

Abstract
Background
Nociceptin/Orphanin FQ (N/OFQ), the endogenous peptide agonist for the opioid receptor-like (ORL1) receptor (also known as NOP or the nociceptin receptor), has been shown to block the acquisition and expression of ethanol-induced conditioned place preference (CPP). Here, we report the characterization of a novel small-molecule NOP ligand AT-312 (1-(1-((cis)-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methanol) in receptor binding and GTPγS functional assays in vitro. We then investigated the effect of AT-312 on the rewarding action of ethanol in mice using the CPP paradigm. Further, using mice lacking the NOP receptor and their wild-type controls, we also examined the involvement of NOP in the effect of AT-312. Motivational effects of AT-312 alone were also assessed in the CPP paradigm.
Methods
Female mice lacking NOP and/or their wild-type controls received conditioning in the presence or absence of the NOP agonist [AT-312 (1, 3 and 10 mg/kg) or the control NOP agonist SCH221510 (10 mg/kg)] followed by saline/ethanol for 3 consecutive days (twice daily) and tested for CPP in a drug-free state on the next day.
Results
Our in vitro data showed that AT-312 is a high affinity, selective NOP full agonist with 17-fold selectivity over the mu opioid receptor and >200-fold selectivity over the kappa opioid receptor. The results of our in vivo studies showed that AT-312 reduced ethanol CPP at the lowest dose (1 mg/kg) tested but completely abolished ethanol CPP at higher doses (3 or 10 mg/kg) compared to their vehicle-treated control group. AT-312 (3 mg/kg) did not alter ethanol-induced CPP in mice lacking NOP, confirming that AT-312 reduced ethanol CPP through its action at the NOP receptor. AT-312 (3 mg/kg) did not induce reward or aversion when administered alone, showing that the novel small molecule NOP agonist shows efficacy in blocking ethanol-induced CPP via the NOP receptor.
Conclusions
Together, these data suggest that small molecule NOP agonists have the potential to reduce alcohol reward and may be promising as medications to treat alcohol addiction.
Keywords: NOP agonist, AT-312, Alcohol reward, NOP knockout mouse, Ethanol-induced conditioned place preference
Introduction
Alcoholism and alcohol-related disorders are major public health issues and place an enormous burden on society and economy (Esser et al., 2017; Esser et al., 2014). When alcohol-related accidents are factored in, alcohol is among the top three causes of death in the US (Mokdad et al., 2004). Of the estimated 18 million alcohol-dependent individuals in the population, only about 1 million actually receive/seek adequate treatment, which mainly involves psychosocial support in conjunction with limited pharmacotherapy. While no single medication or strategy has been shown to be very effective, it is generally accepted that having pharmacotherapy as an adjunct to behavioral interventions is the best approach for treating alcohol dependence and maintaining abstinence. For this however, the current repertoire of pharmacotherapeutic options needs to be significantly expanded. Only three pharmacotherapeutic agents are currently approved for the treatment of alcohol dependence in the US, oral and intramuscular naltrexone (NTX), acamprosate, and disulfiram. Of these, NTX, an opioid receptor antagonist, has shown limited efficacy in reducing craving after stopping alcohol drinking; acamprosate, whose mechanism of action is unclear, improves abstinence rates, whereas disulfiram, produces an aversive reaction to alcohol. These are still not widely adopted by physicians who treat alcohol-dependent patients, mostly due to lack of confidence about their efficacy and a range of unpleasant side effects that limit patient compliance. There still remains a need for new approaches and treatments for alcohol dependence.
Unlike other drugs of abuse, alcohol does not act at one receptor target, but dysregulates many neurotransmitter systems, ion channels, and neurocircuitry in several brain areas, particularly the ventral tegmental area, nucleus accumbens, central amygdala and bed nucleus of stria terminalis (Gilpin and Koob, 2008; Koob and Volkow, 2010).
Among these, the endogenous opioid system is well known to play a key role in the rewarding and reinforcing effects of alcohol (Altshuler et al., 1980; Froehlich et al., 1990; Gianoulakis, 2004; Hubbell et al., 1986; Marfaing-Jallat et al., 1983; Weiss et al., 1990). Indeed, as stated above, NTX, approved for use in the US as an anti-alcohol pharmacotherapy, decreases alcohol consumption and craving in humans, and decreases the rewarding properties of ethanol in animal models (Altshuler et al., 1980; Benjamin et al., 1993; Farren and O’Malley, 1997; Froehlich et al., 1990; Gianoulakis et al., 1996; Hubbell et al., 1991; Ji et al., 2008; Kornet et al., 1991; Marfaing-Jallat et al., 1983; Myers et al., 1986; O’Malley et al., 2002; Oslin et al., 1997; Samson and Doyle, 1985; Volpicelli et al., 1992; Volpicelli et al., 1986; Weiss et al., 1990). The mu, delta and kappa opioid receptors and their respective endogenous ligands β-endorphins, enkephalins and dynorphin have all been shown to be involved in various stages of alcohol addiction cycle (Hall et al., 2001; Oswald and Wand, 2004; Roberts et al., 2000).
A growing body of evidence suggests that the fourth member of the opioid receptor-ligand family, the nociceptin opioid receptor NOP (previously called the opioid receptor-like (ORL1) receptor) and its endogenous neuropeptide ligand, nociceptin/orphanin FQ (N/OFQ) are involved in alcohol reward and reinforcement (Ciccocioppo et al., 1999; Kuzmin et al., 2007; Kuzmin et al., 2003; Ciccocioppo et al., 2002). Similarly to other members of the opioid receptor family, the NOP receptor is widely distributed in areas of the brain implicated in motivational behaviors as well as negative affect, such as the ventral tegmental area, nucleus accumbens, lateral hypothalamus and the central amygdala (Neal et al., 1999a; Neal et al., 1999b). The endogenous ligand of the NOP, N/OFQ, acts to alter neurotransmitter release, particularly dopamine, GABA, and glutamate, all of which are also implicated in alcohol reward (Di Giannuario et al., 1999; Kallupi et al., 2014; Lutfy et al., 2001; Murphy et al., 1996; Murphy and Maidment, 1999; Murphy et al., 2004; Sakoori and Murphy, 2004). N/OFQ is also considered to have an ‘anti-opioid’ action in the brain [for a review, see (Mogil and Pasternak, 2001)]. Exogenous administration of N/OFQ has been shown to suppress basal and drug-stimulated dopamine release in the NAc (Di Giannuario et al., 1999; Lutfy et al., 2001; Murphy et al., 1996; Murphy and Maidment, 1999; Murphy et al., 2004; Sakoori and Murphy, 2004), and the rewarding properties of several common drugs of abuse [reviewed in (Lutfy and Zaveri, 2016)]. In particular, intracerebroventricular (i.c.v) administration of N/OFQ has been shown to block acquisition of conditioned place preference (CPP) induced by morphine (Ciccocioppo et al., 2000; Murphy et al., 1999), cocaine (Sakoori and Murphy, 2004), amphetamines (Kotlinska et al., 2003), and alcohol (Ciccocioppo et al., 1999; Kuzmin et al., 2007; Kuzmin et al., 2003). A small molecule NOP agonist Ro 64-6198, given systemically, was also shown to block both the acquisition and expression of alcohol CPP in mice (Kuzmin et al., 2003) and alcohol self-administration in rats (Kuzmin et al., 2007). Another potent NOP agonist MT-7716 was shown to decrease alcohol intake in alcohol-preferring Marchigian Sardinian (msP) rats and attenuate alcohol withdrawal symptoms in alcohol-dependent Wistar rats (Ciccocioppo et al., 2014b). Recently, SR-8993, a selective NOP agonist was reported to reduce anxiety associated with alcohol withdrawal as well as home cage and limited access alcohol drinking in Wistar rats (Aziz et al., 2016). Interestingly the level of N/OFQ is altered following restraint stress in the amygdala (Ciccocioppo et al., 2014a). These studies suggest that NOP agonists may be potentially promising treatment agents for alcoholism and alcohol use disorders.
