

Abstract
Recent progress in the synthesis and characterization of metal–organic frameworks (MOFs) has opened the door to an increasing number of possible catalytic applications. The great versatility of MOFs creates a large chemical space, whose thorough experimental examination becomes practically impossible. Therefore, computational modeling is a key tool to support, rationalize, and guide experimental efforts. In this outlook we survey the main methodologies employed to model MOFs for catalysis, and we review selected recent studies on the functionalization of their nodes. We pay special attention to catalytic applications involving natural gas conversion.
Short abstract
We discuss computational design of catalytic materials by taking advantage of the capability of metal−organic frameworks (MOFs) to combine the advantages of homogeneous and heterogeneous catalysts.
1. The Challenge of Catalyst Design
The discovery and design of superior catalysts is a fundamental challenge of modern chemistry.1 The use of quantum mechanical calculations for catalyst design is poised to revolutionize the field. The catalysts that are most suitable for both experimental structural characterization and computational modeling are well-defined sites with single-atom or well-defined-cluster catalysts in a uniform mesoscale environment; ideally they should be isolated from each other on supports to minimize cluster–cluster interactions, sintering, and agglomeration, all of which can decrease catalytic activity. Furthermore, one would like to ensure uniformity of critical factors like cluster size, shape, composition, and arrangement of atoms. There are remarkably few experimental examples of uniform, well-defined, supported subnanometer cluster catalysts due to the challenges of synthesis and the limits of stability. The primary examples are supported metal clusters, and a few highly selective and active catalysts of this type have been described.2−5 Homogeneous catalysts are often more active, selective, and well-defined than heterogeneous ones, but they may lack stability and are less widely used than heterogeneous catalysts with a major reason being challenges in separation of products, catalysts, and additives, including the recovery of catalysts for subsequent catalytic cycles.
Metal–organic frameworks (MOFs) are highly tunable nanoporous or microporous (and sometimes mesoporous) network compounds composed of inorganic nodes connected by organic linkers,6−18 and they offer tremendous potential for rational materials design.19−24 MOF-supported catalysts can in principle exploit the advantages of homogeneous catalysis without most of its disadvantages by isolating highly active catalytic sites in a robust and recyclable solid framework having crystalline or near-crystalline periodicity. Taking advantage of this structural control, applications of MOFs for catalysis have started to emerge;7,25−29 but the large variety of possible nodes and organic linkers that may connect them renders the potential number of MOF-supported cluster catalysts enormous, so there are many possibilities that have not been explored yet. It is impractical to synthesize even a modest fraction of the possibilities, much less to experimentally characterize their structures, physical properties, and catalytic efficacy. Computational modeling offers the potential to sort out the myriad of possibilities and prioritize synthetic planning, characterization, and kinetics testing at affordable cost.
MOFs can play several roles in catalysis by serving as catalytic species, by encapsulating them, or by acting as support materials for metals and anchored inorganometallic complexes.30 Postsynthetic modification allows one to precisely tailor and design MOFs for specific applications. Although much work has been devoted to the functionalization of linkers,31−34 less attention has been paid to nodes.35 Common techniques that target node functionalization (Figure 1) are solvent-assisted ligand incorporation (SALI),36 atomic layer deposition in MOFs (AIM),37 solvothermal deposition in MOFs (SIM),38,39 and metal exchange (ME).38,40 Whereas SALI incorporates organic groups on the nodes, AIM, SIM, and ME allow synthesis of a variety of metal-functionalized nodes.
Figure 1.

Methodologies for postsynthetic functionalization of MOF nodes.
Zr-based MOFs are especially appealing materials41,42 due to their outstanding chemical and thermal stability.43,44 We focus here on NU-1000 (Figure 2),37 which is composed of 1,3,6,8-tetrakis(p-benzoate)pyrene linkers and [Zr6(μ3-O)4(μ3-OH)4(OH)4(OH2)4]8+ nodes,45 where aquo and hydroxo groups are available for further functionalization. These types of nodes resemble refractory metal-oxide support materials, but they do not suffer from activity-degrading sintering.
Figure 2.

Porous structure of NU-1000 MOF viewed along two crystal axes. Zr atoms are green, O atoms red, C atoms gray, and H atoms white.
In the quest for improved catalyst design, the computational modeling of nanoporous materials plays a prominent role.46−51 Modeling catalysis on MOFs, like modeling traditional heterogeneous catalysis,52−63 poses higher-level challenges than modeling molecular64−69 catalysis because one must model not only the local catalytic site but also the structure of the support and its possible local and long-range influence on reactivity and selectivity. Theoretically informed surface catalysis design has made significant progress, and interaction energies of molecules and atoms with metal surfaces can now be described with sufficient accuracy to predict trends in reactivity for transition metals and alloys.52,53,64−68,70−72 Similar progress has been made in homogeneous catalysis.52,53,64−68,73−75 In both these instances, the chief tool is generally Kohn–Sham density functional theory76 (KS-DFT), and by using KS-DFT each elementary step in a catalytic reaction (or in a family of reactions) can be described in a detail that is often not available from experiment alone.
Achieving energy-efficient liquefaction of natural gas is a grand challenge for society, and it ranks high among the targets for catalytic design. This involves either catalytic oligomerization of abundant C1, C2, and C3 hydrocarbons to longer congeners or selective oxidation to alcohols or other fuel molecules, while avoiding overoxidation to water and carbon dioxide. As the energy economy of North America has been transformed by the exploitation of rich shale-oil deposits, this challenge is one of enormous potential economic significance, and it provides a rich application area to study a number of fundamental aspects of heterogeneous catalytic processes that will have significance beyond this specifically targeted chemistry.
In this outlook we discuss the current progress, ongoing challenges, and future prospects of the computational modeling of functionalized MOF nodes in catalysis. This contribution is focused on NU-1000 nodes, but the majority of the considerations are more broadly applicable, especially to other Zr- and Hf-based materials.42 After a brief summary of the available methodologies and models, we discuss catalytic transformations and their challenges for computational modeling.
2. Computational Methods and Models
Modeling of functionalized MOFs for catalysis has four major goals: (i) understanding the structure and possible postmodification structural changes of these materials and their relation to reactivity; (ii) understanding the energetics and details of the chemical dynamics at catalytic sites, including modeling of potential energy surfaces for competing mechanisms; (iii) discovering and understanding underlying structure–function relationships that can lead to further catalyst discovery; and (iv) predicting novel materials with superior catalytic properties. Classical molecular mechanics simulations of macroscopic properties of MOFs77−79 are useful for goal (i), but since catalysis involves bond breaking and formation, a quantum mechanical treatment46−48 is most appropriate for (ii), (iii), and (iv) and for the reactivity aspect of (i). A variety of quantum mechanical methods are available, and each method has advantages and drawbacks.
2.1. Electronic Structure Methods
One of the main challenges of modern electronic structure theory is to find the balance between affordability and reliability for complex and large-scale systems.46,80 Kohn–Sham DFT is widely applied in materials science owing to its relatively low computational cost for large systems and its semiquantitative accuracy. A key issue in applying KS-DFT is which density functional to use. While many KS-DFT validation studies have been carried out for molecular species81,82 and surfaces,72,83−86 less attention has been paid specifically to MOFs, although Nazarian et al.87 computed structural parameters, mechanical properties, and partial charges of a wide variety of MOFs using several density functionals. Validation is especially important because the best choice of density functional depends on the application and may be different for molecular structures and bond energies, barrier heights and transition-state structures, relative energies of spin energies, van der Waals interactions, and lattice constants, although progress is being made in developing more universally accurate density functionals.82,88,89
The MOFs with most promise for catalysis contain transition metals, and when the 3d orbitals of these metals are partially filled, this can produce degenerate or near-degenerate electron configurations that are challenging for both KS-DFT and wave function theory (WFT); in general the latter can handle such systems reliably only by using high-level excitation operators (e.g., for quadruple excitations), which is very demanding of computational resources, or by a multireference method, which is also demanding. One important approach is multiconfiguration self-consistent field (MCSCF) followed by second order perturbation theory, e.g., the CASPT2 method.90,91 Novel methods that combine the advantages of WFT and DFT, namely, MC-PDFT, have been developed to provide highly accurate results at lower computational costs.92 However, in the present outlook, we focus mainly on KS-DFT, which is a more mature approach.
Among the density functionals that have proven useful in the KS-DFT context, we single out the Minnesota functional M06-L,93,94 which has shown a good performance in reproducing experimental trends and being able to accurately predict spin-state energetics in MOFs in good agreement with high-level methods, such as the CASPT2 method.95,96 The M06-L functional has been validated for many applications relevant to catalysis97−119 (including some cases very similar to the cases discussed in the next section but also including studies relevant to the broader applicability for transition metal catalysis and barrier heights) and MOFs.120,121
2.2. Modeling the Framework
Both periodic and cluster models are used for MOFs. The periodic approach describes the unit cell of an infinite crystal with periodic boundary conditions (PBC) (Figure 3). Such models are needed to get realistic structural information and to study collective properties, cavities, and pores; they are also useful when taking into account the rearrangement (mobility and sintering) of catalytic sites on length scales that make cluster modeling unwieldy. For moderate length scales, periodic structures can be cropped and capped to create finite-sized cluster models, and this allows more accurate modeling of local effects should levels of theory beyond KS-DFT be desirable to explore. Note that the accuracy depends on the choice of the electronic structure method, and the accuracy for periodic calculations cannot be as high as for clusters because the highest-level post-SCF correlation methods are not available for periodic calculations. Cluster calculations can be particularly useful for metal-functionalized nodes of MOFs, where chemical reactions occur on the node and diffusion of reactants through the pores is not usually rate limiting.
Figure 3.
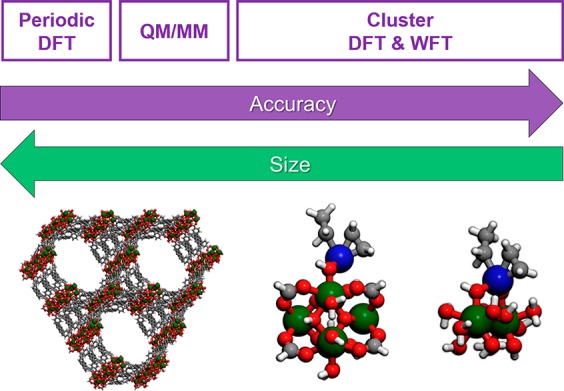
Modeling MOFs: from periodic DFT to WFT cluster models. Zr atoms are green, O atoms red, C atoms gray, H atoms white, and generic deposited metal atoms are indicated in blue for some models.
