundation award (A.G.M), an American Cancer Society Scholar Award RSG-15-046 (A.G.M.) and Children's Leukemia Research Association (A.G.M.).
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Miller CA, Wilson RK, Ley TJ. Genomic landscapes and clonality of de novo AML. N Engl J Med. 2013;369(15):1473. doi: 10.1056/NEJMc1308782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Milne TA, Kim J, Wang GG, Stadler SC, Basrur V, Whitcomb SJ, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell. 2010;38(6):853–63. doi: 10.1016/j.molcel.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muntean AG, Chen W, Jones M, Granowicz EM, Maillard I, Hess JL. MLL fusion protein-driven AML is selectively inhibited by targeted disruption of the MLL-PAFc interaction. Blood. 2013;122(11):1914–22. doi: 10.1182/blood-2013-02-486977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muntean AG, Tan J, Sitwala K, Huang Y, Bronstein J, Connelly JA, et al. The PAF complex synergizes with MLL fusion proteins at HOX loci to promote leukemogenesis. Cancer Cell. 2010;17(6):609–21. doi: 10.1016/j.ccr.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32(4):676–80. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 6.Hanks S, Perdeaux ER, Seal S, Ruark E, Mahamdallie SS, Murray A, et al. Germline mutations in the PAF1 complex gene CTR9 predispose to Wilms tumour. Nat Commun. 2014;5:4398. doi: 10.1038/ncomms5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moniaux N, Nemos C, Schmied BM, Chauhan SC, Deb S, Morikane K, et al. The human homologue of the RNA polymerase II-associated factor 1 (hPaf1), localized on the 19q13 amplicon, is associated with tumorigenesis. Oncogene. 2006;25(23):3247–57. doi: 10.1038/sj.onc.1209353. [DOI] [PubMed] [Google Scholar]
- 8.Zeng H, Xu W. Ctr9, a key subunit of PAFc, affects global estrogen signaling and drives ERalpha-positive breast tumorigenesis. Genes Dev. 2015;29(20):2153–67. doi: 10.1101/gad.268722.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J, Guermah M, Roeder RG. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell. 2010;140(4):491–503. doi: 10.1016/j.cell.2009.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller CL, Jaehning JA. Ctr9, Rtf1, and Leo1 are components of the Paf1/RNA polymerase II complex. Mol Cell Biol. 2002;22(7):1971–80. doi: 10.1128/MCB.22.7.1971-1980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wade PA, Werel W, Fentzke RC, Thompson NE, Leykam JF, Burgess RR, et al. A novel collection of accessory factors associated with yeast RNA polymerase II. Protein Expr Purif. 1996;8(1):85–90. doi: 10.1006/prep.1996.0077. [DOI] [PubMed] [Google Scholar]
- 12.Jaehning JA. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim Biophys Acta. 2010;1799(5-6):379–88. doi: 10.1016/j.bbagrm.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomson BN, Arndt KM. The many roles of the conserved eukaryotic Paf1 complex in regulating transcription, histone modifications, and disease states. Biochim Biophys Acta. 2013;1829(1):116–26. doi: 10.1016/j.bbagrm.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He N, Chan CK, Sobhian B, Chou S, Xue Y, Liu M, et al. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc Natl Acad Sci U S A. 2011;108(36):E636–45. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim M, Ahn SH, Krogan NJ, Greenblatt JF, Buratowski S. Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J. 2004;23(2):354–64. doi: 10.1038/sj.emboj.7600053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavri R, Zhu B, Li G, Trojer P, Mandal S, Shilatifard A, et al. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125(4):703–17. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 17.Qiu H, Hu C, Wong CM, Hinnebusch AG. The Spt4p subunit of yeast DSIF stimulates association of the Paf1 complex with elongating RNA polymerase II. Mol Cell Biol. 2006;26(8):3135–48. doi: 10.1128/MCB.26.8.3135-3148.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, et al. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 2009;137(3):459–71. doi: 10.1016/j.cell.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Oss SB, Shirra MK, Bataille AR, Wier AD, Yen K, Vinayachandran V, et al. The Histone Modification Domain of Paf1 Complex Subunit Rtf1 Directly Stimulates H2B Ubiquitylation through an Interaction with Rad6. Mol Cell. 2016 doi: 10.1016/j.molcel.2016.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell. 2003;11(3):721–9. doi: 10.1016/s1097-2765(03)00091-1. [DOI] [PubMed] [Google Scholar]