In the present study, we characterized a novel small-molecule NOP ligand AT-312 (1-(1-((cis)-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methanol), for its selectivity and affinity toward the NOP and classical opioid receptors using radioligand binding assays and determined its efficacy in the GTP(γ)S functional assay conducted in Chinese hamster ovary (CHO) cells transfected with the human opioid receptors. We also determined its bioavailability and brain penetration, which showed appreciable plasma exposure and a brain-to-plasma ratio greater than 1 after systemic (subcutaneous, s.c.) administration (Table 2). Using this route of administration, we further determined its efficacy in reducing the rewarding action of ethanol in the CPP paradigm, a widely used animal model of drug reward (Bardo and Bevins, 2000). To demonstrate that the effect of AT-312 in reducing alcohol CPP in mice is due to its activity at the NOP receptor, we compared its efficacy in reducing CPP induced by ethanol in mice lacking the NOP receptor and their wild-type littermates/controls. We also investigated the effect of a known NOP agonist SCH 221510 on alcohol reward in this same paradigm as a control.
Table 2.
In vivo pharmacokinetic profile of AT-312 after subcutaneous administration in mice
| PK parameters | Dose, route (10 mg/kg, s.c.) |
|---|---|
| Plasma Cmax | 1263 nM |
| Plasma tmax | 1 h |
| Brain Cmax * | 5465 nM |
| Brain tmax | 1 h |
| Brain-to-plasma ratio at Cmax * | 4.33 |
| Brain-to-plasma ratio AUC* | 4.68 |
Total brain concentrations; AUC (Area under the curve)
MATERIALS AND METHODS
Cells
Human NOP, mu, delta, and kappa opioid receptors were individually expressed in Chinese hamster ovary cells stably transfected with the human receptor cDNA, as we have described previously (Zaveri et al., 2001; Toll et al., 2009). The HORL, HDOR, HKOR-FLAG19 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum, in the presence of 0.4 mg/ml G418 and 0.5% penicillin/streptomycin, in 150-mm tissue culture dishes. The HKOR-CN cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum, in the presence of 0.5% penicillin/streptomycin and no G418. The HMOR cells in 50% F12/DMEM with 10% fetal bovine serum, in the presence of 0.4 mg/ml G418 and 0.5% penicillin/streptomycin. Kappa-CN cells were used for KOP radioligand binding assays, while Kappa-FLG19 cells were used in KOP [35S]GTPγS functional assays.
Animals
Female mice lacking NOP (Nishi et al., 1997) and their wild-type littermates/controls (2-6 months old), fully backcrossed on C57BL/6J mouse strain, bred in house, were used throughout. We used female mice because they exhibit a robust CPP response compared to male mice using the current 3-day conditioning paradigm (Nguyen et al., 2012; Tseng et al., 2013). Mice were housed 2-4 per cage with free access to laboratory chow and tap water and kept under a 12 h light/12 h dark cycle in a temperature- and humidity-controlled room. The light was on 6 AM and off at 6 PM. All experiments were conducted during the light cycle between the hours of 10:00 AM to 5:00 PM and were according to the National Institute of Health for the proper use of animals in research and approved by the Institutional Animal Care and Use Committee at Western University of Health Sciences (Pomona, California, USA).
Drugs
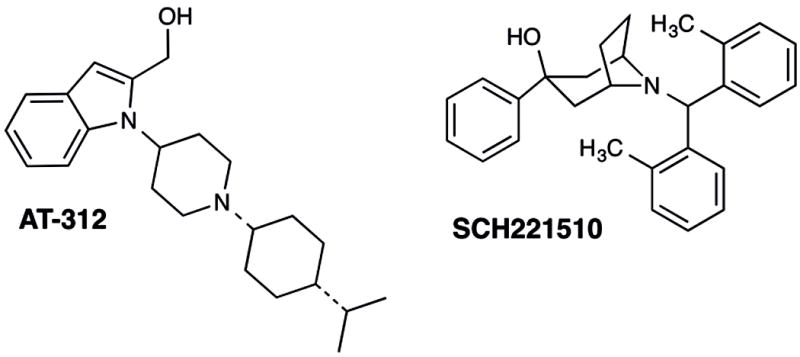
AT-312 (1-(1-((cis)-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methanol) (Figure 1) was synthesized at Astraea Therapeutics, and was of >99% chemical purity as fully characterized by nuclear magnetic resonance spectroscopy, LC-MS and elemental analysis. SCH221510 (Figure 1) was purchased from Tocris. These test compounds were dissolved in 1-2% DMSO and then diluted to the desired concentration with 0.5% aqueous hydroxypropylcellulose (HPC) and injected subcutaneously (s.c.) in a volume of 0.1 ml/10g of body weight. Controls received 0.1 ml/10g of body weight of the appropriate vehicle (1-2% DMSO in 0.5% of HPC).
Figure 1. Structures of NOP agonists AT-312 and SCH221510.
In vitro Characterization
Membrane preparation
The cell lines are grown to confluency, then harvested for membrane preparation. The membranes are prepared in 50 mM Tris buffer (pH 7.4). Cells are scraped and centrifuged at 500 × g for 12 mins. The cell pellet is homogenized in 50 mM Tris with a Fisher Scientific PowerGen 125 rotor-stator type homogenizer, centrifuged at 20,000 × g for 25 mins, washed and recentrifuged once more at 20,000 × g for 25 mins, and aliquoted at a concentration of 3 mg/ml protein per vial and stored in a -80 °C freezer till further use.
Receptor Binding
Compounds were dissolved at 10 mM stock in 100% DMSO. The assay was performed in a 96-well polystyrene plate with triplicates of six concentrations of each test compound (1μM – 0.01 nM), adding 100 μl of compound and 100 μl of tritiated ligands [3H]DAMGO (51.0 Ci/mmole, Kd 0.59 nM for MOP), [3H]DPDPE (42.0 Ci/mmole, Kd 1.11 nM for DOP), [3H]U69593 (41.7 Ci/mmole, Kd 1.05 nM for KOP), and [3H]N/OFQ (130 Ci/mmole, Kd 0.12 nM for NOP). Nonspecific binding was determined using 1.0 μM of the unlabeled nociceptin for NOP, 10 μM unlabeled DAMGO for MOP, 1.0 μM unlabeled DPDPE for DOP, and 10 μM unlabeled U69,593 for KOP. Assays were initiated by addition of 800 μl of membrane per well. Samples were incubated for 60 min at 25°C in a total volume of 1.0 ml. In NOP receptor experiments, 1 mg/ml BSA was added to the compound dilution buffer. The incubation was terminated by rapid filtration through 0.5% PEI-soaked glass fiber filter mats (GF/C Filtermat A, Perkin-Elmer) on a Tomtec Mach III cell harvester and washed 5 times with 0.5 ml of ice-cold 50 nM Tris-HCl, pH 7.4 buffer. The filters were dried overnight and soaked with scintillation cocktail before counting on a Wallac Beta plate 1205 liquid scintillation counter. Radioactivity was determined as counts per minutes (cpm). IC50 values were determined using at least six concentrations of test compound, and calculated using Graphpad/Prism (ISI, San Diego, CA). Ki values were determined by the method of Cheng and Prusoff (Cheng and Prusoff, 1973).
[35S]GTPγS binding Assay
[35S]GTPγS binding was conducted as we have described previously (Toll et al., 2009; Traynor and Nahorski, 1995; Zaveri et al., 2001). Cells were scraped from tissue culture dishes into 20 mM Hepes, 1 mM EDTA, then centrifuged at 500 × g for 10 min. Cells were re-suspended in this buffer and homogenized using a Polytron Homogenizer. The homogenate was centrifuged at 27,000 × g for 15 min, and the pellet resuspended in Buffer A, containing: 20 mM Hepes, 10 mM MgCl2, 100 mM NaCl, pH 7.4. The suspension was recentrifuged at 27,000 × g and suspended once more in Buffer A. For the binding assay, membranes (8-15 × g protein) were incubated with [35S]GTPγS (50 pM), GDP (10 μM), and the appropriate compound, in a total volume of 1.0 ml, for 60 min at 25°C. Samples were filtered over glass fiber filters and counted as described for the binding assays. Statistical analysis was conducted using Prism.
