

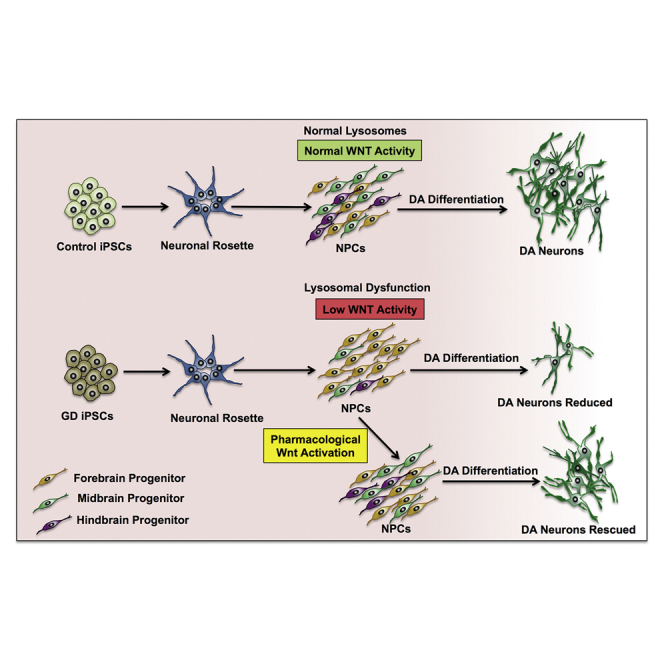
Summary
Gaucher’s disease (GD) is an autosomal recessive disorder caused by mutations in the GBA1 gene, which encodes acid β-glucocerebrosidase (GCase). Severe GBA1 mutations cause neuropathology that manifests soon after birth, suggesting that GCase deficiency interferes with neuronal development. We found that neuronopathic GD induced pluripotent stem cell (iPSC)-derived neuronal progenitor cells (NPCs) exhibit developmental defects due to downregulation of canonical Wnt/β-catenin signaling and that GD iPSCs’ ability to differentiate to dopaminergic (DA) neurons was strikingly reduced due to early loss of DA progenitors. Incubation of the mutant cells with the Wnt activator CHIR99021 (CHIR) or with recombinant GCase restored Wnt/β-catenin signaling and rescued DA differentiation. We also found that GD NPCs exhibit lysosomal dysfunction, which may be involved in Wnt downregulation by mutant GCase. We conclude that neuronopathic mutations in GCase lead to neurodevelopmental abnormalities due to a critical requirement of this enzyme for canonical Wnt/β-catenin signaling at early stages of neurogenesis.
Keywords: Gaucher’s disease, GBA1, glucocerebrosidase, neuronal progenitors, dopaminergic development, iPSCs, Wnt/β-catenin, lysosomal storage disease, neurodegeneration
Graphical Abstract
Highlights
-
•
Neuronopathic GBA1 mutations attenuate canonical Wnt signaling in iPSC-derived NPCs
-
•
GD NPC differentiation to DA neurons impaired due to early loss of DA progenitors
-
•
GBA1-mediated lysosomal alterations may be involved in Wnt signal downregulation
-
•
The Wnt pathway may be a potential new therapeutic target for neuronopathic GD
In this article, Feldman and colleagues describe a new mechanism linking severe GBA1 mutations to neurodevelopmental defects through Wnt/β-catenin downregulation. Using GD iPSCs as a model, the authors show that the ability of neuronopathic GD NPCs to differentiate to DA neurons is strikingly reduced due to early loss of DA progenitors and that lysosomal dysfunction may be directly involved in canonical Wnt downregulation.
Introduction
Gaucher’s disease (GD) is an inherited disorder caused by biallelic mutations in the GBA1 gene, which encodes the lysosomal enzyme β-glucocerebrosidase (GCase) (Cox, 2010). Reduced GCase activity results in accumulation of glucosylceramide and glucosylsphingosine in liver, spleen, bone marrow, and nervous system (Brady et al., 1966, Farfel-Becker et al., 2014, Thomas et al., 2014). Clinically, patients with severe mutations develop a broad range of neurological manifestations that vary in onset and severity (Sidransky, 2012). Neuronopathic GD subtypes (types 2 and 3 GD) are characterized by neuronal loss and degeneration in various areas of the brain including cerebral cortex, hypothalamus, cerebellum, and midbrain (Kaye et al., 1986, Wong et al., 2004). Type 3 GD exhibits a subacute, slow, progressive course, while type 2 GD develops rapid and extensive neuronal loss leading to death in early childhood due to neurodegeneration that starts during gestation (Pastores and Hughes, 1993, Orvisky et al., 2000, Stone et al., 2000, Weiss et al., 2015). GBA1 mutation is also the most frequent genetic risk factor for Parkinson's disease (PD) (Lwin et al., 2004, Swan and Saunders-Pullman, 2013).
Using GD mouse models and patient-derived induced pluripotent stem cells (iPSCs), it has been shown that GBA1 mutant neurons exhibit lysosomal alterations, defective autophagic clearance, accumulation of protein aggregates, and increased vulnerability to cell death (Sun and Grabowski, 2010, Sun et al., 2010, Mazzulli et al., 2011, Schondorf et al., 2014, Awad et al., 2015). We further showed that autophagy lysosomal pathway (ALP) alterations in GD are due to deregulation of transcription factor EB (TFEB) (Awad et al., 2015), the master regulator of lysosomal biogenesis and autophagy (Settembre et al., 2011, Settembre et al., 2012). In addition to the essential role of the ALP in the survival of post-mitotic neurons, this system plays a direct role in neuronal development and differentiation through subcellular remodeling (Song et al., 2008, Aburto et al., 2012). Moreover, recent reports showing that the endolysosomal compartment modulates canonical Wnt/β-catenin signaling (Taelman et al., 2010, Dobrowolski et al., 2012) further suggest that this compartment may also regulate neuronal development through direct interaction with neurodevelopmental signaling cascades.
The canonical Wnt/β-catenin pathway is a highly conserved developmental pathway that plays a key role in neuronal development (Loh et al., 2016, Noelanders and Vleminckx, 2016). Wnt ligands are secreted glycoproteins that bind to Frizzled receptor and LRP5/6 co-receptors on target cells (Mikels and Nusse, 2006). Wnt receptor binding prevents β-catenin association with its destruction complex, which consists of glycogen synthase kinase 3β (GSK3β), adenomatous polyposis coli, and axis inhibition protein (AXIN). This prevents β-catenin phosphorylation by GSK3β and its subsequent degradation by the proteasome (Verheyen and Gottardi, 2010, Nusse and Clevers, 2017). It has been proposed that sequestration of GSK3β into the endolysosomal compartment stabilizes β-catenin, allowing its translocation to the nucleus (Niehrs and Acebron, 2010). In the nucleus, β-catenin associates with TCF/LEF transcription factors to activate Wnt target genes, many of which regulate the survival, proliferation, and differentiation of neuronal stem/progenitor cells (Willert and Nusse, 1998, Grigoryan et al., 2008). Wnt/β-catenin signaling is important for brain development as well as maintaining neuronal functions during adulthood (Bengoa-Vergniory and Kypta, 2015, Noelanders and Vleminckx, 2016). Several Wnt family members also play a critical role in embryonic midbrain dopaminergic (DA) neurogenesis by regulating the survival, proliferation, and fate commitment of DA precursors (Joksimovic and Awatramani, 2014, Arenas et al., 2015). The importance of Wnt signaling in midbrain DA neurogenesis is highlighted by the use of chemical Wnt activators for efficient generation of midbrain DA neurons from cultured PSCs (Kriks et al., 2011). In agreement with this role of Wnt in DA development, genome-wide analysis of gene expression identified Wnt signaling as an over-represented pathway in iPSC-derived DA population (Momcilovic et al., 2014).
