

Abstract
Objective
Macrophage pro-inflammatory responses induced by modified low-density lipoproteins (modLDL) contribute to atherosclerotic progression. How modLDL causes macrophages to become pro-inflammatory is still enigmatic. Macrophage foam cell formation induced by modLDL requires glycerolipid synthesis. Lipin-1, a key enzyme in the glycerolipid synthesis pathway, contributes to modLDL-elicited macrophage pro-inflammatory responses in vitro. The objective of this study was to determine if macrophage-associated lipin-1 contributes to atherogenesis and to assess its role in modLDL-mediated signaling in macrophages.
Approach and Results
We developed mice lacking lipin-1 in myeloid-derived cells and used adeno-associated viral vector 8 expressing the gain-of-function mutation of mouse proprotein convertase subtilisin/kexin type 9 (AAV8-PCSK9) to induce hypercholesterolemia and plaque formation. Mice lacking myeloid-associated lipin-1 had reduced atherosclerotic burden compared to control mice despite similar plasma lipid levels. Stimulation of bone marrow-derived macrophages with modLDL activated a persistent PKCα/βII-ERK1/2-cJun signaling cascade that contributed to macrophage pro-inflammatory responses that was dependent on lipin-1 enzymatic activity.
Conclusions
Our data demonstrate that macrophage-associated lipin-1 is atherogenic, likely through persistent activation of a PKCα/βII-ERK1/2-cJun signaling cascade that contributes to foam cell pro-inflammatory responses. Taken together these results suggest modLDL-induced foam cell formation and modLDL-induced macrophage pro-inflammatory responses are not independent consequences of modLDL stimulation, but rather are both directly influenced by enhanced lipid synthesis.
Keywords: Lipin-1, macrophage, atherosclerosis, inflammation
Subject code: Animal models of human disease, inflammation, lipids and cholesterol, metabolism, cardiovascular disease, atherosclerosis
Graphical abstract
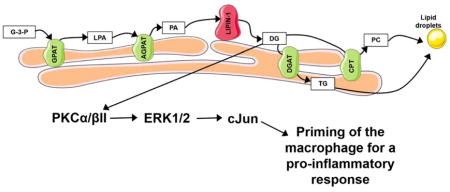
INTRODUCTION
Macrophage responses to modified low-density lipoproteins (e.g. oxidized LDL) contribute to atherosclerosis progression. Internalization of modified LDL (modLDL) by macrophages results in the generation of a lipid-laden phenotype (foam cell) and generation of pro-inflammatory mediators. The foam cell phenotype results from both enhanced modLDL uptake via scavenger receptor recognition and reduced ability to process cholesterol and deliver it to high-density lipoproteins.1 Uptake of modLDLs is also known to induce production of pro-inflammatory cytokines, such as TNF-α, IL-6, IL-1, PGE2, CCL2, etc., that promote atherosclerosis.2 However, the mechanism through which foam cells become pro-inflammatory has been elusive. ModLDLs can be recognized by macrophages in vitro as “pathogens” via pattern recognition receptors (PRRs) such as TLR2, TLR4, and NLRP3, resulting in acute signaling responses that elicit pro-inflammatory mediator production.3–6 However, in vivo the induction of macrophage pro-inflammatory responses is clearly more complex as hematopoietic deletion of TLR2, TLR4, or NLRP3 does not reduce atherosclerosis burden.7, 8 We and others have demonstrated that glycerolipid synthesis can not only contribute to foam cell formation but can also mediate macrophage pro-inflammatory responses.9–11 Together, these data suggest glycerolipid synthesis may provide an additional mechanism by which modLDLs influence macrophage pro-inflammatory responses.
ModLDLs are engulfed by macrophages and broken down by lysosomal acid lipases within endosomes, resulting in the generation of free cholesterol and free fatty acids.12 Excess free cholesterol is esterified at the endoplasmic reticulum (ER) and then stored in lipid droplets, which give foam cells their phenotypic appearance.1 Although the composition of foam cell lipid droplets is dominated by cholesterol esters, they also contain triacylglycerol (TAG) and phosphatidylcholine (PC).12, 13 One lipid synthetic pathway that contributes to lipid droplet generation in macrophages is the glycerolipid synthetic pathway, which produces TAG and PC.14 Glycerolipid synthesis consists of four enzymatic steps in which glycerol-3-phosphate (G3P) is converted into TAG or PC at the ER (Graphical abstract). Sequential esterification of G3P with acyl-CoA charged long-chain-fatty-acids by glycerol phosphate acyl-transferase and lysophosphate-acyl transferase enzymes produces phosphatidic acid (PA). PA is dephosphorylated by the phosphatidic acid phosphatase (PAP), lipin-1, to generate diacylglycerol (DAG). DAG can then be converted into TAG by diglyceride acyltransferase (DGAT) enzymes or PC via the activity of CDP-DAG:cholinephosphotransferase.15 The inhibition of long-chain-fatty-acid-CoA ligase (with triacin C), the genetic deletion of DGAT1, or genetic reduction of lipin-1 all result in reduced foam cell formation in response to modLDLs.10, 11, 16 Thus, these studies provide evidence that glycerolipid synthesis contributes to foam cell formation.