In vivo Pharmacology
Experiment 1: To determine the effect of NOP agonists on ethanol-induced CPP
We used an unbiased CPP paradigm, widely used as an animal model of drug reward (Bardo and Bevins, 2000), to determine the effect of AT-312 on the rewarding action of ethanol. The details of the CPP apparatus and paradigm have been provided elsewhere (Nguyen et al., 2012; Tseng et al., 2013). Briefly, mice were tested for preconditioning place preference on day 1. On this day, mice were placed in the central neutral chamber and allowed to freely explore the conditioning chambers through this smaller central chamber. The amount of time that mice spent in each CPP chamber was recorded. On days 2-4, mice were conditioned with ethanol in the presence and absence of the NOP agonist. In the morning on each day, mice were treated with vehicle or one of the doses of AT-312 (1, 3 or 10 mg/kg, s.c.; n = 6-9 mice per group) followed, 15 min later, by ethanol (2 g/kg, i.p.) and then immediately confined to the drug-paired chamber (DPCh) for 15 min. In the afternoon, mice received vehicle followed by saline and were conditioned in the vehicle-paired chamber (VPCh). The order of conditioning were reversed for some mice to counterbalance the treatment and chamber assignment as well as the use of wild-type versus knockout mice for the morning and afternoon conditioning. Mice were then tested under a drug-free state for postconditioning place preference on day 5, as described for day 1. SCH221510 has been previously reported by Varty and colleagues (Varty et al., 2008) as a NOP agonist. Thus, we used this compound as the control NOP agonist and determined its effect on the rewarding action of alcohol. To this end, mice were tested for preconditioning place preference on day 1, conditioned with ethanol in the presence or absence of SCH221210 (10 mg/kg) on days 2-4 and then tested for CPP on day 5, as described above.
Experiment 2: To characterize the role of the NOP receptor in the inhibitory action of AT-312 on alcohol CPP
Mice lacking NOP and their wild-type controls were tested for preconditioning place preference, received conditioning with ethanol (2 g/kg, i.p.) in the presence or absence of AT-312 (3 mg/kg, s.c.; n = 7 mice per treatment per each genotype) on days 2-4 and then were tested for postconditioning place preference on day 5. On each test day, the amount of time that mice spent in the CPP chambers was recorded, as described above.
Experiment 3: To assess the motivational effect of AT-312 in the place conditioning paradigm
Mice were tested for baseline place preference on day 1, received conditioning on days 2-4 and were tested for postconditioning place preference on day 5. On each conditioning day, mice were treated with vehicle or AT-312 (3 mg/kg, s.c.; n = 5 mice per treatment) followed by saline and placed in the vehicle-paired (VPCh) or drug-paired chamber (DPCh). In the afternoon, mice were treated with the alternate treatment and conditioned to the opposite chamber. The amount of time that mice spent in the CPP chamber was recorded on each test day (days 1 and 5), as described above.
Experiment 4: To determine the effect of pentobarbital on ethanol-induced CPP
Mice were tested for preconditioning place preference on day 1, treated with saline or pentobarbital (25 mg/kg, s.c.; n = 6 mice per treatment) 15 min before ethanol (2 g/kg, i.p.) on each conditioning day and were confined to the drug paired chamber (DPCh). Animals were treated with saline 15 min before saline and confined to the vehicle-paired chambers (VPCh). These treatments were given either in the morning or afternoon in a counterbalanced manner. Each conditioning session lasted for 15 min and was conducted on days 2-4. Mice were then tested for CPP on day 5, as described above.
Data Analysis
Data are presented as mean (±S.E.M.) of the amount of time that mice spent in the drug-paired chamber (DPCh) or DPCh vs. vehicle-paired chamber (VPCh) on preconditioning test day (day 1, D1) and postconditioning test day (day 5, D5) and were analyzed using repeated measures two- or three-way analysis of variance (ANOVA). The Bonferroni’s post-hoc test was used to reveal significant changes between different groups. P<0.05 was considered significant.
RESULTS
In vitro NOP Receptor Binding affinity and Opioid Receptor Selectivity of AT-312
The chemical structure of AT-312 is shown in Fig. 1. The receptor binding affinity of AT-312 was determined using radioligand displacement assays conducted in membranes from CHO cells stably expressing the human NOP, MOP, DOP and KOP receptors. As shown in Table 1, AT-312 showed high binding affinity for the NOP receptor, yielding a subnanomolar Ki value of 0.34 ± 0.13 nM in competition with [3H]N/OFQ as the radioligand. In similar experiments using [3H]DAMGO, [3H]U69593 and [3H]DPDPE at the MOP, KOP and DOP receptors respectively, AT-312 showed binding selectivity of 17-fold versus MOP, 216-fold versus KOP and 378-fold versus DOP receptors. The NOP agonist SCH221510 tested in the same assays showed NOP binding Ki of 13.7 nM, about 40-fold lower affinity at NOP than AT-312. Also, the NOP binding affinity of SCH221510 was only 5-fold selective versus MOP, 3.6-fold versus KOP and 29-fold versus DOP receptors. AT-312 therefore, exhibits significantly higher binding affinity and selectivity for NOP compared to the positive control SCH221510. The high affinity of AT-312 for the NOP receptor is similar to that observed for other reported NOP agonists Ro 64-6198 and MT-7716 (Zaveri, 2016).
Table 1.
In vitro pharmacological profile of NOP agonists in binding and functional assays at the opioid receptors*
| Receptor Binding Ki (nM) | [35S] GTPγS NOP | [35S] GTPγS MOP | [35S] GTPγS KOP | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| NOP | MOP | KOP | DOP | EC50 (nM) | % Stim | EC50 (nM) | % Stim | EC50 (nM) | % Stim | |
|
| ||||||||||
| N/OFQ | 0.08 ± 0.03 | 133 ± 30 | 247 ± 3.4 | ND | 4.0 ± 0.1 | 100 | >10,000 | >10,000 | ||
| DAMGO | 2.96 ± 0.54 | 32.6 ± 4.06 | 100 | |||||||
| DPDPE | 1.11 ± 0.07 | |||||||||
| U69,593 | 1.05 ± 0.02 | 60.14 ± 7.45 | 100 | |||||||
| AT-312 | 0.34 ± 0.13 | 5.99 ± 0.97 | 73.5 ± 28.3 | 128.7± 57.4 | 29.9 ± 1.4 | 102.3 ± 0.75 | 81.5 ± 15.9 | 24.6 ± 2.4 | >10,000 | - |
| SCH221510 | 13.7 ± 2.30 | 65.4 ± 11.3 | 49.7 ± 11.3 | 403.7 ± 109.7 | 18.9 ± 5.9 | 95.1 ± 7.8 | 139.3 ± 4.6 | 76.8 ± 13.1 | 142.0 ± 15.6 | 82.72 ± 0.22 |
GTP(γ)S functional assays only carried out if binding affinity Ki<100 nM. The functional efficacy at the delta opioid receptor was therefore not determined for AT-312 and SCH221510. Values are the Mean ± SEM of three independent experiments run in triplicate. Functional activity was determined by stimulation of [35S]GTPγS binding to cell membranes, % stimulation was obtained as a percentage of stimulation of the standard agonists N/OFQ (for NOP), DAMGO (for MOP) and U69,593 (for KOP) taken as 100%.
As observed for other piperidinyl NOP ligands from our own compound library as well as those reported in the literature, AT-312 did not show appreciable affinity for the DOP receptor. Affinity profiling in a panel of 68 receptors and ion channels showed that, at a concentration of 100 nM, AT-312 did not bind to any non-opioid off-target receptors, whereas at 10 μM, it inhibited the specific binding of radioligands at the α1 adrenergic receptor by 60%, dopamine D4 receptor (86%), dopamine D3 receptor (100%), muscarinic M1 and M2 receptors (85%), NK2 receptor (92%), Ca+2 channel (L-type) (73%) and Na+ channel (site 2) (94%) and norepinephrine transporter (65%). Overall, AT-312 appears to be a selective NOP receptor ligand.
