

Abstract
The histamine H1-receptor (H1R) is an important mediator of allergy and inflammation. H1R antagonists have particular clinical utility in allergic rhinitis and urticaria. Here we have developed six novel fluorescent probes for this receptor that are very effective for high resolution confocal imaging, alongside bioluminescence resonance energy transfer approaches to monitor H1R ligand binding kinetics in living cells. The latter technology exploits the opportunities provided by the recently described bright bioluminescent protein NanoLuc when it is fused to the N-terminus of a receptor. Two different pharmacophores (mepyramine or the fragment VUF13816) were used to generate fluorescent H1R antagonists conjugated via peptide linkers to the fluorophore BODIPY630/650. Kinetic properties of the probes showed wide variation, with the VUF13816 analogues having much longer H1R residence times relative to their mepyramine-based counterparts. The kinetics of these fluorescent ligands could also be monitored in membrane preparations providing new opportunities for future drug discovery applications.
Introduction
G protein-coupled receptors (GPCR) are one of the major targets for currently approved drugs, with circa 30% acting at the GPCR superfamily1. Furthermore, there remains huge potential for innovation within this protein family since only 30% of the non-olfactory GPCRs have been successfully targeted2. The development of new therapeutics has been hampered in recent years, however, by the failure of many drugs in late-stage clinical trials as a consequence of a lack of appropriate clinical efficacy3. The increasing number of crystal structures available for GPCRs has facilitated the application of rational design efforts to the drug discovery process4,5 but since these receptors are highly dynamic proteins that can adopt a wide range of conformations, there is a need to study these receptors in their natural cellular environment6.
An important, but often overlooked, property of a drug candidate is the rate at which it binds to, and dissociates from, its target receptor7. Drugs with similar affinity can display markedly different binding kinetics, and optimising a drug’s binding kinetics to clinical need is thought to be one way to reduce drug discovery attrition rates8,9. The use of isolated membranes from homogenized cells in combination with radiolabelled ligands has been the most frequently used method to measure ligand binding kinetics to a GPCR. However, intracellular signalling proteins can have marked allosteric influences on the binding of ligands to GPCRs10–12 and one consequence of allosteric interactions is that they change ligand binding kinetics13. As a result of this, there may be differences in the binding kinetics of compounds measured in whole cells compared to those measurements made in isolated membranes. One way to study ligand-binding kinetics of receptors in their natural cellular environment is through the use of fluorescently labelled agonists and antagonists14,15.
Fluorescent ligands for GPCRs have been used to study various aspects of receptor pharmacology and function including ligand binding16,17, endogenous receptor localisation18–20, receptor organisation within the cell membrane21,22 and ligand binding kinetics23–25. However, fluorescent ligands often require optimisation for use in a specific application. For example, in the case of the histamine H1 receptor (H1R), Rose et al. described a fluorescent ligand that was successfully used for fluorescence correlation spectroscopy, but showed high levels of non-specific membrane binding and cytoplasmic uptake that made confocal imaging difficult26. Fluorescent ligands for GPCRs are normally composed of an agonist or antagonist for the receptor of interest chemically linked to a fluorescent molecule such as one of the boron-dipyrromethene (BODIPY) or cyanine dyes27. For fluorescent small molecule ligands, such as those for Class A GPCRs, addition of the linker and fluorophore results in a significant increase in molecular weight and complexity and these molecules should therefore be treated as new pharmacological entities. For instance, we have shown that the linker region can influence the fluorescent ligand properties, with peptide-based linkers improving both affinity and imaging capabilities of Xanthine Amine Congener based ligands for the adenosine A3 receptor28.
The H1R is a Class A GPCR which couples predominantly to Gq/11 proteins, leading to phospholipase C activation and release of intracellular calcium29. It is expressed in a wide variety of cell types including, smooth muscle, endothelium, immune cells and neurons30, and its activation by the endogenous ligand histamine is a key mediator of allergy and inflammation. A wide range of clinically approved antagonists for the H1R have been successfully used for many years in the treatment of allergy related conditions such as allergic rhinitis and urticaria31. The crystal structure of an antagonist bound H1R has been recently solved32 and therefore to gain further understanding of the dynamic regulation of ligand binding to this receptor in live cells, improvements to the currently available fluorescent ligands26 needed to be made. By using the strategy previously described for the adenosine A3 receptor28, where peptide linkers were introduced between the fluorophore and pharmacophore, we aimed to generate broad utility fluorescent ligands for the H1R that are suitable for use in a range of different fluorescent techniques including confocal microscopy, but particularly for assessing receptor binding kinetics using bioluminescence resonance energy transfer (BRET).
Results
Pharmacological Characterization of Fluorescent Ligands
Six fluorescent antagonists for the H1R were synthesized as part of this study (Figure S1); three based on the commonly used H1R antagonist mepyramine and three based on the recently described fragment-like antagonist VUF1381633,34 (Fig. 1a). The di- or tripeptide linkers were designed based on the predicted docking pose of VUF1381634 in the H1R crystal structure32 (Fig. 1b). The binding mode of VUF13816 has been experimentally validated by combined structure-activity relationship and site-directed mutagenesis studies35 and suggests that the basic amine nitrogen atom that interacts with the anionic D1073.32 residue in TM helix III provides a good point to attach a peptide linker that: i) has the appropriate length to cross the extracellular vestibule to place the fluorophore in the membrane bilayer, and ii) is compatible with the H1R binding surface (Fig. 1b). 6-(((4,4-Difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy)acetyl)aminohexanoic acid (BODIPY630/650-X) was preferred due to previous success with this fluorophore for Class A GPCR fluorescent ligands28,36,37 as it has favourable excitation/emission wavelength for planned techniques. BODIPY630/650 is known to have an increased quantum yield in a lipid environment38 and as the modelling suggests the fluorophore would reside within the membrane bilayer this would increase its brightness for imaging studies and reduce background fluorescence in aqueous solution.
Figure 1.

Chemical structure and molecular modelling of fluorescent ligands for the H1R. (a) Chemical structures of fluorescent mepyramine (10, 11, 12) and VUF13816 (23, 24, 25) derivatives (b) Modelled binding pose of 23 in the H1R, based on the experimentally validated binding mode of VUF1381634,35 in the H1R crystal structure (PDB code: 3RZE)32.
To confirm that the newly synthesized fluorescent ligands retained the ability to bind to the human H1R in HEK293T cells, competition radioligand binding studies were performed. All six compounds retained affinity at the H1R (Fig. 2, Table 1) demonstrating that the peptide linker and appended fluorophore were well tolerated and did not lead to a reduction in affinity compared to the parent pharmacophores. No significant difference in the affinity of the mepyramine-based compounds (10–12) was observed despite the different peptide linkers, with all three compounds displaying affinities in the 10–25 nM range. This was also true for the VUF13816-based compounds (23–25), although they exhibited slightly higher affinity (4–6 nM) than those based on mepyramine. Each of the compounds was also tested for their ability to antagonize histamine-stimulated calcium release in CHO cells expressing the H1R (Fig. 2). All six compounds produced a rightward-shift in the histamine mediated concentration response curves. As seen previously for the H1R, a suppression of the maximal calcium response was observed in the presence of each of the compounds, which is related to the non-equilibrium kinetics of the assay26,39. Taking this into account EC25, rather than EC50, values were used to estimate the pKB of the fluorescent compounds (Table 1). The values obtained in the calcium assay were in close agreement with those from the radioligand binding experiments, with the VUF13816 based conjugates again displaying higher affinity than the mepyramine based compounds.
Figure 2.

Pharmacological characterisation and live cell confocal imaging of fluorescent ligands at the H1R. (a and b) Inhibition of specific [3H]mepyramine binding to cell homogenates transiently expressing the human H1R by mepyramine (a) and VUF13816 (b) based fluorescent antagonists. Data points represent mean ± SEM of triplicate determinations. Graphs shown are representative of three experiments performed. (c and d) CHO-K1 cells stably expressing the human H1R were preincubated with 100 nM of the indicated fluorescent antagonist for 30 min at 37 °C prior to the addition of increasing concentrations of histamine and changes in intracellular calcium monitored. Data were normalized to basal (in the absence of histamine or antagonist) and 100 µM histamine for each experiment. The data shown represent the mean ± SEM of four experiments performed in triplicate. (e and f) Live cell confocal images of CHO cells expressing H1-YFP incubated with 50 nM (e) 10 or (f) 23 at 37 °C in the absence (top panels) or presence (bottom panels) of 10 µM mepyramine. Single equatorial images were taken of YFP (left hand panels) and BY630/650 (right hand panels). YFP and BY630/650 images are shown in greyscale to avoid issues with colour rendering. For each compound, images in the presence and absence of mepyramine were obtained using identical settings for laser power, detector offset and gain. Data shown are representative of images obtained in three independent experiments. Scale bars = 20 µm.
Table 1.
Binding affinities and kinetic parameters of fluorescent antagonists at the human H1R.
| Compound | Radioligand binding | Calcium assay | NanoBRET saturation | NanoBRET kinetics | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pKi | n | pKD | n | pKD | n | kon (× 106 M−1min−1) | koff (min−1) | Tr (min) | pKD | n | |
| 10 | 7.6 ± 0.1 | 3 | 7.5 ± 0.2 | 4 | 8.1 ± 0.1 | 6 | 46.4 ± 5.9 | 0.214 ± 0.029 | 4.99 ± 0.78 | 8.3 ± 0.01 | 4 |
| 11 | 8.0 ± 0.1 | 3 | 7.9 ± 0.1 | 4 | 7.2 ± 0.1 | 4 | 19.8 ± 2.2 | 0.126 ± 0.015 | 8.40 ± 0.91 | 8.2 ± 0.06 | 5 |
| 12 | 7.7 ± 0.1 | 3 | 7.8 ± 0.1 | 4 | 7.3 ± 0.2 | 5 | 3.3 ± 0.6 | 0.134 ± 0.011 | 7.68 ± 0.58 | 7.4 ± 0.09 | 6 |
| 23 | 8.4 ± 0.1 | 3 | 8.6 ± 0.2 | 4 | 8.1 ± 0.1 | 6 | 1.6 ± 0.3 | 0.019 ± 0.002 | 54.08 ± 5.33 | 7.9 ± 0.10 | 4 |
| 24 | 8.3 ± 0.1 | 3 | 8.1 ± 0.3 | 4 | 7.6 ± 0.1 | 5 | 2.9 ± 0.6 | 0.027 ± 0.002 | 37.81 ± 3.44 | 8.0 ± 0.09 | 4 |
| 25 | 8.2 ± 0.2 | 3 | 7.8 ± 0.3 | 4 | 7.5 ± 0.1 | 5 | 2.6 ± 0.4 | 0.030 ± 0.006 | 39.53 ± 9.40 | 8.0 ± 0.10 | 4 |
pKi values were calculated from inhibition of [3H]mepyramine binding to membranes from HEK293T cells transiently expressing human H1R. Calcium assay pKD values were estimated from a shift in histamine concentration response curves to 100 nM of each fluorescent compound in CHO cells expressing H1R. NanoBRET saturation pKD values were calculated from the saturation curve of the fluorescent ligands binding to HEK293T cells expressing Nluc-H1R. The kinetic parameters, kon, koff and pKD values were obtained using varying concentrations of fluorescent ligand and measuring the NanoBRET signal over time in HEK293T cells expressing Nluc-H1R. The non-specific signal was determined using a high concentration (10 μM) of doxepin as a competitor. The residence time (Tr) was calculated as the mean of the reciprocal of the koff values from each individual experiment. All values represent mean ± SEM from n separate experiments performed in triplicate.