Glycerolipid synthesis has been shown to influence immune cell function and enhance inflammation.17–19 Our lab and others have demonstrated that lipin-1, a key regulatory enzyme in the glycerolipid synthetic pathway, mediates DAG production and contributes to pro-inflammatory responses in LPS-, oxLDL-, and acetylated LDL- (acLDL) stimulated macrophages.9, 10 Lipin-1 is a bi-functional enzyme with both enzymatic PAP activity and transcriptional cofactor activity.14, 20 However, only lipin-1 enzymatic activity is required for glycerolipid synthesis.21 The contribution of lipin-1 to macrophage pro-inflammatory responses led us to hypothesize that macrophage-associated lipin-1 enzymatic activity influences atherosclerosis development. To evaluate the role of macrophage-associated lipin-1 enzymatic activity in atherosclerosis, we developed a mouse model with a myeloid-specific deletion of lipin-1 enzymatic activity (lipin-1mEnzyKO) and induced atherosclerosis using the adeno-associated viral vector 8 expressing a gain-of-function mutation (D377Y) of mouse proprotein convertase subtillsin/kexin type 9 (referred to as AAV8-PCSK9) and high-fat diet.22 Additionally, our in vitro studies provide evidence as to the molecular mechanisms by which macrophage-associated lipin-1 enzymatic activity promotes atherosclerosis.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
Myeloid-associated lipin-1 is atherogenic
To investigate the potential contribution of macrophage-associated lipin-1 on atherosclerosis, we generated mice lacking lipin-1 enzymatic activity in myeloid-derived cells. Lpin1 floxed (lipin-1flox/flox) mice were crossed with LysM-Cre mice to generate myeloid-specific lipin-1 enzymatic knockout (lipin-1mEnzyKO) mice. Due to the location of the flox sites in these lipin-1flox/flox mice, the original translational start site is lost, but an alternative start site is enforced and the resulting truncated lipin-1 protein lacks enzymatic activity while transcriptional coregulatory function is retained.23
Equal amounts of lipin-1 were present in non-myeloid-derived tissue from the heart and lung in lipin-1mEnzyKO and lipin-1flox/flox littermate control mice (Figure 1A). BMDMs derived from lipin-1mEnzyKO mice have a smaller lipin-1 band compared to the lipin-1 band detected in the lipin-1flox/flox BMDMs, the smaller band represents the truncated form of lipin-123 (Figure 1A). We first wanted to verify that BMDMs from lipin-1mEnzyKO mice responded to oxLDL in a manner similar to the lipin-1-depleted RAW264.7 macrophage cell line.10 RAW264.7 macrophages lacking lipin-1 have reduced foam cell formation and DAG production, while cholesterol uptake and scavenger receptor expression remains unaltered in response to oxLDL as compared to control RAW264.7 macrophages.10 Consistent with this, LC-MS/MS analysis showed lipin-1mEnzyKO BMDMs had reduced DAG production 24 and 48 hours after oxLDL stimulation as compared to lipin-1flox/flox control BMDMs (Figure 1B). Furthermore, analysis of cholesterol esters showed no difference in cholesterol content between the lipin-1mEnzyKO and lipin-1flox/flox control BMDMs (Figure 1C). Nile red analysis also demonstrated a reduction in lipid content of lipin-1mEnzyKO BMDMs compared to lipin-1flox/flox control BMDMs (Figure 1D). Similar cholesterol content suggests oxLDL engulfment is unaltered in the lipin-1mEnzyKO BMDMs, which was supported by similar levels of scavenger receptor gene expression as measured by qRT-PCR analysis (Supplemental Figure IA). Together these data demonstrate that lipin-1mEnzyKO BMDMs have reduced foam cell formation and DAG production but continue to engulf oxLDLs.
Figure 1.
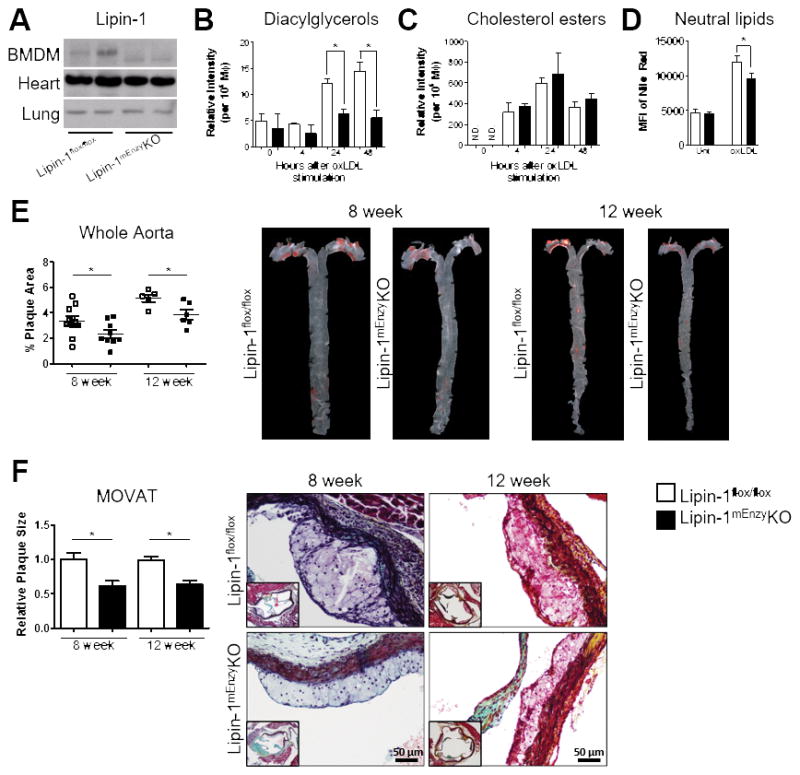
To elicit atherosclerosis, lipin-1flox/flox and lipin-1mEnzyKO mice were retro-orbitally injected with 3×1010 vector genomes of AAV8-PCSK9 and placed on high fat diet for 8 or 12 weeks. Both lipin-1flox/flox and lipin-1mEnzyKO mice gained weight in response to high fat diet at similar rates (Supplemental Figure IIA&B). Serum lipid analysis was performed at the time of euthanasia to determine the state of hypercholesterolemia. Comparable levels of total cholesterol (TC), triglycerides (TG), high-density lipoprotein (HDL) cholesterol, and low-density lipoprotein (LDL) cholesterol were observed between the lipin-1mEnzyKO and lipin-1flox/flox mice (Supplemental Figure IIC&D). To ensure the lipin-1mEnzyKO did not have alterations in immune composition, we analyzed splenocytes in lipin-1mEnzyKO and lipin-1flox/flox mice at time of euthanasia and demonstrated equivalent percentages and numbers of granulocytes, macrophages, dendritic cells, T cells, B cells, and NK cells between the mouse strains (Table 1). We also determined atherosclerotic burden in lipin-1flox/flox and lipin-1mEnzyKO mice. Oil Red O staining of en face aortas revealed less plaque development in the aortas of lipin-1mEnzyKO mice compared to the corresponding lipin-1flox/flox controls (Figure 1E). Additionally, MOVAT staining and subsequent analysis of the aortic root demonstrated reduced plaque area in lipin-1mEnzyKO mice compared to the corresponding lipin-1flox/flox mice (Figure 1F). Immunofluorescent analysis of the aortic root showed no significant difference in plaque composition (Supplemental Figure IIE&F), suggesting that monocyte recruitment is similar between lipin-1mEnzyKO and lipin-1flox/flox mice.
TABLE 1.
Analysis of Splenocyte Population in Mice*
| Cell Type† | Marker‡ | Mouse Strain | |||
|---|---|---|---|---|---|
|
| |||||
| Lipin-1flox/flox | Lipin-1mEnzyKO | ||||
|
| |||||
| Cell % | Cell # | Cell % | Cell # | ||
|
| |||||
| Macrophages | CD11b+ | 10.2 (2.4)§ | 12.5 (3.5)|| | 8.8 (1.8) | 11.1 (2.3) |
| Dendritic Cells | CD11c+ | 0.2 (0.04) | 0.2 (0.04) | 0.2 (0.05) | 0.2 (0.07) |
| T Helper Cells | CD3+CD4+ | 8.8 (3.0) | 10.5 (2.3) | 10.0 (2.4) | 12.7 (3.4) |
| Cytotoxic T Cells | CD3+CD8+ | 2.9 (1.8) | 3.3 (1.9) | 3.8 (2.0) | 5.0 (2.6) |
| NK cells | CD161 (NK1.1) | 0.8 (0.2) | 1.0 (0.2) | 0.8 (0.2) | 1.0 (0.1) |
| B Cells | CD19+ | 51.1 (2.4) | 62.5 (10.5) | 51.9 (5.3) | 66.8 (13.4) |
Mice were injected with 3×1010 AAV8-PCSK9 vector genomes then placed on high-fat diet for 8 weeks. Splenocytes were isolated and immune cells were quantified by flow cytometric analysis.