In vitro Functional Efficacy of AT-312
The intrinsic efficacy of AT-312 at the NOP and traditional opioid receptors was determined using the GTPγS binding assay conducted in membranes of CHO cells stably transfected with the NOP and classical opioid receptors. Table 1 shows the in vitro functional efficacy profile of AT-312 and SCH221510. AT-312 is a full agonist at the NOP receptor, showing potency (EC50) of 30 nM and 100% agonist stimulation compared to the endogenous NOP agonist N/OFQ. In contrast, it showed only a partial agonist efficacy of 25% at the MOP receptor and significantly lower potency compared to the MOP opioid full agonist DAMGO (Table 1). AT-312 had no agonist efficacy at the KOP receptor. In these experiments, SCH221510 was also found to be a full agonist at the NOP receptor, with comparable potency as that of AT-312 (Table 1). However, it also showed significant agonist stimulation at the KOP receptor, in contrast to AT-312.
AT-312, a novel NOP agonist, dose-dependently blocked the development of ethanol-induced CPP
The novel NOP agonist AT-312 dose-dependently reduced the rewarding action of ethanol (Fig. 2). Repeated measures ANOVA of the amount of time that mice spent in the drug-paired chamber (DPCh) on pre- and postconditioning days revealed a significant effect of treatment (F3,26 = 21.35; P< 0.01) but no effect of time (F1,26 = 4.48; P = 0.08) and no significant interaction between the two factors (F3,26 = 10.38; P = 0.08). The post-hoc test showed that the amount of time that mice spent in the DPCh was significantly (P<0.05) increased following ethanol conditioning in the vehicle-treated control group (Fig. 2, compare D5 vs. D1 for the mice treated with vehicle before ethanol on the conditioning days and this response was reduced by AT-312 in a dose-dependent manner (Fig. 2). In particular, the two higher doses of AT-312 (3 and 10 mg/kg) blocked ethanol-induced CPP [compare the amount of time between the vehicle-treated group on day 5 vs. the AT-312 (3 mg/kg) group (P<0.01) as well as against AT-312 (10 mg/kg) on this day (P<0.001)]. Together, these results suggest that AT-312 dose-dependently abolished the rewarding action of alcohol.
Figure 2. The effect of AT-312, a novel NOP agonist, on ethanol-induced CPP in C57BL/6J mice.
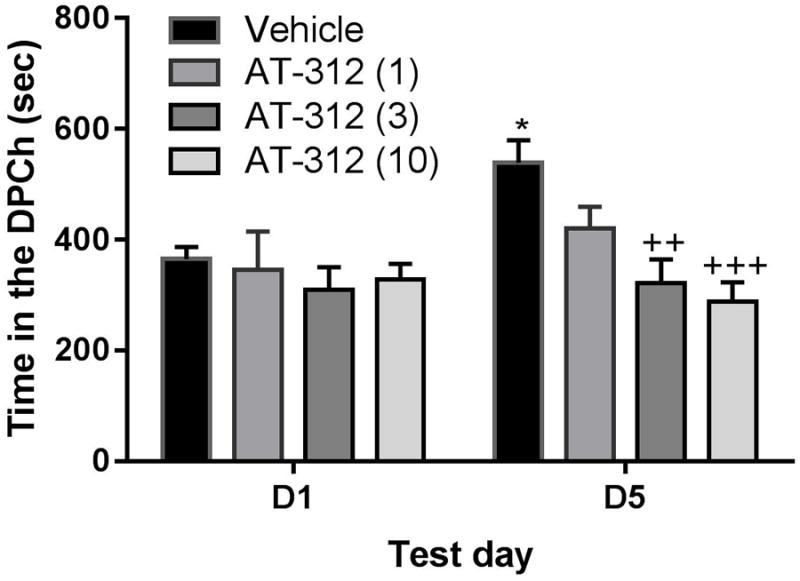
Data are mean (±S.E.M.) of the amount of time that mice spent in the drug-paired chamber (DPCh) before (D1) (preconditioning test day) and after (D5) conditioning (post-conditioning test day). Mice treated with vehicle or AT-312 (1, 3 or 10 mg/kg, s.c.) 15 min before ethanol on the conditioning days. *P<0.05, indicates a significant increase in the amount of time in the DPCh on D5 vs. D1; ++P<0.01, +++P<0.001, significantly different from the control group on D5.
AT-312 reduced the rewarding action of ethanol in wild-type but not in NOP knockout mice
The amount of time that mice lacking NOP and their wild-type littermates/controls spent in the ethanol-paired chamber on the preconditioning (D1) and postconditioning (D5) test days is shown in Figure 3. Three-way ANOVA revealed a significant effect of time (F1,1 =26; P<0.0001), a significant effect of context (F1,1 = 8.27; P<0.01) but no effect of genotype (F1,1 = 1.58; P>0.05). However, there was a significant interaction between time, context and genotype (F1,1 = 6.39; P<0.02). The post-hoc test showed that conditioning with ethanol induced a significant (P<0.05) CPP in both wild-type and knockout mice pretreated with vehicle prior to ethanol on the conditioning days, as evidenced by a significant increase in the amount of time that vehicle-treated control mice of either genotype spent in the ethanol-paired on day 5 compared to day 1 (Fig. 3, left panel; compare each bar on D5 vs. D1 for each genotype). The CPP response was significantly (P<0.001) reduced by AT-312 (10 mg/kg) in wild-type mice (Fig. 3, compare wild-type mice (NOP+/+) treated with AT-312 vs. vehicle on D5). On the other hand, mice lacking NOP spent the same amount of time in the DPCh on day 5 regardless of whether they were injected with the NOP agonist or vehicle (Fig. 3, compare NOP-/- treated with AT-312 group vs. vehicle-treated NOP-/- as well as against AT-312-treated NOP+/+ on D5). This result suggests that AT-312 exerts its inhibitory effect on the rewarding action of ethanol via the NOP receptor. AT-312 reduced locomotor activity in wild-type mice but this response was absent in mice lacking NOP (data not shown).
Figure 3. The action of AT-312, a novel NOP agonist, on ethanol-induced CPP in mice lacking NOP and their wild-type littermates/controls.
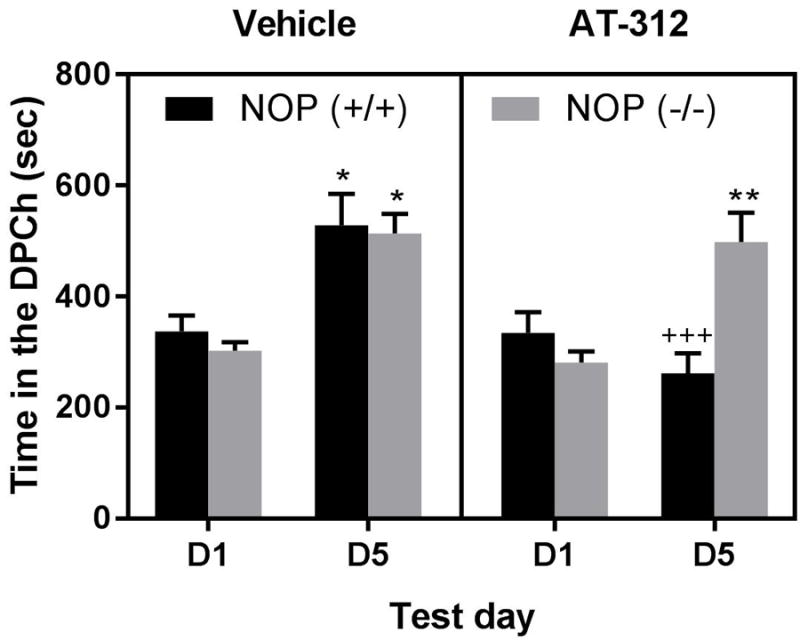
Data are mean (±S.E.M.) of the amount of time that mice spent in the drug-paired chamber (DPCh) before (D1) (preconditioning test day) and after (D5) conditioning (post-conditioning test day). Mice lacking NOP [NOP (-/-)] and their wild-type littermates [NOP (+/+)] were treated with vehicle (left panel) or AT-312 (10 mg/kg, right panel) 15 min before ethanol on conditioning days. ***P<0.001; *P<0.05 DPCh vs. VPCh
AT-312 given alone did not have any motivational effect in the place conditioning paradigm
Figure 4 shows the amount of time that mice, treated with vehicle in both conditioning chambers (Vehicle) and those that received vehicle in one chamber and AT-312 (3 mg/kg) in the other chamber, spent in the drug-paired chamber (DPCh). Two-way ANOVA revealed no significant effect of treatment (F1,8 = 0.75; P>0.05), no significant effect of time (F1,8 = 0.45; P>0.05) and no significant interaction between the two factors (F1,8 = 1.69; P>0.05), showing that AT-312 at this dose (3 mg/kg) did not possess motivational effects of its own.