Confocal Microscopy
Peptide linkers were used between the pharmacophore and fluorophore component of the fluorescent ligands in an attempt to reduce the compounds’ lipophilicities and ability to cross cell membranes (compared to the equivalent alkyl linker), thus optimising their properties for use in confocal imaging. Imaging studies were performed on CHO cells expressing H1R linked to yellow fluorescent protein (H1-YFP), which was predominantly expressed at the cell surface. Exposure of H1-YFP cells to 50 nM of 10, 11 or 12 for 30 min at 37 °C resulted in clear membrane localisation of the BODIPY630/650 fluorescence emission for each of the ligands (Fig. 2 and Figure S2). To confirm the specificity of binding to the H1R, H1-YFP expressing cells were pre-treated with 10 µM mepyramine prior to the addition of the fluorescent mepyramine derivatives and subsequent imaging. Under these conditions, very little fluorescence was observed for 10 and 12. However, some residual cell surface fluorescence was observed for 11. To illustrate the improvement in the imaging properties obtained with the peptide linkers, cells were also exposed to a derivative with a non-peptidic linker, mepyramine-X-BODIPY630/65026, in the presence and absence of mepyramine. In contrast to the ligands with peptidic linkers, very little cell surface binding of mepyramine-X-BODIPY630/650 could be discerned due to high levels of intracellular accumulation of the fluorescent ligand. This was not prevented by the presence of mepyramine, indicating significant non-specific binding and cellular uptake (Figure S3). The three VUF13816-based compounds (23–25) were also imaged in the presence and absence of mepyramine (Fig. 2 and S3). These fluorescent compounds also displayed clear cell surface binding which showed a similar binding pattern to the H1R-YFP fluorescence. For each of the compounds clear reduction in cell surface fluorescence was observed following pre-treatment with the unlabelled antagonist mepyramine indicating the majority of the membrane fluorescence was specific binding to the H1R. Some residual binding was observed with 23, which may be due to it being the highest affinity of the fluorescent ligands tested.
Saturation and competition binding studies using NanoBRET in whole cells
Bioluminescence resonance energy transfer (BRET) is a proximity (<10 nm) assay which can be used to measure the binding of fluorescent ligands to a receptor of interest expressing a bioluminescent protein (NanoLuc) tag on the N-terminus40,41. We used our fluorescent ligands in conjunction with a NanoLuc tagged H1R (Nluc-H1R) to determine their binding kinetics at the receptor. Initially, to confirm that the NanoLuc tag had no steric hindrance effects on the binding of ligands to the H1R, we performed association binding experiments using [3H]mepyramine to determine its kinetic parameters at both HA-H1R and Nluc-H1R (Fig. 3). The association and dissociation rates of [3H]mepyramine at Nluc-H1R and HA-H1R, and consequently its affinity, were not significantly different (Table 2). All six of the fluorescent ligands used in this study showed a clear concentration-dependent and saturable increase in BRET signal when incubated with HEK293T cells stably expressing Nluc-H1 (Fig. 4 and S4). Low levels of non-specific BRET were observed for all fluorescent ligands, as determined by co-incubation with a high concentration (10 µM) of unlabelled mepyramine. Due to the wide concentration range of fluorescent ligands used in these studies, specific binding was plotted versus ligand concentration on a log scale and fitted with a sigmoidal function to determine the KD value as the concentration at which 50% specific binding is achieved. In addition, the wide concentration range of fluorescent ligand used also appeared to cause an increase in the non-specific binding at the highest concentration of fluorescent ligand used (300 nM). This was almost certainly due to a direct competition of fluorescent and unlabelled ligand for the receptor with the extent of the shift in the log concentration-binding curve effectively reflecting the affinity of mepyramine for the H1R. The KD values obtained for each of the six fluorescent H1R antagonists in Nluc-H1R-HEK293T cells are shown in Table 1.
Figure 3.
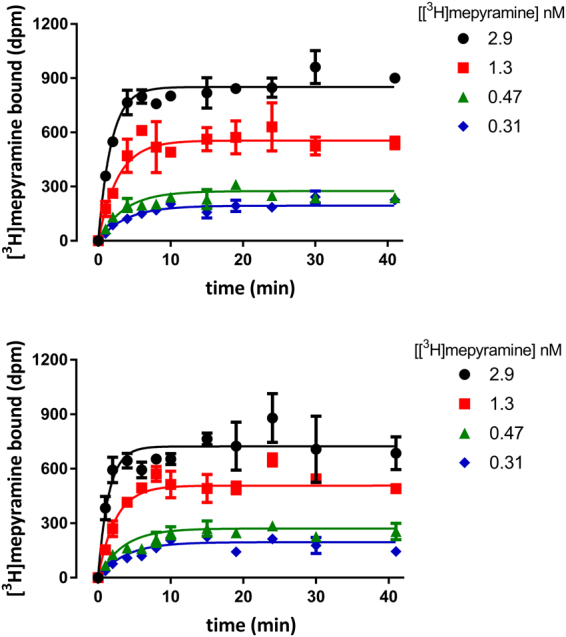
Association binding of [3H]mepyramine at HA-H1R and Nluc-H1R. Specific [3H]mepyramine binding over time to cell homogenate transiently expressing the human H1R was measured for cells expressing the HA-tagged receptor (a) or the Nluc-tagged receptor (b). Data points represent mean ± SEM of duplicate determinations. Graphs shown are representative of 12 (a) or 4 (b) experiments performed.
Table 2.
Kinetic parameters of [3H]mepyramine at HA-H1R and Nluc-H1R.
| kon (×106 M−1 min−1) | koff (min−1) | pKD | n | |
|---|---|---|---|---|
| HA-H1R | 122 ± 7 | 0.21 ± 0.01 | 8.7 ± 0.0 | 12 |
| Nluc-H1R | 147 ± 16 | 0.24 ± 0.02 | 8.8 ± 0.0 | 4 |
Kinetic parameters for [3H]mepyramine-receptor binding were determined by monitoring [3H]mepyramine binding to a homogenate of HEK293T cells expressing either HA-H1R or Nluc-H1R. All values represent mean ± SEM from n separate experiments performed in duplicate.
Figure 4.

Saturation and competition NanoBRET studies with Fluorescent Ligands 10 and 23. (a and b) Saturation binding curves from BRET experiments in Nluc-H1R HEK cells treated with increasing concentrations of (a) 10 or (b) 23 in the absence (black circles) or presence (red squares) of 10 µM mepyramine. The data shown are representative of six independent experiments performed in triplicate. (c–f) Inhibition of BRET signal in Nluc-H1R HEK293T cells treated with 25 nM 10 (c,d) or 25 nM 23 (e,f) and increasing concentrations of the unlabelled ligands; tripolidine, loratadine, mepyramine and promethazine (c,e) or levocetirizine, fexofenadine, olopatadine and doxepin (d,f). Data were normalized to maximal BRET signal in the absence of competitor. The data shown represent the mean ± SEM of at least three experiments (as detailed in Table 3) performed in triplicate.
To further study the suitability of these novel fluorescent ligands to monitor ligand binding at the H1R we used the mepyramine- and VUF13816-based fluorescent ligands with the highest affinity for the receptor (10 and 23) to perform competition binding assays (Fig. 4). All eight compounds caused a concentration-dependent decrease in BRET ratio with both fluorescent ligands and this allowed pKi values of eight diverse H1R antagonists to be determined (Table 3). In general, the pKi values obtained using 10 or 23 were comparable (R2 = 0.91) with doxepin displaying the highest affinity for the H1R and loratadine the lowest. Of the eight compounds tested, two compounds showed a small degree of probe-dependence; mepyramine was found to have a slightly higher affinity when using 10 as the tracer whereas olopatadine was slightly higher affinity in competition with 23 (p < 0.05, unpaired t-test).
Table 3.
Affinities of unlabelled antagonists at Nluc-H1R measured in whole cells and membranes.
| Compound | 25 nM 10 | 25 nM 23 | ||||
|---|---|---|---|---|---|---|
| Whole cells | Membranes | Whole cells | ||||
| pKi | n | pKi | n | pKi | n | |
| Promethazine | 8.12 ± 0.02† | 5 | 8.67 ± 0.07† | 4 | 7.94 ± 0.12 | 4 |
| Mepyramine | 7.62 ± 0.05* | 5 | 7.77 ± 0.14 | 4 | 7.28 ± 0.12* | 5 |
| Loratadine | 6.48 ± 0.05† | 5 | 6.21 ± 0.09† | 4 | 6.36 ± 0.10 | 4 |
| Triprolidine | 7.55 ± 0.11 | 4 | 7.77 ± 0.09 | 4 | 7.34 ± 0.13 | 5 |
| Doxepin | 8.36 ± 0.10† | 5 | 9.06 ± 0.15† | 4 | 8.30 ± 0.13 | 5 |
| Olopatadine | 7.73 ± 0.10*† | 5 | 8.40 ± 0.13† | 4 | 8.05 ± 0.04* | 4 |
| Fexofenadine | 6.78 ± 0.18 | 5 | 7.00 ± 0.11 | 4 | 6.70 ± 0.10 | 4 |
| Levocetirizine | 7.28 ± 0.06† | 3 | 7.66 ± 0.08† | 4 | 7.38 ± 0.10 | 5 |
pKi values of eight H1R antagonists in HEK293T cells or membranes expressing Nluc-H1R were determined by competition binding with 25 nM 10 (whole cells and membranes) or 25 nM 23 (whole cells only). All values represent mean ± SEM from n separate experiments performed in triplicate. *p < 0.05 pKi measured using 10 versus 23 (Student’s unpaired t test). †p < 0.05 pKi measured using 10 in whole cells versus membranes (Student’s unpaired t test).
Suitability of NanoBRET for kinetic studies
The saturation and competition measurements described above were performed as end point readings with the assumption that equilibrium had been reached. If this was correct, then the pKi values obtained should be independent of the kinetics of the fluorescent and non-fluorescent compounds used. To test this, we first used the NanoBRET assay to directly determine the kinetic constants (kon and koff) of the fluorescent ligands. As the BRET measurements are dependent on the luminescence from the NanoLuc protein, and since kinetic measurements may need to be taken for up to 2 hours, it was necessary to confirm that the BRET signal was stable over this time. To test this, Nluc-H1R HEK293T cells were treated with 25 nM 10 in the presence and absence of 10 μM doxepin for 60 min prior to the addition of 0.5 µM furimazine (Nluc substrate). Luminescence and fluorescence emissions were monitored every minute for 90 min and then an additional 0.5 µM furimazine was added and luminescence and fluorescence monitored for a further 30 min. As seen in Figure S5, fluorescence and luminescence signals decayed with time after the addition of the substrate (due to consumption of the substrate by the bioluminescence reaction) and exhibited an instant increase after the addition of additional furimazine which subsequently decayed again with time. Due to the ratiometric nature of the BRET signal, however, the resulting BRET ratio was found to be very stable 15 min following the addition of substrate, even after the second substrate addition (Figure S5).
Kinetic studies in whole cells
The kinetics of the six fluorescent ligands was therefore evaluated in Nluc-H1R HEK293T cells. The specific binding of these compounds at each time point was calculated by subtracting non-specific binding from the equivalent time-matched total signal (Fig. 5). The values for the non-specific signal were obtained in the presence of a high concentration (10 μM) of doxepin as a competitor. The kinetic KD, kon and koff values obtained from the BRET data, as well as the calculated residence time for the six fluorescent H1R antagonists in intact cells, are shown in Table 1. These kinetic studies showed that the residence time of the fluorescent ligands based on mepyramine were approximately four fold less than those of the VUF13816 based fluorescent ligands. In addition, we observed that the kinetic KD values were comparable with those obtained from the saturation binding experiments in the majority of the cases with the exception of 11, where the kinetic KD was tenfold lower than that obtained in the saturation studies. The direct measurement of kinetic parameters at a GPCR in an intact cell environment is limited to compounds that are either fluorescently or radiolabelled. To overcome this, the theoretical framework developed by Motulsky and Mahan42 allow the kinetic parameters of unlabelled ligands to be quantified if the kinetics of the labelled ligand are known. To demonstrate the usefulness of these novel ligands in kinetic screening of unlabelled compounds in the NanoBRET assay, we used 10 to determine the kinetic parameters of two unlabelled antagonists, promethazine and loratadine (Fig. 6 and Table 4). Kinetic data obtained in the presence of a range of competitor concentrations allowed their kon and koff to be determined (Table 4).
Figure 5.

NanoBRET association kinetic studies with mepyramine and VUF13816 based fluorescent ligands. Nluc-H1R HEK293 cells were treated with the indicated concentrations of 10 (a), 11 (b), 12 (c), 23 (d), 24 (e) and 25 (f) and BRET monitored at room temperature every min for 60 min (a–c) or 90 min (d–f). The data shown are representative examples from four (10, 23, 24, 25), five (11) or six (12) independent experiments performed in triplicate.
Figure 6.