In this study we used GD iPSCs to investigate whether, in addition to deregulating lysosomal functions (Awad et al., 2015), neuronopathic GBA1 mutations would affect the developmental potential of neuronal stem cells. We found that severe biallelic mutations in GBA1 resulted in a dramatic decrease in the survival of DA progenitors due to interference with Wnt/β-catenin signaling. Consistent with mutant GBA1 interference with Wnt signaling, GD NPCs also exhibited reduced expression of hindbrain progenitor markers and an increased expression of forebrain progenitor markers. This mechanism highlights the requirement for normal GCase activity during early stages of neuronal development and points to the Wnt/β-catenin pathway as a potential therapeutic target for neuronopathic GD.
Results
Generation of GD iPSC-Derived Neuronal Progenitor Cells
In this study we used previously described control and GD iPSC lines that were derived from two neuronopathic type 2 GD patients harboring the biallelic mutations L444P/RecNciI and W184R/D409H (GD2), two neuronopathic type 3 patients with L444P/L444P mutations (GD3), and a non-neuronopathic type 1 GD patient carrying N370S/N370S mutations (GD1) (Panicker et al., 2012, Panicker et al., 2014, Sgambato et al., 2015). To generate neuronal progenitor cells (NPCs) from control and GD mutant iPSC lines, we followed our previously described protocol (Awad et al., 2015). In brief, iPSC-derived neuronal rosettes expressing neuronal stem cell markers SOX1 and PAX6 (Figure S1A) were manually picked and expanded in culture. The resulting NPC population from both control and GD mutant iPSC lines expressed similar levels of MUSASHI and SOX1 (Figure S1B). As shown in Figures S1C and S1D, GCase protein levels and enzymatic activity were decreased in neuronopathic NPCs, and incubation with recombinant GCase (rGCase) resulted in a significant increase in GCase activity in mutant cells.
Defective Dopaminergic Differentiation of Neuronopathic GD NPCs
Because of the well-established link between GBA1 mutations and PD, we evaluated the ability of GD NPCs to undergo dopaminergic differentiation using a protocol that recapitulates DA neurogenesis from primitive NPCs, which were picked and expanded from neuronal rosettes (Figure 1A) (Swistowski et al., 2009, Swistowski and Zeng, 2012). Following this protocol, freshly generated NPCs from both control and GD iPSC gave rise to DA neurons, as shown by tyrosine hydroxylase (TH) staining (Figure 1B). However, quantitative image analysis showed a small reduction in the percentage of TH-positive neurons generated from neuronopathic GD NPCs compared with control cells (Figure 1C). We also noticed that upon propagation in culture, while control GD NPCs retained their ability to generate DA neurons the GD NPC population exhibited a striking decline in its ability to differentiate to TH-positive neurons (Figure 2A). As shown in Figure 2B, quantitation of TH-positive neurons from immunofluorescence images showed a significant decrease in the percentage of TH-positive neurons generated from GD3 and GD2 NPCs compared with control cells. In contrast, we did not detect a significant difference in TH expression between control and non-neuronopathic GD1 NPCs after passage (Figure S2). When we examined the expression of additional DA markers, namely PITX3, VMAT2, LMX1, GIRK2, and FOX2A, there was also decreased expression of these DA markers in GD2 DA cultures compared with controls (Figure 2C). Consistent with our previous report (Awad et al., 2015), GD NPCs’ ability to differentiate to TUJ1 (neuron-specific class III β-tubulin)-positive neurons was not diminished compared with control NPCs (Figures 2A and 2C) (Awad et al., 2015). We then examined the expression level of DA differentiation gene regulatory network by qRT-PCR at days 0, 15, and 30 of DA differentiation. We found that in control DA cultures there was a significant induction of LMX1B and NURR1, two transcription factors that are critical for DA differentiation (Figure 3A). Concomitantly, control DA cultures exhibited significant upregulation of the DA markers TH, VMAT2, AADC, and DBH over the time course of DA differentiation (Figure 3A). In contrast, there was no significant induction of LMX1B, NURR1, or DA markers in GD2 and GD3 cultures over time, indicating downregulation of the DA gene regulatory network in GD NPCs. Next, we investigated whether incubation with rGCase would restore GD NPCs’ ability to differentiate to DA neurons. Treatment of GD NPCs with rGCase during the DA differentiation period did not upregulate DA marker expression as determined by qRT-PCR analysis (data not shown). However, when treatment of the GD cultures with rGCase started at the embryoid body (EB) stage, before neuroectodermal induction, and was continued throughout NPC generation and DA differentiation, there was an increase in the abundance of TH-expressing neurons (Figure 3B) and a significant upregulation of DA marker expression (Figure 3C). These results suggest that neuronopathic GD NPCs exhibit altered DA neurogenesis due to early defects in the neuronal progenitor population.
Figure 1.

Differentiation of Early-Passage NPCs to DA Neurons
(A) Schematic representation of the dopaminergic differentiation protocol from iPSCs used in this study (Swistowski and Zeng, 2012). EBs grown in adherent cultures gave rise to neural tube-like rosette structures. Neuronal rosettes were then manually picked and expanded in NPC medium. NPCs were differentiated to DA neurons by culturing in medium supplemented with SHH and FGF8 for 10 days, followed by prolonged culture in medium supplemented with BDNF and GDNF, as described in Supplemental Experimental Procedures.
(B) Representative immunofluorescence images from control, GD2, and GD3 dopaminergic differentiation cultures generated from freshly generated NPCs. Neurons were co-labeled with anti-TUJ1 (red) and anti-tyrosine hydroxylase (TH, green) antibodies. Magnification 20×; scale bar, 100 μm.
(C) Quantitative analysis of TH expression in control, GD2, and GD3 dopaminergic neurons from freshly generated NPCs. Bar graph represents the percentage of TH-positive neurons relative to control counted in three different fields per experiment ± SEM. n = 3 per group (compiled data are repeats from two GD2 and one GD3 patient). p > 0.05 between control and both GD3 and GD2 as assessed by one-way ANOVA.
Figure 2.

Defective Differentiation of Neuronopathic GD NPCs to DA Neurons
(A) Representative confocal z stack images from control, GD2, and GD3 dopaminergic neuronal cultures differentiated as described in the legend to Figure 1A. Neurons were co-labeled with anti-TUJ1 (red) and anti-TH (green) antibodies. Magnification 40×; scale bar, 50 μm.
(B) Quantitative analysis of TH expression in control, GD2, and GD3 DA neurons. Bar graph represents the percentage of TH-positive neurons relative to control counted in three different fields per experiment. n = 5 per group ± SEM (compiled data are repeats from two GD2 and two GD3 patients). ∗∗p < 0.005 between control and both GD2 and GD3 as assessed by one-way ANOVA.