Characterization of cell type
Cell differentiation (CD) marker used to define cellular population
Cellular percentage (marker/total cells) +/− (SD) n=7
Cell number (splenocyte count x cell percentage) +/− (SD) n=7
Our data suggests lipin-1 enzymatic activity is responsible for the decreased severity of atherosclerosis seen in the lipin-1mEnzyKO mice. However, as truncated proteins are known to undergo enhanced degradation in vivo, it is possible that the loss of lipin-1 transcriptional coregulatory activity contributes to changes in atherosclerosis severity in our mouse model. However, this is unlikely, as this lipin-1flox/flox mouse has been used in other studies to investigate lipin-1 transcriptional coregulatory activity in hepatocytes.25 Furthermore, lipin-1 has been demonstrated to enhance the activity of transcription factors such as PPARγ, PPARα, and PGC-1α, which are known to induce an atheroprotective response.26–30 The known interactions of lipin-1 transcriptional coregulatory activity suggest the degradation of the truncated lipin-1 protein in our mice would enhance atherosclerosis severity, which is in contrast to our data. We also evaluated PPARγ-regulated gene expression by qRT-PCR to determine if lipin-1 transcriptional co-regulator activity was altered in our lipin-1mEnzyKO BMDs and observed no differences between lipin-1mEnzyKO and lipin-1flox/flox BMDMs after oxLDL stimulation (Supplemental Figure IB). These data suggest lipin-1 transcriptional coregulatory activity is either not active or is unaltered in lipin-1mEnzyKO BMDMs in response to oxLDL.
These data demonstrate that lipin-1mEnzyKO mice have less severe atherosclerosis, even when plasma cholesterol levels are similar to control mice, suggesting that myeloid-associated lipin-1 enzymatic activity positively contributes to the development of atherosclerosis in vivo.
ModLDL stimulation of BMDMs results in activation of a PKCα/βII-ERK1/2-cJun signaling cascade
Having demonstrated that myeloid-associated lipin-1 enzymatic activity is pro-atherogenic in vivo (Figure 1), we sought to determine the molecular mechanism by which macrophage-associated lipin-1 enzymatic activity may contribute to atherosclerosis. Lipin-1 mediates oxLDL- and LPS-induced DAG production and LPS-induced MAPK and AP-1 activity in macrophages.9, 10 ModLDL stimulation of macrophages induces the activation of DAG responsive proteins (PKCα/βII and PKCδ), MAPKs (JNK1/2, ERK1/2 and p38), and AP-1.31 Furthermore, the engulfment, breakdown, and storage of cholesterol and lipids from modLDLs is an ongoing process in the macrophage as long as modLDLs are present. These observations led us to hypothesize that lipin-1 enzymatic activity is responsible for persistent activation of a PKC-MAPK-AP-1 signal transduction pathway in macrophages in response to modLDL-stimulation. We first set out to determine if there is persistent activation of a PKC-MAPK-AP-1 signal transduction pathway in oxLDL-stimulated macrophages.
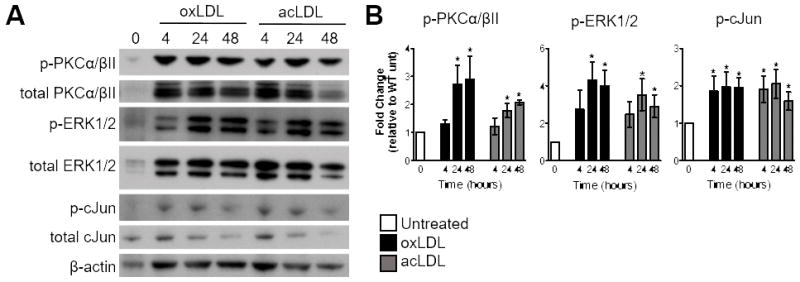
The phosphorylation status of PKCs, MAPKs, and the AP-1 subunits were monitored via Western blot analysis as a correlate of protein activity at 4, 24, and 48 hours after oxLDL-stimulation. Phosphorylation of PKCα/βII, PKCδ, PKCθ, and PKCμ were evaluated in wildtype (WT) BMDMs stimulated with oxLDL. We failed to detect total or phosphorylated PKCθ and PKCμ in WT BMDMs. Although phosphorylation of PKCδ was detected, the level of phosphorylation remained unchanged over the 48 hour period (Figure 2A). PKCα/βII phosphorylation was elevated at 4, 24, and 48 hours after oxLDL stimulation (Figure 2A&B). Analysis of MAPK phosphorylation demonstrated increased phosphorylation of ERK1/2 at 4, 24, and 48 hours after oxLDL stimulation (Figure 2C&D). Sustained phosphorylation of JNK1/2 and p38 was not observed at these time points (Figure 2C). Finally, phosphorylation of the AP-1 subunits, cJun and cFos, was evaluated. The phosphorylation of both cJun and cFos was elevated after 4 hours of oxLDL-stimulation (Figure 2E). However, only cJun remained phosphorylated at 24 and 48 hours after treatment, whereas cFos returned to basal levels (Figure 2E&F). The phosphorylation of cJun initiates its translocation to the nucleus where it can either bind to cFos or homodimerize to form AP-1.32, 33 To determine if cJun was localizing to the nucleus of oxLDL-stimulated BMDMs, we performed immunocytochemistry and observed nuclear cJun in WT BMDMs at 4, 24, and 48 hours after oxLDL stimulation (Figure 2G&H). To determine if these three proteins were linked in a signaling cascade, we used the small molecule inhibitors Gö6376 (PKCα/βII) and U0126 (inhibits MEK1/2 directly upstream of ERK1/2) to determine if PKCα/βII and ERK1/2 were upstream of cJun. Due to the fact that PKCs and MAPKs directly contribute to oxLDL internalization by macrophages, inhibitors were added 24 hours after oxLDL treatment as to not confound the data interpretation.34 Both small molecules inhibited the phosphorylation of cJun (Supplemental Figure IIIA&B), suggesting that we identified a linked PKCα/βII-ERK1/2-cJun signaling pathway. To ensure the signaling responses observed in oxLDL-stimulated macrophages were not due to LPS contamination, WT macrophages were stimulated with 1.2 pg/mL LPS (maximum calculated endotoxin present in the modLDL preparations), 60 pg/mL (50 times the maximum endotoxin present in the modLDL preparations), or 10 ng/mL of LPS (amount of LPS used in Figure 5). Persistent phosphorylation of the PKCα/βII-ERK1/2-cJun pathway was not observed in any of the LPS treatments (Supplemental Figure IV).
Figure 2.