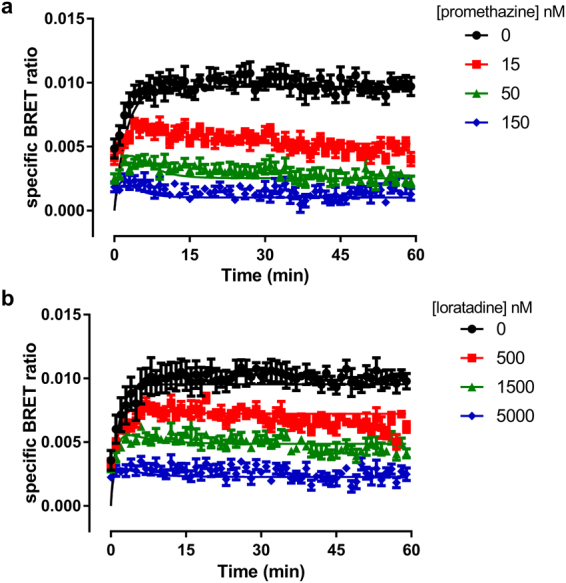
Use of fluorescent ligand 10 to determine the kinetics of unlabelled ligands. Nluc-H1R HEK293 cells were treated with 10 nM 10 and the indicated concentrations of promethazine (a) and loratidine (b) and BRET monitored at room temperature every min for 60 min. The data shown are representative examples from five independent experiments performed in triplicate.
Table 4.
Kinetic parameters of promethazine and loratadine at Nluc-H1R determined using NanoBRET.
| kon (×105 M−1 min−1) | koff (min−1) | pKD | |
|---|---|---|---|
| Promethazine | 337.6 ± 147.1 | 0.18 ± 0.05 | 8.18 ± 0.09 |
| Loratadine | 2.2 ± 0.4 | 0.35 ± 0.13 | 5.83 ± 0.20 |
Kinetic parameters were obtained by monitoring the binding of 10 nM 10 in the presence of increasing concentrations of unlabelled ligand and calculated as detailed in Methods. All values represent mean ± SEM from 5 experiments performed in triplicate.
BRET studies performed in cell membranes
One of the main advantages of the NanoBRET technique is its ability to determine ligand binding parameters in living cells. To compare the effects of an intact cellular environment on the affinity and kinetics properties of our fluorescent ligands, we also performed BRET studies in membrane preparations from Nluc-H1 HEK293T cells. In initial optimisation studies, very low levels of specific binding were observed when measuring the kinetics of conjugate 10. To overcome this, we performed experiments in the presence of the detergent saponin (1 mg/ml), in order to disrupt membrane vesicles, which greatly improved the specific BRET ratios obtained (Figure S6). Since saponin was found to have no effect no competition binding curves and on the affinity of doxepin (Figure S6), all further studies in membranes were performed in the presence of saponin.
Saturation binding curves for 10 obtained in cell membranes from Nluc-H1R HEK293T cells obtained in the presence and absence of 10 μM of unlabelled mepyramine are shown in Fig. 7. As in cells, compound 10 showed a clear concentration-dependent and saturable increase in BRET signal, with a pKD value of 7.54 ± 0.04 (n = 5). We also used 10 to perform competition binding assays in cell membranes with the same eight H1R antagonists as used previously (Fig. 7). The competition-binding pKi values obtained (Table 3) for unlabelled antagonists were comparable with those obtained on intact cells (R2 = 0.96) although some minor differences were seen for individual compounds, with a trend for compounds to show higher affinity in membranes compared to whole cells. The affinity of promethazine, doxepin, olopatadine and levocetirizine was significantly higher (p < 0.05, unpaired t-test) in membranes than whole cells, whilst that for loratadine was lower (p < 0.05, unpaired t-test). Finally, we obtained the kinetic constants of 10 in membranes from cells expressing Nluc-H1 (Fig. 7). The kon (19.64 ± 4.12 × 106 M−1.min−1) and the koff (0.065 min−1) determined in membranes were both signifcantly slower (p < 0.05, unpaired t-test) than those obtained in intact cells, but there was no difference in the calculated affinity (pKD = 8.45 ± 0.12, n = 4, p > 0.05, unpaired t-test).
Figure 7.

NanoBRET saturation, competition and kinetic binding studies with fluorescent ligand 10 in cell membranes. (a). Saturation BRET binding curves from membranes of Nluc-H1R HEK293 cells treated with increasing concentrations of 10 in absence (black circles) or presence (red squares) of 10 µM mepyramine. The data shown are representative of five independent experiments performed in triplicate. (b and c) Inhibition of BRET signal in membranes of Nluc-H1R HEK293T cells treated with 25 nM 10 and increasing concentrations of unlabelled ligands; tripolidine, loratadine, mepyramine and promethazine (b) or levocetirizine, fexofenadine, olopatadine and doxepin (c). Data were normalized to the maximal BRET ratio in each experiment. Data points are combined mean ± SEM from four separate experiments performed in triplicate (d). Membranes of HEK293T cells stably expressing Nluc-H1R treated with various concentrations of 10 were monitored by BRET at room temperature every min for 30 min. All experiments using membranes expressing Nluc-H1R were performed in the presence of 1 mg/ml saponin. The data shown are representative of four independent experiments performed in triplicate.
Discussion
The recent development of fluorescent ligands has enabled many aspects of receptor pharmacology and function to be studied14,43. Here we have increased the range and improved the properties of fluorescent ligands available to study the H1R by synthesising six new ligands based on two different orthosteric binding moieties (mepyramine and VUF13816). We have created a series of new, high affinity fluorescent H1R ligands suitable for use in both confocal imaging and, in particular, in NanoBRET assays for monitoring ligand binding in living cells.
To date, there have been two reports on fluorescent ligands for the H1R based on the prototypic antagonist mepyramine25,26, but in the case of mepyramine-X-BODIPY630/650 its lipophilic nature meant it was unsuitable for confocal microscopy26. Using a previous strategy to develop new fluorescent ligands for the adenosine A3 receptor28,44, here we replaced the six carbon linker between the mepyramine and the BODIPY630/650 fluorophore26 with di- or tripeptides. Introduction of these peptide linkers resulted in some loss in affinity compared to the previously described fluorescent mepyramine (mepyramine-X-BODIPY630/650; pKD = 8.926), but more importantly improved their imaging properties. All three new mepyramine fluorescent ligands allowed clear visualisation of membrane binding by confocal microscopy that could be prevented by a high concentration of unlabelled mepyramine. This was in direct contrast to mepyramine-X-BODIPY630/650, where intracellular accumulation of the fluorescent ligand masked the detection of specific membrane binding by confocal microscopy. Intracellular accumulation of fluorescent ligands has been previously observed as problematic with ligands for the β1 adrenoceptor17, cannabinoid CB1 receptor20 and µ opioid receptor37.
We also synthesised a second series of fluorescent ligands based on a small molecule (VUF13816, MW = 267) that was first identified in a virtual screen of the H1R crystal structure34. Incorporation of peptide linkers and the BODIPY630/650-X fluorophore onto this molecule resulted in three fluorescent ligands that retained similar affinity at the H1R to the parent compound (pKi = 8.2034). VUF13816 is structurally different from any other H1R ligand, although molecular modelling indicated that it makes similar contacts in the receptor to doxepin34. There was little difference in the affinity of the three VUF13816-based fluorescent ligands, even though they contained more diverse peptide linkers than the mepyramine-based ligands. As with the mepyramine based derivatives, the VUF13816-based compounds were also suitable for confocal imaging. The success of these ligands, in particular those based on VUF13816, validates the combined virtual screening and ligand design as a more structured route to fluorescent ligand design.
Both the mepyramine and VUF13816 based fluorescent ligands used in the study had similar affinity values to the parent compounds. However, for small molecule ligands for Class A GPCRs, addition of linker and fluorophore significantly increases the size and molecular complexity of the ligand. These compounds should therefore be treated as new pharmacological entities27, as often either increases28,36 or decreases45,46 in affinity are seen. Studies examining the influence of both the linker and fluorophore on the affinity of a fluorescent ligand have demonstrated that they can influence affinity in multiple different ways, with both the linker and flurophore playing a role in determining the final affinity of a compound27,28,36,38,47.
To further extend the use of the new fluorescent ligands to examine receptor binding kinetics, we examined their utility in a recently described NanoBRET proximity assay40. This assay utilises the extremely bright luciferase NanoLuc48 that is fused to the N-terminus of H1R. The distance constraints of energy transfer49,50 mean that BRET can only occur when the fluorescent ligand is in very close proximity to the receptor (i.e. <10 nm). Association kinetic binding assays using [3H]mepyramine indicated that the N/uc-H1R showed similar binding kinetics and affinity for mepyramine, suggesting that the N terminal tag did not significantly affect the receptor binding properties. The affinity of all six fluorescent ligands was measured using NanoBRET saturation assays, and the values obtained were consistent with those from radioligand binding and calcium assays. The affinities of eight unlabelled antagonists were also determined in competition binding assays using fluorescent ligands 10 and 23 as tracers. Two compounds, olopatadine and mepyramine, showed small but significant differences in their affinity depending on the fluorescent ligand used. Similar probe dependence has been observed previously for the adenosine A3 receptor40 and may reflect potential allosteric interactions across a dimeric interface24; the H1R has been previously shown to form homodimers51.
The NanoBRET binding assay has the advantage that it is performed in intact cells that preserve the potential for allosteric interactions with intracellular signalling proteins10–12. In order to investigate the influence of an intact cellular environment on the affinity of unlabelled antagonists, NanoBRET assays were also performed in cell membranes. There was a general trend for the affinities obtained in membranes to be slightly higher than those deduced in intact cells. This may be due to the presence of phosphate in the buffer used for the membrane binding assays which has been shown to be a positive modulator of ligand binding at the H1R32.
It is becoming increasingly clear that determining binding kinetics is important in the development of new therapeutics9,52,53, and certain kinetic profiles are desirable depending on the disease being targeted54,55. The determination of binding kinetics at a GPCR is often achieved through radioligand binding assays, where every time point requires a separate binding reaction that needs to be terminated before measurements can be made. This is time consuming and has led to the development of assays that allow continuous monitoring of binding, such as assays using a scintillation proximity assay for radioligands56,57. Greater resolution can be obtained, however, with proximity techniques such as the BRET assay used here or using fluorescence resonance energy transfer23,25. In the present study, the kinetic parameters of six fluorescent antagonists were determined. In equilibrium NanoBRET binding assays all of the fluorescent ligands showed similar affinities. However, the use of kinetics revealed that the VUF13816 based compounds had a slower off rate than their mepyramine counterparts. As a consequence, the VUF13816 based compounds had a residence time at the receptor that was circa four times longer than the mepyramine based compounds. The binding kinetics of compound 10 were also determined in membrane preparations where there were differences in both the association (19.64 × 106 M−1.min−1 and 46.4 × 106 M−1.min−1; cells and membranes, respectively) and dissociation rate constants (0.065 min−1 and 0.214 min−1; cells and membranes, respectively). Measuring the kinetics of a labelled compounds limits this to a small set of ligands for a given receptor, to overcome this the method developed by Motulsky and Mahan can be employed to determine the kinetics of unlabelled compounds. This was successfully achieved using the NanoBRET assay in combination with compound 10 for two unlabelled H1R antagonists, promethazine and loratadine. It was found that both unlabelled ligand displayed similar koff rates but different kon rates which is reflected in their affinity at equilibrium. The NanoBRET assay has also recently been used to resolve the kinetics of unlabelled ligands at the free fatty acid receptor 2 in cell membranes58. The ability to also perform these measurements in whole cells opens up the potential to assess the effect of cellular environments on rate constants as it has been shown that for some receptors that an intact cell environment can alter the binding kinetics59.
This study demonstrates the broad utility of fluorescent ligands for studying the H1R. The use of fluorescent ligands and the NanoBRET assay allows multiple aspects of receptor pharmacology to be studied in ways that are difficult with traditional assays. These can provide important insights in the function of GPCRs and into the characteristics of lead compounds for drug discovery.