The size of the clusters to be studied varies and depends on the material, the reaction under investigation, and the methodology. Taking NU-1000 as an example, three approaches have been adopted, namely, 2-node, 1-node, and small cluster models (Figure 3). Selected examples will be discussed in the following section. The 2-node cluster approach122 contains two Zr6 nodes connected through four pyrene-like linkers and is useful to describe the small 12 Å pore of the material (Figure 2). The 1-node model45 is constructed by one node and eight linkers truncated to any of biphenyl-4-carboxylate, benzoate, acetate, or formate groups, depending on the perceived importance of fully reproducing the linker steric environment. Selected atoms of linkers are generally kept fixed during geometry optimizations of cluster models to mimic the rigid conditions of the framework. This is our most used model for reaction mechanisms and computational screening. Finally, a very small cluster123 can be constructed containing only two Zr atoms and retaining only their respective first coordination spheres. While the size of the 2-node and 1-node structures renders them affordable for the most part only to KS-DFT, the very small cluster is specifically designed to also allow high-level WFT calculations. One should keep in mind that errors due to truncation of the periodic system cannot be separated from those due to the inexactness of the density functional or incompleteness of the wave function method.
For clusters, one could also consider combined quantum mechanics and molecular mechanics (QM/MM) methods,124−131 with QM methods for the chemically reactive portion of the cluster and a set of structural and interaction parameters for the MM part. The QM/MM accuracy depends on the choice of the QM methodology and MM parameters and also on the treatment of electrostatics at the QM–MM boundary.132−134
2.3. Dynamics
Once the structure of the functionalized MOF nodes is understood, one can try to develop design principles and predict novel catalysts. To achieve these goals it is necessary to employ accurate, but computationally affordable, electronic structure methods in conjunction with methods that efficiently scan the energy landscape, taking into account dynamical aspects of the system over a range of time scales. A variety of active site structures can be simultaneously present on a functionalized MOF, and they can yield several chemical species as products of disparate plausible pathways. The most useful catalytic processes are those specific for individual products. Hence a major goal is the understanding and prediction of selectivity. The correct prediction of selectivity requires a reliable computation of kinetic and thermodynamic quantities.67,68 Two general kinds of approaches are available, which we may classify as molecular dynamics simulations and catalogue-based approaches.
In molecular dynamics (MD) simulations, one explores configurations, local structures, defects, and branching probabilities without a catalogue.20,135,136 The time scales that need to be sampled are usually too large for conventional molecular dynamics, so one must use rare-event sampling of some sort, such as weighted-histogram analysis, hyperdynamics,137 temperature-accelerated dynamics,138 or metadynamics.139 These methods can allow one to study time scales many orders of magnitude larger than straight molecular dynamics, in some instances requiring the selection of appropriate collective variables from which to construct bias potentials.
Catalogue-based approaches include kinetic Monte Carlo and microkinetic modeling. These methods140−153 involve three or four steps: (i) create a catalogue of microscopic states, local structures, and possible transformations, energy transfer events, site-to-site diffusion or hopping processes, and reactions; (ii) characterize the rate constants for each of the elementary processes atomistically; (iii, sometimes omitted) develop a model for the environmental dependence of such elementary processes when they occur in a complex cluster, fluid, or material; and (iv) gather the rate constants for elementary reaction steps into a nonlinear coupled set of kinetic equations, called a master equation,154 and simulate the solution of the master equation by Monte Carlo methods or solve the master equation by numerical integration. The master equation should take account of reaction conditions, in particular concentrations, flows, partial pressures, chemical potentials, and temperature. This kind of complete treatment has been achieved for some catalytic processes, but not so far for MOFs. Rather, for MOFs we are still at the stage of calculating the energetics (energies, enthalpies, and free energies) and kinetics of the elementary steps without yet arriving at full modeling with the inclusion of reaction conditions. Such full modeling is challenging, but it is a realistic near-term goal because the regular nature of MOF crystals—with well-defined, isolated sites in a uniform environment as emphasized in the very first paragraph of this article—not only makes consideration of well-defined elementary steps possible (and thereby enables the catalogue-based approach) but also is the central feature that motivated the use of MOFs for catalysis in the first place. The periodic nature of MOFs can also facilitate the modeling of well-defined cell-to-cell transport phenomena. However, the first and critical step for modeling dynamics is to calculate accurately the energetics of reactions and their reaction rates for the various chemical reactions in the catalytic mechanism.
The simplest indicator of whether a reaction will proceed is provided by the reaction energy, which is the potential energy difference of the product from the reactant structure on the Born–Oppenheimer potential energy surface, and by the classical barrier height, which is the potential energy difference of the transition-state (TS) structure from the reactant. A TS structure is a first order saddle point on the potential energy surface which may be thought of either as the highest-energy point on the lowest-energy reaction path from reactants to products or as the lowest-energy point of the conventional transition state, which is a hypersurface in coordinate space separating reactants from products. Reaction energies and barrier heights can be converted to enthalpies of reaction at 0 K (or enthalpies of activation at 0 K) by adding the difference of zero point energies of the products (or TS structure) from those of the reactants. For highest accuracy, zero point energies are calculated from scaled vibrational frequencies.155 Enthalpies at 0 K can then be turned into standard-state free energies of reaction and standard-state free energies of activation by adding thermal enthalpic and entropic effects. Low-frequency modes are usually very anharmonic, and one can obtain more accurate entropies by raising frequencies calculated to be lower than 50 cm–1 up to 50 or 100 cm–1 when entropies are computed.49,88,156−160 Standard-state free energies of reaction can be used to calculate equilibrium constants. All the quantities mentioned in this paragraph are straightforward to calculate by KS-DFT.
A higher-level estimate of the reaction rate than the free energy of activation is provided by transition state theory (TST).161 In quasiclassical TST the rate constant is obtained from the free energy of activation described above. In conventional TST, the transition state passes through the saddle point; in variational TST, it is found by maximizing the free energy of activation with respect to the location of the transition state.162 Quasiclassical theory is classical in that it does not include tunneling, but it does use quantized vibrational states to calculate partition functions.
In full variational TST, one adds a transmission coefficient that is greater than unity and that increases the calculated rate constant by accounting for quantum mechanical tunneling,163 which is mainly important when the reaction coordinate involves a hydrogen, proton, or hydride transfer.
A key take-home message of this outlook is that density functional theory and transition state theory, taken together, are accurate enough to understand many chemical mechanisms, even in systems as complicated as MOFs.
3. Structures of Bare and Functionalized MOF Nodes
Characterization is essential for understanding the kinetic relevance of different catalytic sites and for building correlations to predict catalytic power of new or potential materials, and theory has become a major tool for characterizing the local and complex structure of catalytic nodes.164 This section collects several characterization studies where the synergy between experiment and theory led to a precise description of the local structure of the materials. After initial studies with bare Zr-based nodes, selected examples of metal-functionalized nodes are highlighted.
The starting point involves the characterization of metal-oxide MOF nodes, whose proton topologies are not straightforward to define.165,166 Planas et al.45 used KS-DFT to assign the proton topology of Zr6 nodes of NU-1000 by means of cluster and periodic calculations. Among the potential tautomers, the so-called mix-staggered (mix-S) configuration was predicted to be the thermodynamically most stable structure (Figure 2 left). This node contains four μ3-O and four μ3-OH groups and displays eight faces that are connected to eight organic linkers and four faces with terminal −OH2 and −OH ligands with hydrogen-bonding motifs. Interrogation of the computed NU-1000 proton topology via diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was consistent with this assignment. Troya has recently assigned a similar mixed topology for the Zr-based node of MOF-808.167
This kind of OH/H2O termination is closely related to the defect sites of other Zr-based MOFs.168 For instance, the UiO family contains the same Zr6O4(μ3-OH)4 nodes connected to 12 linkers, e.g., benzene-1,4-dicarboxylic acid (UiO-66) and biphenyl-4,4′-dicarboxylic acid (UiO-67). However, all Zr atoms in “pristine” UiO MOFs are saturated with linker functionality and a priori there are no accessible sites for reactivity. Therefore, all catalytic activity has been associated with nodes in which linkers are missing from the framework. Based on X-ray diffraction studies, Trickett et al.169 initially claimed that the missing linker termination in UiO-66 was charge balanced by a hydroxide group hydrogen-bonded to μ3-OH of the node. Later, Ling and Slater135 performed simulations on defective UiO-66 and showed that the OH group was actually bound to Zr, as previously reported for NU-1000.45 They also highlighted the dynamic fluxionality of defects where hydroxides transform into waters via rapid proton transfers. In the case of bound water molecules, as opposed to hydroxides, the binding free energy is sufficiently small that site occupations may depend on the relative humidity of the environment. The different protons on the MOF node have been experimentally observed to exhibit characteristic pKa values that may influence the activity of the MOF nodes for catalysis of hydrolysis reactions.170−172
The nanoscale nature of metal-oxide nodes provides new challenges associated with structural behavior. Valenzano et al.173,174 initially reported periodic calculations for the transformation of pristine Zr6O4(μ3-OH)4 to dehydrated Zr6O6 UiO nodes, and Vandichel et al.175 studied different mechanistic pathways for the dehydration of defect sites of UiO-66. Very recently, Platero-Prats et al.176 reported local structural transitions of Zr-based MOF nodes. For the [M6(μ3-O)4(μ3-OH)4(OH)4(H2O)4]8+ node of NU-1000 under dehydration conditions (150 °C), one would expect the formation of symmetric [M6O8]8+ cores (M = Zr, Hf). However, a combination of in situ pair distribution function (PDF) experiments and KS-DFT calculations revealed the formation of metastable node distortions. To further probe such structures, ab initio MD simulations were performed on a hydrated NU-1000 node. While low and intermediate temperature simulations display proton transfers and terminal water dissociations, at high temperatures the dynamic features of μ3-OH groups become prominent. Reoptimization of selected high-temperature snapshots for fully dehydrated nodes yields dislocations where oxygen atoms change their coordination mode from μ3 to μ2 and nonbridging. These structural transitions are related to transitions between polymorphic forms of bulk oxides,177 although milder conditions are required for such transitions in the nanoscale M6O8 nodes of MOFs.
Synthetic strategies have been developed that lead to a partial dehydration and functionalization of nodes. Yang et al.178 reported the fine-tuning of defective UiO-66 and NU-1000 node faces through the intermediacy of alkoxy groups (Figure 4). Treatment of the pristine node 1 with methanol affords the methoxy derivative 2, which becomes the hydroxo species 3 upon hydration. While cluster KS-DFT calculations predict exergonic processes from 1 to 2, the coordination of one methanol molecule to the vacant site in 2 is uphill. Computed C–H and C–O vibrational frequencies of 2 were in line with experimental IR spectra. Binding of different capping agents to defective UiO-66 nodes has also been evaluated using cluster and periodic KS-DFT.179−181
Figure 4.
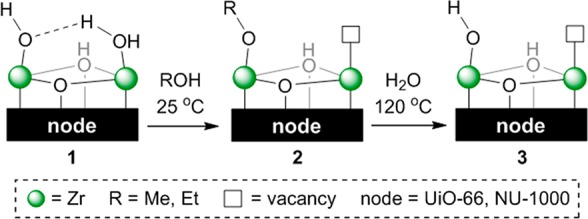
Fine-tuning of node faces via alkoxy intermediates.