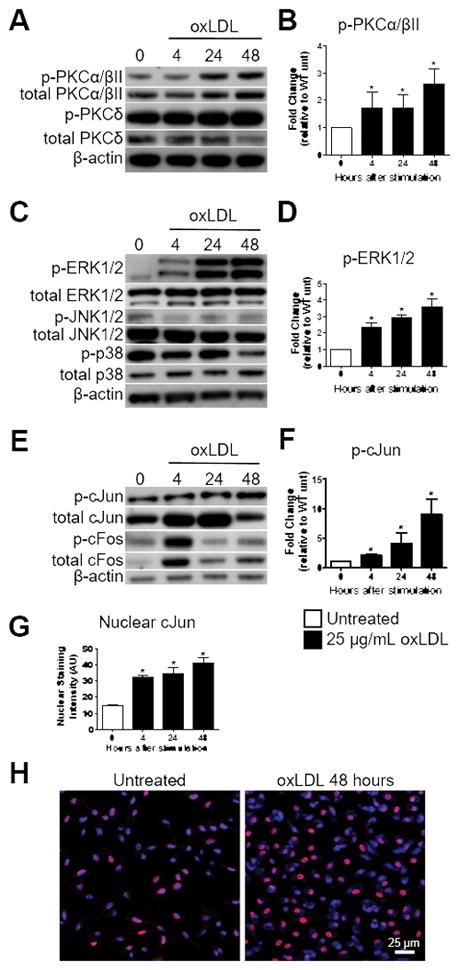
Figure 5.
To ensure that persistent activation of the PKCα/βII-ERK1/2-cJun pathway was not due to a single type of LDL modification, WT macrophages were stimulated with 25 μg/mL acLDL or 25 μg/mL oxLDL. Stimulation with either oxLDL or acLDL resulted in sustained phosphorylation of PKCα/βII, ERK1/2, and cJun at 24 and 48 hours (Figure 3A&B). Together these results suggest that modLDL stimulation of macrophages induces persistent activation of a PKCα/βII-ERK1/2-cJun signaling axis.
Figure 3.
OxLDL stimulation of BMDMs activates the lipin-1-dependent signaling cascade PKCα/βII-ERK1/2-cJun
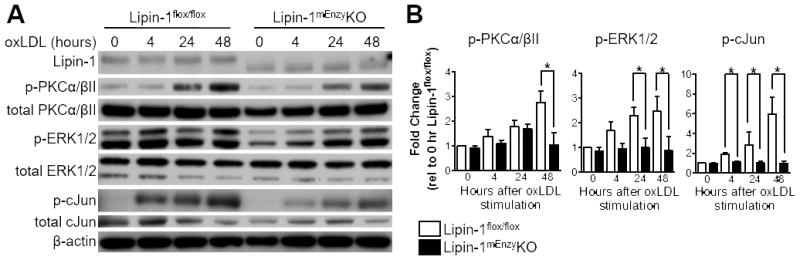
To determine if the PKCα/βII-ERK1/2-cJun pathway is lipin-1 dependent, the phosphorylation status of PKCα/βII, ERK1/2, and cJun was evaluated in BMDMs generated from lipin-1mEnzyKO and lipin-1flox/flox mice. BMDMs were stimulated with 25 μg/mL oxLDL and protein was collected at 4, 24, and 48 hours for Western blot analysis. Lipin-1mEnzyKO BMDMs had reduced phosphorylation of PKCα/βII, ERK1/2, and cJun at the 24 and 48 hour time points compared to the lipin-1flox/flox BMDMs (Figure 4A&B), suggesting the persistent phosphorylation events of these proteins are dependent upon lipin-1 enzymatic activity. These data demonstrate PKCα/βII-ERK1/2-cJun is a lipin-1-dependent signaling cascade activated in response to oxLDL-stimulation.
Figure 4.
Lipin-1 contributes to oxLDL-elicited TNF-α production in BMDMs
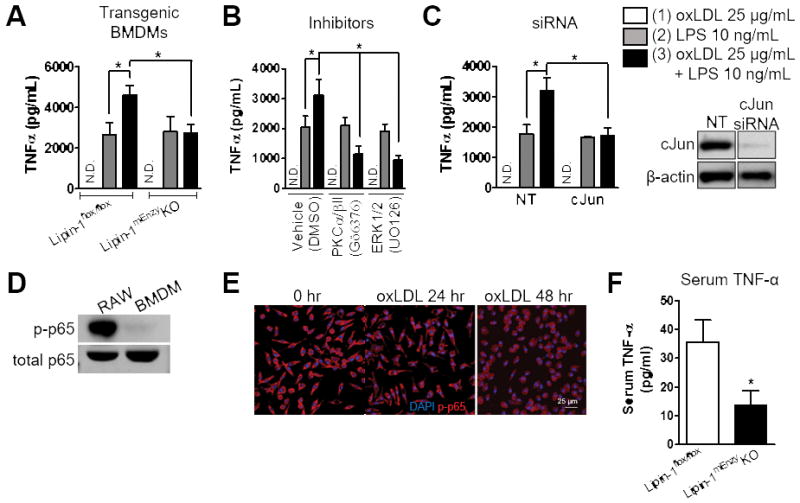
We previously demonstrated oxLDL elicits TNF-α production by RAW264.7 macrophages in a lipin-1-dependent manner.10 To determine if lipin-1 enzymatic activity regulates TNF-α production in BMDMs in response to oxLDL, lipin-1mEnzyKO and lipin-1flox/flox BMDMs were stimulated with 25 μg/mL of oxLDL and TNF-α production in supernatants was analyzed by ELISA 48 hours after stimulation. This late time point (48 hours) was chosen as it corresponds to the time of maximal TNF-α production by oxLDL-stimulated RAW264.7 macrophages.10 However, stimulation of BMDMs with oxLDL alone failed to elicit TNF-α from either the lipin-1mEnzyKO or lipin-1flox/flox BMDMs (Figure 5A). Based upon previous findings in RAW264.7 macrophages, we reasoned the lack of TNF-α production by BMDMs might be a result of very low basal NF-κB activity.10 In contrast to untreated RAW264.7 macrophages, there was no NF-κB (p65) phosphorylation in untreated WT BMDMs as detected by Western blot analysis (Figure 5D). Additionally, immunocytochemistry confirmed that NF-κB (p65) was not present in the nucleus of untreated or oxLDL-stimulated WT BMDMs (Figure 5E).
The presence of pre-existing NF-κB activity in the RAW264.7 macrophage cell line is noteworthy because oxLDL alone does not induce NF-κB activity in BMDMs, which is required for TNF-α production.35 Additionally, AP-1 transcriptional activity is not sufficient for TNF-α production, but can enhance the activity of other transcription factors in macrophages, including NF-κB, to augment TNF-α production.36 Thus, we hypothesized that lipin-1-mediated activation of cJun (AP-1) triggered by oxLDL treatment may prime macrophages to be more responsive to a subsequent NF-κB activating stimulus, such as LPS. To examine the effect of an NF-κB stimulus on oxLDL-stimulated macrophages, we treated WT (lipin-1flox/flox) BMDMs with either (1) 25 μg/mL oxLDL, (2) 10 ng/mL LPS, or (3) 25 μg/mL oxLDL for 24 hours, then 10 ng/mL LPS for an additional 24 hours. Supernatants were collected 48 hours after oxLDL addition and evaluated by ELISA for TNF-α production. LPS stimulation resulted in TNF-α production in WT BMDMs and stimulation of WT BMDMs with oxLDL and LPS resulted in augmented TNF-α production (Figure 5A).