(C) Representative immunofluorescence images for DA marker expression in control and GD2 DA neuronal cultures. Neurons were co-stained with antibodies to TUJ1 (red) and antibodies to PITX1, VMAT2, LMX1, GIRK2, or FOX2A (green) as indicated. Magnification 20×; scale bar, 100 μm.
Figure 3.

Downregulation of Dopaminergic Marker Gene Expression in Neuronopathic GD DA Neurons
(A) qRT-PCR analysis showing fold expression of DA marker gene expression in control, GD2, and GD3 DA neuronal cultures at days 0, 15, and 30 of dopaminergic differentiation according to the diagram in Figure 1A. Dopaminergic marker genes examined were NURR1, LMX1B, TH, VMAT2, AADC, and DBH. ∗∗p < 0.005 and ∗p < 0.05 between the indicated groups as assessed by one-way ANOVA. Error bars denote SEM; n = 3–4 per group (compiled data are repeats from two GD2 and one GD3 patient).
(B) Representative immunofluorescence images from control and GD2 DA neuronal cultures that were either left untreated or were treated with 0.24 U/mL rGCase at the EB stage. rGCase incubation was continued throughout the NPC generation and DA differentiation period. Neurons were co-labeled with anti-TUJ1 (red) and anti-tyrosine hydroxylase (TH, green) antibodies. Magnification 20×; scale bar, 100 μm.
(C) qRT-PCR analysis of DA marker gene expression in GD2 DA neurons that were either left untreated or were treated with 0.24 U/mL rGCase at the EB stage, with treatment continued throughout the NPC generation and DA differentiation period. Data represent fold change relative to control. ∗∗p < 0.005 and ∗p < 0.05 assessed by one-way ANOVA. Error bars denote SEM; n = 3 (compiled data are repeats from one GD2 patient).
Pharmacological Wnt/β-Catenin Activation Rescues Neuronopathic GD Dopaminergic Differentiation
During embryonic development, the Wnt/β-catenin signaling pathway is known to regulate dopaminergic differentiation by controlling the expression of DA gene regulatory network (Joksimovic and Awatramani, 2014). To determine whether exogenous Wnt/β-catenin activation would restore DA differentiation in GD NPCs, we treated the mutant progenitors with a pharmacological Wnt activator. To this end, we modified our protocol for DA differentiation outlined in Figure 1A by adding the Wnt activator CHIR99021 (CHIR) during the first 10 days of incubation of NPCs, during the Sonic Hedgehog (SHH)/fibroblast growth factor 8 (FGF8) incubation period. Immunofluorescence images showed that adding CHIR markedly increased TH expression in GD NPC cultures compared with those differentiated in the absence of CHIR (Figure 4A). Quantitation of TH-positive neurons showed a significant increase in the percentage of TH-positive neurons generated from GD2 NPC cultures differentiated in the presence of CHIR compared with those differentiated in its absence. However, TH expression was not fully restored to control levels (Figure 4B). qRT-PCR analysis showed that the expression of other DA markers was also significantly upregulated in GD cultures treated with CHIR (Figure 4C). The rescue of DA differentiation by pharmacological Wnt activation in the mutant cells (Figures 4A–4C) suggests that the defective DA differentiation observed may be due to impaired Wnt/β-catenin signaling in GD NPCs.
Figure 4.

Wnt Activation by CHIR Enhances Dopaminergic Differentiation of Neuronopathic GD NPCs
(A) Representative immunofluorescence images for control and GD2 dopaminergic neurons that were differentiated from NPCs in the presence or absence of 3 μM CHIR for the first 10 days of the differentiation protocol shown in Figure 1A. Neuronal cultures were labeled with antibodies against MAP2 (red) and TH (green). Rightmost panel is an enlargement of a small area from each corresponding TH panel showing TH expression in DA neurons differentiated in the presence or absence of CHIR. Magnification 20×; scale bar, 100 μm.
(B) Quantitative analysis of TH expression in GD2 DA neurons that were differentiated in the presence or absence of 3 μM CHIR for the first 10 days of the differentiation protocol shown in Figure 1A. Bar graph represents the percentage of TH-positive neurons relative to total cell number counted in three different fields ± SEM, n = 5 per group (compiled data are repeats from two GD2 patients). ∗∗p < 0.005 between control and GD2, and ∗p < 0.05 between GD2 and GD2+CHIR, as assessed by unpaired Student’s t test.
(C) qRT-PCR analysis of DA marker gene expression in GD2 DA neuronal cultures that were either left untreated or were treated with CHIR for the first 10 days of the differentiation protocol shown in Figure 1A. DA markers examined were AADC, DAT, LMX1B, TH, and DBH. Data represent fold change relative to control. ∗p < 0.05 between GD2 and GD2 + CHIR as assessed by unpaired Student's t test. Error bars denote SEM; n = 3 per group (compiled data are repeats from one GD2 patient).
Decreased Survival of Neuronopathic GD DA Progenitors in the Absence of Exogenous Wnt/β-Catenin Activation
We then determined whether neuronopathic GBA1 mutations interfere with the generation of DA progenitors, and further examined the requirement for exogenous Wnt/β-catenin activation in this process. To this end, we used an efficient DA differentiation protocol that has been optimized for enrichment of midbrain DA progenitors directly from iPSCs, which results in high yields of TH-positive neurons (Kriks et al., 2011). Significantly, CHIR is one of the key components in the DA differentiation media (flow diagram in Figure 5A). When we followed this protocol, both control and GD2 mutant iPSCs efficiently differentiated into DA neurons as shown by TH expression (Figure 5B), and the yield of TH-positive neurons was >85% in both control and GD2 cultures. This indicates that when a pharmacological Wnt activator is used to enrich for DA progenitors, GD iPSCs are capable of efficiently differentiating to TH-positive neurons.
Figure 5.

Decreased Survival of Neuronopathic GD NPCs in the Absence of Exogenous Wnt Activation
(A) Schematic representation of the protocol used for directed differentiation of iPSCs to DA neurons as described by Kriks et al. (2011).
(B) Representative immunofluorescence images for TH expression (green) in control and GD2 DA neurons generated by directed differentiation of iPSCs as shown in (A). Nuclei were labeled with DAPI (blue). Magnification 20×; scale bar, 100 μm.
(C) Control and GD iPSC were differentiated in the presence or absence of CHIR as indicated in the text. Images show the culture dishes containing control and GD2 iPSC cultures at day 13 of differentiation following the protocol described in (A).
(D) Control and GD2 iPSCs were differentiated in the presence or absence of CHIR and rGCase as indicated in the text. Panels show representative phase-contrast images for control and GD2 iPSC cultures at day 13 of DA differentiation following the protocol described in (A). Insets are an enlargement of a small area in each condition. Magnification 10×; scale bar, 100 μm.
To further investigate the requirement for exogenous Wnt activation during DA differentiation from GD iPSCs, we carried out directed differentiation of control and GD iPSCs to DA neurons in the presence or absence of CHIR. All of the protocol steps were precisely followed except for the omission of CHIR from day 3 of differentiation (Figure 5A). We found that initially both control and GD iPSC cultures grew similarly, but after 10 days from the beginning of CHIR treatment, in GD iPSC cultures not incubated with CHIR the majority of the cells died and detached from the culture dishes (Figure 5C). In striking difference, in the control iPSC cultures without CHIR, no significant loss of cells was observed and the cells continued to grow, albeit to a lower density than in the presence of CHIR (Figure 5D). We then determined whether rGCase would be able to functionally replace CHIR. To this end, we carried out directed differentiation of GD iPSC to DA neurons in the presence of rGCase without CHIR, and followed survival of the cultured cells. Interestingly, rGCase treatment of GD2 cultures grown without CHIR enhanced DA progenitor survival, and no noticeable loss of cells was observed (Figure 5D). These results lend strong support to the idea that defective DA neurogenesis in neuronopathic GD NPCs is due to decreased survival of DA progenitors because of interference of mutant GCase with canonical Wnt signaling.