Methods
Experimental Section
Chemistry
Chemicals and solvents of an analytical grade were purchased from commercial suppliers and used without further purification. 1,2-Diaminoethane trityl resin was purchased from Sigma Aldrich. 6-(((4,4-Difluoro-5-(2-thienyl)-4-bora-3a,4a-diaza-s-indacene-3-yl)styryloxy)acetyl)aminohexanoic acid, succinimidyl ester (BODIPY 630/650-X-SE) was purchased from Molecular Probes® (Invitrogen, UK). Unless otherwise stated, reactions were carried out at ambient temperature. Reactions were monitored by thin layer chromatography on commercially available pre-coated aluminium-backed plates (Merck Kieselgel 60 F254). Visualization was by examination under UV light (254 and 366 nm), or staining with KMnO4 dip. Flash chromatography was performed using Merck Kieselgel 60, 230–400 mesh (Merck KgaA, Darmstadt, Germany) on a Biotage Flashmaster II system. 1H NMR spectra were recorded on a Bruker-AV 400 at 400.13 MHz or a Bruker AV(II) 500 at 503.13 MHz. 13C NMR spectra were recorded on a Bruker AV(II) 500 with a dual (CH) cryoprobe at 125.8 MHz. Solvents used for NMR analysis (reference peaks listed) were DMSO-d6 ((CHD2)2SO at δH 2.50 ppm, (CD3)2SO at 39.52 ppm) and CDCl3 (CHCl3 at δH 7.26 ppm, CDCl3 at 77.16). Chemical shifts (δ) are recorded in parts per million (ppm). Coupling constants (J) are recorded in Hz and the significant multiplicities described by singlet (s), doublet (d), triplet (t), quadruplet (q), broad (br), and multiplet (m). Spectra were assigned using appropriate COSY, DEPT, HSQC and HMBC sequences. High resolution mass spectra (HRMS) – time of flight, electrospray (TOF ES +/−) were recorded on a Waters 2795 separation module/micromass LCT platform. RP-HPLC was performed using a Waters 2767 sample manager, Waters 2525 binary gradient module, and visualized at 254 nm and 366 nm with a Waters 2487 dual wavelength absorbance detector. Spectra were analyzed using MassLynx. Analytical reversed-phase high-performance liquid chromatography (RP-HPLC) was used to ensure that the purity of compounds tested in biological systems was ≥ 95% (the retention times (Rt) of these compounds are reported). Analytical RP-HPLC was performed using a YMC-Pack C8 column (150 mm × 4.6 mm × 5 μm) at a flow rate of 1 mL/min, and using a method of 0–2 min 10% solvent B in solvent A, 2–25 min gradient of 10% to 90% solvent B in solvent A, 25–27 min held at 90% B in solvent A, 27–29 min gradient of 90% to 10% solvent B in solvent A, 29–33 min held at 10% solvent B in solvent A (solvent A = 0.05% TFA in H2O, solvent B = 0.05% TFA in 9:1 v:v CH3CN:H2O).
Methyl 3-((2-((4-methoxybenzyl)(pyridin-2-yl)amino)ethyl)(methyl)amino)propanoate (2)
Mepyramine (1) was purchased as the dimaleate salt. Prior to demethylation, mepyramine dimaleate salt was dissolved in dichloromethane and sequentially washed with saturated hydrogen bicarbonate and water. The dichloromethane layer was dried over magnesium sulfate, filtered and the solvent evaporated under reduced pressure to give mepyramine free base as a colourless oil. Mepyramine (free base) (22.7 g, 79.5 mmol) was dissolved in 300 mL of 1,2-dichloroethane and a small amount (approx. 20–30 mg) of sodium hydrogen bicarbonate was added. The solution was cooled to 0 °C and 1-chloroethyl chloroformate (25 g, 175 mmol) was added in 5 equal portions over 2 h. The reaction mixture was then heated at 60 °C for 3 h. The solution was cooled to rt, filtered, and the filtrate evaporated under reduced pressure. Methanol (350 mL) was added to the orange residue obtained and the resultant solution refluxed for 2 h. The solvent was removed under reduced pressure, the residue treated with 2 M aq. sodium hydroxide solution and extracted three times with ethyl acetate. The ethyl acetate portions were combined, dried over magnesium sulfate, filtered and evaporated under reduced pressure to give the crude product as an orange oil. Purification by flash silica column chromatography (0.5% to 5% 7 M ammonia in methanol / ethyl acetate) gave N1-(4-methoxybenzyl)-N2-methyl-N1-(pyridin-2-yl)ethane-1,2-diamine (1.428 g, 5.27 mmol, 7%) as a colourless oil. 1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3 H, NHCH3), 2.80 (t, J = 6.6 Hz, 2 H, CH2CH2), 3.67 (t, J = 6.6 Hz, 2 H, CH2CH2), 3.76 (s, 3 H, OCH3), 4.70 (s, 2 H, CH2Ar), 6.48 (d, J = 8.6 Hz, 1 H, Ar pyridyl), 6.52 (m, 1 H, Ar pyridyl), 6.82 (d, J = 8.7 Hz, 2 H, Ar benzyl), 7.14 (d, J = 8.7 Hz, 2 H, Ar benzyl), 7.37 (m, 1 H, Ar pyridyl), 8.15 (m, 1 H, Ar pyridyl). 13C NMR (100 MHz, CDCl3) δ 36.65, 48.28, 49.92, 51.51, 55.32, 106.03, 111.95, 114.04, 128.10, 130.69, 137.29, 148.04, 158.53, 158.70. HRMS calculated for C16H22N3O 272.1757 (M + H)+, found 272.1740. To a stirred solution of N1-(4-methoxybenzyl)-N2-methyl-N1-(pyridin-2-yl)ethane-1,2-diamine (1.428 g, 5.27 mmol) in 1,2-dichloroethane (8 mL) was added methyl acrylate (2.2 mL, approx. 26 mmol, approx. 5 equiv), and the mixture stirred at 80 °C for 3 h. The solvent was removed under reduced pressure to give an orange oil. Purification by flash silica column chromatography (0.1% to 0.5% 7 M ammonia in methanol / ethyl acetate) gave 2 (1.602 g, 4.49 mmol, 85%) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 2.27 (s, 3 H, NCH3), 2.45 (t, J = 7.2 Hz, 2 H, CH2CH2), 2.57 (t, J = 7.3 Hz, 2 H, CH2CH2), 2.73 (t, J = 7.2 Hz, 2 H, CH2CH2), 3.62 (t, J = 7.3 Hz, 2 H, CH2CH2), 3.65 (s, 3 H, CO2CH3), 3.78 (s, 3 H, OCH3), 4.69 (s, 2 H, CH2Ar), 6.44 (d, J = 8.8 Hz, 1 H, Ar pyridyl), 6.52 (m, 1 H, Ar pyridyl), 6.84 (d, J = 8.6 Hz, 2 H, Ar benzyl), 7.16 (d, J = 8.6 Hz, 2 H, Ar benzyl), 7.37 (m, 1 H, Ar pyridyl), 8.16 (m, 1 H, Ar pyridyl). 13C NMR (100 MHz, CDCl3) δ 32.61, 42.49, 46.49, 51.51, 51.64, 53.27, 54.66, 55.33, 105.83, 111.76, 114.01, 128.23, 130.89, 137.24, 148.12, 158.23, 158.71, 173.09. HRMS calculated for C20H28N3O3 358.2125 (M + H)+, found 358.2139.
3-((2-((4-Methoxybenzyl)(pyridin-2-yl)amino)ethyl)(methyl)amino)propanoic acid (3)
To a stirred solution of 2 (1.602 g, 4.49 mmol) in THF at 0 °C was added dropwise a solution of 0.2 M aq. lithium hydroxide (8.98 mmol, 45 mL). The solution was stirred at 0 °C, monitored by LCMS, and after 2 h the reaction was complete. The solution was adjusted to pH 7 using 1 M aq. HCl, gently evaporated under reduced pressure to constant volume (to remove the THF), and then freeze-dried to afford a white solid. Chloroform (approx. 100 mL) was added, the mixture sonicated, and then filtered. The filtrate was evaporated to dryness under reduced pressure, to yield approximately 2 g of white solid. To further desalt the sample, the solid was re-dissolved in chloroform and washed with water, the water layer extracted three times with chloroform, the chloroform extracts combined, dried, and evaporated to dryness to give 3 as a white solid (1.696 g). Assuming one equivalent of lithium chloride, this gave 4.4 mmol, 90% yield. 1H NMR (400 MHz, CDCl3) δ 2.45 (t, J = 6.1 Hz, 2 H, CH2CH2), 2.53 (s, 3 H, NCH3), 2.89 (m, 4 H, CH2CH2, CH2CH2), 3.76 (s, 3 H, OCH3), 3.86 (t, J = 6.6 Hz, 2 H, CH2CH2), 4.60 (s, 2 H, CH2Ar), 6.48 (d, J = 8.5 Hz, 1 H, Ar pyridyl), 6.56 (m, 1 H, Ar pyridyl), 6.83 (d, J = 8.4 Hz, 2 H, Ar benzyl), 7.11 (d, J = 8.7 Hz, 2 H, Ar benzyl), 7.37 (m, 1 H, Ar pyridyl), 8.13 (m, 1 H, Ar pyridyl), 10.36 (br s, 1 H, CO2H). 13C NMR (100 MHz, CDCl3) δ 30.43, 40.70, 44.89, 52.00, 53.38, 53.65, 55.36, 106.42, 112.62, 114.24, 128.05, 129.82, 137.58, 147.98, 157.89, 158.93, 174.20. HRMS calculated for C19H26N3O3 344.1969 (M + H)+, found 344.1951.
General Procedure A (Supplementary Figure 1a): Solid-phase synthesis of 7, 8, 9, 20, 21 and 22
1,2-Diaminoethane trityl resin (substitution 1.2–1.7 mmol/g depending on batch) (200–350 mg, assume 0.34–0.60 mmol) was swelled in DMF for 4 h, drained, then a solution of the first Fmoc-amino acid (3 equiv), HBTU (3 equiv) and DIPEA (6 equiv) in DMF (approx. 3 mL) was added and left for 1–2 h. The resin was drained, washed with DMF, and double coupled using the same Fmoc-amino acid/HBTU/DIPEA coupling procedure. The resin was drained, washed with DMF, and a solution of acetic anhydride (500 µL) and DIPEA (500 µL) in DMF was added to cap any unreacted sites. The resin loading was estimated by determining the Fmoc-content of the resin and calculated to be 0.5–0.7 mmol/g across different resin batches and experiments. The resin was then treated with three repeated cycles of 20% v/v piperidine in DMF for 10 min, washed with DMF, then the second Fmoc-amino acid (3 equiv rel. to resin loading) was coupled using HBTU (3 equiv) and DIPEA (6 equiv) in DMF (approx. 3 mL). The Fmoc groups was again cleaved using 20% v/v piperidine/DMF as described above, and then any additional amino acids (or Fmoc-4-aminobutyric acid) were added as required using the same HBTU coupling/Fmoc cleavage standard solid-phase peptide synthesis procedure described above. After the last required amino acid was coupled and Fmoc deprotected, either the mepyramine derivative 3 or VUF13816 derivative 16 (1.1 equiv rel to resin loading) was coupled to the resin-bound peptide using HATU (1.1 equiv) and DIPEA (4 equiv) in DMF. The resin was washed with DMF then DCM, dried under nitrogen and vacuum, and the product cleaved from the resin using 5% TFA/DCM. The filtrate solvent was evaporated, and if the peptide contained t-butyl side chain protecting group(s), this residue was dissolved in 1:1 v/v DCM/TFA and stirred for 2–4 hours to allow for amino acid side-chain deprotection.
(S)-N-(2-aminoethyl)-2-((S)-2-(3-((2-((4-methoxybenzyl)(pyridin-2-yl)amino)ethyl)(methyl)amino)propanamido)propanamido)propanamide (7)
1,2-Diaminoethane trityl resin (560 mg) was reacted according to General Procedure A, and the crude resin-cleaved residue purified using preparative RP-HPLC to give 7 (166 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.20 (m, 6 H, ala CH3), 2.67 (t, J = 7.4 Hz, 2 H, COCH2), 2.83–2.87 (m, 5 H, NCH3, CH2NH2), 3.27–3.32 (m, 4 H, NHCH2, MeNCH2), 3.39 (m, 2 H, MeNCH2), 3.72 (s, 3 H, OMe), 3.90 (t, J = 6.5 Hz, 2 H, ArNCH2), 4.19 (m, 1 H, ala CH), 4.30 (m, 1 H, ala CH), 4.65 (s, 2 H, ArCH2), 6.66–6.73 (m, 2 H, ArH), 6.90 (d, J = 8.7 Hz, 2 H, ArH PMB), 7.15 (d, J = 8.7 Hz, 2 H, ArH PMB), 7.54 (m, 1 H, ArH), 7.92 (br s, 2 H, NH2), 8.08 (t, J = 5.5 Hz, 1 H, NHCH2), 8.12–8.15 (m, 2 H, ala NH, ArH), 8.40 (d, J = 7.8 Hz, 1 H, ala NH). 13C NMR (125 MHz, DMSO-d6) δ 17.98, 18.09, 29.35, 36.46, 38.43, 43.47, 48.28, 48.38, 50.91, 51.87, 53.36, 55.08, 107.22, 112.68, 114.08, 127.99, 129.54, 138.40, 146.77, 157.10, 158.44, 168.90, 171.89, 172.76. HRMS calculated for C27H42N7O4 528.3293 (M + H)+, found 528.3271.