We next wanted to determine if the identified lipin-1-dependent PKCα/βII-ERK1/2-cJun signaling axis was responsible for augmented TNF-α production in BMDMs stimulated with both oxLDL and LPS. Lipin-1mEnzyKO and lipin-1flox/flox BMDMs were treated with either (1) 25 μg/mL oxLDL, (2) 10 ng/mL LPS or (3) 25 μg/mL oxLDL for 24 hours followed by addition of 10 ng/mL LPS for an additional 24 hours. LPS alone induced TNF-α production in both the lipin-1mEnzyKO and lipin-1flox/flox BMDMs (Figure 5A). However, pre-loading lipin-1flox/flox BMDMs with oxLDL augmented LPS-elicited TNF-α production, which was not observed in the lipin-1mEnzyKO BMDMs (Figure 5A). Next, WT BMDMs were stimulated with either (1) oxLDL, (2) LPS, or (3) oxLDL and LPS with inhibitors of PKCα/βII (Gö6376) and ERK1/2 (U0126). Inhibitors were added at 24 hours along with LPS stimulation. Inhibition of PKCα/βII and ERK1/2 ablated oxLDL-augmented LPS-induced TNF-α production (Figure 5B). Finally, to determine if the augmented TNF-α production by lipin-1flox/flox BMDMs stimulated with (3) oxLDL and LPS was due to elevated cJun activity, cJun was depleted using siRNA and TNF-α production was examined following treatment. WT BMDMs were transfected with either non-target siRNA or cJun-targeted siRNA (Figure 5C) and left untreated or stimulated with 25 μg/mL oxLDL for 24 hours prior to 10 ng/mL LPS treatment for an additional 24 hours. Depletion of cJun prevented the augmentation of LPS-elicited TNF-α production, similar to what was seen in the lipin-1mEnzyKO BMDMs in Figure 5A (Figure 5C).
Inhibition of the macrophages ability to produce TNF-α in vivo results in reduced serum TNF-α levels in atherosclerotic mice.37 Therefore, we wanted to determine if there was a reduction in serum TNF-α in the lipin-1mEnzyKO mice compared to lipin-1flox/flox mice. Serum from lipin-1mEnzyKO and lipin-1flox/flox mice was collected after 8 weeks on high fat diet and analyzed by ELISA for TNF-α production. The lipin-1mEnzyKO mice had a significant reduction in serum TNF-α compared to lipin-1flox/flox mice (Figure 5F). Although this data is not definitive, this begins to provide evidence that lipin-1 enzymatic activity is contributing to the pro-inflammatory responses in vivo that promote atherosclerosis. These data suggest that oxLDL induces activation of cJun (AP-1) via lipin-1 enzymatic activity, which effectively primes the macrophage for enhanced responses to a pro-inflammatory stimulus above what would be exhibited by the pro-inflammatory stimulus alone.
DISCUSSION
Using the lipin-1mEnzyKO mouse model with the viral model of atherosclerosis, we demonstrated the loss of myeloid-associated lipin-1 enzymatic activity results in reduced atherosclerosis in mice fed high fat diet for 8 and 12 weeks. Furthermore, we demonstrated that macrophage-associated lipin-1 enzymatic activity contributes to foam cell formation and regulates a DAG-dependent signaling cascade that results in persistent AP-1 activation in response to modLDL. Persistent activation of AP-1 primes the macrophage to be hyper-responsive to LPS-induced TNF-α production. Although the glycerolipid synthetic pathway is likely not the only lipid synthesis pathway contributing to foam cell formation, it is a critical pathway that influences modLDL-elicited pro-inflammatory responses. Taken together, our data highlights the dynamic interaction between modLDL-elicited lipid synthetic events and their contribution to macrophage pro-inflammatory mediator production that promotes atherosclerosis.
The combination of our in vitro and in vivo data suggest that the reduced plaque development observed in the lipin-1mEnzyKO mice is due primarily to monocytes and macrophages. However, LysM-Cre was used to knockout lipin-1 enzymatic activity in these mice and LysM expression is not restricted to monocytes and macrophages. Analysis of LysM-Cre-mediated gene deletion demonstrates gene excision in dendritic cells and granulocytes, as well as monocytes and macrophages.38, 39 The contribution of dendritic cells to atherosclerosis is unclear because tools to specifically deplete this population long term are currently lacking. However, the few studies using CD11cDTR mice, in which dendritic cells are transiently depleted, demonstrate no alteration in plaque size or composition.40 The contribution of lipin-1 to dendritic cell function is completely unknown, and will need to be looked at in the future. Neutrophils clearly contribute to atherosclerosis progression as they can be identified in both early and advanced murine and human plaques, and antibody depletion of neutrophils has been shown to contribute to plaque burden in mice.41–43 However, lipin-1 is not readily detected in neutrophils, suggesting lipin-1 has little contribution to neutrophil function.9 Thus, we propose that the loss of lipin-1 enzymatic activity in monocytes and macrophages is most likely responsible for the reduction in atherosclerosis observed in the lipin-1mEnzyKO mice.
Our studies identified a novel mechanism through which lipin-1, a regulatory enzyme in the glycerolipid synthesis pathway, initiates a DAG-mediated PKCα/βII-ERK1/2-cJun signaling cascade that contributes to macrophage pro-inflammatory responses to modLDL. Glycerolipid synthesis is a universal pathway required for the storage of incoming lipids. We demonstrate that stimulation of macrophages with both oxLDL and acLDL results in sustained phosphorylation of the PKCα/βII-ERK1/2-cJun pathway up to 48 hours. The initiation of glycerolipid synthesis by any modLDL would then be capable of activating the DAG-dependent PKCα/βII-ERK1/2-cJun signaling pathway leading to persistent macrophage pro-inflammatory responses. Acute macrophage pro-inflammatory responses have been investigated as a result of modLDLs being recognized by toll-like receptors or the inflammasome.3–6 However, our data highlights another molecular mechanism by which modLDLs can elicit signaling that contribute to macrophage pro-inflammatory responses that may be independent of receptor-mediated recognition of modLDLs. Our data investigating PKCα/βII supports this proposed alternative mechanism, as PKCβ is required for modLDL uptake in vitro.44 However, the lipin-1mEnzyKO macrophages do not have an oxLDL uptake deficiency even with a loss of PKCβ phosphorylation, suggesting that late activity of PKCα/βII (24 and 48 hours after oxLDL stimulation) is dependent on lipin-1, while early activation (up to 4 hours) is not. This supports the idea that there are multiple signals that contribute to modLDL-induced pro-inflammatory responses in a temporal manner. Our studies identified ERK1/2 and cJun as downstream proteins of PKCα/βII. Although JNK1/2 is the classical kinase associated with phosphorylation of cJun, ERK1/2 can also phosphorylate cJun specifically at Ser63 and Ser72.45, 46 The ability of ERK1/2 to phosphorylate cJun may also be temporally regulated. Studies using total knockout of JNK1/2 in mice show prevention of the early steps of atherosclerosis and thus reduction in plaque burden.47 However, inhibition of JNK1/2 during late stage atherosclerosis in humans showed minimal effects on levels of pro-inflammatory mediators in serum.48 We hypothesize that targeting JNK1/2 in the human study was ineffective because ERK1/2 may be responsible for chronic activation of cJun in macrophages during later stages of atherosclerosis. Overall, our data supports the idea that there is a temporal difference in signaling responses that is determined by macrophage interactions with modLDLs. Early signaling seems to be dependent on receptor interactions and later signaling events are dependent on prolonged lipid synthetic events.