- 21.Daser A, Rabbitts TH. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol. 2005;15(3):175–88. doi: 10.1016/j.semcancer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 22.Aplan PD. Chromosomal translocations involving the MLL gene: molecular mechanisms. DNA Repair (Amst) 2006;5(9-10):1265–72. doi: 10.1016/j.dnarep.2006.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaudhary K, Deb S, Moniaux N, Ponnusamy MP, Batra SK. Human RNA polymerase II-associated factor complex: dysregulation in cancer. Oncogene. 2007;26(54):7499–507. doi: 10.1038/sj.onc.1210582. [DOI] [PubMed] [Google Scholar]
- 24.Collins CT, Hess JL. Role of HOXA9 in leukemia: dysregulation, cofactors and essential targets. Oncogene. 2016;35(9):1090–8. doi: 10.1038/onc.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Girardot M, Hirasawa R, Kacem S, Fritsch L, Pontis J, Kota SK, et al. PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G + C-rich regions of the mouse genome. Nucleic Acids Res. 2014;42(1):235–48. doi: 10.1093/nar/gkt884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Migliori V, Muller J, Phalke S, Low D, Bezzi M, Mok WC, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19(2):136–44. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 27.Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. 2004;24(21):9630–45. doi: 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mongiardi MP, Savino M, Bartoli L, Beji S, Nanni S, Scagnoli F, et al. Myc and Omomyc functionally associate with the Protein Arginine Methyltransferase 5 (PRMT5) in glioblastoma cells. Sci Rep. 2015;5:15494. doi: 10.1038/srep15494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang B, Dong S, Li Z, Lu L, Zhang S, Chen X, et al. Targeting protein arginine methyltransferase 5 inhibits human hepatocellular carcinoma growth via the downregulation of beta-catenin. J Transl Med. 2015;13:349. doi: 10.1186/s12967-015-0721-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu F, Cheng G, Hamard PJ, Greenblatt S, Wang L, Man N, et al. Arginine methyltransferase PRMT5 is essential for sustaining normal adult hematopoiesis. J Clin Invest. 2015;125(9):3532–44. doi: 10.1172/JCI81749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015;11(6):432–7. doi: 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- 32.Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y, et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest. 2016;126(10):3961–80. doi: 10.1172/JCI85239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Chitnis N, Nakagawa H, Kita Y, Natsugoe S, Yang Y, et al. PRMT5 is required for lymphomagenesis triggered by multiple oncogenic drivers. Cancer Discov. 2015;5(3):288–303. doi: 10.1158/2159-8290.CD-14-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pal S, Baiocchi RA, Byrd JC, Grever MR, Jacob ST, Sif S. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. EMBO J. 2007;26(15):3558–69. doi: 10.1038/sj.emboj.7601794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, et al. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126(4):755–66. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 36.Li L, Schmitt A, Reinhardt P, Greiner J, Ringhoffer M, Vaida B, et al. Reconstitution of CD40 and CD80 in dendritic cells generated from blasts of patients with acute myeloid leukemia. Cancer Immun. 2003;3:8. [PubMed] [Google Scholar]
- 37.Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, et al. PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell. 2015;162(5):1003–15. doi: 10.1016/j.cell.2015.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat Med. 2015;21(4):335–43. doi: 10.1038/nm.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Cuellar MP, Buttner C, Bartenhagen C, Dugas M, Slany RK. Leukemogenic MLL-ENL Fusions Induce Alternative Chromatin States to Drive a Functionally Dichotomous Group of Target Genes. Cell Rep. 2016;15(2):310–22. doi: 10.1016/j.celrep.2016.03.018. [DOI] [PubMed] [Google Scholar]