The OH and OH/H2O motifs of metal-oxide nodes can be functionalized with a wide variety of metal precursors, in analogy to the functionalization of hydroxylated bulk surfaces.182−184 To date there are reports on gas-phase and solution functionalization of Zr- and Hf-based MOF nodes with Mg(II),185 Al(III),37,41,186 Si(IV),187 Ca(II),121 V(V),188 Fe(II),189 Fe(III),190 Co(II),38,39,189,191−193 Ni(II),38,39,194 Cu(II),38,122,195 Zn(II),37,38,196 Zr(IV),197 Nb(V),198 Mo(VI),170,199 Rh(I),200 In(III),186 Ir(I),201,202 and Au(I).203 In many cases, modeling has been used to understand the structures that have been synthesized.
Mixed-metal structures can also be synthetically accessed as recently shown for Co(II)Al(III)-functionalized NU-1000 nodes.204,205 Precoating of MOF nodes with aluminum-oxide clusters and subsequent functionalization with Ir(I) precursors presents a major challenge to structural characterization, with extensive calculations suggesting that AlxOy clusters and Ir may attach to different faces of individual nodes.206 Transition metal complexes can also be attached to MOF nodes via SALI.207
Another possibility is that the framework itself can act as a host for metal nanoparticles.208,209 However, calculations of these complex systems are scarce210,211 due to their high computational cost, so we do not comment on this further here. Rather we discuss some selected cases where the synergistic combination of experiments and theory was crucial for the structural determination of the functionalized MOFs.
The OH/H2O groups that direct metal functionalization are directed into the 30 Å hexagonal pores and 12 Å small windows (c pores) of NU-1000 (Figure 2). In this regard, Gallington et al.196 reported the regioselective deposition of Zn(II)O species into the small pores. A typical atomic layer deposition (ALD) cycle involves treatment with a metal precursor, ZnEt2 in this case, followed by exposure to water vapor. Difference envelope densities (DED) reveal electron density on node faces within the small window, while no significant densities were found in the large hexagonal pores. Periodic KS-DFT calculations reported in the same article found lower energies for Zn complexes located in the small pore compared to those place in the hexagonal cavity. Further energetic analysis revealed that this preference was dictated by damped dispersion interactions between the Zn alkyl and the linkers at the preferential adsorption sites prior to hydrolysis.
Rimoldi et al.41 functionalized the nodes of NU-1000 using a mild Al(III) precursor. A joint experimental and computational effort pointed toward the presence of distributed AlxOy clusters confined within the small pores of NU-1000. Since spectroscopic evidence suggests similarities between the nanoconfined clusters and bulk γ-Al2O3, one Al4 (4) and two Al8 models (5, 6) were constructed from periodic γ-Al2O3 structures (Figure 5). The computational models qualitatively reproduce the OH stretching regions in the DRIFTS and the Al–O and Al–Al scattering signals in extended X-ray absorption fine structure (EXAFS) data. The structure 5 labeled Al8-flat (Figure 5b) best fits the DRIFTS spectrum, but the presence of different clusters cannot be completely excluded.
Figure 5.

(a) Al4, (b) Al8-flat, and (c) Al8-vertical clusters deposited on an NU-1000 node. Zr atoms are green, O atoms red, C atoms gray, H atoms white, and Al atoms magenta.
Ikuno et al.122 performed an ALD of Cu(II) precursors on NU-1000 nodes. The combination of EXAFS analysis with computational models suggests the presence of Cu-hydroxo-like species as products. Calculations on clusters with two nodes and periodic calculations predict a stable [Cu3(OH)4] fragment connecting two consecutive nodes within the small pore of NU-1000 (7, Figure 6). Further DED analysis by Platero-Prats et al.195 agreed with the bridging motif inside the small pore. A related structure has been elucidated for Ni(II)-functionalized MOF nodes, where KS-DFT-optimized α-Ni(OH)2 clusters tethered in the small pore best match experimental data.212
Figure 6.
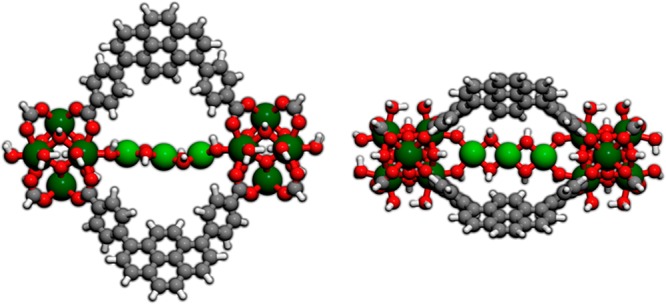
Side (left) and top (right) views of the 2-node cluster model 7 of Cu-NU-1000. Zr atoms are green, Cu atoms light green, O atoms red, C atoms gray, and H atoms white.
Thus, modeling of MOFs contributes to an understanding of proton topologies, defect sites, and metal-functionalized nodes that form a foundation for follow-on catalytic studies.
4. Catalysis with Functionalized MOF Nodes
One approach to the modeling of these materials begins with the optimization of crystal structures by static periodic KS-DFT calculations from which cluster models (see section 2.2) are extracted in order to explore the potential energy surface associated with catalysis at a moderate cost. A full understanding of catalytic cycles based on computed relative energies and free energies sets the foundation for a rational design of metal-functionalized materials. This section highlights computational studies on the transformation of natural-gas-derived substrates, with particular emphasis on studies that show the promise of using theory to understand reaction mechanisms and identify chemical descriptors.
4.1. Ethylene Conversion Catalyzed by Noble Metals
Gates et al. anchored single-site Ir(I) and Rh(I) diethylene complexes on the Zr6-based nodes of NU-1000 and UiO-66 using M(C2H4)(acac) precursors (acac = acetylacetonate).200−202,206 Both metal-supported materials have shown catalytic activity under relevant C2H4/H2 flow conditions for ethylene hydrogenation and dimerization. KS-DFT calculations shed light on the competitive mechanism between these two reactions (Figure 7).200,206 Electronic effects of the support can indeed modulate the catalyst performance,200−202,206 and an interesting goal of computational work is to find correlations between simple descriptors and catalytic selectivity that may be exploited in computational design; correlations with experimental observables such as stretching frequencies for metal–carbonyl compounds would be particularly interesting.
Figure 7.

General reaction mechanisms of ethylene hydrogenation and dimerization.
Thus, Bernales et al.200 used this vibrational frequency descriptor to explore other supports such as Hf6-based NU-1000 and dealuminated zeolite Y (DAY, SiO2/Al2O3 ratio = 30), where the zeolite is known to have a higher selectivity toward dimerization. The CO vibrational frequencies for the Hf6 analogue were predicted to be similar to those for Zr6-based NU-1000, whereas those in DAY zeolite were predicted to be higher, in agreement with experimental trends in selectivity. A comparison between MOFs and DAY zeolite was performed, showing the impact of the support’s electronic structure on the catalytic performance through activation energies. For both hydrogenation and dimerization reactions, lower barriers were calculated for Rh-DAY zeolite for every step along the reaction coordinate. However, the substantially higher selectivity for dimerization exhibited by the DAY-supported catalyst has been attributed to spillover effects rather than a direct electronic effect on the Rh(I) center, and this is not captured in the stretching frequency descriptor.213−216
The good performance of the aluminum-containing DAY zeolite inspired work involving a treatment of MOF nodes with Al oxides followed by deposition of Ir(I) to further tune the electronic effects of the support. Yang et al.206 showed that incorporation of Al clusters weakened the electron donor tendency of the MOF and increased the catalytic activity for ethylene hydrogenation and selectivity for dimerization. KS-DFT calculations were in agreement with the observed results, and the calculated free energies of activation for the rate-determining step, the insertion of ethylene into the metal–hydride bond (from 9 to 10, Figure 7), were 9.8 and 11.4 kcal mol–1 for Al-NU-1000 and bare NU-1000 supports, respectively.206 Unfortunately, no quantity (such as partial atomic charges on the metal or supporting atoms) was found to correlate with the CO stretching descriptor or catalyst performance, limiting the further insight that can be used for design purposes in these systems.
4.2. Ethylene Conversion Catalyzed by First-Row Transition Metals
Li et al.194 reported a Ni(II)-decorated NU-1000 (synthesized via ALD) that is also catalytically active for ethylene hydrogenation and oligomerization (Figure 8). The hydrogenation reaction mechanism was initially studied at the KS-DFT level using a cluster model with formate linkers and a single Ni atom supported on the surface of the node. The computed activation enthalpy for the rate-determining step is 4.9 kcal mol–1, slightly below the lower error bar of the experimental value of 8.3 kcal mol–1, the latter being a phenomenological enthalpy of activation encompassing all elementary steps. As mentioned before, it is not possible to separate the source of discrepancy between experiment and theory, since it can be related to the computational model, methodology, complexity of the kinetics with respect to the number of steps affecting the rate constant, or all of the above.
Figure 8.

(a) Cossee–Arlman dimerization mechanism. (b) Orbital descriptor.
Later on, Bernales et al. reported a computational study of ethylene dimerization including the reported Ni(II)-modified NU-1000 material194 as well as a hypothetical Co(II) analogue.123,194 In ref (123) computations were used to characterize the structure resulting from ALD, to examine three plausible catalytic pathways, to calculate reaction energetics, to explain the mechanism of activation by Et2AlCl, to calculate transition states and activation energies for steps that are assumed to be rate-determining for ethylene insertion into metal–methyl bonds, and to explain the greater reactivity of the Ni(II) by transition-state stabilization associated with an empty 3d orbital that hybridizes more readily with relevant carbon 2p orbitals in low-spin Ni than in high-spin Co. Cluster KS-DFT calculations (with acetate linkers) suggested a Cossee–Arlman reaction mechanism as the most plausible pathway (Figure 8a). It was found that the electron configuration of the metals had a strong impact on the preferential coordination adopted by the support and the reactants, enhancing the activity of Ni (15.5 kcal mol–1) compared to the Co (24.1 kcal mol–1) for the rate-determining C–C bond formation step (from 16 to 17). Additionally, CASSCF orbitals of key transition structures demonstrated the crucial role of the metal in assisting the bond formation (Figure 8b). These calculations motivated further experiments, which indeed showed that the Co(II) analogue material was catalytically active for ethylene dimerization, but less effective than Ni. Subsequent periodic KS-DFT studies with pore-spanning polynuclear Ni clusters showed similar trends to those found for single Ni atoms sited on node faces,217 suggesting that the local reactivity of these catalysts can indeed be captured with cluster models extracted from periodic systems.