The persistent activation of the PKCα/βII-ERK1/2-cJun signaling axis primes macrophages to be hyper-responsive to secondary pro-inflammatory stimuli, such as LPS. Many of the comorbidities that contribute to atherosclerosis progression are associated with increased endotoxin levels in the plasma, called endotoxemia. As expected, chronic bacterial infections, such as periodontitis and chlamydial infections, increase plasma endotoxin levels and non-infectious comorbidities such as obesity, consumption of a high-fat diet (both in mice and humans), type 2 diabetes mellitus, and alcoholism also result in increased serum endotoxin levels.49–55 Enhanced macrophage pro-inflammatory responses induced via cooperation of AP-1 and NF-κB have previously been demonstrated to enhance vascular inflammation when compared to NF-κB (via LPS stimulation) alone.36 Our data provide a molecular mechanism by which hypercholesterolemia and endotoxemia promote pro-inflammatory responses that mediate atherosclerosis. We have provided evidence that oxLDL elicits persistent DAG generation that leads to AP-1 activation via a PKCα/βII-ERK1/2-cJun signaling axis in macrophages, and interruption of this pathway reduces enhanced pro-inflammatory responses (TNF-α) (Figure 5). In vivo, the loss of lipin-1 from myeloid-derived cells reduced circulating TNF-α levels in response to high-fat diet. Serum TNF-α levels often correspond to degree of plaque inflammation and growth.56 This supports the hypothesis that macrophage-associated lipin-1 contributes to modLDL-elicited pro-inflammatory responses. Combining our in vitro observations of the contribution of lipin-1 to oxLDL-elicited pro-inflammatory responses with our in vivo data demonstrating that myeloid-associated lipin-1 contributes to atherosclerotic progression, suggests that lipid synthetic events required to store incoming lipids from modLDLs also mediate pro-inflammatory responses in macrophages that contribute to atheroma progression. We suggest hypercholesterolemia in vivo results in prolonged enhancement of glycerolipid synthesis within macrophages, priming the macrophage with active AP-1, resulting in macrophage foam cells that are hyper-responsive to additional pro-inflammatory stimuli (such as LPS).
A better understanding of the fundamental mechanisms of intrinsic plaque inflammation may provide additional therapeutic targets to reduce atherosclerosis better than statin therapy alone. Interestingly, the statins most effective at reducing cardiovascular disease risk are those that also exhibit anti-inflammatory properties.57 We currently lack mechanisms to target plaque intrinsic inflammation without causing global immune-suppression. Macrophage foam cell formation is largely unique to atherosclerosis, and our results suggest that modLDL-induced foam cell formation and modLDL-induced macrophage pro-inflammatory responses are linked via enhanced lipid synthesis. Thus, understanding of modLDL-elicited lipid synthetic events that contribute to macrophage pro-inflammatory responses may lead to an ideal therapeutic target to reduce plaque intrinsic inflammation without causing global immune-suppression to reduce cardiovascular disease.
Supplementary Material
HIGHLIGHTS.
Macrophage-associated lipin-1 is pro-atherogenic.
ModLDLs elicit activation of a lipin-1-dependent PKCα/βII-ERK1/2-cJun signaling cascade in macrophages.
Lipin-1 primes the macrophage to be hyper-responsive to additional pro-inflammatory stimuli.
Acknowledgments
SOURCES OF FUNDING
This work was supported by the National Heart, Lung, and Blood Institute 1 RO1 HL131844 and the National Institute of General Medicine Science, which funds the Louisiana Clinical and Translational Science Center 1 U54 GM104940 (MDW), American Heart Association Predoctoral Fellowships 17PRE33661114 (AEV) and 17PRE33440111 (ACF), Malcolm Feist Predoctoral Fellowships (AEV and ACF), NIH RO1 HL098435 (AWO), HL133497 (AWO), and HL119225 (BNF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- AAV8
adeno-associated viral vector 8
- ABCA1
ATP binding cassette subfamily A member 1
- ABCG1
ATP binding cassette subfamily G member 1
- AcLDL
acetylated low-density lipoprotein
- AP-1
activator protein-1
- BMDM
bone marrow-derived macrophage
- CCL2
C-C motif chemokine ligand 2
- cFos
fos proto-oncogene, AP-1 transcription factor subunit
- cJun
jun proto-oncogene, AP-1 transcription factor subunit
- DAG
diacylglycerol
- DGAT
diglyceride acyltransferase
- ER
endoplasmic reticulum
- ERK
extracellular receptor kinase
- G3P
glycerol-3-phosphate
- IL-1
interleukin 1
- IL-6
interleukin 6
- IL-10
interleukin 10
- LXRα
liver x receptor alpha
- JNK
c-Jun NH2-terminal kinase
- HDL
high-density lipoprotein
- LC-MS/MS
liquid chromatography tandem mass spectrometry
- LDL
low-density lipoprotein
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- oxLDL
oxidized low-density lipoprotein
- PA
phosphatidic acid
- PAP
phosphatidic acid phosphohydrolase
- PC
phosphatidylcholine
- PGC-1α
PPAR-gamma coactivator-1 alpha
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PGE2
prostaglandin E2
- PKC
protein kinase C
- PPARα
peroxisome proliferator-activated receptor alpha
- PPAR
peroxisome proliferator-activated receptor gamma
- p38
protein 38
- TAG
triacylglycerol
- TC
total cholesterol
- TG
triglyceride
- TLR4
toll-like receptor 4
- TNF-α
tumor necrosis factor alpha
- WT
wildtype
Footnotes
DISCLOSURES
None.