Skewed Anterior-Posterior Axial Identity of GD NPCs
We also examined the anterior-posterior (A-P) axial identity of the NPC population, which has been shown to be regulated by endogenous Wnt activity both in vivo and in vitro (Nordstrom et al., 2002, Blauwkamp et al., 2012, Moya et al., 2014, Imaizumi et al., 2015). Immunofluorescence analysis using antibodies to regional specific markers showed that GD2 NPCs exhibited a marked decrease in both the hindbrain/spinal cord NPC marker HOXB4 and the midbrain NPC marker EN1, consistent with low Wnt activity (Figure S3A). On the other hand, expression of the forebrain NPC marker, FOXG1, which is not dependent on high Wnt activity, was markedly increased in GD2 compared with control NPCs (Figure S3A). qRT-PCR analysis of A-P gene expression confirmed the downregulation of posterior hindbrain markers (HOXB4 and HOXC4) and the upregulation of anterior forebrain markers (FOXG1 and SIX3) in GD NPCs (Figure S3B). Incubation of GD NPCs with CHIR resulted in increased HOXB4 and EN1 expression (Figure S3C), showing that exogenous Wnt activation resulted in a GD NPC population with a more posteriorized pattern of regional identity. These results further suggest that GBA1 mutation interferes with endogenous Wnt signaling, which skews the A-P identity of the mutant NPC population.
Downregulation of Canonical Wnt Signaling in Neuronopathic GD NPCs
To further investigate the effect of GBA1 mutations on Wnt/β-catenin signaling, we examined levels of β-catenin, the key mediator of canonical Wnt signaling, in GD versus control NPCs. As shown in Figure 6A, immunofluorescence images showed a noticeable decrease in β-catenin expression in GD2 and GD3 NPCs compared with controls. Incubation with rGCase increased β-catenin expression in GD2 and GD3 NPCs, demonstrating that this phenotype is caused by GCase deficiency. To determine whether the decreased β-catenin level in mutant NPCs was due to decreased protein stability, we treated control and mutant cells with a proteasome inhibitor (PSI). As shown in Figure 6B, PSI treatment did not significantly change total β-catenin levels in control NPCs but markedly increased those in GD2 and GD3 cells. We then examined the levels of active β-catenin (unphosphorylated form) and found that in GD2 NPCs, active β-catenin levels were significantly decreased compared with control NPCs, as assessed by immunofluorescence staining and immunoblot analysis (Figures 6C and 6E). Western blot analysis showed that incubation with PSI also restored active β-catenin expression in GD2 NPCs to control levels, suggesting that active β-catenin in the mutant cells is destabilized by proteasomal degradation (Figure 6D). To determine whether active β-catenin levels in mutant cells can be restored by pharmacological Wnt/β-catenin activators, we treated control and GD NPCs with the GSK3β inhibitor CHIR, which prevents β catenin destruction. As shown in Figure 6E, treatment with CHIR stabilized active β-catenin in GD NPCs, increasing its expression to a level similar to that in control cells. rGCase treatment also upregulated active β-catenin levels in GD NPCs (Figure 6E). We conclude that in neuronopathic GD NPCs, Wnt/β-catenin signaling is likely downregulated due to increased β-catenin degradation by the proteasome, and that both exogenous Wnt activation and rGCase treatment can stabilize β-catenin.
Figure 6.

Decreased β-Catenin Level in Neuronopathic GD NPCs
(A) Representative immunofluorescence images of control, GD3, and GD2 NPCs stained with anti-β-catenin antibody (green) and with DAPI (blue). Cells were either left untreated or were treated with 0.24 U/mL rGCase for 5 days. Magnification 20×; scale bar, 100 μm.
(B) Representative immunofluorescence images of control, GD3, and GD2 NPCs stained with anti-β-catenin antibodies (green) and with DAPI (blue). Cells were either left untreated or were treated with proteasome inhibitor (PSI) for 18 hr. Magnification 40×; scale bar, 75 μm.
(C) Representative immunofluorescence images of control and GD2 NPCs stained with anti-active β-catenin antibodies (green) and with DAPI (blue). Magnification 20×; scale bar, 100 μm.
(D) Representative Western blot showing active β-catenin level in control and GD2 NPCs that were either left untreated or were treated with PSI for 18 hr as indicated. β-Actin was used as a loading control.
(E) Representative Western blot showing active β-catenin level in control and GD2 NPCs. Cells were either left untreated, treated with rGCase for 5 days, or treated with the Wnt activator CHIR for 3 days as indicated. β-Actin was used as a loading control. Bar graph below represents quantitation of active β-catenin level in NPCs with and without treatment. Data represent average ± SEM, n = 3–4 per group (compiled data are repeats from one GD2 patient). ∗p < 0.05 between untreated GD2 and all groups as assessed by one-way ANOVA.
Decreased Level of pGSK3β(S9) in Neuronopathic GD NPCs
GSK3β, the major negative regulator of Wnt signaling, is a constitutively active kinase that phosphorylates β-catenin, triggering its degradation by the proteasome. The activity of GSK3β is negatively regulated by inhibitory phosphorylation on Ser 9, which stabilizes β-catenin by preventing its degradation (Doble and Woodgett, 2003, Verheyen and Gottardi, 2010). To investigate the mechanism of excess β-catenin degradation in GD NPCs, we examined the expression of both total GSK3β and pGSK3β(S9). Western blot analysis showed no noticeable difference in total GSK3β protein level between control and mutant NPCs (Figure 7A, upper panel). However, both the levels and subcellular localization of inhibitory pGSK3β(S9) were different in GD NPCs compared with control cells. Western blot analysis showed a significant decrease in pGSK3β(S9) levels in neuronopathic GD NPCs compared with control cells (Figure 7A, middle and lower panels). As shown in Figure 7B, immunofluorescence analysis using an antibody against pGSK3β(S9) also showed marked decrease in inhibitory pGSK3β(S9) signal in GD2 NPCs compared with controls. While control NPCs exhibited a bright, punctate pGSK3β(S9) signal clustered in the supranuclear region, the pGSK3β(S9) signal in GD2 NPCs was weak and diffuse in the cytoplasm (Figure 7B, insets). Treatment of GD2 NPCs with rGCase upregulated pGSK3β(S9) signal intensity and restored its punctate appearance in these cells (Figure 7B). We conclude that mutant GBA1 interferes with both the activity and subcellular distribution of GSK3β.
Figure 7.

Decreased pGSK3β(S9) Levels in Neuronopathic GD NPCs
(A) Representative Western blot showing levels of total GSK3β (upper panel) and pGSK3β(S9) (middle panel) in control and neuronopathic GD NPCs. β-Actin was used as a loading control. Bar graph below represents quantitation of pGSK3β(S9) levels in control, GD2, and GD3 NPCs. Data represent average ± SEM, n = 3–4 per group (compiled data are repeats from two GD2 and two GD3 patients). ∗∗p < 0.005 between control and GD2, ∗p < 0.05 between control and GD3.