Figure 4. Motivational effect of AT-312, a novel NOP agonist, in the place conditioning paradigm.
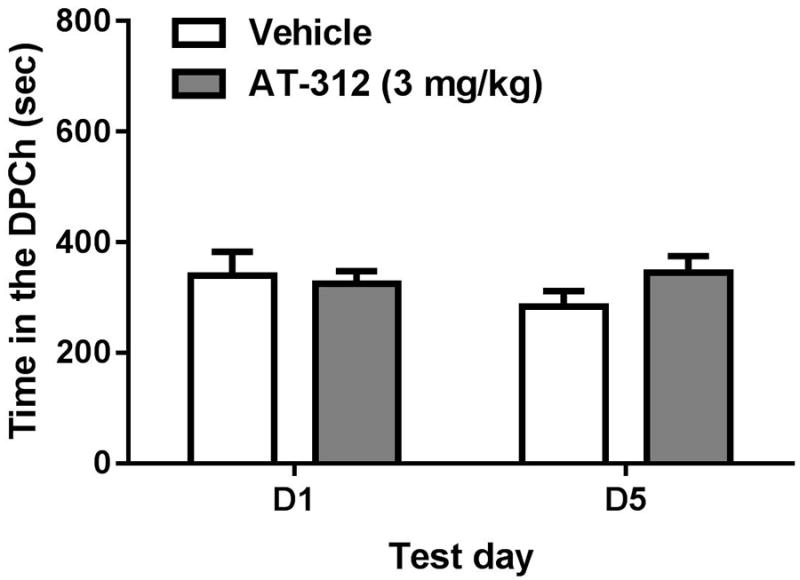
Data are mean (±S.E.M.) of the amount of time that mice spent in the drug-paired chamber (DPCh) on test days before (D1) and after (D5) conditioning. Mice received vehicle or AT-312 (3 mg/kg, right panel) 15 min before saline on the conditioning days.
Ethanol-induced CPP was reduced in mice treated with SCH221510, a NOP agonist
We also determined the effect of a known NOP agonist on ethanol-induced CPP. Considering that this compound was less selective toward the NOP compared to AT-312, we determined the effect of a relatively higher dose (10 mg/kg) of this compound on the rewarding action of ethanol. Figure 5 shows the amount of time that mice treated with vehicle or the NOP agonist spent in the ethanol-paired chamber (DPCh). Two-way repeated measures ANOVA revealed a significant effect of time that mice spent in the ethanol-paired chamber on day 5 vs. day 1 (F1,14 = 20.44; P<0.02) but no significant effect of treatment (F1,14 = 4.53; P>0.05) and no significant interaction between the two factors (F1,14 = 3.47; P>0.05). The Bonferroni post-hoc test showed that the amount of time that mice spent in the ethanol-paired chamber was increased in vehicle-pretreated mice, suggesting that ethanol induced a significant CPP in the control group (Fig. 5, compare the amount of time that vehicle-pretreated mice spent in the DPCh on D5 vs. D1). However, this response was reduced in mice treated with SCH221510.
Figure 5. Effects of SCH221510 (SCH; 10 mg/kg) on ethanol CPP in C57BL/6J mice.

Data are mean (±S.E.M.) of the amount of time that mice spent in the drug-paired chamber (DPCh) on test days before (D1) and after (D5) conditioning. Mice were treated with vehicle or SCH221510 15 min before ethanol on the conditioning days. *P<0.05, significant difference in the amount of time between D5 vs D1 for the vehicle-treated group
Pentobarbital reduced motor activity but failed to alter the rewarding action of ethanol
Figure 6 illustrates the amount of time that mice spent in the DPCh on the pre- and postconditioning test days. Three-way repeated measure ANOVA revealed a significant effect of context (DPCh vs. VPCh; F1,1 = 11.61; P<0.002) but no significant effect of time (F1,1 = 0.21; P>0.05) and no significant effect of treatment (F1,1 = 0.50; P>0.05). Although there was a significant context × time interaction (F1,1 = 13.40; P<0.0001, there was no treatment × context (F1,1 = 0.001; P>0.05) or time × context × treatment (F1,1 = 0.001; P>0.05) interaction. The post hoc test showed that ethanol induced a comparable CPP response in both groups, showing that pentobarbital did not alter the rewarding action of ethanol. Interestingly, pentobarbital induced a robust motor sedative effect and potentiated the sedative effect of alcohol on each conditioning day (data not shown).
Figure 6. Effects of pentobarbital (Pento; 25 mg/kg) on ethanol CPP in C57BL/6J mice.

Data are mean (±S.E.M.) of the amount of time that mice spent in the drug-paired (DPCh) and vehicle-paired chamber (VPCh) on test days before (D1) and after (D5) conditioning. Mice were treated with vehicle or pentobarbital 15 min before ethanol on the conditioning days. *P<0.05, significant difference in the amount of time that mice spent in the DPCh vs. VPCh on D5.
DISCUSSION
The main findings of the present study are that the novel NOP agonist AT-312 reduced the acquisition of CPP induced by ethanol, and that this effect was absent in mice lacking the NOP receptor. Similar, albeit less potent effects were also observed on alcohol reward in mice treated with the control NOP agonist SCH221510. The current results also demonstrate that AT-312 did not have motivational effects of its own at a dose (3 mg/kg) that completely abolished ethanol CPP in wild-type mice. Together, these results are consistent with previous studies with NOP agonists N/OFQ and Ro 64-6198, and confirm that NOP agonists can reduce acquisition of ethanol CPP in mice via selective action at the NOP receptor.
AT-312 is a selective and high affinity NOP full agonist, belonging to a novel class of NOP ligands structurally unrelated to NOP agonists Ro 64-6198, MT-7716, SR-8993 and others that have shown efficacy in reducing the rewarding effects of alcohol in various animal models and paradigms (Zaveri, 2016). A growing body of evidence suggests that NOP may be a potential target to reduce the rewarding and reinforcing actions of alcohol and other addictive drugs [(see recent reviews (Lutfy and Zaveri, 2016; Witkin et al., 2014; Zaveri, 2011; Zaveri, 2016)]. Consistent with existing literature, this novel NOP agonist AT-312 dose-dependently reduced the rewarding action of alcohol and appeared to be more potent than the known NOP agonist, SCH221510. Although further studies are needed to define the mechanism for the greater effect of AT-312 compared to SCH221510, we speculate that it may be due to its higher affinity toward NOP. Our in vitro studies show that AT-312 exhibits at least 20-fold binding selectivity toward the NOP versus the MOP receptor and is a full agonist at NOP but a weak partial agonist at the MOP receptor, with no appreciable agonist efficacy at the KOP receptor. In comparison, SCH221510 has only a four-fold binding selectivity versus the KOP receptor and has significant agonist efficacy at both the MOP and KOP receptors in the same assays (Table 1). However, further studies are needed to assess the contribution of each receptor in the inhibitory effects of the two NOP agonists.
Considering that AT-312 displayed higher affinity for and acted as a partial agonist at the mu opioid receptor, one may argue that the inhibitory action of the drug may be due to its interaction with the MOP receptor or both receptor systems rather than NOP only. In order to address this issue, we used mice lacking NOP and their wild-type controls and tested if the inhibitory effect of AT-312 is mediated via the NOP receptor. We rationalized that if AT-312 inhibits the rewarding action of alcohol via the NOP receptor, the drug would fail to alter the rewarding action of alcohol in mice lacking the NOP receptor. Consistent with this hypothesis, we observed that while the novel NOP agonist significantly reduced the rewarding action of alcohol in wild-type mice, the drug failed to alter ethanol-induced CPP in mice lacking NOP. This result suggests that AT-312 reduces the rewarding action of ethanol via the NOP receptor. However, further research is needed to assess the contribution of MOP receptor partial agonist activity in this response. Nevertheless, it is noteworthy to state that buprenorphine, a MOP partial agonist, was found to reduce alcohol consumption via its interaction with NOP (Ciccocioppo et al., 2007) and also found to reduce cocaine self-administration due to its agonist activity at the NOP and the MOP receptors (Kallupi et al., 2017).