References
- 1.Mori M, Itabe H, Higashi Y, Fujimoto Y, Shiomi M, Yoshizumi M, Ouchi Y, Takano T. Foam cell formation containing lipid droplets enriched with free cholesterol by hyperlipidemic serum. J Lipid Res. 2001;42:1771–1781. [PubMed] [Google Scholar]
- 2.Kleemann R, Zadelaar S, Kooistra T. Cytokines and atherosclerosis: A comprehensive review of studies in mice. Cardiovasc Res. 2008;79:360–376. doi: 10.1093/cvr/cvn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein e. Proc Natl Acad Sci U S A. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. Cd36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtiss LK, Black AS, Bonnet DJ, Tobias PS. Atherosclerosis induced by endogenous and exogenous toll-like receptor (tlr)1 or tlr6 agonists. J Lipid Res. 2012;53:2126–2132. doi: 10.1194/jlr.M028431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J. Atherosclerosis in apoe-deficient mice progresses independently of the nlrp3 inflammasome. Cell Death Dis. 2011;2:e137. doi: 10.1038/cddis.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meana C, Pena L, Lorden G, Esquinas E, Guijas C, Valdearcos M, Balsinde J, Balboa MA. Lipin-1 integrates lipid synthesis with proinflammatory responses during tlr activation in macrophages. J Immunol. 2014;193:4614–4622. doi: 10.4049/jimmunol.1400238. [DOI] [PubMed] [Google Scholar]
- 10.Navratil AR, Vozenilek AE, Cardelli JA, Green JM, Thomas MJ, Sorci-Thomas MG, Orr AW, Woolard MD. Lipin-1 contributes to modified low-density lipoprotein-elicited macrophage pro-inflammatory responses. Atherosclerosis. 2015;242:424–432. doi: 10.1016/j.atherosclerosis.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Namatame I, Tomoda H, Arai H, Inoue K, Omura S. Complete inhibition of mouse macrophage-derived foam cell formation by triacsin c. J Biochem. 1999;125:319–327. doi: 10.1093/oxfordjournals.jbchem.a022289. [DOI] [PubMed] [Google Scholar]
- 12.McGookey DJ, Anderson RG. Morphological characterization of the cholesteryl ester cycle in cultured mouse macrophage foam cells. J Cell Biol. 1983;97:1156–1168. doi: 10.1083/jcb.97.4.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bozza PT, Melo RC, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: Contribution to allergy and host defense. Pharmacol Ther. 2007;113:30–49. doi: 10.1016/j.pharmthera.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Reue K, Brindley DN. Thematic review series: Glycerolipids. Multiple roles for lipins/phosphatidate phosphatase enzymes in lipid metabolism. J Lipid Res. 2008;49:2493–2503. doi: 10.1194/jlr.R800019-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibellini F, Smith TK. The kennedy pathway--de novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life. 2010;62:414–428. doi: 10.1002/iub.337. [DOI] [PubMed] [Google Scholar]
- 16.Harris CA, Haas JT, Streeper RS, Stone SJ, Kumari M, Yang K, Han X, Brownell N, Gross RW, Zechner R, Farese RV., Jr Dgat enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes. J Lipid Res. 2011;52:657–667. doi: 10.1194/jlr.M013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang YL, Morales-Rosado J, Ray J, Myers TG, Kho T, Lu M, Munford RS. Toll-like receptor agonists promote prolonged triglyceride storage in macrophages. J Biol Chem. 2014;289:3001–3012. doi: 10.1074/jbc.M113.524587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fagone P, Sriburi R, Ward-Chapman C, Frank M, Wang J, Gunter C, Brewer JW, Jackowski S. Phospholipid biosynthesis program underlying membrane expansion during b-lymphocyte differentiation. J Biol Chem. 2007;282:7591–7605. doi: 10.1074/jbc.M608175200. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz M, Jove M, Schluter A, Casasnovas C, Villarroya F, Guilera C, Ortega FJ, Naudi A, Pamplona R, Gimeno R, Fourcade S, Portero-Otin M, Pujol A. Altered glycolipid and glycerophospholipid signaling drive inflammatory cascades in adrenomyeloneuropathy. Hum Mol Genet. 2015;24:6861–6876. doi: 10.1093/hmg/ddv375. [DOI] [PubMed] [Google Scholar]
- 20.Reue K, Zhang P. The lipin protein family: Dual roles in lipid biosynthesis and gene expression. FEBS Lett. 2008;582:90–96. doi: 10.1016/j.febslet.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han GS, Carman GM. Characterization of the human lpin1-encoded phosphatidate phosphatase isoforms. J Biol Chem. 2010;285:14628–14638. doi: 10.1074/jbc.M110.117747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–1689. doi: 10.1161/CIRCRESAHA.114.302937. [DOI] [PubMed] [Google Scholar]
- 23.Mitra MS, Chen Z, Ren H, Harris TE, Chambers KT, Hall AM, Nadra K, Klein S, Chrast R, Su X, Morris AJ, Finck BN. Mice with an adipocyte-specific lipin 1 separation-of-function allele reveal unexpected roles for phosphatidic acid in metabolic regulation. Proc Natl Acad Sci U S A. 2013;110:642–647. doi: 10.1073/pnas.1213493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kent C. Eukaryotic phospholipid biosynthesis. Annu Rev Biochem. 1995;64:315–343. doi: 10.1146/annurev.bi.64.070195.001531. [DOI] [PubMed] [Google Scholar]
- 25.Schweitzer GG, Chen Z, Gan C, McCommis KS, Soufi N, Chrast R, Mitra MS, Yang K, Gross RW, Finck BN. Liver-specific loss of lipin-1-mediated phosphatidic acid phosphatase activity does not mitigate intrahepatic tg accumulation in mice. J Lipid Res. 2015;56:848–858. doi: 10.1194/jlr.M055962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarthy C, Lieggi NT, Barry D, Mooney D, de Gaetano M, James WG, McClelland S, Barry MC, Escoubet-Lozach L, Li AC, Glass CK, Fitzgerald DJ, Belton O. Macrophage ppar gamma co-activator-1 alpha participates in repressing foam cell formation and atherosclerosis in response to conjugated linoleic acid. EMBO Mol Med. 2013;5:1443–1457. doi: 10.1002/emmm.201302587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim HE, Bae E, Jeong DY, Kim MJ, Jin WJ, Park SW, Han GS, Carman GM, Koh E, Kim KS. Lipin1 regulates ppargamma transcriptional activity. Biochem J. 2013;453:49–60. doi: 10.1042/BJ20121598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic pgc-1alpha/pparalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 29.Ricote M, Huang J, Fajas L, Li A, Welch J, Najib J, Witztum JL, Auwerx J, Palinski W, Glass CK. Expression of the peroxisome proliferator-activated receptor gamma (ppargamma) in human atherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci U S A. 1998;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soskic SS, Dobutovic BD, Sudar EM, Obradovic MM, Nikolic DM, Zaric BL, Stojanovic SD, Stokic EJ, Mikhailidis DP, Isenovic ER. Peroxisome proliferator-activated receptors and atherosclerosis. Angiology. 2011;62:523–534. doi: 10.1177/0003319711401012. [DOI] [PubMed] [Google Scholar]