- 40.Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, et al. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science. 2015;350(6266):1383–6. doi: 10.1126/science.aad2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, et al. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell. 2016;29(1):32–48. doi: 10.1016/j.ccell.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vu LP, Perna F, Wang L, Voza F, Figueroa ME, Tempst P, et al. PRMT4 blocks myeloid differentiation by assembling a methyl-RUNX1-dependent repressor complex. Cell Rep. 2013;5(6):1625–38. doi: 10.1016/j.celrep.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell. 2014;25(1):21–36. doi: 10.1016/j.ccr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dey P, Ponnusamy MP, Deb S, Batra SK. Human RNA polymerase II-association factor 1 (hPaf1/PD2) regulates histone methylation and chromatin remodeling in pancreatic cancer. PLoS One. 2011;6(10):e26926. doi: 10.1371/journal.pone.0026926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Penheiter KL, Washburn TM, Porter SE, Hoffman MG, Jaehning JA. A posttranscriptional role for the yeast Paf1-RNA polymerase II complex is revealed by identification of primary targets. Mol Cell. 2005;20(2):213–23. doi: 10.1016/j.molcel.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 46.Huang Y, Sitwala K, Bronstein J, Sanders D, Dandekar M, Collins C, et al. Identification and characterization of Hoxa9 binding sites in hematopoietic cells. Blood. 2012;119(2):388–98. doi: 10.1182/blood-2011-03-341081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351(6278):1214–8. doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016;15(3):574–87. doi: 10.1016/j.celrep.2016.03.043. [DOI] [PubMed] [Google Scholar]
- 49.Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat. 2010;31(3):295–307. doi: 10.1002/humu.21188. [DOI] [PubMed] [Google Scholar]
- 50.Bandyopadhyay S, Harris DP, Adams GN, Lause GE, McHugh A, Tillmaand EG, et al. HOXA9 methylation by PRMT5 is essential for endothelial cell expression of leukocyte adhesion molecules. Mol Cell Biol. 2012;32(7):1202–13. doi: 10.1128/MCB.05977-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tee WW, Pardo M, Theunissen TW, Yu L, Choudhary JS, Hajkova P, et al. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010;24(24):2772–7. doi: 10.1101/gad.606110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang DB, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-kappaB. Proc Natl Acad Sci U S A. 2013;110(33):13516–21. doi: 10.1073/pnas.1311784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tarighat SS, Santhanam R, Frankhouser D, Radomska HS, Lai H, Anghelina M, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia. 2016;30(4):789–99. doi: 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang H, Zhao X, Zhao L, Liu L, Li J, Jia W, et al. PRMT5 competitively binds to CDK4 to promote G1-S transition upon glucose induction in hepatocellular carcinoma. Oncotarget. 2016 doi: 10.18632/oncotarget.12351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Youn MY, Yoo HS, Kim MJ, Hwang SY, Choi Y, Desiderio SV, et al. hCTR9, a component of Paf1 complex, participates in the transcription of interleukin 6-responsive genes through regulation of STAT3-DNA interactions. J Biol Chem. 2007;282(48):34727–34. doi: 10.1074/jbc.M705411200. [DOI] [PubMed] [Google Scholar]
- 56.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13(1):37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 57.Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9(10):1208–15. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- 58.Shia WJ, Okumura AJ, Yan M, Sarkeshik A, Lo MC, Matsuura S, et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood. 2012;119(21):4953–62. doi: 10.1182/blood-2011-04-347476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang L, Pal S, Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol. 2008;28(20):6262–77. doi: 10.1128/MCB.00923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pawlak MR, Scherer CA, Chen J, Roshon MJ, Ruley HE. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol Cell Biol. 2000;20(13):4859–69. doi: 10.1128/mcb.20.13.4859-4869.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yadav N, Lee J, Kim J, Shen J, Hu MC, Aldaz CM, et al. Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci U S A. 2003;100(11):6464–8. doi: 10.1073/pnas.1232272100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen L, Chen W, Mysliwski M, Serio J, Ropa J, Abulwerdi FA, et al. Mutated Ptpn11 alters leukemic stem cell frequency and reduces the sensitivity of acute myeloid leukemia cells to Mcl1 inhibition. Leukemia. 2015;29(6):1290–300. doi: 10.1038/leu.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, et al. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011;29(1):79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen L, Sun Y, Wang J, Jiang H, Muntean AG. Differential regulation of the c-Myc/Lin28 axis discriminates subclasses of rearranged MLL leukemia. Oncotarget. 2016;7(18):25208–23. doi: 10.18632/oncotarget.8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.