4.3. Propane Oxidative Dehydrogenation
Li et al.192 reported Co-functionalized NU-1000 materials that, upon activation in a flow of O2 at 230 °C, are able to selectively catalyze the oxidative dehydrogenation (ODH) of propane to propylene. This reaction was studied by KS-DFT with the dual purpose of characterizing potential active species (oxidized Co-NU-1000) and elucidating the reaction mechanism. A dehydrated cluster model was employed to properly represent the catalyst under experimental conditions. Based on the weak contribution of Co···Co scattering signals in EXAFS, the initial model considered a single Co deposited on a dehydrated NU-1000 node. Structural changes, namely, the loss of water molecules and the detection of Co(III) species, were observed and modeled. The proposed active species promoted the conversion of propane to propylene with computed activation enthalpies of 12.6 and 15.8 kcal mol–1 for the first and second hydrogen atom abstractions, respectively. It was found that the second hydrogen abstraction (22) was 7.3 kcal mol–1 more favorable than the isopropyl migration step (21), which would otherwise lead to undesired side products (Figure 9). Ongoing work involves the assessment of polynuclear clusters as kinetically competent catalytic sites and the evaluation of the oxidation states and multiconfigurational electronic structures of these active species.
Figure 9.
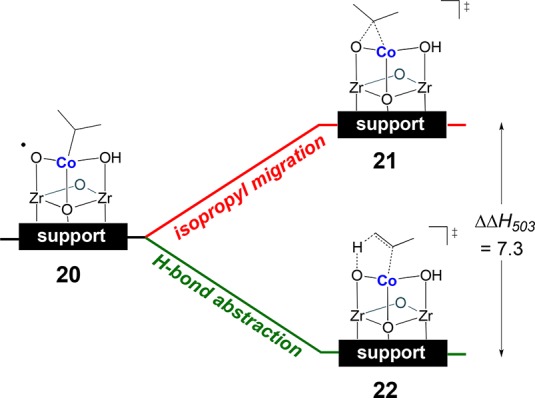
Competing pathways and transition structures for oxidative dehydrogenation: isopropyl migration versus H-bond abstraction.
4.4. Prospecting for Chemical Descriptors for C–H Bond Activation and Beyond
With the mechanistic insights gained from modeling many of the systems above, which can involve single metal atoms, homometallic clusters, heterometallic clusters, or inorganometallic nodes, theory can be employed to investigate a wider variety of catalysts. It can correlate their activities to properties that change along the reaction path, used in this case as chemical descriptors. Indeed, such correlations have been found for Cu-based218 and Co-based219 homo- and heterobimetallic catalysts, where spin densities and partial atomic charges on oxyl groups proved to be robust descriptors to predict trends in homolytic C–H bond breaking processes. These results can be used for in silico design of not-yet-synthesized catalysts that can provide lower activation energies. Catalyst screenings of different processes are also underway.220
5. Conclusions
Using quantum mechanical methods to support and guide experimental work (synthesis, characterization, and catalytic kinetics) on catalyst design at tailored MOF nodes is opening many new prospects for advances in catalysis science, and here we have highlighted some of these in the context of metal-functionalized MOFs with inorganometallic Zr-based nodes. These materials pose grand challenges for theory due to the large sizes of the basic structural units, the complexity of the catalytic active sites, and the inherently multiconfigurational character of the electronic configurations of many transition-metal-containing species. Theory and computation provide structural and kinetic information that complements experimental methods and can be used to suggest experiments as yet unperformed. Theory is of particular use when details of structures of catalytic sites, prereaction complexes, intermediates, and transition states are experimentally inaccessible, and theory can rationalize observations in terms of electronic and geometric structures that explain reactivity and selectivity, and it has made some successful predictions of catalytic activity. We foresee the use of quantum mechanical modeling interacting closely with experimental investigations as the ultimate tool for realizing the tunability potential of MOFs for improved catalysis.
This work is a prime illustration of interdisciplinary research since it lies at the intersection of physical chemistry, inorganic chemistry, organic chemistry, and materials science. Although great advances have been achieved in this field, the journey to in silico design of catalytic MOFs has just begun.
Acknowledgments
The authors are grateful to the many collaborators who are listed in relevant cited references. This work was supported as part of the Inorganometallic Catalyst Design Center, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (DE- SC0012702). The calculations were performed using Minnesota Supercomputing Institute computers, EMSL computers (Proposal ID: 50124) and NERSC computers.
Author Contributions
† V.B. and M.A.O. contributed equally.
The authors declare no competing financial interest.
References
- Corma A. Heterogeneous Catalysis: Understanding for Designing, and Designing for Applications. Angew. Chem., Int. Ed. 2016, 55, 6112–6113. 10.1002/anie.201601231. [DOI] [PubMed] [Google Scholar]
- Gates B. C. Supported Metal Clusters: Synthesis, Structure, and Catalysis. Chem. Rev. 1995, 95, 511–522. 10.1021/cr00035a003. [DOI] [Google Scholar]
- Liu J. Catalysis by Supported Single Metal Atoms. ACS Catal. 2017, 7, 34–59. 10.1021/acscatal.6b01534. [DOI] [Google Scholar]
- Liu L.; Díaz U.; Arenal R.; Agostini G.; Concepción P.; Corma A. Generation of Subnanometric Platinum with High Stability During Transformation of a 2D Zeolite into 3D. Nat. Mater. 2017, 16, 132–138. 10.1038/nmat4757. [DOI] [PubMed] [Google Scholar]
- Cui X.; Junge K.; Dai X.; Kreyenschulte C.; Pohl M.-M.; Wohlrab S.; Shi F.; Brückner A.; Beller M. Synthesis of Single Atom Based Heterogeneous Platinum Catalysts: High Selectivity and Activity for Hydrosilylation Reactions. ACS Cent. Sci. 2017, 3, 580–585. 10.1021/acscentsci.7b00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa H.; Cordova K. E.; O’Keeffe M.; Yaghi O. M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- Lee J.; Farha O. K.; Roberts J.; Scheidt K.; Nguyen S. T.; Hupp J. T. Metal–Organic Framework Materials as Catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. 10.1039/b807080f. [DOI] [PubMed] [Google Scholar]
- Hendon C. H.; Rieth A. J.; Korzyński M. D.; Dincă M. Grand Challenges and Future Opportunities for Metal–Organic Frameworks. ACS Cent. Sci. 2017, 3, 554–563. 10.1021/acscentsci.7b00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. I. Porous Molecular Solids and Liquids. ACS Cent. Sci. 2017, 3, 544–553. 10.1021/acscentsci.7b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook T. R.; Zheng Y. R.; Stang P. J. Metal–Organic Frameworks and Self-Assembled Supramolecular Coordination Complexes: Comparing and Contrasting the Design, Synthesis, and Functionality of Metal–Organic Materials. Chem. Rev. 2013, 113, 734–777. 10.1021/cr3002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y.; Yue Y.; Qian G.; Chen B. Luminescent Functional Metal–Organic Frameworks. Chem. Rev. 2012, 112, 1126–1162. 10.1021/cr200101d. [DOI] [PubMed] [Google Scholar]
- Hu Z.; Deibert B. J.; Li J. Luminescent Metal–Organic Frameworks for Chemical Sensing and Explosive Detection. Chem. Soc. Rev. 2014, 43, 5815–5840. 10.1039/C4CS00010B. [DOI] [PubMed] [Google Scholar]
- Kreno L. E.; Leong K.; Farha O. K.; Allendorf M.; Van Duyne R. P.; Hupp J. T. Metal–Organic Framework Materials as Chemical Sensors. Chem. Rev. 2012, 112, 1105–1125. 10.1021/cr200324t. [DOI] [PubMed] [Google Scholar]
- Li J. R.; Kuppler R. J.; Zhou H. C. Selective Gas Adsorption and Separation in Metal–Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1477–1504. 10.1039/b802426j. [DOI] [PubMed] [Google Scholar]
- Ma L.; Abney C.; Lin W. Enantioselective Catalysis with Homochiral Metal–Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1248–1256. 10.1039/b807083k. [DOI] [PubMed] [Google Scholar]
- Murray L. J.; Dincă M.; Long J. R. Hydrogen Storage in Metal–Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1294–1314. 10.1039/b802256a. [DOI] [PubMed] [Google Scholar]
- Sumida K.; Rogow D. L.; Mason J. A.; McDonald T. M.; Bloch E. D.; Herm Z. R.; Bae T.; Long J. R. Carbon Dioxide Capture in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 724–781. 10.1021/cr2003272. [DOI] [PubMed] [Google Scholar]
- Tranchemontagne D. J.; Mendoza-Cortés J. L.; O’Keeffe M.; Yaghi O. M. Secondary Building Units, Nets and Bonding in the Chemistry of Metal–Organic Frameworks. Chem. Soc. Rev. 2009, 38, 1257–1283. 10.1039/b817735j. [DOI] [PubMed] [Google Scholar]