(S)-N-(2-aminoethyl)-3-hydroxy-2-((10 S,13 S)-1-(4-methoxyphenyl)-5,10,13-trimethyl-8,11-dioxo-2-(pyridin-2-yl)-2,5,9,12-tetraazatetradecan-14-amido)propanamide (8)
1,2-Diaminoethane trityl resin (222 mg) was reacted according to the General Procedure A to give the resin-cleaved crude 8 (120 mg) as a yellow oil. Analytical RP-HPLC and MS analysis showed approximately 50% of the sample to be the desired product and 50% the product but with the p-methoxybenzyl group cleaved. Therefore, a small amount of the crude 8 (10 mg) was purified by preparative RP-HPLC to give a pure sample (4.4 mg, 7.2 µumol) free from any p-methoxybenzyl cleaved side-product. HRMS calculated for C30H47N8O6 615.3613 (M + H)+, found 615.3643. This purified material was used to couple to BODIPY 630/650-X-SE.
N-((S)-1-(((S)-1-((2-aminoethyl)amino)-1-oxopropan-2-yl)amino)-1-oxopropan-2-yl)-4-(3-((2-((4-methoxybenzyl)(pyridin-2-yl)amino)ethyl)(methyl)amino)propanamido)butanamide (9)
1,2-Dichloroethane trityl resin (180 mg) was reacted according to the General Procedure A to give the resin-cleaved crude 9 (105 mg) as a white solid. The crude residue was purified using preparative HPLC to give 9 (65 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.20 (m, 6 H, ala CH3), 1.62 (m, 2 H, CH2), 2.13 (t, J = 7.4 Hz, 2 H, COCH2), 2.60 (t, J = 7.1 Hz, 2 H, COCH2), 2.83–2.87 (m, 5 H, NCH3, CH2NH2), 3.04 (m, 2 H, NHCH2), 3.27–3.33 (m, 4 H, NHCH2, MeNCH2), 3.39 (m, 2 H, MeNCH2), 3.72 (s, 3 H, OMe), 3.90 (t, J = 6.8 Hz, 2 H, ArNCH2), 4.14–4.26 (m, 2 H, ala CH), 4.65 (s, 2 H, ArCH2), 6.66–6.71 (m, 2 H, ArH), 6.89 (d, J = 8.7 Hz, 2 H, ArH PMB), 7.15 (d, J = 8.7 Hz, 2 H, ArH PMB), 7.53 (m, 1 H, ArH), 7.87 (br s, 2 H, NH2), 8.00–8.08 (m, 3 H, ala NH, ala NH, NHCH2), 8.12 (m, 1 H, ArH), 8.19 (t, J = 5.4 Hz, 1 H, NHCH2). 13C NMR (125 MHz, DMSO-d6) δ 17.84, 17.85, 25.19, 29.30, 32.47, 36.43, 38.22, 38.44, 43.44, 48.35, 50.90, 52.06, 53.45, 55.07, 107.11, 112.67, 114.07, 127.96, 129.57, 138.26, 146.95, 157.21, 158.40, 168.96, 171.97, 172.27, 171.78. HRMS calculated for C31H49N8O5 613.3820 (M + H)+, found 613.3790.
General Procedure B: Synthesis of 10, 11, 12, 23, 24, 25 (Supplementary Figure 1b)
The amine congener (as the TFA salt) was dissolved in methanol and Amberlyst 21 resin was added, the solution filtered and the filtrate evaporated to give the neutralized free amine congener. This free amine (1–5 equiv) was dissolved in DMF and BODIPY 630/650-X-SE (1 equiv) was added and the reaction stirred at rt for 2 h in the dark. The solvent was removed under reduced pressure and the residue purified by semi-preparative RP-HPLC.
6-(2-(4-((E)-2-(5,5-difluoro-7-(thiophen-2-yl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)vinyl)phenoxy)acetamido)-N-((10 S,13 S)-1-(4-methoxyphenyl)-5,10,13-trimethyl-8,11,14-trioxo-2-(pyridin-2-yl)-2,5,9,12,15-pentaazaheptadecan-17-yl)hexanamide (10)
According to General Procedure B, the neutralized amine of 7 (3.2 mg, 6 µmol) was reacted with BODIPY 630/650-X-SE (4 mg, 6 µmol) to give a blue solid. This was dissolved in methanol, filtered through a small column containing Amberlyst 21 resin, and the methanol evaporated to give 10 (3.6 mg, 3.4 µmol, 57% yield) as a blue solid. HRMS calculated for C56H68BF2N10O7S 1073.5049 (M + H)+, found 1073.5050. Analytical RP-HPLC Rt = 18.8 min.
6-(2-(4-((E)-2-(5,5-difluoro-7-(thiophen-2-yl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)vinyl)phenoxy)acetamido)-N-((10 S,13 S,16 S)-16-(hydroxymethyl)-1-(4-methoxyphenyl)-5,10,13-trimethyl-8,11,14,17-tetraoxo-2-(pyridin-2-yl)-2,5,9,12,15,18-hexaazaicosan-20-yl)hexanamide (11)
According to General Procedure B, the neutralized amine of 8 (2.2 mg, 3.6µmol) was reacted with BODIPY 630/650-X-SE (1 mg, 1.5 µmol) to give a blue solid. This was dissolved in methanol, filtered through a small column containing Amberlyst 21 resin, and the methanol evaporated to give 11 (1.0 mg, 0.86 µmol, 57% yield) as a blue solid. HRMS calculated for C59H73BF2N11O9S 1160.5369 (M + H)+, found 1160.5326. Analytical RP-HPLC Rt = 17.2 min.
5,5-difluoro-3-((E)-4-(((15 R,18 R)-1-(4-methoxyphenyl)-5,15,18-trimethyl-8,13,16,19,24,31-hexaoxo-2-(pyridin-2-yl)-2,5,9,14,17,20,23,30-octaazadotriacontan-32-yl)oxy)styryl)-7-(thiophen-2-yl)-5H-4λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-5-uide (12)
According to General Procedure B, the neutralized amine of 9 (5 mg, 8.2 µmol) was reacted with BODIPY 630/650-X-SE (1 mg, 1.5 µmol) to give a blue solid. This was dissolved in methanol, filtered through a small column containing Amberlyst 21 resin, and the methanol evaporated to give 12 (0.7 mg, 0.6 µmol, 40% yield) as a blue solid. HRMS calculated for C60H75BF2N11O8S 1158.5576 (M + H)+, found 1158.5603. Analytical RP-HPLC Rt = 18.6 min.
tert-Butyl 4-(2-benzylphenoxy)piperidine-1-carboxylate (14)
Potassium hydroxide (403 mg, 7.2 mmol), 2-benzylphenol (445 mg, 2.4 mmol) and tert-butyl 4-bromopiperidine-1-carboxylate (3.1 g, 11.8 mmol) were dissolved in methanol (5 mL) and refluxed for 2 days. More potassium hydroxide (201 mg, 3.6 mmol) and tert-butyl 4-bromopiperidine-1-carboxylate (1.5 g, 5.7 mmol) were added and the reaction refluxed for a further day. The solvent was evaporated under reduced pressure and the residue dissolved in ethyl acetate. This was washed three times with 1 M NaOH aq. solution, once with saturated aq. brine, the ethyl acetate layer dried over MgSO4, filtered, and evaporated to give the crude product. This was purified using flash silica column chromatography (0 to 16% ethyl acetate in petroleum ether) to give 14 (246 mg, 0.67 mmol, 28% yield) as a colourless oil. 1H NMR (400 MHz, CDCl3) δ 1.15 (s, 9 H, C(CH3)3), 1.74 (m, 2 H, CH2), 1.86 (m, 2 H, CH2), 3.41 (m, 2 H, CH2), 3.50 (m, 2 H, CH2), 4.02 (s, 2 H, ArCH2), 4.52 (m, 1 H, CH), 6.87–6.94 (m, 2 H, ArH), 7.17–7.30 (m, 7 H, ArH). 13C NMR (100 MHz, CDCl3) δ 28.55, 30.47, 36.57, 40.45, 71.41, 79.62, 112.55, 120.62, 125.90, 127.48, 128.30, 128.88, 130.61, 131.13, 141.22, 154.94. HRMS calculated for C23H30NO3+ 368.2220 (M + H)+, found 368.2239.
Methyl 3-(4-(2-benzylphenoxy)piperidin-1-yl)propanoate (15)
14 (246 mg, 0.67 mmol) was dissolved in dichloromethane (4 mL) and trifluoroacetic acid (1 mL) was added. The mixture was stirred for 1 h at rt, then the solvent removed under reduced pressure, and the residue dissolved in ethyl acetate. The organic layer was washed three times was saturated aq. sodium bicarbonate, dried over MgSO4, and evaporated to give 4-(2-benzylphenoxy)piperidine (178 mg, 0.67 mmol, quantitative) as a colourless oil. This was used without purification in the next reaction. HRMS calculated for C18H22NO+ 268.1696 (M + H)+, found 268.1699.
4-(2-benzylphenoxy)piperidine (170 mg, 0.64 mmol) was dissolved in DCM and neutralized by washing with saturated aq. sodium carbonate. The DCM layer was dried and the solvent removed under reduced pressure. This residue was dissolved in 1,2-dichloroethane (1 mL) and methyl acrylate (350 µl, 3.84 mmol) was added. The reaction was heated to 60 °C and stirred for 1.5 h. The solvent was removed under reduced pressure and the residue purified by silica flash column chromatography (petroleum ether, gradient of 0 to 30% solution of [5% 7 M ammonia in methanol in 95% ethyl acetate]) to give 15 (200 mg, 0.57 mmol, 89% yield) as a pale yellow oil. 1H NMR (400 MHz, CDCl3) δ 1.83 (m, 2 H, CH2 pip), 1.95 (m, 2 H, CH2 pip), 2.36 (m, 2 H, CH2 pip), 2.52 (t, J = 7.7 Hz, 2 H, CH2 ethyl), 2.61 (m, 2 H, CH2 pip), 2.70 (t, J = 7.7 Hz, 2 H, CH2 ethyl), 3.72 (s, 3 H, OMe), 4.00 (s, 2 H, ArCH2), 4.39 (m, 1 H, CH), 6.85–6.91 (m, 2 H, ArH), 7.12–7.30 (m, 7 H, ArH). 13C NMR (100 MHz, CDCl3) δ 30.76, 32.36, 36.49, 50.11, 51.76, 53.66, 71.62, 112.59, 120.41, 125.84, 127.41, 128.30, 128.98, 130.60, 131.00, 141.36, 155.14, 173.14. HRMS calculated for C22H28NO3+ 354.2064 (M + H)+, found 354.2034.
3-(4-(2-Benzylphenoxy)piperidin-1-yl)propanoic acid (16)
To a stirred solution of 15 (200 mg, 0.57 mmol) in THF (5 mL) at 0 °C was added dropwise a solution of lithium hydroxide in water (0.2 M, 1.14 mmol, 5.7 mL). The solution was stirred at 0 °C for 2 h, neutralized to pH = 7 using 1 M aq. hydrochloric acid, and the solvent removed under reduced pressure. The material was redissolved in water and freeze-dried overnight. Chloroform was added to the dried sample, and the sample filtered, the white solid washed with chloroform, the chloroform washings combined, dried over magnesium sulfate, and evaporated under reduced pressure to give 16 (180 mg, assuming one equiv. of LiCl salt, 0.47 mmol, 83%) as a pale yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 1.60 (m, 2 H, CH2 pip), 1.85 (m, 2 H, CH2 pip), 2.28–2.34 (m, 4 H, CH2), 2.50 (m, 2 H, CH2 ethyl), 2.54 (m, 2 H, CH2 pip), 3.88 (s, 2 H, ArCH2), 4.41 (m, 1 H, CH), 6.84 (m, 1 H, ArH), 6.95 (d, J = 7.9 Hz, 1 H, ArH), 7.11–7.25 (m, 7 H, ArH). 13C NMR (100 MHz, DMSO-d6) δ 30.14, 32.51, 35.72, 49.35, 53.60, 71.11, 112.93, 120.18, 125.70, 127.48, 128.15, 128.62, 129.97, 130.60, 141.15, 154.54, 174.29. HRMS calculated for C21H26NO3+ 340.1907 (M + H)+, found 340.1941.