Given that ethanol induced aversion in mice pretreated with the highest dose of AT-312 (Fig. 2), one may argue that the NOP agonist may have reduced ethanol-induced CPP by inducing aversion. However, the lower dose of AT-312 (3 mg/kg), which also completely blocked ethanol induced CPP in wild-type but not knockout mice (Fig. 3), failed to induce aversion when given with alcohol (Fig. 3) or alone (Fig. 4). This is in accord with an earlier report showing that NOP agonists may be devoid of any motivational effects (Devine et al., 1996). Such a property could be useful to treat drug reward since the NOP agonist would not alter basal hedonic homeostasis, which may be advantageous for patient compliance and other normal daily functions.
NOP agonists are known to reduce motor activity (Devine et al., 1996). Consistent with this, we found that AT-312 suppressed motor activity in wild-type mice and this response was absent in mice lacking NOP receptor, showing that this effect of AT-312 was also mediated via the NOP receptor. It is generally accepted that drugs that reduce locomotor activity can confound behavioral responding. This may affect the outcome of the CPP response if one tests animals in the presence of a sedative drug. However, we tested the animals for CPP under a drug-free state on the postconditioning test days and found no significant differences in locomotor activity between the vehicle-conditioned and drug-conditioned groups. Additionally, we believe that not all drugs that reduce motor activity during conditioning block the CPP response. Interestingly, alcohol is sedative in mice; yet, it induces a robust CPP response. We further demonstrated this by conducting an experiment using pentobarbital, which is a known sedative hypnotic and examined its effect on ethanol-induced CPP. While pentobarbital significantly reduced locomotor activity during conditioning, it failed to alter the CPP response on the test day (Fig. 6), suggesting that the ability of AT-312 to reduce the rewarding action of ethanol may not be simply due to its sedative effect during conditioning.
The rewarding action of alcohol and other drugs of abuse has been linked to their ability to increase extracellular dopamine in the nucleus accumbens (Di Chiara and Imperato, 1988). Although the mechanism of inhibitory action of AT-312 is not clear at this time, we speculate that the NOP agonist reduces the ability of alcohol to elevate extracellular dopamine levels in the nucleus accumbens. Indeed, previous studies have shown that intracerebroventricular administration of N/OFQ reduced elevation of accumbal dopamine induced by morphine (Di Giannuario et al., 1999) and cocaine (Lutfy et al., 2001; Sakoori and Murphy, 2004). The NOP agonist also attenuated the rewarding action of morphine (Ciccocioppo et al., 2000; Murphy et al., 1999), cocaine (Sakoori and Murphy, 2004) and ethanol (Kuzmin et al., 2003). Thus, we propose that the NOP agonist reduces dopaminergic neurotransmission by acting in the ventral tegmental area and/or nucleus accumbens to reduce the rewarding action of alcohol. However, further studies are needed to identify the neuroanatomical sites of action of the NOP agonist in this regard.
A recent report shows that NOP receptor knockout rats exhibit reduced alcohol consumption compared to their wild-type controls although saccharine intake was not different between the rats of the two genotypes (Kallupi et al., 2017). A similar reduction in ethanol self-administration was observed in rats treated with a novel orally bioavailable NOP antagonist (Rorick-Kehn et al., 2016). Interestingly, we did not observe reduced ethanol-induced CPP in mice lacking NOP although these authors found decreased ethanol self-administration in NOP knockout rats (Kallupi et al., 2017) or in wild-type rats treated with the NOP antagonist (Rorick-Kehn et al., 2016). A parsimonious explanation of such discordant effects is that the two studies measured two different responses. Notably, Kuzmin and colleagues also found that male mice lacking N/OFQ tended to show a stronger response to ethanol (Kuzmin et al., 2003). Alternately, the NOP system has been implicated in feeding, and N/OFQ has hyperphagic effects [for a review, see (Witkin et al., 2014)]. Thus, it is possible that food and drink consumption could be reduced in animals lacking the NOP receptor or its endogenous agonist. However, saccharin consumption was not altered in rats lacking NOP receptors (Kallupi et al., 2017). Nevertheless, it is possible that NOP system is involved in consumption of food and drinks with caloric values and thus one would expect a difference in outcomes of the two studies. It is of interest to note that the novel bioavailable NOP antagonist reduced consumption of highly palatable food to the regular chow level (Statnick et al., 2016).
The other explanation for the discrepant results could be species differences in the current and earlier studies. Interestingly, we found enhanced cocaine-induced CPP in mice lacking NOP (Marquez et al., 2008), whereas these authors reported reduced cocaine-induced CPP in NOP knockout rats (Kallupi et al., 2017). The sex of animals may have contributed to this discrepant data since we used female mice in this study. Our earlier studies have shown that female mice exhibit greater ethanol-induced CPP than male mice, hence were used here (Nguyen et al., 2012). On the other hand, these other studies used male rats to study the role of NOP receptors in alcohol self-administration (Kallupi et al., 2017; Rorick-Kehn et al., 2016).
In summary, we found that a novel NOP agonist, AT-312, reduced the rewarding effects of ethanol in the CPP paradigm. The inhibitory effect of the NOP agonist was absent in NOP knockout mice, showing that the action of AT-312 was via the NOP receptor. AT-312 did not possess any motivational effects of its own at a dose that robustly reduced ethanol-induced CPP. Thus, the NOP receptor may be a potential target for the development of pharmacotherapy to treat alcohol use disorders.
Acknowledgments
Support
These studies were supported by the National Institutes of Health grant R01DA027811 (NTZ), NIAAA Contract HHSN275201300005C and HHSN275201500005C (NTZ) to Astraea Therapeutics, LLC (Mountain View, California, USA) and in part by a Tobacco Related Disease Research Program (TRDRP) 24RT-0023 (KL).
Footnotes
Author Contributions
PM, WEP and MEM conducted experiments, AH conducted genotyping and took care of the mouse breeding colony, NZ developed the compound, supervised its in vitro characterization and wrote the manuscript, and KL designed the CPP experiments, analyzed the behavioral data and wrote the manuscript.
Conflict of Interest
The authors declare no conflicts of interest. NTZ, WEP and MEM are employees of Astraea Therapeutics.