(B) Representative immunofluorescence images for pGSK3β(S9) (green) and DAPI (blue) staining in control and GD2 NPCs. Cells were either left untreated or were treated with rGCase for 5 days. Magnification 40×; scale bar, 75 μm. Inset is an enlargement of an image area from untreated NPCs showing pGSK3β(S9) punctuated appearance in control but not in GD2 NPCs. Magnification 40×; scale bar, 25 μm.
Decreased Co-localization of pGSK3β(S9) with Lysosomes in Neuronopathic GD NPCs
It has been proposed that the endolysosomal compartment is involved in the positive regulation of Wnt/β-catenin signaling through GSK3β sequestration. Upon Wnt activation, GSK3β has been shown to be sequestered into endolysosomal vesicles known as microvesicular bodies (MVBs), thus protecting β-catenin from degradation (Taelman et al., 2010). We previously demonstrated lysosomal depletion and autophagy block in neuronopathic but not non-neuronopathic GD iPSC-derived differentiated neurons. These alterations were linked to downregulation of TFEB (Awad et al., 2015), the master regulator of lysosomal biogenesis and autophagy (Settembre et al., 2011). As shown in Figures S4A and S4B, immunoblot analysis using antibodies against the lysosomal marker LAMP1 and TFEB showed that both LAMP1 and TFEB levels were significantly lower in neuronopathic GD NPCs compared with control and non-neuronopathic (GD1) NPCs. Thus, neuronopathic GBA1 mutations caused lysosomal depletion and TFEB downregulation in early neuronal progenitors (Figure S4) as well as in differentiated neurons (Awad et al., 2015). To determine whether lysosomal alterations in GD NPCs were involved in Wnt deregulation, we examined co-localization of pGSK3β(S9) with LAMP1 by immunofluorescence imaging. We found that in control NPCs, the punctuated pGSK3β(S9) fluorescence signal overlapped with LAMP1 staining almost completely (Figure S5A), indicating that in control NPCs, pGSK3β(S9) co-localizes with lysosomes. In striking contrast, GD2 NPCs exhibited weak LAMP1 signal and decreased pGSK3β(S9) co-localization with LAMP1 (Figure S5A). Treatment with rGCase upregulated LAMP1 expression, restored the pGSK3β(S9) punctuated pattern, and increased pGSK3β(S9)/LAMP1 signal co-localization (Figure S5A). Figure S5B shows the quantitation of fluorescence signal intensity, which reflected a significant decrease in LAMP1 (Figure S5B, upper panel) and decreased pGSK3β(S9)/LAMP1 co-localization signal in GD NPCs compared with control cells (Figure S5B, lower panel). GD NPC treatment with rGCase significantly increased LAMP1 fluorescence intensity and co-localization of pGSK3β(S9) with LAMP1 (Figure S5B).
Treatment with the mTOR (mammalian target of rapamycin) inhibitor Torin has been shown to upregulate lysosomal biogenesis thorough TFEB activation (Settembre et al., 2012). As shown in Figure S6A, Torin treatment markedly upregulated LAMP1 expression in GD2 NPCs, and to a lesser extent in control cells. We then examined whether enhancing lysosomal biogenesis through mTOR inhibition would prevent β-catenin degradation in mutant cells. As shown in Figure S6B, Torin treatment induced a marked increase in active β-catenin levels in GD2 NPCs and had a smaller effect in control cells. Thus, increasing lysosomal biogenesis in the mutant cells either by Torin treatment (Figure S6) or by incubation with recombinant GCase (Figure S5) appeared to be sufficient to stabilize active β-catenin.
Taken together, our data suggest that a dysfunctional endolysosomal compartment in GD NPCs may contribute to β-catenin destabilization, thus interfering with canonical Wnt signaling.
Discussion
In this study we report that the ability of rosette-derived neuronopathic GD NPCs to differentiate to DA neurons was markedly reduced due to downregulation of Wnt/β-catenin signaling. This phenotype was rescued by incubation of the mutant NPCs with CHIR, a potent Wnt/β-catenin activator. Similar results were obtained using a different protocol, which has been optimized to obtain highly enriched populations of DA neurons directly from iPSCs (Kriks et al., 2011). In the presence of CHIR, a critical component of this DA differentiation protocol, the yield of TH-positive neurons from GD iPSC was above 85%. However, omission of CHIR resulted in a marked loss of DA progenitors in GD but not control cultures, demonstrating that the mutant DA progenitors require exogenous Wnt/β-catenin activation for their survival. We also found that rGCase was able to functionally substitute for CHIR, suggesting that normal GCase activity is required for canonical Wnt signaling and DA progenitor survival. A susceptibility of DA neuronal development to the deleterious effects of GBA1 mutations has been reported in some in vivo models of GBA1 deficiency but not in others. Zebrafish with a truncation in the ortholog of human GBA1 display a decrease in motor activity and a reduction in DA neuronal cell count, and this effect is independent from α-synuclein neurotoxicity, as zebrafish lack this gene (Keatinge et al., 2015); similarly, mutations or deletion of GBA1 Drosophila orthologs cause dopaminergic neuronal loss, defective locomotion, and a shorter life span (Maor et al., 2016, Sanchez-Martinez et al., 2016); however, in a GBA−/− medaka model of neuronopathic GD, the mutant fish exhibited brain infiltration of Gaucher-like cells, microgliosis, and progressive neuronal loss that included but was not limited to TH-positive cells (Uemura et al., 2015); and in mice with a targeted deletion of GBA1 in mature, DAT-expressing midbrain dopaminergic neurons (DAT-GBA1-KO mice) there was no reduction in the number of TH-positive neurons in the substantia nigra of the mutant mice (Soria et al., 2017). Thus, the effect of GBA1 mutation on DA neuronal numbers and survival seems to be model dependent, and our results using GD iPSC model will need to be validated in future studies.
Analysis of postmortem brains from neuronopathic GD patients shows widespread neurodegeneration in the cerebral cortex, hypothalamus, midbrain, and cerebellum (Kaye et al., 1986, Wong et al., 2004). There is also prominent gliosis, infiltration of Gaucher macrophages, and neuroinflammation (Kaye et al., 1986, Wong et al., 2004, Farfel-Becker et al., 2011, Vitner et al., 2012), indicative of the involvement of multiple cell types in the neuropathology of GD. As the Wnt/β-catenin pathway regulates neuronal progenitors in different areas of the brain (Chenn and Walsh, 2002, Nordstrom et al., 2002, Mutch et al., 2010, Munji et al., 2011, Blauwkamp et al., 2012), it is possible that Wnt downregulation by mutant GBA1 may interfere not only with DA neurogenesis but also with development of other regional neuronal subtypes. This idea is supported by our finding that in the GBA1 mutant NPC population, not only were midbrain DA progenitors depleted but also hindbrain/spinal cord progenitors, which are also dependent on high Wnt activity. On the other hand, forebrain progenitors, which require low Wnt activity, were highly abundant in mutant NPCs. Furthermore, exogenous Wnt activation by CHIR resulted in phenotypic rescue, with reversal of the mutant NPC population to a more posteriorized pattern of regional identity. These findings are consistent with previous reports demonstrating that establishment of the A-P axis in the developing brain is controlled by a gradient of Wnt signaling activity (Nordstrom et al., 2002, Blauwkamp et al., 2012, Moya et al., 2014, Imaizumi et al., 2015). We should note, however, that while iPSC models of diseases that are manifested in utero or shortly after birth are powerful tools to identify potential early developmental abnormalities, the results of this study cannot be extrapolated to GD pathogenesis in vivo and only point to future areas of inquiry. Thus, whether the Wnt downregulation uncovered in this neuronopathic GD iPSC model reflects pathological mechanisms of GBA1-associated neurodegeneration will require further analysis.