(S)-N-(2-aminoethyl)-2-((S)-2-((S)-2-(3-(4-(2-benzylphenoxy)piperidin-1-yl)propanamido)propanamido)propanamido)propanamide (20)
1,2-Diaminoethane trityl resin (250 mg) was reacted according to the General Procedure A, and the crude resin-cleaved residue purified using preparative RP-HPLC to give 20 (46 mg, 0.065 mmol) as a white solid. Now with a bulky group containing stereocentres attached via N-piperidyl substitution the compound is a mixture of conformational isomers (cis/trans across piperidyl ring) which shows some peak resolution in the 1H and 13C NMR spectra, however the analytical RP-HPLC shows one sharp peak. Where the isomer is resolved in the NMR, this is denoted with an *asterisk for the minor isomer. NMR was assigned using COSY and HSQC spectra. 1H NMR (400 MHz, DMSO-d6) δ 1.20–1.23 (m, 9 H, ala CH3), 1.74 (m, 1 H, CHH pip), 1.95 (m, 2 H, CH2 pip), 2.21 (m, 1 H, CHH pip), 2.58–2.70 (m, 4 H, CH2 pip, COCH2), 2.84 (m, 2 H, H2NCH2), 3.16 (m, 2 H, pipNCH2), 3.27–3.31 (m, 4 H, NHCH2, CH2 pip), 3.87* (s, 2 H, ArCH2), 3.98 (s, 2 H, ArCH2), 4.18 (m, 1 H, ala CH), 4.25 (m, 1 H, ala CH), 4.32 (m, 1 H, ala CH), 4.55* (m, 1 H, CH pip), 4.72 (m, 1 H, CH pip), 6.91 (m, 1 H, ArH), 6.98 (d, J = 8.4 Hz, 1 H, ArH), 7.05* (d, J = 8.6 Hz, 1 H, ArH), 7.15–7.30 (m, 7 H, ArH), 7.84 (br s, 2 H, NH2), 7.92 (d, J = 7.3 Hz, 1 H, ala NH), 8.07 (t, J = 5.4 Hz, 1 H, NHCH2), 8.17 (d, J = 7.3 Hz, 1 H, ala NH), 8.39 (d, J = 7.0 Hz, 1 H, ala NH), 9.62* (br s, 2 H, NH2). 13C NMR (125 MHz, DMSO-d6) δ 17.81, 18.05, 18.19, 26.40, 28.18, 29.40, 29.62*, 35.57*, 35.97*, 36.42, 38.42, 47.29*, 47.38, 48.16, 48.29, 49.94, 50.03*, 51.28*, 52.08, 65.40, 69.98*, 112.24, 113.16*, 120.55, 120.91*, 125.76*, 125.83, 127.54*, 127.74, 128.17*, 128.36, 128.60, 129.51, 130.13*, 130.64*, 131.16, 140.95*, 141.37, 153.87, 154.22*, 168.56, 168.63*, 171.81, 172.08, 172.69. HRMS calculated for C32H47N6O5+ 595.3602 (M + H)+, found 595.3596.
(S)-N1-(2-aminoethyl)-2-(2-(2-(3-(4-(2-benzylphenoxy)piperidin-1-yl)propanamido)acetamido)acetamido)succinamide (21)
1,2-Diaminoethane trityl resin (250 mg) was reacted according to the General Procedure A, and the crude resin-cleaved residue purified using preparative RP-HPLC to give 21 (40 mg, 0.055 mmol) as a white solid. 1H NMR (400 MHz, DMSO-d6) δ 1.75 (m, 1 H, CHH pip), 1.96 (m, 2 H, CH2 pip), 2.12 (m, 1 H, CHH pip), 2.52 (m, 2 H, asn CH2), 2.64–2.71 (m, 4 H, CH2 pip, COCH2), 2.87 (m, 2 H, H2NCH2), 3.18 (m, 2 H, CH2 pip), 3.27–3.37 (m, 4 H, NHCH2, pipNCH2), 3.74–3.80 (m, 4 H, gly CH2), 3.87* (s, 2 H, ArCH2), 3.98 (s, 2 H, ArCH2), 4.50 (m, 1 H, asn CH), 4.56* (m, 1 H, CH pip), 4.73 (m, 1 H, tyr CH), 6.91 (m, 1 H, ArH), 6.99 (m, 1 H, ArH), 7.04* (m, 1 H, ArH), 7.16–7.28 (m, 7 H, ArH), 7.48 (s, 2 H, asn NH2), 7.83 (br s, 2 H, CH2NH2), 8.04 (t, J = 6.0 Hz, 1 H, NHCH2), 8.16 (d, J = 6.8 Hz, 1 H, asn NH), 8.25 (t, J = 5.7 Hz, 1 H, gly NH), 8.48 (br m, 1 H, gly NH), 9.71* (br s, 2 H, NH2). 13C NMR (125 MHz, DMSO-d6) δ 26.40, 28.21, 28.49*, 29.43, 29.65*, 35.61, 35.97, 36.41, 37.13, 38.69, 42.00, 42.07, 47.39, 48.61*, 49.79, 50.02*, 51.29*, 52.09, 54.93*, 65.43, 70.02*, 112.27, 113.18*, 113.64*, 116.03*, 118.41*, 120.56, 120.79*, 120.90*, 125.84, 127.58*, 127.74, 128.19*, 128.37, 128.61, 129.54, 130.14*, 130.64*, 131.15, 140.96*, 141.37, 153.89, 154.22*, 168.68, 169.28, 169.34, 171.60, 171.72. HRMS calculated for C31H44N7O6+ 610.3348 (M + H)+, found 610.3373.
(S)-N1-((S)-1-(((2 S,3 S)-1-((2-aminoethyl)amino)-3-hydroxy-1-oxobutan-2-yl)amino)-1-oxopropan-2-yl)-2-(3-(4-(2-benzylphenoxy)piperidin-1-yl)propanamido)succinamide (22)
1,2-Diaminoethane trityl resin (250 mg) was reacted according to the General Procedure A, and the crude resin-cleaved residue purified using preparative RP-HPLC to give 22 (55 mg, 0.07 mmol) as a white solid. HRMS calculated for C34H50N7O7+ 668.3766 (M + H)+, found 668.3737.
N-((5 S,8 S,11 S)-15-(4-(2-benzylphenoxy)piperidin-1-yl)-5,8,11-trimethyl-4,7,10,13-tetraoxo-3,6,9,12-tetraazapentadecyl)-6-(2-(4-((E)-2-(5,5-difluoro-7-(thiophen-2-yl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)vinyl)phenoxy)acetamido)hexanamide (23)
According to General Procedure B, the neutralized amine of 20 (2.4 mg, 4 µmol) was reacted with BODIPY 630/650-X-SE (1.25 mg, 1.9 µmol) to give a blue solid. This was dissolved in methanol, filtered through a small column containing Amberlyst 21 resin, and the methanol evaporated to give 23 (1.4 mg, 1.2 µmol, 65%) as a blue solid. HRMS calculated for C61H73BF2N9O8S 1140.5358 (M + H)+, found 1140.5352. Analytical RP-HPLC Rt = 20.9 min.
(S,E)-2-(2-(2-(3-(4-(2-benzylphenoxy)piperidin-1-yl)propanamido)acetamido)acetamido)-N1-(2-(6-(2-(4-(2-(5,5-difluoro-7-(thiophen-2-yl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1′-f][1,3,2]diazaborinin-3-yl)vinyl)phenoxy)acetamido)hexanamido)ethyl)succinamide (24)
According to General Procedure B, the neutralized amine of 21(3 mg, 4.9 µmol) was reacted with BODIPY 630/650-X-SE (1.25 mg, 1.9 µmol) to give a blue solid. This was dissolved in methanol, filtered through a small column containing Amberlyst 21 resin, and the methanol evaporated to give 24 (1.7 mg, 1.4 µmol, 73%) as a blue solid. HRMS calculated for C60H70BF2N10O9S 1155.5104 (M + H)+, 1155.5100. Analytical RP-HPLC Rt = 20.4 min.
(S)-2-(3-(4-(2-benzylphenoxy)piperidin-1-yl)propanamido)-N1-((15 S,18 S)-1-(4-((E)-2-(5,5-difluoro-7-(thiophen-2-yl)-5H-4λ4,5λ4-dipyrrolo[1,2-c:2′,1’-f][1,3,2]diazaborinin-3-yl)vinyl)phenoxy)-15-((S)-1-hydroxyethyl)-2,9,14,17-tetraoxo-3,10,13,16-tetraazanonadecan-18-yl)succinamide (25)
According to General Procedure B, the neutralized amine of 22 (3 mg, 4.5 µmol) was reacted with BODIPY 630/650-X-SE (1.25 mg, 1.9 µmol) to give a blue solid. This was dissolved in methanol, filtered through a small column containing Amberlyst 21 resin, and the methanol evaporated to give 25 (0.7 mg, 0.6 µmol, 37%) as a blue solid. HRMS calculated for C63H76BF2N10O10S 1213.5522 (M + H)+, found 1213.5541. Analytical RP-HPLC Rt = 20.5 min.
Computational modelling
Construction of the 23-H1R model
The H1R crystal structure (PDB-code 3RZE)32 was prepared by modeling missing atoms (K442, R481) and missing residues (F168-V174 in extracellular loop 2). The fused T4-lysozyme was removed and the C-terminus of C221 and N-terminus of L405 were linked and three residues upstream and downstream were minimized. All non-amino acids were removed except for the E-isomer of Doxepin (PDB-code 5EH). The complex was inserted in a POPC layer and waters and ions were added according to a previously described protocol60. Subsequently the system was equilibrated and simulated for 12.5 ns using GROMACS 4.6.161 using the Amber ff99SB-ILDN force field61,62. The final MD snapshot (with an RMSD of 1.6 Å compared to the crystal structure) was used for PLANTS63 docking of VUF13816 with increasing segments of the AAA linker. Finally the BY630 fluorophore was manually positioned between the protein and the membrane (near the final part of the linker) and connected to the linker. The full 23 ligand was subsequently minimized while keeping the membrane and the protein rigid.
General Pharmacology Methods
Materials
G418, Lipofectamine and Optimem were obtained from Life Technologies (Paisley, UK) and fetal calf serum from PAA Laboratories (Wokingham, UK). Furimazine was obtained from Promega (Southampton, UK). Fexofenadine and doxepin were from Tocris Bioscience (Bristol, UK). All other chemicals and reagents were obtained from Sigma-Aldrich (Gillingham, UK). GF/C plates, Microscint-O and [3H]mepyramine were from Perkin Elmer (Waltham, MA,USA). 25-kDa linear polyethylenimine for transfection was from Poly- sciences (Warrington, PA, USA).
Generation of cell lines and cell culture
The cDNA clone for the human H1R was obtained from the Missouri S&T cDNA Resource Centre (www.cDNA.org) and the H1-YFP was as used in Rose et al.26. Nluc-H1 was generated by amplifying the full length sequence H1R (with the methionine start signal removed) and fusing it in-frame with the membrane signal sequence of 5HT3A receptor and Nluc. Chinese hamster ovary (CHO; obtained from ATCC, Manassas, VA, USA) cells were transfected with cDNA encoding the H1R or H1-YFP and human embryonic kidney (HEK293T; obtained from ATCC) cells were transfected with Nluc-H1 using Lipofectamine (Life Technologies, Paisley, UK) according to the manufacturer’s instructions. Cells were cultured in medium containing 1 mg/ml G418 to select for successfully transfected cells. Clonal cell lines were subsequently generated for both the H1R and H1-YFP transfected cells. CHO cell lines were maintained in Dulbecco’s modified Eagles medium nutrient mix F12 (DMEM/F12) supplemented with 10% fetal calf serum and 2 mM L-glutamine at 37 °C in a humidified atmosphere of air/CO2 (19:1). HEK293T cells were maintained in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal calf serum. HEK293T cells were grown in the presence of 1% penicillin/streptomycin prior to transient transfection for [3H]-mepyramine ligand binding studies.