References
- Altshuler HL, Phillips PE, Feinhandler DA. Alteration of ethanol self-administration by naltrexone. Life Sci. 1980;26:679–688. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- Aziz AM, Brothers S, Sartor G, Holm L, Heilig M, Wahlestedt C, Thorsell A. The nociceptin/orphanin FQ receptor agonist SR-8993 as a candidate therapeutic for alcohol use disorders: validation in rat models. Psychopharmacology (Berl) 2016;233:3553–3563. doi: 10.1007/s00213-016-4385-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology (Berl) 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- Benjamin D, Grant ER, Pohorecky LA. Naltrexone reverses ethanol-induced dopamine release in the nucleus accumbens in awake, freely moving rats. Brain Res. 1993;621:137–140. doi: 10.1016/0006-8993(93)90309-b. [DOI] [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Angeletti S, Sanna PP, Weiss F, Massi M. Effect of nociceptin/orphanin FQ on the rewarding properties of morphine. Eur J Pharmacol. 2000;404:153–159. doi: 10.1016/s0014-2999(00)00590-2. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, de Guglielmo G, Hansson AC, Ubaldi M, Kallupi M, Cruz MT, Oleata CS, Heilig M, Roberto M. Restraint stress alters nociceptin/orphanin FQ and CRF systems in the rat central amygdala: significance for anxiety-like behaviors. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014a;34:363–372. doi: 10.1523/JNEUROSCI.2400-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccocioppo R, Economidou D, Rimondini R, Sommer W, Massi M, Heilig M. Buprenorphine reduces alcohol drinking through activation of the nociceptin/orphanin FQ-NOP receptor system. Biological psychiatry. 2007;61:4–12. doi: 10.1016/j.biopsych.2006.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccocioppo R, Panocka I, Polidori C, Regoli D, Massi M. Effect of nociceptin on alcohol intake in alcohol-preferring rats. Psychopharmacology (Berl) 1999;141:220–224. doi: 10.1007/s002130050828. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Polidori C, Antonelli L, Salvadori S, Guerrini R, Massi M. Pharmacological characterization of the nociceptin receptor which mediates reduction of alcohol drinking in rats. Peptides. 2002;23:117–125. doi: 10.1016/s0196-9781(01)00587-3. [DOI] [PubMed] [Google Scholar]
- Ciccocioppo R, Stopponi S, Economidou D, Kuriyama M, Kinoshita H, Heilig M, Roberto M, Weiss F, Teshima K. Chronic treatment with novel brain-penetrating selective NOP receptor agonist MT-7716 reduces alcohol drinking and seeking in the rat. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2014b;39:2601–2610. doi: 10.1038/npp.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine DP, Reinscheid RK, Monsma FJ, Jr, Civelli O, Akil H. The novel neuropeptide orphanin FQ fails to produce conditioned place preference or aversion. Brain Res. 1996;727:225–229. doi: 10.1016/0006-8993(96)00476-3. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giannuario A, Pieretti S, Catalani A, Loizzo A. Orphanin FQ reduces morphine-induced dopamine release in the nucleus accumbens: a microdialysis study in rats. Neurosci Lett. 1999;272:183–186. doi: 10.1016/s0304-3940(99)00579-0. [DOI] [PubMed] [Google Scholar]
- Esser MB, Clayton H, Demissie Z, Kanny D, Brewer RD. Current and Binge Drinking Among High School Students - United States 1991-2015. MMWR Morbidity and mortality weekly report. 2017;66:474–478. doi: 10.15585/mmwr.mm6618a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser MB, Hedden SL, Kanny D, Brewer RD, Gfroerer JC, Naimi TS. Prevalence of alcohol dependence among US adult drinkers, 2009-2011. Preventing chronic disease. 2014;11:E206. doi: 10.5888/pcd11.140329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farren CK, O’Malley S. Sequential use of naltrexone in the treatment of relapsing alcoholism. The American journal of psychiatry. 1997;154:714. doi: 10.1176/ajp.154.5.714a. [DOI] [PubMed] [Google Scholar]
- Froehlich JC, Harts J, Lumeng L, Li TK. Naloxone attenuates voluntary ethanol intake in rats selectively bred for high ethanol preference. Pharmacol Biochem Behav. 1990;35:385–390. doi: 10.1016/0091-3057(90)90174-g. [DOI] [PubMed] [Google Scholar]
- Gianoulakis C. Endogenous opioids and addiction to alcohol and other drugs of abuse. Curr Top Med Chem. 2004;4:39–50. doi: 10.2174/1568026043451573. [DOI] [PubMed] [Google Scholar]
- Gianoulakis C, de Waele JP, Thavundayil J. Implication of the endogenous opioid system in excessive ethanol consumption. Alcohol. 1996;13:19–23. doi: 10.1016/0741-8329(95)02035-7. [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Koob GF. Neurobiology of alcohol dependence: focus on motivational mechanisms. Alcohol Res Health. 2008;31:185–195. [PMC free article] [PubMed] [Google Scholar]
- Hall FS, Sora I, Uhl GR. Ethanol consumption and reward are decreased in mu-opiate receptor knockout mice. Psychopharmacology (Berl) 2001;154:43–49. doi: 10.1007/s002130000622. [DOI] [PubMed] [Google Scholar]
- Hubbell CL, Czirr SA, Hunter GA, Beaman CM, LeCann NC, Reid LD. Consumption of ethanol solution is potentiated by morphine and attenuated by naloxone persistently across repeated daily administrations. Alcohol. 1986;3:39–54. doi: 10.1016/0741-8329(86)90070-4. [DOI] [PubMed] [Google Scholar]
- Hubbell CL, Marglin SH, Spitalnic SJ, Abelson ML, Wild KD, Reid LD. Opioidergic, serotonergic, and dopaminergic manipulations and rats’ intake of a sweetened alcoholic beverage. Alcohol. 1991;8:355–367. doi: 10.1016/0741-8329(91)90573-f. [DOI] [PubMed] [Google Scholar]
- Ji D, Gilpin NW, Richardson HN, Rivier CL, Koob GF. Effects of naltrexone, duloxetine, and a corticotropin-releasing factor type 1 receptor antagonist on binge-like alcohol drinking in rats. Behav Pharmacol. 2008;19:1–12. doi: 10.1097/FBP.0b013e3282f3cf70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallupi M, Scuppa G, de Guglielmo G, Calo G, Weiss F, Statnick MA, Rorick-Kehn LM, Ciccocioppo R. Genetic Deletion of the Nociceptin/Orphanin FQ Receptor in the Rat Confers Resilience to the Development of Drug Addiction. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2017;42:695–706. doi: 10.1038/npp.2016.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallupi M, Varodayan FP, Oleata CS, Correia D, Luu G, Roberto M. Nociceptin/orphanin FQ decreases glutamate transmission and blocks ethanol-induced effects in the central amygdala of naive and ethanol-dependent rats. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2014;39:1081–1092. doi: 10.1038/npp.2013.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornet M, Goosen C, Van Ree JM. Effect of naltrexone on alcohol consumption during chronic alcohol drinking and after a period of imposed abstinence in free-choice drinking rhesus monkeys. Psychopharmacology (Berl) 1991;104:367–376. doi: 10.1007/BF02246038. [DOI] [PubMed] [Google Scholar]
- Kotlinska J, Rafalski P, Biala G, Dylag T, Rolka K, Silberring J. Nociceptin inhibits acquisition of amphetamine-induced place preference and sensitization to stereotypy in rats. Eur J Pharmacol. 2003;474:233–239. doi: 10.1016/s0014-2999(03)02081-8. [DOI] [PubMed] [Google Scholar]
- Kuzmin A, Kreek MJ, Bakalkin G, Liljequist S. The nociceptin/orphanin FQ receptor agonist Ro 64-6198 reduces alcohol self-administration and prevents relapse-like alcohol drinking. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2007;32:902–910. doi: 10.1038/sj.npp.1301169. [DOI] [PubMed] [Google Scholar]
- Kuzmin A, Sandin J, Terenius L, Ogren SO. Acquisition, expression, and reinstatement of ethanol-induced conditioned place preference in mice: effects of opioid receptor-like 1 receptor agonists and naloxone. J Pharmacol Exp Ther. 2003;304:310–318. doi: 10.1124/jpet.102.041350. [DOI] [PubMed] [Google Scholar]
- Lutfy K, Do T, Maidment NT. Orphanin FQ/nociceptin attenuates motor stimulation and changes in nucleus accumbens extracellular dopamine induced by cocaine in rats. Psychopharmacology (Berl) 2001;154:1–7. doi: 10.1007/s002130000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutfy K, Zaveri NT. The Nociceptin Receptor as an Emerging Molecular Target for Cocaine Addiction. Prog Mol Biol Transl Sci. 2016;137:149–181. doi: 10.1016/bs.pmbts.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfaing-Jallat P, Miceli D, Le Magnen J. Decrease in ethanol consumption by naloxone in naive and dependent rats. Pharmacol Biochem Behav. 1983;18(Suppl 1):537–539. doi: 10.1016/0091-3057(83)90232-0. [DOI] [PubMed] [Google Scholar]