GBA1 mutations are the highest known risk factor for the development of PD, and 7% of all cases of PD have mutations in GBA1 (Lwin et al., 2004, Swan and Saunders-Pullman, 2013). Although the mechanisms involved in development of GBA1-associated PD are not well understood, there is evidence that mutant GBA1 increases the levels and aggregation of α-synuclein in midbrain DA neurons (Choi et al., 2011, Mazzulli et al., 2011, Mazzulli et al., 2016, Aflaki et al., 2016, Fernandes et al., 2016), and this is believed to be a leading cause of DA neuronal loss in PD. Our results showed that in contrast to controls, DA differentiation of GD NPCs in the absence of CHIR did not result in upregulation of LMX1b and NURR1, two Wnt-regulated transcription factors that are essential for the differentiation and maintenance of DA neurons (Zetterstrom et al., 1997, Kadkhodaei et al., 2009, Doucet-Beaupre et al., 2015, Laguna et al., 2015). Thus, our results raise the question of whether the mechanism described in this study might be a contributing factor in GBA1-associated PD. However, the Wnt alterations and developmental phenotype reported here were observed in neuronal progenitors carrying severe biallelic mutations, and no iPSCs from GD carriers were included in our study. Therefore, without further analysis our results cannot be extrapolated to pathogenic mechanisms in PD cases harboring heterozygous GBA1 mutations.
It has been reported that the endolysosomal compartment positively regulates Wnt signaling by sequestering GSK3β inside MVBs, thus diminishing the cytosolic availability of the β-catenin destruction complex (Taelman et al., 2010). In GD NPCs we detected reduced co-localization of pGSK3β with lysosomes, perhaps as a consequence of the lysosomal depletion caused by mutant GBA1. We also found that increasing lysosomal biogenesis by either mTOR inhibition or by incubation with rGCase increased the levels of active β-catenin in the mutant NPCs. Thus, reduced sequestration of GSK3β by a defective endolysosomal compartment in the mutant cells may result in increased β-catenin degradation and attenuation of the canonical Wnt signal. A similar mechanism of Wnt signal downregulation was recently reported in studies showing that diminished TFEB levels in AMPK-deficient EBs compromise the endolysosomal system, which in turn blunts Wnt signaling (Young et al., 2016). In our studies, treatment of the GD mutant cells with rGCase restored the endolysosomal compartment, increased GSK3β co-localization with lysosomes, and rescued Wnt activation and DA generation. Further analysis is required to ascertain the contribution of lysosomal alterations in GD NPCs to Wnt signal attenuation and impaired neurogenesis.
In summary, this study describes a mechanism by which severe mutations in GBA1 cause neurodevelopmental defects by interfering with Wnt/β-catenin signaling. Our finding that pharmacological Wnt activation can restore the developmental potential of GD NPCs suggests that early therapeutic intervention, before neuronal progenitors have been significantly depleted, may be a promising strategy to ameliorate neuronopathic GD.
Experimental Procedures
All control and GD iPSC lines used in this study have been previously described (Panicker et al., 2012, Panicker et al., 2014). The GD iPSC lines were derived from two acute neuronopathic type 2 GD patients harboring the biallelic mutations L444P/RecNciI and W184R/D409H (GD2), two neuronopathic type 3 GD patients with L444P/L444P mutations (GD3), and a non-neuronopathic type 1 GD patient with N370S/N370S mutations (GD1). Differentiation of NPCs to DA neurons was performed based on the protocol by Swistowski and Zeng (2012). For differentiation of DA neurons directly from iPSCs, we followed the protocol described by Kriks et al. (2011). All the work with human iPSCs described in this study was carried out with approval from the Institutional Review Board and Embryonic Stem Cell Research Oversight committees. Detailed information on all experimental procedures is described in Supplemental Experimental Procedures.
Statistical Analysis
Data presented were compiled from independent experiments using iPSCs that were derived from two type 2 GD patients and two type 3 GD patients, referred to as GD2 and GD3, respectively (as indicated in each corresponding figure legend). Results are expressed as mean ± SEM. Data were analyzed using one-way ANOVA followed by Tukey’s or Bonferroni’s post test to determine statistical differences between multiple groups. Two-tailed unpaired Student’s t tests were used for comparison between two groups when appropriate. p values of <0.05 were considered statistically significant. The confidence level for significance was 95%. Data were analyzed using Prism software version 4.0c (GraphPad Software).
Author Contributions
O.A. directed the project, planned and carried out experiments, analyzed data, and wrote the manuscript. T.S.P. and E.T.Z. provided reagents, planned experiments, and analyzed data. L.M.P., R.M.D., and M.P.S. planned and carried out experiments and analyzed data. R.A.B., A.V., and T.P. carried out experiments and analyzed data. R.A.F. directed the project, planned experiments, analyzed data, and wrote the manuscript.
Acknowledgments
This work was supported by grants from the Maryland Stem Cell Research Fund (MSCRF) 2015-MSCRFI-1662 (to R.A.F.), the Children's Gaucher Research Fund (CGRF) (to R.A.F.), the Michael J. Fox Foundation for Parkinson's Research, Priority Target Award 11577 (to O.A.), and the NIH (NIH/NEI R01EY023962 to E.T.Z. and T.S.P., NIH/NICHD R01HD082098 to E.T.Z., and RPB Stein Innovation Award to E.T.Z.). A.V. and T.P. were supported by the PRISM Program/OSR and the Scholars/MPowering Program of the University of Maryland School of Medicine, respectively.
Published: November 30, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2017.10.029.