- 31.Lin CS, Lin FY, Ho LJ, Tsai CS, Cheng SM, Wu WL, Huang CY, Lian CH, Yang SP, Lai JH. Pkcdelta signalling regulates sr-a and cd36 expression and foam cell formation. Cardiovasc Res. 2012;95:346–355. doi: 10.1093/cvr/cvs189. [DOI] [PubMed] [Google Scholar]
- 32.Grondin B, Lefrancois M, Tremblay M, Saint-Denis M, Haman A, Waga K, Bedard A, Tenen DG, Hoang T. C-jun homodimers can function as a context-specific coactivator. Mol Cell Biol. 2007;27:2919–2933. doi: 10.1128/MCB.00936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Halazonetis TD, Georgopoulos K, Greenberg ME, Leder P. C-jun dimerizes with itself and with c-fos, forming complexes of different dna binding affinities. Cell. 1988;55:917–924. doi: 10.1016/0092-8674(88)90147-x. [DOI] [PubMed] [Google Scholar]
- 34.Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM, Jr, Hajjar DP, Nicholson AC. Induction of cd36 expression by oxidized ldl and il-4 by a common signaling pathway dependent on protein kinase c and ppar-gamma. J Lipid Res. 2000;41:688–696. [PubMed] [Google Scholar]
- 35.Ohlsson BG, Englund MC, Karlsson AL, Knutsen E, Erixon C, Skribeck H, Liu Y, Bondjers G, Wiklund O. Oxidized low density lipoprotein inhibits lipopolysaccharide-induced binding of nuclear factor-kappab to dna and the subsequent expression of tumor necrosis factor-alpha and interleukin-1beta in macrophages. J Clin Invest. 1996;98:78–89. doi: 10.1172/JCI118780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, Gonen A, Butler S, Witztum JL, Glass CK, Miller YI. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappa b and activator protein-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2010;107:56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu M, Zhou H, Zhao J, Xiao N, Roychowdhury S, Schmitt D, Hu B, Ransohoff RM, Harding CV, Hise AG, Hazen SL, DeFranco AL, Fox PL, Morton RE, Dicorleto PE, Febbraio M, Nagy LE, Smith JD, Wang J, Li X. Myd88-dependent interplay between myeloid and endothelial cells in the initiation and progression of obesity-associated inflammatory diseases. J Exp Med. 2014;211:887–907. doi: 10.1084/jem.20131314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using lysmcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 39.Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-cre deleting strains using rosa-eyfp reporter mice. J Immunol Methods. 2014;408:89–100. doi: 10.1016/j.jim.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rombouts M, Cools N, Grootaert MO, de Bakker F, Van Brussel I, Wouters A, De Meyer GR, De Winter BY, Schrijvers DM. Long-term depletion of conventional dendritic cells cannot be maintained in an atherosclerotic zbtb46-dtr mouse model. PLoS One. 2017;12:e0169608. doi: 10.1371/journal.pone.0169608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–1845. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
- 42.Rotzius P, Thams S, Soehnlein O, Kenne E, Tseng CN, Bjorkstrom NK, Malmberg KJ, Lindbom L, Eriksson EE. Distinct infiltration of neutrophils in lesion shoulders in apoe−/− mice. Am J Pathol. 2010;177:493–500. doi: 10.2353/ajpath.2010.090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ionita MG, van den Borne P, Catanzariti LM, Moll FL, de Vries JP, Pasterkamp G, Vink A, de Kleijn DP. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol. 2010;30:1842–1848. doi: 10.1161/ATVBAHA.110.209296. [DOI] [PubMed] [Google Scholar]
- 44.Harja E, Chang JS, Lu Y, Leitges M, Zou YS, Schmidt AM, Yan SF. Mice deficient in pkcbeta and apolipoprotein e display decreased atherosclerosis. Faseb j. 2009;23:1081–1091. doi: 10.1096/fj.08-120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pulverer BJ, Kyriakis JM, Avruch J, Nikolakaki E, Woodgett JR. Phosphorylation of c-jun mediated by map kinases. Nature. 1991;353:670–674. doi: 10.1038/353670a0. [DOI] [PubMed] [Google Scholar]
- 46.Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-jun. Embo j. 2003;22:3876–3886. doi: 10.1093/emboj/cdg388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osto E, Matter CM, Kouroedov A, Malinski T, Bachschmid M, Camici GG, Kilic U, Stallmach T, Boren J, Iliceto S, Luscher TF, Cosentino F. C-jun n-terminal kinase 2 deficiency protects against hypercholesterolemia-induced endothelial dysfunction and oxidative stress. Circulation. 2008;118:2073–2080. doi: 10.1161/CIRCULATIONAHA.108.765032. [DOI] [PubMed] [Google Scholar]
- 48.Meijer CA, Le Haen PA, van Dijk RA, Hira M, Hamming JF, van Bockel JH, Lindeman JH. Activator protein-1 (ap-1) signalling in human atherosclerosis: Results of a systematic evaluation and intervention study. Clin Sci (Lond) 2012;122:421–428. doi: 10.1042/CS20110234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lehr HA, Sagban TA, Ihling C, Zahringer U, Hungerer KD, Blumrich M, Reifenberg K, Bhakdi S. Immunopathogenesis of atherosclerosis: Endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemic diet. Circulation. 2001;104:914–920. doi: 10.1161/hc3401.093153. [DOI] [PubMed] [Google Scholar]
- 50.Kalayoglu MV, Libby P, Byrne GI. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. Jama. 2002;288:2724–2731. doi: 10.1001/jama.288.21.2724. [DOI] [PubMed] [Google Scholar]
- 51.Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. 2007;27:1433–1439. doi: 10.1161/ATVBAHA.106.138743. [DOI] [PubMed] [Google Scholar]
- 52.Yoshimatsu M, Terasaki Y, Sakashita N, Kiyota E, Sato H, van der Laan LJ, Takeya M. Induction of macrophage scavenger receptor marco in nonalcoholic steatohepatitis indicates possible involvement of endotoxin in its pathogenic process. Int J Exp Pathol. 2004;85:335–343. doi: 10.1111/j.0959-9673.2004.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pendyala S, Walker JM, Holt PR. A high-fat diet is associated with endotoxemia that originates from the gut. Gastroenterology. 2012;142:1100–1101. e1102. doi: 10.1053/j.gastro.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jayashree B, Bibin YS, Prabhu D, Shanthirani CS, Gokulakrishnan K, Lakshmi BS, Mohan V, Balasubramanyam M. Increased circulatory levels of lipopolysaccharide (lps) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol Cell Biochem. 2014;388:203–210. doi: 10.1007/s11010-013-1911-4. [DOI] [PubMed] [Google Scholar]
- 55.Rao R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology. 2009;50:638–644. doi: 10.1002/hep.23009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Puz P, Lasek-Bal A, Ziaja D, Kazibutowska Z, Ziaja K. Inflammatory markers in patients with internal carotid artery stenosis. Arch Med Sci. 2013;9:254–260. doi: 10.5114/aoms.2013.34533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antonopoulos AS, Margaritis M, Lee R, Channon K, Antoniades C. Statins as anti-inflammatory agents in atherogenesis: Molecular mechanisms and lessons from the recent clinical trials. Curr Pharm Des. 2012;18:1519–1530. doi: 10.2174/138161212799504803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.