- Lu W.; Wei Z.; Gu Z. Y.; Liu T. F.; Park J.; Park J.; Tian J.; Zhang M.; Zhang Q.; Gentle T.; Bosch M.; Zhou H.-C.; Daturi M.; Ramos-Fernandez E. V.; Llabrés i Xamena F. X.; Van Speybroeck V.; Gascon J. Tuning the Structure and Function of Metal–Organic Frameworks via Linker Design. Chem. Soc. Rev. 2014, 43, 5561–5593. 10.1039/C4CS00003J. [DOI] [PubMed] [Google Scholar]
- Brozek C. K.; Michaelis V. K.; Ong T. C.; Bellarosa L.; López N.; Griffin R. G.; Dincă M. Dynamic DMF Binding in MOF-5 Enables the Formation of Metastable Cobalt-Substituted MOF-5 Analogues. ACS Cent. Sci. 2015, 1, 252–260. 10.1021/acscentsci.5b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoufi A.; Benhamed K.; Boukli-Hacene L.; Maurin G. Electrically Induced Breathing of the MIL-53(Cr) Metal–Organic Framework. ACS Cent. Sci. 2017, 3, 394–398. 10.1021/acscentsci.6b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapustin E. A.; Lee S.; Alshammari A. S.; Yaghi O. M. Molecular Retrofitting Adapts a Metal–Organic Framework to Extreme Pressure. ACS Cent. Sci. 2017, 3, 662–667. 10.1021/acscentsci.7b00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Duan H.-B.; Khan S. I.; Garcia-Garibay M. A. Diffusion-Controlled Rotation of Triptycene in a Metal–Organic Framework (MOF) Sheds Light on the Viscosity of MOF-Confined Solvent. ACS Cent. Sci. 2016, 2, 608–613. 10.1021/acscentsci.6b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmer C. E.; Leaf M.; Lee C. Y.; Farha O. K.; Hauser B. G.; Hupp J. T.; Snurr R. Q. Large-Scale Screening of Hypothetical Metal–Organic Frameworks. Nat. Chem. 2012, 4, 83–89. 10.1038/nchem.1192. [DOI] [PubMed] [Google Scholar]
- Corma A.; García H.; Llabrés i Xamena F. X. Engineering Metal Organic Frameworks for Heterogeneous Catalysis. Chem. Rev. 2010, 110, 4606–4655. 10.1021/cr9003924. [DOI] [PubMed] [Google Scholar]
- Chughtai A. H.; Ahmad N.; Younus H. A.; Laypkov A.; Verpoort F. Metal–Organic Frameworks: Versatile Heterogeneous Catalysts for Efficient Catalytic Organic Transformations. Chem. Soc. Rev. 2015, 44, 6804–6849. 10.1039/C4CS00395K. [DOI] [PubMed] [Google Scholar]
- Rogge S. M. J.; Bavykina A.; Hajek J.; Garcia H.; Olivos-Suarez A. I.; Sepúlveda-Escribano A.; Vimont A.; Clet G.; Bazin P.; Kapteijn F.; et al. Metal–Organic and Covalent Organic Frameworks as Single-Site Catalysts. Chem. Soc. Rev. 2017, 46, 3134–3184. 10.1039/C7CS00033B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E. D.; Brozek C. K.; Comito R. J.; Dincă M. Selective Dimerization of Ethylene to 1-Butene with a Porous Catalyst. ACS Cent. Sci. 2016, 2, 148–153. 10.1021/acscentsci.6b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H. D.; Dincă M.; Román-Leshkov Y. Heterogeneous Epoxide Carbonylation by Cooperative Ion-Pair Catalysis in Co(CO)4–-Incorporated Cr-MIL-101. ACS Cent. Sci. 2017, 3, 444–448. 10.1021/acscentsci.7b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvekens P.; Vermoortele F.; De Vos D. Metal–Organic Frameworks as Catalysts: The Role of Metal Active Sites. Catal. Sci. Technol. 2013, 3, 1435–1445. 10.1039/c3cy20813c. [DOI] [Google Scholar]
- Cohen S. M. Postsynthetic Methods for the Functionalization of Metal–Organic Frameworks. Chem. Rev. 2012, 112, 970–1000. 10.1021/cr200179u. [DOI] [PubMed] [Google Scholar]
- Tanabe K. K.; Cohen S. M. Postsynthetic Modification of Metal–Organic Frameworks—A Progress Report. Chem. Soc. Rev. 2011, 40, 498–519. 10.1039/C0CS00031K. [DOI] [PubMed] [Google Scholar]
- Marshall R. J.; Forgan R. S. Postsynthetic Modification of Zirconium Metal-Organic Frameworks. Eur. J. Inorg. Chem. 2016, 2016, 4310–4331. 10.1002/ejic.201600394. [DOI] [Google Scholar]
- Cohen S. M. The Postsynthetic Renaissance in Porous Solids. J. Am. Chem. Soc. 2017, 139, 2855–2863. 10.1021/jacs.6b11259. [DOI] [PubMed] [Google Scholar]
- Islamoglu T.; Goswami S.; Li Z.; Howarth A. J.; Farha O. K.; Hupp J. T. Postsynthetic Tuning of Metal–Organic Frameworks for Targeted Applications. Acc. Chem. Res. 2017, 50, 805–813. 10.1021/acs.accounts.6b00577. [DOI] [PubMed] [Google Scholar]
- Deria P.; Mondloch J. E.; Tylianakis E.; Ghosh P.; Bury W.; Snurr R. Q.; Hupp J. T.; Farha O. K. Perfluoroalkane Functionalization of NU-1000 via Solvent-Assisted Ligand Incorporation: Synthesis and CO2 Adsorption Studies. J. Am. Chem. Soc. 2013, 135, 16801–16804. 10.1021/ja408959g. [DOI] [PubMed] [Google Scholar]
- Mondloch J. E.; Bury W.; Fairen-Jimenez D.; Kwon S.; DeMarco E. J.; Weston M. H.; Sarjeant A. A.; Nguyen S. T.; Stair P. C.; Snurr R. Q.; Farha O. K.; Hupp J. T. Vapor-Phase Metalation by Atomic Layer Deposition in a Metal–Organic Framework. J. Am. Chem. Soc. 2013, 135, 10294–10297. 10.1021/ja4050828. [DOI] [PubMed] [Google Scholar]
- Klet R. C.; Wang T. C.; Fernandez L. E.; Truhlar D. G.; Hupp J. T.; Farha O. K. Synthetic Access to Atomically Dispersed Metals in Metal–Organic Frameworks via a Combined Atomic-Layer-Deposition-in-MOF and Metal-Exchange Approach. Chem. Mater. 2016, 28, 1213–1219. 10.1021/acs.chemmater.5b04887. [DOI] [Google Scholar]
- Yuan S.; Chen Y.-P.; Qin J.; Lu W.; Wang X.; Zhang Q.; Bosch M.; Liu T.-F.; Lian X.; Zhou H.-C. Cooperative Cluster Metalationand Ligand Migration in Zirconium Metal–Organic Frameworks. Angew. Chem., Int. Ed. 2015, 54, 14696–14700. 10.1002/anie.201505625. [DOI] [PubMed] [Google Scholar]
- Zhao C.; Dai X.; Yao T.; Chen W.; Wang X.; Wang J.; Yang J.; Wei S.; Wu Y.; Li Y. Ionic Exchange of Metal–Organic Frameworks to Access Single Nickel Sites for Efficient Electroreduction of CO2. J. Am. Chem. Soc. 2017, 139, 8078–8081. 10.1021/jacs.7b02736. [DOI] [PubMed] [Google Scholar]
- Rimoldi M.; Bernales V.; Borycz J.; Vjunov A.; Gallington L. C.; Platero-Prats A. E.; Kim I. S.; Fulton J. L.; Martinson A. B. F.; Lercher J. A.; Chapman K. W.; Cramer C. J.; Gagliardi L.; Hupp J. T.; Farha O. K. Atomic Layer Deposition in a Metal-Organic Framework: Synthesis, Characterization, and Performance of a Solid Acid. Chem. Mater. 2017, 29, 1058–1068. 10.1021/acs.chemmater.6b03880. [DOI] [Google Scholar]
- Rimoldi M.; Howarth A. J.; DeStefano M. R.; Lin L.; Goswami S.; Li P.; Hupp J. T.; Farha O. K. Catalytic Zirconium/Hafnium-Based Metal–Organic Frameworks. ACS Catal. 2017, 7, 997–1014. 10.1021/acscatal.6b02923. [DOI] [Google Scholar]
- Howarth A. J.; Liu Y.; Li P.; Li Z.; Wang T. C.; Hupp J. T.; Farha O. K. Chemical, Thermal and Mechanical Stabilities of Metal–Organic Frameworks. Nat. Rev. Mater. 2016, 1, 15018. 10.1038/natrevmats.2015.18. [DOI] [Google Scholar]
- Bai Y.; Dou Y.; Xie L. H.; Rutledge W.; Li J. R.; Zhou H. C. Zr-Based Metal–Organic Frameworks: Design, Synthesis, Structure, and Application. Chem. Soc. Rev. 2016, 45, 2327–2367. 10.1039/C5CS00837A. [DOI] [PubMed] [Google Scholar]
- Planas N.; Mondloch J. E.; Tussupbayev S.; Borycz J.; Gagliardi L.; Hupp J. T.; Farha O. K.; Cramer C. J. Defining the Proton Topology of the Zr6-Based Metal–Organic Framework NU-1000. J. Phys. Chem. Lett. 2014, 5, 3716–3723. 10.1021/jz501899j. [DOI] [PubMed] [Google Scholar]
- Odoh S. O.; Cramer C. J.; Truhlar D. G.; Gagliardi L. Quantum-Chemical Characterization of the Properties and Reactivities of Metal–Organic Frameworks. Chem. Rev. 2015, 115, 6051–6111. 10.1021/cr500551h. [DOI] [PubMed] [Google Scholar]
- Coudert F. X.; Fuchs A. H. Computational Characterization and Prediction of Metal–Organic Framework Properties. Coord. Chem. Rev. 2016, 307, 211–236. 10.1016/j.ccr.2015.08.001. [DOI] [Google Scholar]
- Evans J. D.; Fraux G.; Gaillac R.; Kohen D.; Trousselet F.; Vanson J. M.; Coudert F. X. Computational Chemistry Methods for Nanoporous Materials. Chem. Mater. 2017, 29, 199–212. 10.1021/acs.chemmater.6b02994. [DOI] [Google Scholar]
- Van Speybroeck V.; Hemelsoet K.; Joos L.; Waroquier M.; Bell R. G.; Catlow C. R. A. Advances in Theory and their Application within the Field of Zeolite Chemistry. Chem. Soc. Rev. 2015, 44, 7044–7111. 10.1039/C5CS00029G. [DOI] [PubMed] [Google Scholar]
- Corma A. State of the Art and Future Challenges of Zeolites as Catalysts. J. Catal. 2003, 216, 298–312. 10.1016/S0021-9517(02)00132-X. [DOI] [Google Scholar]
- Smit B.; Maesen T. L. Molecular Simulations of Zeolites: Adsorption, Diffusion, and Shape Selectivity. Chem. Rev. 2008, 108, 4125–4184. 10.1021/cr8002642. [DOI] [PubMed] [Google Scholar]
- Nørskov J. K.; Abild-Pedersen F.; Studt F.; Bligaard T. Density Functional Theory in Surface Chemistry and Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 937–943. 10.1073/pnas.1006652108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørskov J. K.; Bligaard T.; Rossmeisl J.; Christensen C. H. Towards the Computational Design of Solid Catalysts. Nat. Chem. 2009, 1, 37–46. 10.1038/nchem.121. [DOI] [PubMed] [Google Scholar]