[3H]mepyramine saturation and competition binding
All [3H]mepyramine binding studies were performed on HEK293T cells transiently transfected to express the human H1R. This was achieved as follows, two million cells were seeded per 60 cm2 dish and transfected the following day with pcDEF3-HA-hH1R or pcDEF3-Nluc-hH1R using 5 μg DNA, 30 μg 25-kDa linear polyethylenimine in 150 mM NaCl per dish as described by Nijmeijer et al.64. Two days after transfection, cells were removed from the dish and washed in ice-cold phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4 and 2 mM KH2PO4). Cells pellets were obtained by centrifugation at 1900 × g for 10 min at 4 °C and stored at −20 °C until further experimentation. Cell pellets (expressing HA-hH1R) were reconstituted in HEPES buffered saline solution (HBSS; 145 mM NaCl, 5 mM KCl, 2 mM sodium pyruvate, 10 mM D-glucose, 10 mM HEPES, 1 mM MgSO4 and 1.3 mM CaCl2; pH = 7.4 at 37 °C) and dounce homogenized by plunging the pestle 10 times (Tamson, Bleiswijk, The Netherlands). For association binding experiments cells pellets (expressing either HA-hH1R or Nluc-hH1R) were instead reconstituted in phosphate buffer as described in Bosma et al.65 For saturation binding 1–4 μg of the cell homogenate was incubated (37 °C, 4 hours) with increasing concentrations of [3H]mepyramine (1–130 nM) both in the absence and presence of 10 µM mianserin to differentiate between total and non-specific binding. In competition binding experiments cell homogenate was incubated (37 °C, 4 hours) with 4 nM [3H]mepyramine and increasing concentrations competitor as required. Association binding experiments were performed according to the method described previously65, briefly, cell homogenate was incubation at 25 °C for between 1 and 41 min using 4 different concentrations of [3H]mepyramine (0–5 nM) both in the absence and presence of 10 µM mianserin to differentiate between total and non-specific binding. All ligand binding was performed under gentle agitation. Incubations were terminated by three rapid wash steps over GF/C filter plates using ice-cold 50 mM Tris-HCl buffer at pH7.4. Filter plates were dried at 52 °C before adding 25 μL Microscint-O per well. Finally, bound [3H]mepyramine was measured using a Wallac Microbeta counter (Perkin Elmer).
Intracellular calcium mobilization assay
CHO H1R cells were grown to confluence in black-walled, clear-bottom 96-well plates. On the day of the experiment, media was replaced with 100 µl HBSS containing 2.5 mM probenecid, 2.3 µM Fluo-4AM (Life Technologies), 0.023% Pluronic F-127, 0.5 mM Brilliant Black BN and where required 100 nM of fluorescent antagonist. Cells were incubated in this Fluo-4 containing buffer for 45 min at 37 °C in the dark. Plates were then loaded onto a multi-well fluorometric imaging plate reader (FlexStation; Molecular Devices, Sunnyvale, CA) and Fluo-4 fluorescence was measured (excitation, 485 nm; emission, 520 nm) every 1.52 s for 200 s. HBSS or HBSS containing the required concentration of histamine was added after 15 s.
Confocal Imaging
CHO H1R and CHO H1-YFP cells were grown to approximately 80% confluency on 8-well Labtek chambered coverglasses (Nunc Nalgene). On the day of the experiment, cells were washed twice in HBSS and then incubated in the presence or absence of 1 µM mepyramine for 30 min at 37 °C. Cells were then incubated with the required concentration of fluorescent ligand for 30 min prior to the collection of single equatorial confocal images. Images were obtained using a Zeiss LSM710 confocal microscope (Carl Zeiss GmbH, Jena, Germany) fitted with a 63× plan-Apochromat NA1.3 Ph3 oil-immersion lens within the School of Life Sciences Imaging Unit. For YFP, a 488 nm argon laser was used to excite the fluorophore and emission was detected using a BP505-30 filter. For the BY630 fluorophore, a 633 nm helium-neon laser was used for excitation and emission was detected using a 650 nm long pass filter. For all experiments a pinhole of 1 Airy Unit was used and fixed laser power, gain and offset for the BY630 containing compounds were kept the same for samples within each experiment.
Cell Membrane Preparation for NanoBRET assays
HEK293T cell stably expressing Nluc-H1 were grown to confluence in 500 cm2 dishes. Normal growth media was replaced with ice-cold PBS and the cells were removed from the dish by scraping. The cells were then transferred to a 50 ml tube and centrifuged at 250 × g for 5 min. The supernatant was removed and the resulting pellets stored at −80 °C. Thawed pellets were resuspended in PBS and homogenized using an electrical homogenizer in 10 × 2 s bursts. Unbroken cells and nuclei were removed by centrifugation (1200 × g for 10 min) and the supernatant subsequently centrifuged at 41,415 g for 30 min. The membrane pellet was resuspended in PBS and homogenized by 20 passes of a glass-on-Teflon homogenizer. Protein concentration was determined using a BCA protein assay and membranes stored at −80 °C until required.
NanoBRET binding assays
For whole cell NanoBRET assays, HEK293T cells stably expressing Nluc-H1 were seeded in white Thermo Scientific 96-well microplates and grown for 24 h prior to experimentation in normal growth medium. Immediately before experimentation, media was replaced with HBSS. For NanoBRET assay in membranes, membranes were diluted to the required concentration (10 µg/well) in binding buffer (50 mM Na2HPO4, 50 mM KH2PO4, pH 7.4) containing saponin (1 mg/ml) and placed in white Thermo Scientific 96-well microplates for 10 min prior to addition of compounds. For saturation and competition experiments in both whole cells and membranes the required concentrations of fluorescent ligand and competing ligand were added simultaneously. Plates were then incubated for 1 h at 37 °C (no CO2). After 1 h, 10 µM furimazine (Promega) was added to each well and fluorescence and luminescence measured after 5 min. For association and competition association kinetic experiments, 10 µM furimazine was added to each well and incubated at room temperature in the dark for 15 min to allow the luminescence signal to reach equilibrium. For association kinetics experiments, the required concentration of fluorescent ligand in the presence or absence of 10 µM doxepin was added simultaneously. For competition association kinetic experiments, 10 nM 10 was added in the presence or absence of the indicated concentration of unlabelled ligand. Non-specific binding was determined by the addition of 10 nM 10 and 10 µM doxepin. The plates were then read immediately with each well being read once per minute for 60 min. For all experiments fluorescence and luminescence was read sequentially using a PHERAstar FS plate reader (BMG Labtech) at room temperature. Filtered light emissions were measured at 460 nm (80-nm bandpass) and at >610 nm (longpass) and the raw BRET ratio was calculated by dividing the >610-nm emission by the 460-nm emission.
Data analysis
All data were analysed and presented using Prism 6 (GraphPad Software, San Diego, CA).
Total and non-specific saturation binding curves were fitted simultaneously using equation (1):
| 1 |
where Bmax is the maximal response, [B] is the concentration of fluorescent ligand in nM, KD is the equilibrium dissociation constant in nM, M is the slope of the non-specific binding component and C is the intercept with the Y-axis.
We fitted the both the radioligand and fluorescence competition binding curves using equation (2):
| 2 |
where [L] is the concentration of [3H]mepyramine, 10 or 23 in nM and KD is the equilibrium dissociation constant of the labelled ligand in nM (24 nM for [3H]mepyramine, 9.2 nM for 10 in whole cells, 28.8 nM for 10 in membranes and 9.8 nM for 23). The IC50 is calculated as in equation (3):
| 3 |
where [A] is the concentration of unlabelled competing drug and IC50 is the molar concentration of this competing ligand required to inhibit 50% of the specific binding of the concentration [L] of the labelled ligand.
For the calcium mobilization experiments, estimated affinity values (pKB) were calculated from the shift of the agonist concentration response curves in the presence of the fluorescent antagonists using equation (4):
| 4 |
Where DR (dose ratio) is the ratio of the agonist concentration required to stimulate an identical response in the presence and absence of antagonist, [B]. As there was a decrease in observed maximal efficacy of histamine in the presence of all six fluorescent antagonists, the EC25 value was used to determine the DR39.
From association kinetic data, non-specific binding was determined for each concentration of fluorescent ligand at each time point by the addition of 10 µM doxepin and was subtracted from total binding. Then kon, koff and KD values were obtained from the data using equation (5):
| 5 |
Where KD is the equilibrium dissociation constant and koff is the dissociation rate constant of the ligand in min−1. kon is the association rate constant in M−1 min−1 and is calculated as follows in equation (6):
| 6 |
Where [L] is the ligand concentration in M and kobs is calculated from global fitting of the data to the following monoexponential association function (equation (7)):
| 7 |
Here Ymax equals levels of binding at infinite time (t), and kobs is the rate constant for the observed rate of association.
The binding kinetics of unlabelled ligands was quantified in a competition association assay based on the theoretical framework proposed by Motulsky and Mahan42. The kon and koff of the unlabelled ligands were obtained using equations (8–14):
| 8 |
| 9 |
| 10 |
| 11 |
| 12 |
| 13 |
| 14 |
Where k1 is the kon of the labelled ligand (M−1min−1), k2 is the koff of the labelled ligand (min−1), L is the concentration of the labelled ligand in nM, I is the concentration of the unlabelled competitior in nM, X is the time (min) and Y is the specific binding of the labelled ligand (NanoBRET ratio). From these equations the kon (k3, M−1min−1), koff (k4, min−1) of the unlabelled ligand and the Bmax (total binding given as maximal BRET ratio) were calculated.
Statistical significance was determined by Student’s unpaired t test where p < 0.05 was deemed to be statistically significant throughout this study.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
This work was supported by the Medical Research Council [grant number MR/N020081/1]; and the Innovative Medicines Initiative Joint Undertaking under grant agreement 115366, resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007-2013) and European Federation of Pharmaceutical Industries and Associations (EFPIA) companies’ in-kind contribution.
Author Contributions
Conceived the study: Hill, Briddon, Stoddart, Kellam, Leurs. Chemical synthesis of fluorescent ligands: Vernall, Kellam. Molecular docking studies: de Graaf, Kooistra. Participated in research design: Hill, Briddon, Stoddart, Kellam, Leurs, Vischer, Bosma. Conducted experiments: Stoddart, Vernall, Bouzo-Lorenzo, Bosma. Performed data analysis: Stoddart, Vernall, Bouzo-Lorenzo, Bosma. Wrote or contributed to the writing of the manuscript: Stoddart, Hill, Bouzo-Lorenzo, Bosma, Briddon, de Graaf, Kooistra, Leurs, Vernall, Kellam.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-19714-2.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Barrie Kellam, Email: barrie.kellam@nottingham.ac.uk.