Supplemental Information
References
- Aburto M.R., Hurle J.M., Varela-Nieto I., Magarinos M. Autophagy during vertebrate development. Cells. 2012;1:428–448. doi: 10.3390/cells1030428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aflaki E., Borger D.K., Moaven N., Stubblefield B.K., Rogers S.A., Patnaik S., Schoenen F.J., Westbroek W., Zheng W., Sullivan P. A new glucocerebrosidase chaperone reduces alpha-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and Parkinsonism. J. Neurosci. 2016;36:7441–7452. doi: 10.1523/JNEUROSCI.0636-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas E., Denham M., Villaescusa J.C. How to make a midbrain dopaminergic neuron. Development. 2015;142:1918–1936. doi: 10.1242/dev.097394. [DOI] [PubMed] [Google Scholar]
- Awad O., Sarkar C., Panicker L.M., Miller D., Zeng X., Sgambato J.A., Lipinski M.M., Feldman R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 2015;24:5775–5788. doi: 10.1093/hmg/ddv297. [DOI] [PubMed] [Google Scholar]
- Bengoa-Vergniory N., Kypta R.M. Canonical and noncanonical Wnt signaling in neural stem/progenitor cells. Cell Mol. Life Sci. 2015;72:4157–4172. doi: 10.1007/s00018-015-2028-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwkamp T.A., Nigam S., Ardehali R., Weissman I.L., Nusse R. Endogenous Wnt signalling in human embryonic stem cells generates an equilibrium of distinct lineage-specified progenitors. Nat. Commun. 2012;3:1070. doi: 10.1038/ncomms2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady R.O., Kanfer J.N., Bradley R.M., Shapiro D. Demonstration of a deficiency of glucocerebroside-cleaving enzyme in Gaucher's disease. J. Clin. Invest. 1966;45:1112–1115. doi: 10.1172/JCI105417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A., Walsh C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Choi J.H., Stubblefield B., Cookson M.R., Goldin E., Velayati A., Tayebi N., Sidransky E. Aggregation of alpha-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol. Genet. Metab. 2011;104:185–188. doi: 10.1016/j.ymgme.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T.M. Gaucher disease: clinical profile and therapeutic developments. Biologics. 2010;4:299–313. doi: 10.2147/BTT.S7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doble B.W., Woodgett J.R. GSK-3: tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003;116(Pt 7):1175–1186. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski R., Vick P., Ploper D., Gumper I., Snitkin H., Sabatini D.D., De Robertis E.M. Presenilin deficiency or lysosomal inhibition enhances Wnt signaling through relocalization of GSK3 to the late-endosomal compartment. Cell Rep. 2012;2:1316–1328. doi: 10.1016/j.celrep.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet-Beaupre H., Ang S.L., Levesque M. Cell fate determination, neuronal maintenance and disease state: the emerging role of transcription factors Lmx1a and Lmx1b. FEBS Lett. 2015;589(24 Pt A):3727–3738. doi: 10.1016/j.febslet.2015.10.020. [DOI] [PubMed] [Google Scholar]
- Farfel-Becker T., Vitner E.B., Kelly S.L., Bame J.R., Duan J., Shinder V., Merrill A.H., Jr., Dobrenis K., Futerman A.H. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 2014;23:843–854. doi: 10.1093/hmg/ddt468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfel-Becker T., Vitner E.B., Pressey S.N., Eilam R., Cooper J.D., Futerman A.H. Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum. Mol. Genet. 2011;20:1375–1386. doi: 10.1093/hmg/ddr019. [DOI] [PubMed] [Google Scholar]
- Fernandes H.J., Hartfield E.M., Christian H.C., Emmanoulidou E., Zheng Y., Booth H., Bogetofte H., Lang C., Ryan B.J., Sardi S.P. ER stress and autophagic perturbations lead to elevated extracellular alpha-Synuclein in GBA-N370S Parkinson's iPSC-derived dopamine neurons. Stem Cell Reports. 2016;6:342–356. doi: 10.1016/j.stemcr.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryan T., Wend P., Klaus A., Birchmeier W. Deciphering the function of canonical Wnt signals in development and disease: conditional loss- and gain-of-function mutations of beta-catenin in mice. Genes Dev. 2008;22:2308–2341. doi: 10.1101/gad.1686208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi K., Sone T., Ibata K., Fujimori K., Yuzaki M., Akamatsu W., Okano H. Controlling the regional identity of hPSC-derived neurons to uncover neuronal subtype specificity of neurological disease phenotypes. Stem Cell Reports. 2015;5:1010–1022. doi: 10.1016/j.stemcr.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksimovic M., Awatramani R. Wnt/beta-catenin signaling in midbrain dopaminergic neuron specification and neurogenesis. J. Mol. Cell Biol. 2014;6:27–33. doi: 10.1093/jmcb/mjt043. [DOI] [PubMed] [Google Scholar]
- Kadkhodaei B., Ito T., Joodmardi E., Mattsson B., Rouillard C., Carta M., Muramatsu S., Sumi-Ichinose C., Nomura T., Metzger D. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J. Neurosci. 2009;29:15923–15932. doi: 10.1523/JNEUROSCI.3910-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye E.M., Ullman M.D., Wilson E.R., Barranger J.A. Type 2 and type 3 Gaucher disease: a morphological and biochemical study. Ann. Neurol. 1986;20:223–230. doi: 10.1002/ana.410200208. [DOI] [PubMed] [Google Scholar]
- Keatinge M., Bui H., Menke A., Chen Y.C., Sokol A.M., Bai Q., Ellett F., Da Costa M., Burke D., Gegg M. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015;24:6640–6652. doi: 10.1093/hmg/ddv369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriks S., Shim J.W., Piao J., Ganat Y.M., Wakeman D.R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguna A., Schintu N., Nobre A., Alvarsson A., Volakakis N., Jacobsen J.K., Gomez-Galan M., Sopova E., Joodmardi E., Yoshitake T. Dopaminergic control of autophagic-lysosomal function implicates Lmx1b in Parkinson's disease. Nat. Neurosci. 2015;18:826–835. doi: 10.1038/nn.4004. [DOI] [PubMed] [Google Scholar]
- Loh K.M., van Amerongen R., Nusse R. Generating cellular diversity and spatial form: Wnt signaling and the evolution of multicellular animals. Dev. Cell. 2016;38:643–655. doi: 10.1016/j.devcel.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Lwin A., Orvisky E., Goker-Alpan O., LaMarca M.E., Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004;81:70–73. doi: 10.1016/j.ymgme.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Maor G., Cabasso O., Krivoruk O., Rodriguez J., Steller H., Segal D., Horowitz M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016;25:2712–2727. doi: 10.1093/hmg/ddw129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli J.R., Xu Y.H., Sun Y., Knight A.L., McLean P.J., Caldwell G.A., Sidransky E., Grabowski G.A., Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli J.R., Zunke F., Tsunemi T., Toker N.J., Jeon S., Burbulla L.F., Patnaik S., Sidransky E., Marugan J.J., Sue C.M. Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J. Neurosci. 2016;36:7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikels A.J., Nusse R. Wnts as ligands: processing, secretion and reception. Oncogene. 2006;25:7461–7468. doi: 10.1038/sj.onc.1210053. [DOI] [PubMed] [Google Scholar]