- Ferguson G. A.; Mehmood F.; Rankin R. B.; Greeley J. P.; Vajda S.; Curtiss L. A. Exploring Computational Design of Size-Specific Subnanometer Clusters Catalysts. Top. Catal. 2012, 55, 353–365. 10.1007/s11244-012-9804-4. [DOI] [Google Scholar]
- Tao F. F.; Schneider W. F.; Kamat P. V. In Heterogeneous Catalysis at Nanoscale for Energy Applications; McCalman D. C., Schneider W. F., Eds.; John Wiley & Sons: New York, 2015; Vol. 1. [Google Scholar]
- Salciccioli M.; Stamatakis M.; Caratzoulas S.; Vlachos D. G. A Review of Multiscale Modeling of Metal-Catalyzed Reactions: Mechanism Development for Complexity and Emergent Behavior. Chem. Eng. Sci. 2011, 66, 4319–4355. 10.1016/j.ces.2011.05.050. [DOI] [Google Scholar]
- Vajda S.; White M. G. Catalysis Applications of Size-Selected Cluster Deposition. ACS Catal. 2015, 5, 7152–7176. 10.1021/acscatal.5b01816. [DOI] [Google Scholar]
- López N.; Almora-Barrios N.; Carchini G.; Błoński P.; Bellarosa L.; Garcia-Muelas R.; Novell-Leruth G.; Garcia-Mota M. State-of-the-Art and Challenges in Theoretical Simulations of Heterogeneous Catalysis at the Microscopic Level. Catal. Sci. Technol. 2012, 2, 2405–2417. 10.1039/c2cy20384g. [DOI] [Google Scholar]
- Carter E. A. Challenges in Modeling Materials Properties without Experimental Input. Science 2008, 321, 800–803. 10.1126/science.1158009. [DOI] [PubMed] [Google Scholar]
- Bromley S. T.; de P. R. Moreira I.; Neyman K. M.; Illas F. Approaching Nanoscale Oxides: Models and Theoretical Methods. Chem. Soc. Rev. 2009, 38, 2657–2670. 10.1039/b806400h. [DOI] [PubMed] [Google Scholar]
- Neyman K. M.; Illas F. Theoretical Aspects of Heterogeneous Catalysis: Applications of Density Functional Methods. Catal. Today 2005, 105, 2–16. 10.1016/j.cattod.2005.04.006. [DOI] [Google Scholar]
- Calle-Vallejo F.; Loffreda D.; Koper M. T.; Sautet P. Introducing Structural Sensitivity into Adsorption–Energy Scaling Relations by means of Coordination Numbers. Nat. Chem. 2015, 7, 403–410. 10.1038/nchem.2226. [DOI] [PubMed] [Google Scholar]
- Loffreda D.; Delbecq F.; Vigné F.; Sautet P. Fast Prediction of Selectivity in Heterogeneous Catalysis from Extended Brønsted–Evans–Polanyi Relations: A Theoretical Insight. Angew. Chem., Int. Ed. 2009, 48, 8978–8980. 10.1002/anie.200902800. [DOI] [PubMed] [Google Scholar]
- Kiss G.; Çelebi-Ölçüm N.; Moretti R.; Baker D.; Houk K. N. Computational Enzyme Design. Angew. Chem., Int. Ed. 2013, 52, 5700–5725. 10.1002/anie.201204077. [DOI] [PubMed] [Google Scholar]
- Houk K. N.; Cheong P. H. Y. Computational Prediction of Small-Molecule Catalysts. Nature 2008, 455, 309–313. 10.1038/nature07368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperger T.; Sanhueza I. A.; Schoenebeck F. Computation and Experiment: A Powerful Combination to Understand and Predict Reactivities. Acc. Chem. Res. 2016, 49, 1311–1319. 10.1021/acs.accounts.6b00068. [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Catalysts by Design: The Power of Theory. Acc. Chem. Res. 2017, 50, 561–566. 10.1021/acs.accounts.6b00555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poree C.; Schoenebeck F. A Holy Grail in Chemistry: Computational Catalyst Design: Feasible or Fiction?. Acc. Chem. Res. 2017, 50, 605–608. 10.1021/acs.accounts.6b00606. [DOI] [PubMed] [Google Scholar]
- Thiel W. Computational Catalysis—Past, Present, and Future. Angew. Chem., Int. Ed. 2014, 53, 8605–8613. 10.1002/anie.201402118. [DOI] [PubMed] [Google Scholar]
- Fang P.-P.; Duan S.; Lin X.-D.; Anema J. R.; Li J.-F.; Buriez O.; Ding Y.; Fan F.-R.; Wu D. Y.; Ren B.; Wang Z. L.; Amatore C.; Tian Z.-Q. Tailoring Au-Core Pd-Shell Pt-Cluster Nanoparticles for Enhanced Electrocatalytic Activity. Chem. Sci. 2011, 2, 531–539. 10.1039/C0SC00489H. [DOI] [Google Scholar]
- Wu J.; Yang H. Platinum-Based Oxygen Reduction Electrocatalysts. Acc. Chem. Res. 2013, 46, 1848–1857. 10.1021/ar300359w. [DOI] [PubMed] [Google Scholar]
- Duanmu K.; Truhlar D. G. Validation of Density Functionals for Adsorption Energies on Transition Metal Surfaces. J. Chem. Theory Comput. 2017, 13, 835–842. 10.1021/acs.jctc.6b01156. [DOI] [PubMed] [Google Scholar]
- Hall M. B.; Margl P.; Náray-Szabó G.; Schramm V. L.; Truhlar D. G.; van Santen R. A.; Warshel A.; Whitten J. L. Quantum Catalysis: The Modeling of Catalytic Transition States. ACS Symp. Ser. 1999, 721, 2–17. 10.1021/bk-1999-0721.ch001. [DOI] [Google Scholar]
- Wang Z.; Wang H.-F.; Hu P. Possibility of Designing Catalysts Beyond the Traditional Volcano Curve: A Theoretical Framework for Multi-Phase Surfaces. Chem. Sci. 2015, 6, 5703–5711. 10.1039/C5SC01732G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine T. M.; Kärkäs M. D.; Liao R.-Z.; Siegbahn P. E. M.; Akermark B. A Dinuclear Ruthenium-Based Water Oxidation Catalyst: Use of Non-Innocent Ligand Frameworks for Promoting Multi-Electron Reactions. Chem. - Eur. J. 2015, 21, 10039–10048. 10.1002/chem.201406613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn W.; Becke A. D.; Parr R. G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. 10.1021/jp960669l. [DOI] [Google Scholar]
- Getman R. B.; Bae Y. S.; Wilmer C. E.; Snurr R. Q. Review and Analysis of Molecular Simulations of Methane, Hydrogen, and Acetylene Storage in Metal–Organic Frameworks. Chem. Rev. 2012, 112, 703–723. 10.1021/cr200217c. [DOI] [PubMed] [Google Scholar]
- Fang H.; Demir H.; Kamakoti P.; Sholl D. S. Recent Developments in First-Principles Force Fields for Molecules in Nanoporous Materials. J. Mater. Chem. A 2014, 2, 274–291. 10.1039/C3TA13073H. [DOI] [Google Scholar]
- Addicoat M. A.; Vankova N.; Akter I. F.; Heine T. Extension of the Universal Force Field to Metal–Organic Frameworks. J. Chem. Theory Comput. 2014, 10, 880–891. 10.1021/ct400952t. [DOI] [PubMed] [Google Scholar]
- Yu J.; Xie L. H.; Li J. R.; Ma Y.; Seminario J. M.; Balbuena P. B. CO2 Capture and Separations Using MOFs: Computational and Experimental Studies. Chem. Rev. 2017, 117, 9674–9754. 10.1021/acs.chemrev.6b00626. [DOI] [PubMed] [Google Scholar]
- Cramer C. J.; Truhlar D. G. Density Functional Theory for Transition Metals and Transition Metal Chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. 10.1039/b907148b. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; He X.; Li S. L.; Truhlar D. G. MN15: A Kohn-Sham Global-Hybrid Exchange-Correlation Density Functional with Broad Accuracy for Multi-Reference and Single-Reference Systems and Noncovalent Interactions. Chem. Sci. 2016, 7, 5032–5051. 10.1039/C6SC00705H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzunova E. L.; Göltl F.; Kresse G.; Hafner J. Application of Hybrid Functionals to the Modeling of NO Adsorption on Cu-SAPO-34 and Co-SAPO-34: A Periodic DFT Study. J. Phys. Chem. C 2009, 113, 5274–5291. 10.1021/jp809927k. [DOI] [Google Scholar]
- Valero R.; Gomes J. R. B.; Truhlar D. G.; Illas F. Density Functional Study of CO and NO Adsorption on Ni-Doped MgO(100). J. Chem. Phys. 2010, 132, 104701. 10.1063/1.3340506. [DOI] [PubMed] [Google Scholar]
- Bao J. L.; Yu H. S.; Duanmu K.; Makeev M. A.; Xu X.; Truhlar D. G. Density Functional Theory of the Water Splitting Reaction on Fe(0): Comparison of Local and Nonlocal Correlation Functionals. ACS Catal. 2015, 5, 2070–2080. 10.1021/cs501675t. [DOI] [Google Scholar]
- Wellendorff J.; Silbaugh T. L.; Garcia-Pintos D.; Nørskov J. K.; Bligaard T.; Studt F.; Campbell C. T. A Benchmark Database for Adsorption Bond Energies to Transition Metal Surfaces and Comparison to Selected DFT Functionals. Surf. Sci. 2015, 640, 36–44. 10.1016/j.susc.2015.03.023. [DOI] [Google Scholar]
- Nazarian D.; Ganesh P.; Sholl D. S. Benchmarking Density Functional Theory Predictions of Framework Structures and Properties in a Chemically Diverse Test Set of Metal–Organic Frameworks. J. Mater. Chem. A 2015, 3, 22432–22440. 10.1039/C5TA03864B. [DOI] [Google Scholar]
- Yu H. S.; He X.; Truhlar D. G. MN15-L: A New Local Exchange-Correlation Functional for Kohn–Sham Density Functional Theory with Broad Accuracy for Atoms, Molecules, and Solids. J. Chem. Theory Comput. 2016, 12, 1280–1293. 10.1021/acs.jctc.5b01082. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Jin X.; Yu H. S.; Truhlar D. G.; He X. Revised M06-L Functional for Improved Accuracy on Chemical Reaction Barrier Heights, Noncovalent Interactions, and Solid-State Physics. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 8487–8492. 10.1073/pnas.1705670114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson K.; Malmqvist P. A.; Roos B. O.; Sadlej A. J.; Wolinski K. Second-Order Perturbation Theory with a CASSCF Reference Function. J. Phys. Chem. 1990, 94, 5483–5488. 10.1021/j100377a012. [DOI] [Google Scholar]