Stephen J. Hill, Email: steve.hill@nottingham.ac.uk
References
- 1.Santos R, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discov. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garland SL. Are GPCRs still a source of new targets? J Biomol Screen. 2013;18:947–966. doi: 10.1177/1087057113498418. [DOI] [PubMed] [Google Scholar]
- 3.Waring MJ, et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 2015;14:475–486. doi: 10.1038/nrd4609. [DOI] [PubMed] [Google Scholar]
- 4.Kumari P, Ghosh E, Shukla AK. Emerging Approaches to GPCR Ligand Screening for Drug Discovery. Trends Mol Med. 2015;21:687–701. doi: 10.1016/j.molmed.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Shonberg J, Kling RC, Gmeiner P, Lober S. GPCR crystal structures: Medicinal chemistry in the pocket. Bioorg Med Chem. 2015;23:3880–3906. doi: 10.1016/j.bmc.2014.12.034. [DOI] [PubMed] [Google Scholar]
- 6.McRobb FM, Negri A, Beuming T, Sherman W. Molecular dynamics techniques for modeling G protein-coupled receptors. Curr Opin Pharmacol. 2016;30:69–75. doi: 10.1016/j.coph.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 7.Hoffmann C, et al. Ligand residence time at G-protein-coupled receptors-why we should take our time to study it. Mol Pharmacol. 2015;88:552–560. doi: 10.1124/mol.115.099671. [DOI] [PubMed] [Google Scholar]
- 8.Vauquelin G, Charlton SJ. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br J Pharmacol. 2010;161:488–508. doi: 10.1111/j.1476-5381.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 10.Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kenakin TP. Biased signaling and allosteric machines; new vistas and challenges for drug discovery. Br J Pharmacol. 2012;166:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams, C. & Hill, S.J. GPCR signaling: understanding the pathway to successful drug discovery, in G protein-coupled receptors in drug discovery, Vol. 552. (ed. W.R. Leifert)39–50 (2009). [DOI] [PubMed]
- 13.Christopoulos A, et al. International Union of Basic and Clinical Pharmacology. XC. multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev. 2014;66:918–947. doi: 10.1124/pr.114.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoddart LA, Kilpatrick LE, Briddon SJ, Hill SJ. Probing the pharmacology of G protein-coupled receptors with fluorescent ligands. Neuropharmacology. 2015;98:48–57. doi: 10.1016/j.neuropharm.2015.04.033. [DOI] [PubMed] [Google Scholar]
- 15.Stoddart LA, White CW, Nguyen K, Hill SJ, Pfleger KD. Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Br J Pharmacol. 2016;173:3028–3037. doi: 10.1111/bph.13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoddart LA, et al. Fragment screening at adenosine-A3 receptors in living cells using a fluorescence-based binding assay. Chem Biol. 2012;19:1105–1115. doi: 10.1016/j.chembiol.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gherbi K, Briddon SJ, Hill SJ. Detection of the secondary, low-affinitybeta1 -adrenoceptor site in living cells using the fluorescent CGP 12177 derivative BODIPY-TMR-CGP. Br J Pharmacol. 2014;171:5431–5445. doi: 10.1111/bph.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corriden R, et al. Adenosine-A3 receptors in neutrophil microdomains promote the formation of bacteria-tethering cytonemes. EMBO reports. 2013;14:726–732. doi: 10.1038/embor.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo J, Song L, Liu M, Mahon MJ. Fluorescent ligand-directed co-localization of the parathyroid hormone 1 receptor with the brush-border scaffold complex of the proximal tubule reveals hormone-dependent changes in ezrin immunoreactivity consistent with inactivation. Biochim Biophys Acta Mol Cell Res. 2012;1823:2243–2253. doi: 10.1016/j.bbamcr.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daly CJ, et al. Fluorescent ligand binding reveals heterogeneous distribution of adrenoceptors and ‘cannabinoid-like’ receptors in small arteries. Br J Pharmacol. 2010;159:787–796. doi: 10.1111/j.1476-5381.2009.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corriden R, Kilpatrick LE, Kellam B, Briddon SJ, Hill SJ. Kinetic analysis of antagonist-occupied adenosine-A3 receptors within membrane microdomains of individual cells provides evidence of receptor dimerization and allosterism. FASEB J. 2014;28:4211–4222. doi: 10.1096/fj.13-247270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Briddon SJ, et al. Quantitative analysis of the formation and diffusion of A(1)-adenosine receptor-antagonist complexes in single living cells. Proc Natl Acad Sci USA. 2004;101:4673–4678. doi: 10.1073/pnas.0400420101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein Herenbrink C, et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat Commun. 2016;7:10842. doi: 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.May LT, Bridge LJ, Stoddart LA, Briddon SJ, Hill SJ. Allosteric interactions across native adenosine-A(3) receptor homodimers: quantification using single-cell ligand-binding kinetics. FASEB J. 2011;25:3465–3476. doi: 10.1096/fj.11-186296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiele F, Ayaz P, Fernandez-Montalvan A. A universal homogeneous assay for high-throughput determination of binding kinetics. Anal Biochem. 2014;468C:42–49. doi: 10.1016/j.ab.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 26.Rose RH, Briddon SJ, Hill SJ. A novel fluorescent histamine H1 receptor antagonist demonstrates the advantage of using fluorescence correlation spectroscopy to study the binding of lipophilic ligands. Br J Pharmacol. 2012;165:1789–1800. doi: 10.1111/j.1476-5381.2011.01640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vernall AJ, Hill SJ, Kellam B. The evolving small-molecule fluorescent-conjugate toolbox for Class A GPCRs. Br J Pharmacol. 2014;171:1073–1084. doi: 10.1111/bph.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vernall AJ, et al. Conversion of a non-selective adenosine receptor antagonist into A3-selective high affinity fluorescent probes using peptide-based linkers. Org Biomol Chem. 2013;11:5673–5682. doi: 10.1039/c3ob41221k. [DOI] [PubMed] [Google Scholar]
- 29.Hill SJ, et al. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–278. [PubMed] [Google Scholar]
- 30.Traiffort E, et al. Guinea pig histamine H1 receptor. I. Gene cloning, characterization, and tissue expression revealed by in situ hybridization. J Neurochem. 1994;62:507–518. doi: 10.1046/j.1471-4159.1994.62020507.x. [DOI] [PubMed] [Google Scholar]
- 31.Simons FE, Simons KJ. Histamine and H1-antihistamines: celebrating a century of progress. J Allergy Clin Immunol. 2011;128:1139–1150.e1134. doi: 10.1016/j.jaci.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 32.Shimamura T, et al. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–U82. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kooistra AJ, et al. Function-specific virtual screening for GPCR ligands using a combined scoring method. Sci Rep. 2016;6:28288. doi: 10.1038/srep28288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Graaf C, et al. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuhne S, et al. Identification of ligand binding hot spots of the histamine H1 receptor following structure-based fragment optimization. J Med Chem. 2016;59:9047–9061. doi: 10.1021/acs.jmedchem.6b00981. [DOI] [PubMed] [Google Scholar]
- 36.Vernall AJ, Stoddart LA, Briddon SJ, Hill SJ, Kellam B. Highly potent and selective fluorescent antagonists of the human adenosine A3 receptor based on the 1,2,4-triazolo 4,3-a quinoxalin-1-one scaffold. J Med Chem. 2012;55:1771–1782. doi: 10.1021/jm201722y. [DOI] [PubMed] [Google Scholar]
- 37.Schembri LS, et al. Synthesis, biological evaluation, and utility of fluorescent ligands targeting the mu-opioid receptor. J Med Chem. 2015;58:9754–9767. doi: 10.1021/acs.jmedchem.5b01664. [DOI] [PubMed] [Google Scholar]
- 38.Baker JG, et al. Influence of fluorophore and linker composition on the pharmacology of fluorescent adenosine A1 receptor ligands. Br J Pharmacol. 2010;159:772–786. doi: 10.1111/j.1476-5381.2009.00488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christopoulos A, Parsons AM, Lew MJ, El-Fakahany EE. The assessment of antagonist potency under conditions of transient response kinetics. Eur J Pharmacol. 1999;382:217–227. doi: 10.1016/S0014-2999(99)00550-6. [DOI] [PubMed] [Google Scholar]
- 40.Stoddart LA, et al. Application of BRET to monitor ligand binding to GPCRs. Nat Methods. 2015;12:661–663. doi: 10.1038/nmeth.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soave M, Stoddart LA, Brown A, Woolard J, Hill SJ. Use of a new proximity assay (NanoBRET) to investigate the ligand-binding characteristics of three fluorescent ligands to the human beta1-adrenoceptor expressed in HEK-293 cells. Pharmacol Res Perspect. 2016;4:e00250. doi: 10.1002/prp2.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Motulsky HJ, Mahan LC. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- 43.Sridharan R, Zuber J, Connelly SM, Mathew E, Dumont ME. Fluorescent approaches for understanding interactions of ligands with G protein coupled receptors. Biochim Biophys Acta. 2014;1838:15–33. doi: 10.1016/j.bbamem.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stoddart LA, Vernall AJ, Briddon SJ, Kellam B, Hill SJ. Direct visualisation of internalization of the adenosine A3 receptor and localization with arrestin3 using a fluorescent agonist. Neuropharmacology. 2015;98:68–77. doi: 10.1016/j.neuropharm.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 45.Morishima S, et al. Visualization and tissue distribution of alpha1L-adrenoceptor in human prostate by the fluorescently labeled ligand Alexa-488-silodosin. J Urol. 2010;183:812–819. doi: 10.1016/j.juro.2009.09.078. [DOI] [PubMed] [Google Scholar]
- 46.Lacivita E, et al. Identification of a red-emitting fluorescent ligand for in vitro visualization of human serotonin 5-HT(1A) receptors. Bioorg Med Chem Lett. 2010;20:6628–6632. doi: 10.1016/j.bmcl.2010.09.027. [DOI] [PubMed] [Google Scholar]
- 47.Mitronova GY, et al. high-affinity functional fluorescent ligands for human beta-adrenoceptors. Sci Rep. 2017;7:12319. doi: 10.1038/s41598-017-12468-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hall MP, et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ayoub MA, Pfleger KD. Recent advances in bioluminescence resonance energy transfer technologies to study GPCR heteromerization. Curr Opin Pharmacol. 2010;10:44–52. doi: 10.1016/j.coph.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 50.Cottet M, et al. BRET and time-resolved FRET strategy to study GPCR oligomerization: from cell lines toward native tissues. Front Endocrinol (Lausanne) 2012;3:92. doi: 10.3389/fendo.2012.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakker RA, et al. Domain swapping in the human histamine H1 receptor. J Pharmacol Exp Ther. 2004;311:131–138. doi: 10.1124/jpet.104.067041. [DOI] [PubMed] [Google Scholar]
- 52.Swinney DC, Haubrich BA, Van Liefde I, Vauquelin G. The role of binding kinetics in GPCR drug discovery. Curr Top Med Chem. 2015;15:2504–2522. doi: 10.2174/1568026615666150701113054. [DOI] [PubMed] [Google Scholar]
- 53.Schuetz DA, et al. Kinetics for Drug Discovery: an industry-driven effort to target drug residence time. Drug Discov Today. 2017;22:896–911. doi: 10.1016/j.drudis.2017.02.002. [DOI] [PubMed] [Google Scholar]
- 54.Vauquelin G, Bostoen S, Vanderheyden P, Seeman P. Clozapine, atypical antipsychotics, and the benefits of fast-off D2 dopamine receptor antagonism. Naunyn-Schmiedebergs Arch Pharmacol. 2012;385:337–372. doi: 10.1007/s00210-012-0734-2. [DOI] [PubMed] [Google Scholar]
- 55.Tee, A.K. et al. Long-acting beta2-agonists versus theophylline for maintenance treatment of asthma. The Cochrane database of systematic reviews, Cd001281 (2007). [DOI] [PMC free article] [PubMed]
- 56.Xia L, de Vries H, AP IJ, Heitman LH. Scintillation proximity assay (SPA) as a new approach to determine a ligand’s kinetic profile. A case in point for the adenosine A1 receptor. Purinergic Signal. 2016;12:115–126. doi: 10.1007/s11302-015-9485-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swinney DC, et al. A study of the molecular mechanism of binding kinetics and long residence times of human CCR5 receptor small molecule allosteric ligands. Br J Pharmacol. 2014;171:3364–3375. doi: 10.1111/bph.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen AH, et al. Development and characterization of a fluorescent tracer for the free fatty acid receptor 2 (FFA2/GPR43) J Med Chem. 2017;60:5638–5645. doi: 10.1021/acs.jmedchem.7b00338. [DOI] [PubMed] [Google Scholar]
- 59.Vanderheyden PML, Benachour N. Influence of the cellular environment on ligand binding kinetics at membrane-bound targets. Bioorg Med Chem Lett. 2017;27:3621–3628. doi: 10.1016/j.bmcl.2017.06.051. [DOI] [PubMed] [Google Scholar]
- 60.Cordomi A, Caltabiano G, Pardo L. Membrane Protein Simulations Using AMBER Force Field and Berger Lipid Parameters. J Chem Theory Comput. 2012;8:948–958. doi: 10.1021/ct200491c. [DOI] [PubMed] [Google Scholar]
- 61.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 62.Lindorff-Larsen K, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korb O, Stutzle T, Exner TE. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J Chem Inf Model. 2009;49:84–96. doi: 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- 64.Nijmeijer S, et al. Design and pharmacological characterization of VUF14480, a covalent partial agonist that interacts with cysteine 98(3.36) of the human histamine H(4) receptor. Br J Pharmacol. 2013;170:89–100. doi: 10.1111/bph.12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bosma R, Moritani R, Leurs R, Vischer HF. BRET-based beta-arrestin2 recruitment to the histamine H1 receptor for investigating antihistamine binding kinetics. Pharmacol Res. 2016;111:679–687. doi: 10.1016/j.phrs.2016.07.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.