- Momcilovic O., Liu Q., Swistowski A., Russo-Tait T., Zhao Y., Rao M.S., Zeng X. Genome wide profiling of dopaminergic neurons derived from human embryonic and induced pluripotent stem cells. Stem Cells Dev. 2014;23:406–420. doi: 10.1089/scd.2013.0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya N., Cutts J., Gaasterland T., Willert K., Brafman D.A. Endogenous WNT signaling regulates hPSC-derived neural progenitor cell heterogeneity and specifies their regional identity. Stem Cell Reports. 2014;3:1015–1028. doi: 10.1016/j.stemcr.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munji R.N., Choe Y., Li G., Siegenthaler J.A., Pleasure S.J. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J. Neurosci. 2011;31:1676–1687. doi: 10.1523/JNEUROSCI.5404-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch C.A., Schulte J.D., Olson E., Chenn A. Beta-catenin signaling negatively regulates intermediate progenitor population numbers in the developing cortex. PLoS One. 2010;5:e12376. doi: 10.1371/journal.pone.0012376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C., Acebron S.P. Wnt signaling: multivesicular bodies hold GSK3 captive. Cell. 2010;143:1044–1046. doi: 10.1016/j.cell.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Noelanders R., Vleminckx K. How Wnt signaling builds the brain: bridging development and disease. Neuroscientist. 2016 doi: 10.1177/1073858416667270. [DOI] [PubMed] [Google Scholar]
- Nordstrom U., Jessell T.M., Edlund T. Progressive induction of caudal neural character by graded Wnt signaling. Nat. Neurosci. 2002;5:525–532. doi: 10.1038/nn0602-854. [DOI] [PubMed] [Google Scholar]
- Nusse R., Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169:985–999. doi: 10.1016/j.cell.2017.05.016. [DOI] [PubMed] [Google Scholar]
- Orvisky E., Sidransky E., McKinney C.E., Lamarca M.E., Samimi R., Krasnewich D., Martin B.M., Ginns E.I. Glucosylsphingosine accumulation in mice and patients with type 2 Gaucher disease begins early in gestation. Pediatr. Res. 2000;48:233–237. doi: 10.1203/00006450-200008000-00018. [DOI] [PubMed] [Google Scholar]
- Panicker L.M., Miller D., Awad O., Bose V., Lun Y., Park T.S., Zambidis E.T., Sgambato J.A., Feldman R.A. Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells. 2014;32:2338–2349. doi: 10.1002/stem.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker L.M., Miller D., Park T.S., Patel B., Azevedo J.L., Awad O., Masood M.A., Veenstra T.D., Goldin E., Stubblefield B.K. Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc. Natl. Acad. Sci. USA. 2012;109:18054–18059. doi: 10.1073/pnas.1207889109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastores G.M., Hughes D.A. Gaucher disease. In: Pagon R.A., Adam M.P., Bird T.D., Dolan C.R., Fong C.T., Stephens K., editors. GeneReviews® [Internet] University of Washington; 1993. [Google Scholar]
- Sanchez-Martinez A., Beavan M., Gegg M.E., Chau K.Y., Whitworth A.J., Schapira A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016;6:31380. doi: 10.1038/srep31380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schondorf D.C., Aureli M., McAllister F.E., Hindley C.J., Mayer F., Schmid B., Sardi S.P., Valsecchi M., Hoffmann S., Schwarz L.K. iPSC-derived neurons from GBA1-associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014;5:4028. doi: 10.1038/ncomms5028. [DOI] [PubMed] [Google Scholar]
- Settembre C., Di Malta C., Polito V.A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C., Facchinetti V. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato J.A., Park T.S., Miller D., Panicker L.M., Sidransky E., Lun Y., Awad O., Bentzen S.M., Zambidis E.T., Feldman R.A. Gaucher disease-induced pluripotent stem cells display decreased erythroid potential and aberrant myelopoiesis. Stem Cells Transl. Med. 2015;4:878–886. doi: 10.5966/sctm.2014-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E. Gaucher disease: insights from a rare Mendelian disorder. Discov. Med. 2012;14:273–281. [PMC free article] [PubMed] [Google Scholar]
- Song J.W., Misgeld T., Kang H., Knecht S., Lu J., Cao Y., Cotman S.L., Bishop D.L., Lichtman J.W. Lysosomal activity associated with developmental axon pruning. J. Neurosci. 2008;28:8993–9001. doi: 10.1523/JNEUROSCI.0720-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria F.N., Engeln M., Martinez-Vicente M., Glangetas C., Lopez-Gonzalez M.J., Dovero S., Dehay B., Normand E., Vila M., Favereaux A. Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration. Hum. Mol. Genet. 2017;26:2603–2615. doi: 10.1093/hmg/ddx120. [DOI] [PubMed] [Google Scholar]
- Stone D.L., Tayebi N., Orvisky E., Stubblefield B., Madike V., Sidransky E. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum. Mutat. 2000;15:181–188. doi: 10.1002/(SICI)1098-1004(200002)15:2<181::AID-HUMU7>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Sun Y., Grabowski G.A. Impaired autophagosomes and lysosomes in neuronopathic Gaucher disease. Autophagy. 2010;6:648–649. doi: 10.4161/auto.6.5.12047. [DOI] [PubMed] [Google Scholar]
- Sun Y., Liou B., Ran H., Skelton M.R., Williams M.T., Vorhees C.V., Kitatani K., Hannun Y.A., Witte D.P., Xu Y.H. Neuronopathic Gaucher disease in the mouse: viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010;19:1088–1097. doi: 10.1093/hmg/ddp580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan M., Saunders-Pullman R. The association between β-glucocerebrosidase mutations and parkinsonism. Curr. Neurol. Neurosci. Rep. 2013;13:368. doi: 10.1007/s11910-013-0368-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swistowski A., Peng J., Han Y., Swistowska A.M., Rao M.S., Zeng X. Xeno-free defined conditions for culture of human embryonic stem cells, neural stem cells and dopaminergic neurons derived from them. PLoS One. 2009;4:e6233. doi: 10.1371/journal.pone.0006233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swistowski A., Zeng X. Scalable production of transplantable dopaminergic neurons from hESCs and iPSCs in xeno-free defined conditions. Curr. Protoc. Stem Cell Biol. 2012;Chapter 2 doi: 10.1002/9780470151808.sc02d12s22. Unit2D.12. [DOI] [PubMed] [Google Scholar]
- Taelman V.F., Dobrowolski R., Plouhinec J.L., Fuentealba L.C., Vorwald P.P., Gumper I., Sabatini D.D., De Robertis E.M. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A.S., Mehta A., Hughes D.A. Gaucher disease: haematological presentations and complications. Br. J. Haematol. 2014;165:427–440. doi: 10.1111/bjh.12804. [DOI] [PubMed] [Google Scholar]
- Uemura N., Koike M., Ansai S., Kinoshita M., Ishikawa-Fujiwara T., Matsui H., Naruse K., Sakamoto N., Uchiyama Y., Todo T. Viable neuronopathic Gaucher disease model in Medaka (Oryzias latipes) displays axonal accumulation of alpha-synuclein. PLoS Genet. 2015;11:e1005065. doi: 10.1371/journal.pgen.1005065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verheyen E.M., Gottardi C.J. Regulation of Wnt/beta-catenin signaling by protein kinases. Dev. Dyn. 2010;239:34–44. doi: 10.1002/dvdy.22019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitner E.B., Farfel-Becker T., Eilam R., Biton I., Futerman A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher's disease. Brain. 2012;135(Pt 6):1724–1735. doi: 10.1093/brain/aws095. [DOI] [PubMed] [Google Scholar]
- Weiss K., Gonzalez A.N., Lopez G., Pedoeim L., Groden C., Sidransky E. The clinical management of Type 2 Gaucher disease. Mol. Genet. Metab. 2015;114:110–122. doi: 10.1016/j.ymgme.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K., Nusse R. Beta-catenin: a key mediator of Wnt signaling. Curr. Opin. Genet. Dev. 1998;8:95–102. doi: 10.1016/s0959-437x(98)80068-3. [DOI] [PubMed] [Google Scholar]
- Wong K., Sidransky E., Verma A., Mixon T., Sandberg G.D., Wakefield L.K., Morrison A., Lwin A., Colegial C., Allman J.M. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Young N.P., Kamireddy A., Van Nostrand J.L., Eichner L.J., Shokhirev M.N., Dayn Y., Shaw R.J. AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 2016;30:535–552. doi: 10.1101/gad.274142.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom R.H., Solomin L., Jansson L., Hoffer B.J., Olson L., Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.