- Szalay P. G.; Muller T.; Gidofalvi G.; Lischka H.; Shepard R. Multiconfiguration Self-Consistent Field and Multireference Config- uration Interaction Methods and Applications. Chem. Rev. 2012, 112, 108–181. 10.1021/cr200137a. [DOI] [PubMed] [Google Scholar]
- Gagliardi L.; Truhlar D. G.; Li Manni G.; Carlson R. K.; Hoyer C. E.; Bao J. L. Multiconfiguration Pair-Density Functional Theory: A New Way to Treat Strongly Correlated Systems. Acc. Chem. Res. 2017, 50, 66–73. 10.1021/acs.accounts.6b00471. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101. 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- Xiao D. J.; Bloch E. D.; Mason J. A.; Queen W. L.; Hudson M. R.; Planas N.; Borycz J.; Dzubak A. L.; Verma P.; Lee K.; Bonin0 F.; Crocellà V.; Yano J.; Bordiga S.; Truhlar D. G.; Gagliardi L.; Brown C. M.; Long R. J. Oxidation of Ethane to Ethanol by N2O in a Metal–Organic Framework with Coordinatively Unsaturated Iron(II) Sites. Nat. Chem. 2014, 6, 590–595. 10.1038/nchem.1956. [DOI] [PubMed] [Google Scholar]
- Verma P.; Vogiatzis K. D.; Planas N.; Borycz J.; Xiao D. J.; Long J. R.; Gagliardi L.; Truhlar D. G. Mechanism of Oxidation of Ethane to Ethanol at Iron (IV)–Oxo Sites in Magnesium-Diluted Fe2 (dobdc). J. Am. Chem. Soc. 2015, 137, 5770–5781. 10.1021/jacs.5b00382. [DOI] [PubMed] [Google Scholar]
- Torker S.; Merki D.; Chen P. Gas-Phase Thermochemistry of Ruthenium Carbene Metathesis Catalysts. J. Am. Chem. Soc. 2008, 130, 4808–4814. 10.1021/ja078149z. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Benchmark Data for Interactions in Zeolite Model Complexes and Their Use for Assessment and Validation of Electronic Structure Methods. J. Phys. Chem. C 2008, 112, 6860–6868. 10.1021/jp7112363. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. Benchmark Energetic Data in a Model System for Grubbs II Metathesis Catalysis and Their Use for Assessment and Validation of Electronic Structure Methods. J. Chem. Theory Comput. 2009, 5, 324–333. 10.1021/ct800386d. [DOI] [PubMed] [Google Scholar]
- Wannakao S.; Boekfa B.; Khongpracha P.; Probst M.; Limtrakul J. Oxidative Dehydrogenation of Propane Over a VO2-Exchanged MCM-22 Zeolite: A DFT Study. ChemPhysChem 2010, 11, 3432–3438. 10.1002/cphc.201000586. [DOI] [PubMed] [Google Scholar]
- Wannakao S.; Warakulwit C.; Kongpatpanich K.; Probst M.; Limtrakul J. Methane Activation in Gold Cation-Exchanged Zeolites: A DFT Study. ACS Catal. 2012, 2, 986–992. 10.1021/cs200653q. [DOI] [Google Scholar]
- Averkiev B. B.; Truhlar D. G. Free Energy of Reaction by Density Functional Theory: Oxidative Addition of Ammonia by an Iridium Complex with PCP Pincer Ligands. Catal. Sci. Technol. 2011, 1, 1526–1529. 10.1039/c1cy00227a. [DOI] [Google Scholar]
- Ashworth I. W.; Hillier I. H.; Nelson D. J.; Percy J. M.; Vincent M. A. Olefin Metathesis by Grubbs-Hoveyda Complexes: Computational and Experimental Studies of the Mechanism and Substrate-Dependent Kinetics. ACS Catal. 2013, 3, 1929–1939. 10.1021/cs400164w. [DOI] [Google Scholar]
- Larionov E.; Nakanishi M.; Katayev D.; Besnard C.; Kündig E. P. Scope and Mechanism of Asymmetric C(sp3)-H/C(Ar)-X Coupling Reactions: Computational and Experimental Study. Chem. Sci. 2013, 4, 1995–2005. 10.1039/c3sc00098b. [DOI] [Google Scholar]
- Getsoian A. B.; Bell A. T. The Influence of Functionals on Density Functional Theory Calculations of the Properties of Reducible Transition Metal Oxide Catalysts. J. Phys. Chem. C 2013, 117, 25562–25578. 10.1021/jp409479h. [DOI] [Google Scholar]
- Kumar M.; Chaudhari R. V.; Subramaniam B.; Jackson T. A. Ligand Effects on the Regioselectivity of Rhodium-Catalyzed Hydroformylation: Density Functional Calculations Illuminate the Role of Long-Range Noncovalent Interactions. Organometallics 2014, 33, 4183–4191. 10.1021/om500196g. [DOI] [Google Scholar]
- Kumar M.; Chaudhari R. V.; Subramaniam B.; Jackson T. A. Importance of Long-Range Noncovalent Interactions in the Regioselectivity of Rhodium-Xantphos-Catalyzed Hydroformylation. Organometallics 2015, 34, 1062–1073. 10.1021/om5012775. [DOI] [Google Scholar]
- Roy T.; Barik S.; Kumar M.; Kureshy R. I.; Ganguly B.; Khan N.-U. H.; Abdi S. H. R.; Bajaj H. C. Asymmetric Hydrolytic Kinetic Resolution with Recyclable Polymeric Co(III)-Salen Complexes: A Practical Strategy in the Preparation of (S)-Metoprolol, (S)-Toliprolol and (S)-Alprenolol: Computational Rationale for Enantioselectivity. Catal. Sci. Technol. 2014, 4, 3899–3908. 10.1039/C4CY00594E. [DOI] [Google Scholar]
- Rohmann K.; Hölscher M.; Leitner W. Can Contemporary Density Functional Theory Predict Energy Spans in Molecular Catalysis Accurately Enough to Be Applicable for in Silico Catalyst Design? A Computational/Experimental Case Study for the Ruthenium-Catalyzed Hydrogenation of Olefins. J. Am. Chem. Soc. 2016, 138, 433–443. 10.1021/jacs.5b11997. [DOI] [PubMed] [Google Scholar]
- Meeprasert J.; Namuangruk S.; Boekfa B.; Dhital R. N.; Sakurai H.; Ehara M. Mechanism of Ullmann Coupling Reaction of Chloroarene on Au/Pd Alloy Nanocluster: A DFT Study. Organometallics 2016, 35, 1192–1201. 10.1021/acs.organomet.5b01009. [DOI] [Google Scholar]
- Mondal T.; De S.; Maity B.; Koley D. Exploring the Oxidative-Addition Pathways of Phenyl Chloride in the Presence of PdII Abnormal N-Heterocyclic Carbene Complexes: A DFT Study. Chem. - Eur. J. 2016, 22, 15778–15790. 10.1002/chem.201602735. [DOI] [PubMed] [Google Scholar]
- Özkılıç Y.; Tüzün N. S. A DFT Study on the Binuclear CuAAC Reaction: Mechanism in Light of New Experiments. Organometallics 2016, 35, 2589–2599. 10.1021/acs.organomet.6b00279. [DOI] [Google Scholar]
- Patel P. D.; Laird B. B.; Thompson W. H. A Density Functional Theory Study of Ethylene Epoxidation Catalyzed by Niobium-Doped Silica. J. Mol. Catal. A: Chem. 2016, 424, 1–7. 10.1016/j.molcata.2016.07.052. [DOI] [Google Scholar]
- Jiang Y. Y.; Jiang J. L.; Fu Y. Mechanism of Vanadium-Catalyzed Deoxydehydration of Vicinal Diols: Spin-Crossover-Involved Processes. Organometallics 2016, 35, 3388–3396. 10.1021/acs.organomet.6b00602. [DOI] [Google Scholar]
- Yang K.; Zheng J.; Zhao Y.; Truhlar D. G. Tests of the RPBE, revPBE, τ-HCTHhyb, ωB97X-D, and MOHLYP Density Functional Approximations and 29 Others Against Representative Databases for Diverse Bond Energies and Barrier Heights in Catalysis. J. Chem. Phys. 2010, 132, 164117. 10.1063/1.3382342. [DOI] [PubMed] [Google Scholar]
- Yang H.-C.; Huang Y.-C.; Lan Y.-K.; Luh T.-Y.; Zhao Y.; Truhlar D. G. Carbene Rotamer Switching Explains the Reverse Trans Effect in Forming the Grubbs Second-Generation Olefin Metathesis Catalyst. Organometallics 2011, 30, 4196–4200. 10.1021/om200529m. [DOI] [Google Scholar]
- Dang Y.; Qu S.; Nelson J. W.; Pham H. D.; Wang Z.-X.; Wang X. The Mechanism of a Ligand-Promoted C(sp3)–H Activation and Arylation Reaction via Palladium Catalysis: Theoretical Demonstration of a Pd(II)/Pd(IV) Redox Manifold. J. Am. Chem. Soc. 2015, 137, 2006–2014. 10.1021/ja512374g. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Zhang Q.; Shi J.; Fu Y. Mechanism Study of Mn(I) Complex-Catalyzed Imines and Alkynes Dehydrogenation Coupling Reaction. Huaxue Xuebao 2016, 74, 422–428. 10.6023/A15110736. [DOI] [Google Scholar]
- Xu D.; Qi X.; Duan M.; Yu Z.; Zhu L.; Shan C.; Yue X.; Bai R.; Lan Y. Thiolate-Palladium(IV) or Sulfonium-Palladate(0)? A Theoretical Study on the Mechanism of Palladium-Catalyzed C-S Bond Formation Reactions. Org. Chem. Front. 2017, 4, 943–950. 10.1039/C6QO00841K. [DOI] [Google Scholar]
- Witte J.; Neaton J. B.; Head-Gordon M. Assessing Electronic Structure Approaches for Gas-Ligand Interactions in Metal-Organic Frameworks: The CO2-Benzene Complex. J. Chem. Phys. 2014, 140, 104707. 10.1063/1.4867698. [DOI] [PubMed] [Google Scholar]
- Lownsbury J. M.; Santos-López I. A.; Zhang W.; Campbell C. T.; Yu H. S.; Liu W.-G.; Cramer C. J.; Truhlar D. G.; Wang T.; Hupp J. T.; Farha O. K. Calcium Vapor Adsorption on the Metal–Organic Framework NU-1000: Structure and Energetics. J. Phys. Chem. C 2016, 120, 16850–16862. 10.1021/acs.jpcc.6b05707. [DOI] [Google Scholar]
- Ikuno T.; Zheng J.; Vjunov A.; Sanchez-Sanchez M.; Ortuño M. A.; Pahls D. R.; Fulton J. L.; Camaioni D. M.; Li Z.; Ray D.; Mehdi B. L.; Browning N. D.; Farha O. K.; Hupp J. T.; Cramer C. J.; Gagliardi L.; Lercher J. A. Methane Oxidation to Methanol Catalyzed by Cu-oxo Clusters Stabilized in NU-1000 Metal-Organic Framework. J. Am. Chem. Soc. 2017, 139, 10294–10301. 10.1021/jacs.7b02936. [DOI] [PubMed] [Google Scholar]