- Marquez P, Borse J, Nguyen AT, Hamid A, Lutfy K. The role of the opioid receptor-like (ORL1) receptor in motor stimulatory and rewarding actions of buprenorphine and morphine. Neuroscience. 2008;155:597–602. doi: 10.1016/j.neuroscience.2008.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Pasternak GW. The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacological reviews. 2001;53:381–415. [PubMed] [Google Scholar]
- Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA. 2004;291:1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Lee Y, Maidment NT. Orphanin FQ/nociceptin blocks acquisition of morphine place preference. Brain Res. 1999;832:168–170. doi: 10.1016/s0006-8993(99)01425-0. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Ly HT, Maidment NT. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience. 1996;75:1–4. doi: 10.1016/0306-4522(96)00322-3. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Maidment NT. Orphanin FQ/nociceptin modulation of mesolimbic dopamine transmission determined by microdialysis. Journal of neurochemistry. 1999;73:179–186. doi: 10.1046/j.1471-4159.1999.0730179.x. [DOI] [PubMed] [Google Scholar]
- Murphy NP, Tan AM, Lam HA, Maidment NT. Nociceptin/orphanin FQ modulation of rat midbrain dopamine neurons in primary culture. Neuroscience. 2004;127:929–940. doi: 10.1016/j.neuroscience.2004.05.055. [DOI] [PubMed] [Google Scholar]
- Myers RD, Borg S, Mossberg R. Antagonism by naltrexone of voluntary alcohol selection in the chronically drinking macaque monkey. Alcohol. 1986;3:383–388. doi: 10.1016/0741-8329(86)90058-3. [DOI] [PubMed] [Google Scholar]
- Neal CR, Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Akil H, Watson SJ., Jr Opioid receptor-like (ORL1) receptor distribution in the rat central nervous system: comparison of ORL1 receptor mRNA expression with (125)I-[(14)Tyr]-orphanin FQ binding. J Comp Neurol. 1999a;412:563–605. [PubMed] [Google Scholar]
- Neal CR, Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Watson SJ., Jr Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J Comp Neurol. 1999b;406:503–547. [PubMed] [Google Scholar]
- Nguyen K, Tseng A, Marquez P, Hamid A, Lutfy K. The role of endogenous dynorphin in ethanol-induced state-dependent CPP. Behavioural brain research. 2012;227:58–63. doi: 10.1016/j.bbr.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi M, Houtani T, Noda Y, Mamiya T, Sato K, Doi T, Kuno J, Takeshima H, Nukada T, Nabeshima T, Yamashita T, Noda T, Sugimoto T. Unrestrained nociceptive response and disregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. EMBO J. 1997;16:1858–1864. doi: 10.1093/emboj/16.8.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley SS, Krishnan-Sarin S, Farren C, Sinha R, Kreek MJ. Naltrexone decreases craving and alcohol self-administration in alcohol-dependent subjects and activates the hypothalamo-pituitary-adrenocortical axis. Psychopharmacology (Berl) 2002;160:19–29. doi: 10.1007/s002130100919. [DOI] [PubMed] [Google Scholar]
- Oslin D, Liberto JG, O’Brien J, Krois S, Norbeck J. Naltrexone as an adjunctive treatment for older patients with alcohol dependence. The American journal of geriatric psychiatry : official journal of the American Association for Geriatric Psychiatry. 1997;5:324–332. doi: 10.1097/00019442-199700540-00007. [DOI] [PubMed] [Google Scholar]
- Oswald LM, Wand GS. Opioids and alcoholism. Physiol Behav. 2004;81:339–358. doi: 10.1016/j.physbeh.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, McDonald JS, Heyser CJ, Kieffer BL, Matthes HW, Koob GF, Gold LH. mu-Opioid receptor knockout mice do not self-administer alcohol. J Pharmacol Exp Ther. 2000;293:1002–1008. [PubMed] [Google Scholar]
- Rorick-Kehn LM, Ciccocioppo R, Wong CJ, Witkin JM, Martinez-Grau MA, Stopponi S, Adams BL, Katner JS, Perry KW, Toledo MA, Diaz N, Lafuente C, Jimenez A, Benito A, Pedregal C, Weiss F, Statnick MA. A Novel, Orally Bioavailable Nociceptin Receptor Antagonist, LY2940094, Reduces Ethanol Self-Administration and Ethanol Seeking in Animal Models. Alcoholism, clinical and experimental research. 2016;40:945–954. doi: 10.1111/acer.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakoori K, Murphy NP. Central administration of nociceptin/orphanin FQ blocks the acquisition of conditioned place preference to morphine and cocaine, but not conditioned place aversion to naloxone in mice. Psychopharmacology (Berl) 2004;172:129–136. doi: 10.1007/s00213-003-1643-3. [DOI] [PubMed] [Google Scholar]
- Samson HH, Doyle TF. Oral ethanol self-administration in the rat: effect of naloxone. Pharmacol Biochem Behav. 1985;22:91–99. doi: 10.1016/0091-3057(85)90491-5. [DOI] [PubMed] [Google Scholar]
- Statnick MA, Chen Y, Ansonoff M, Witkin JM, Rorick-Kehn L, Suter TM, Song M, Hu C, Lafuente C, Jimenez A, Benito A, Diaz N, Martinez-Grau MA, Toledo MA, Pintar JE. A Novel Nociceptin Receptor Antagonist LY2940094 Inhibits Excessive Feeding Behavior in Rodents: A Possible Mechanism for the Treatment of Binge Eating Disorder. J Pharmacol Exp Ther. 2016;356:493–502. doi: 10.1124/jpet.115.228221. [DOI] [PubMed] [Google Scholar]
- Toll L, Khroyan TV, Polgar WE, Jiang F, Olsen C, Zaveri NT. Comparison of the antinociceptive and antirewarding profiles of novel bifunctional nociceptin receptor/mu-opioid receptor ligands: implications for therapeutic applications. J Pharmacol Exp Ther. 2009;331:954–964. doi: 10.1124/jpet.109.157446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor JR, Nahorski SR. Modulation by mu-opioid agonists of guanosine-5’-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol. 1995;47:848–854. [PubMed] [Google Scholar]
- Tseng A, Nguyen K, Hamid A, Garg M, Marquez P, Lutfy K. The role of endogenous beta-endorphin and enkephalins in ethanol reward. Neuropharmacology. 2013;73:290–300. doi: 10.1016/j.neuropharm.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varty GB, Lu SX, Morgan CA, Cohen-Williams ME, Hodgson RA, Smith-Torhan A, Zhang H, Fawzi AB, Graziano MP, Ho GD, Matasi J, Tulshian D, Coffin VL, Carey GJ. The anxiolytic-like effects of the novel, orally active nociceptin opioid receptor agonist 8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510. J Pharmacol Exp Ther. 2008;326:672–682. doi: 10.1124/jpet.108.136937. [DOI] [PubMed] [Google Scholar]
- Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Archives of general psychiatry. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- Volpicelli JR, Davis MA, Olgin JE. Naltrexone blocks the post-shock increase of ethanol consumption. Life Sci. 1986;38:841–847. doi: 10.1016/0024-3205(86)90601-6. [DOI] [PubMed] [Google Scholar]
- Weiss F, Mitchiner M, Bloom FE, Koob GF. Free-choice responding for ethanol versus water in alcohol preferring (P) and unselected Wistar rats is differentially modified by naloxone, bromocriptine, and methysergide. Psychopharmacology (Berl) 1990;101:178–186. doi: 10.1007/BF02244123. [DOI] [PubMed] [Google Scholar]
- Witkin JM, Statnick MA, Rorick-Kehn LM, Pintar JE, Ansonoff M, Chen Y, Tucker RC, Ciccocioppo R. The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacology & therapeutics. 2014;141:283–299. doi: 10.1016/j.pharmthera.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri N, Polgar WE, Olsen CM, Kelson AB, Grundt P, Lewis JW, Toll L. Characterization of opiates, neuroleptics, and synthetic analogs at ORL1 and opioid receptors. Eur J Pharmacol. 2001;428:29–36. doi: 10.1016/s0014-2999(01)01282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri NT. The nociceptin/orphanin FQ receptor (NOP) as a target for drug abuse medications. Curr Top Med Chem. 2011;11:1151–1156. doi: 10.2174/156802611795371341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaveri NT. Nociceptin Opioid Receptor (NOP) as a Therapeutic Target: Progress in Translation from Preclinical Research to Clinical Utility. J Med Chem. 2016;59:7011–7028. doi: 10.1021/acs.jmedchem.5b01499. [DOI] [PMC free article] [PubMed] [Google Scholar]