- Bernales V.; League A. B.; Li Z.; Schweitzer N. M.; Peters A. W.; Carlson R. K.; Hupp J. T.; Cramer C. J.; Farha O. K.; Gagliardi L. Computationally Guided Discovery of a Catalytic Cobalt-Decorated Metal–Organic Framework for Ethylene Dimerization. J. Phys. Chem. C 2016, 120, 23576–23583. 10.1021/acs.jpcc.6b07362. [DOI] [Google Scholar]
- Hirao H.; Ng W. K. H.; Moeljadi A. M. P.; Bureekaew S. Multiscale Model for a Metal–Organic Framework: High-Spin Rebound Mechanism in the Reaction of the Oxoiron(IV) Species of Fe-MOF-74. ACS Catal. 2015, 5, 3287–3291. 10.1021/acscatal.5b00475. [DOI] [Google Scholar]
- Choomwattana S.; Maihom T.; Khongpracha P.; Probst M.; Limtrakul J. Structures and Mechanisms of the Carbonyl-ene Reaction between MOF-11 Encapsulated Formaldehyde and Propylene: An ONIOM Study. J. Phys. Chem. C 2008, 112, 10855–10861. 10.1021/jp8021437. [DOI] [Google Scholar]
- Doitomi K.; Xu K.; Hirao H. The Mechanism of an Asymmetric Ring-Opening Reaction of Epoxide with Amine Catalyzed by a Metal–Organic Framework: Insights from Combined Quantum Mechanics and Molecular Mechanics Calculations. Dalton Trans. 2017, 46, 3470–3481. 10.1039/C6DT04745A. [DOI] [PubMed] [Google Scholar]
- Oxford G. A.; Snurr R. Q.; Broadbelt L. J. Hybrid Quantum Mechanics/Molecular Mechanics Investigation of (salen) Mn for use in Metal–Organic Frameworks. Ind. Eng. Chem. Res. 2010, 49, 10965–10973. 10.1021/ie100165j. [DOI] [Google Scholar]
- Zheng M.; Liu Y.; Wang C.; Liu S.; Lin W. Cavity-Induced Enantioselectivity Reversal in a Chiral Metal–Organic Framework Brønsted Acid Catalyst. Chem. Sci. 2012, 3, 2623–2627. 10.1039/c2sc20379k. [DOI] [Google Scholar]
- Yu D.; Yazaydin A. O.; Lane J. R.; Dietzel P. D. C.; Snurr R. Q. A Combined Experimental and Quantum Chemical Study of CO2 Adsorption in the Metal–Organic Framework CPO-27 with Different Metals. Chem. Sci. 2013, 4, 3544–3556. 10.1039/c3sc51319j. [DOI] [Google Scholar]
- Moeljadi A. M. P.; Schmid R.; Hirao H. Dioxygen Binding to Fe-MOF-74: Microscopic Insights from Periodic QM/MM Calculations. Can. J. Chem. 2016, 94, 1144–1150. 10.1139/cjc-2016-0284. [DOI] [Google Scholar]
- Doitomi K.; Hirao H. Hybrid Computational Approaches for Deriving Quantum Mechanical Insights into Metal–Organic Frameworks. Tetrahedron Lett. 2017, 58, 2309–2317. 10.1016/j.tetlet.2017.04.088. [DOI] [Google Scholar]
- Antes I.; Thiel W. In Combined Quantum Mechanical and Molecular Mechanical Methods; Gao J., Thompson M. A., Eds.; ACS Symposium Series Vol. 712, American Chemical Society: Washington, 1998; pp 50−65. [Google Scholar]
- Wang B.; Truhlar D. G. Combined Quantum Mechanical and Molecular Mechanical Methods for Calculating Potential Energy Surfaces: Tuned and Balanced Redistributed-Charge Algorithm. J. Chem. Theory Comput. 2010, 6, 359–369. 10.1021/ct900366m. [DOI] [PubMed] [Google Scholar]
- Kundu A.; Piccini G.; Sillar K.; Sauer J. Ab Initio Prediction of Adsorption Isotherms for Small Molecules in Metal–Organic Frameworks. J. Am. Chem. Soc. 2016, 138, 14047–14056. 10.1021/jacs.6b08646. [DOI] [PubMed] [Google Scholar]
- Ling S.; Slater B. Dynamic Acidity in Defective UiO-66. Chem. Sci. 2016, 7, 4706–4712. 10.1039/C5SC04953A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. L.; Wu S.; Yang P.; He S.; Dou L.; Wang F. F. Ab Initio Molecular Dynamic Simulations on Pd Clusters Confined in UiO-66-NH2. J. Phys. Chem. C 2017, 121, 8857–8863. 10.1021/acs.jpcc.7b00957. [DOI] [Google Scholar]
- Voter A. F. Hyperdynamics: Accelerated Molecular Dynamics of Infrequent Events. Phys. Rev. Lett. 1997, 78, 3908–3911. 10.1103/PhysRevLett.78.3908. [DOI] [Google Scholar]
- Perez D.; Uberuaga B. P.; Shim Y.; Amar J. G.; Voter A. F. Accelerated Molecular Dynamics Methods: Introduction and Recent Developments. Annu. Rep. Comput. Chem. 2009, 5, 79–98. 10.1016/S1574-1400(09)00504-0. [DOI] [Google Scholar]
- Barducci A.; Bonomi M.; Parrinello M. Metadynamics. WIREs Comput. Mol. Sci. 2011, 1, 826–843. 10.1002/wcms.31. [DOI] [Google Scholar]
- Ovesson S.; Lundqvist B. I.; Schneider W. F.; Bogicevic A. NO Oxidation Properties of Pt(111) Revealed by Ab Initio Kinetic Simulations. Phys. Rev. B: Condens. Matter Mater. Phys. 2005, 71, 115406. 10.1103/PhysRevB.71.115406. [DOI] [Google Scholar]
- Getman R. B.; Schneider W. F. DFT-Based Coverage-Dependent Model of Pt-Catalyzed NO Oxidation. ChemCatChem 2010, 2, 1450–1460. 10.1002/cctc.201000146. [DOI] [Google Scholar]
- Matera S.; Reuter K. Transport Limitations and Bistability for In Situ CO Oxidation at RuO2(110): First-Principles Based Multiscale Modeling. Phys. Rev. B: Condens. Matter Mater. Phys. 2010, 82, 085446. 10.1103/PhysRevB.82.085446. [DOI] [Google Scholar]
- Piskorz W.; Zasada F.; Stelmachowski P.; Diwald O.; Kotarba A.; Sojka Z. Computational and Experimental Investigations into N2O Decomposition Over MgO Nanocrystals from Thorough Molecular Mechanism to Ab Initio Microkinetics. J. Phys. Chem. C 2011, 115, 22451–22460. 10.1021/jp2070826. [DOI] [Google Scholar]
- Reuter K. In Modeling and Simulation of Heterogeneous Catalytic Reactions; Deutschmann O., Ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- McEwen J.-S.; Bray J. M.; Wu C.; Schneider W. F. How Low Can You Go? Minimum Energy Pathways for O2 Dissociation on Pt(111). Phys. Chem. Chem. Phys. 2012, 14, 16677–16685. 10.1039/c2cp42225e. [DOI] [PubMed] [Google Scholar]
- Ma R.; Schuette G. F.; Broadbelt L. J. Microkinetic Modeling of CO2 Hydrolysis Over Zn-(1,4,7,10-Tetraazacyclododecane) Catalyst Based on First Principles: Revelation of Rate-Determining Step. J. Catal. 2014, 317, 176–184. 10.1016/j.jcat.2014.06.001. [DOI] [Google Scholar]
- Rozanska X.; Fortrie R.; Sauer J. Size-Dependent Catalytic Activity of Supported Vanadium Oxide Species: Oxidative Dehydrogenation of Propane. J. Am. Chem. Soc. 2014, 136, 7751–7761. 10.1021/ja503130z. [DOI] [PubMed] [Google Scholar]
- Stamatakis M. Kinetic Modelling of Heterogeneous Catalytic Systems. J. Phys.: Condens. Matter 2015, 27, 013001. 10.1088/0953-8984/27/1/013001. [DOI] [PubMed] [Google Scholar]
- Patet R. E.; Nikbin N.; Williams C. L.; Green S. K.; Chang C.-C.; Fan W.; Caratzoulas S.; Dauenhauer P. J.; Vlachos D. G. Kinetic Regime Change in the Tandem Dehydrative Aromatization of Furan Diels–Alder Products. ACS Catal. 2015, 5, 2367–2375. 10.1021/cs5020783. [DOI] [Google Scholar]
- Yang L.; Tsilomelekis G.; Caratzoulas S.; Vlachos D. G. Mechanism of Brønsted Acid-Catalyzed Glucose Dehydration. ChemSusChem 2015, 8, 1334–1341. 10.1002/cssc.201403264. [DOI] [PubMed] [Google Scholar]
- Reuter K. Ab Initio Thermodynamics and First-Principles Microkinetics for Surface Catalysis. Catal. Lett. 2016, 146, 541–563. 10.1007/s10562-015-1684-3. [DOI] [Google Scholar]
- John M.; Alexopoulos K.; Reyniers M.; Marin G. B. Mechanistic Insights into the Formation of Butene Isomers from 1-Butanol in H-ZSM-5: DFT Based Microkinetic Modelling. Catal. Sci. Technol. 2017, 7, 1055–1072. 10.1039/C6CY02474B. [DOI] [Google Scholar]
- Che F.; Ha S.; McEwen J. Hydrogen Oxidation and Water Dissociation over an Oxygen-Enriched Ni/YSZ Electrode in the Presence of an Electric Field: A First-Principles-Based Microkinetic Model. Ind. Eng. Chem. Res. 2017, 56, 1201–1213. 10.1021/acs.iecr.6b04028. [DOI] [Google Scholar]
- Klippenstein S. J.; Pande V.; Truhlar D. G. Chemical Kinetics and Mechanisms of Complex Systems: A Perspective on Recent Theoretical Advances. J. Am. Chem. Soc. 2014, 136, 528–546. 10.1021/ja408723a. [DOI] [PubMed] [Google Scholar]
- Alecu I. M.; Zheng J.; Zhao Y.; Truhlar D. G. Computational Thermochemistry: Scale Factor Databases and Scale Factors for Vibrational Frequencies Obtained from Electronic Model Chemistries. J. Chem. Theory Comput. 2010, 6, 2872–2887. 10.1021/ct100326h. [DOI] [PubMed] [Google Scholar]
- Pratt L. M.; Truhlar D. G.; Cramer C. J.; Kass S. R.; Thompson J. D.; Xidos J. D. Aggregation of Alkyllithiums in Tetrahydrofuran. J. Org. Chem. 2007, 72, 2962–2966. 10.1021/jo062557o. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Computational Characterization and Modeling of Buckyball Tweezers: Density Functional Study of Concave–Convex π···π Interactions. Phys. Chem. Chem. Phys. 2008, 10, 2813–2818. 10.1039/b717744e. [DOI] [PubMed] [Google Scholar]
- Ribeiro R. F.; Marenich A. V.; Cramer C. J.; Truhlar D. G. Use of Solution-Phase Vibrational Frequencies in Continuum Models for the Free Energy of Solvation. J. Phys. Chem. B 2011, 115, 14556–14562. 10.1021/jp205508z. [DOI] [PubMed] [Google Scholar]
- Van Yperen-De Deyne A.; Hendrickx K.; Vanduyfhuys L.; Sastre G.; Van Der Voort P.; Van Speybroeck V.; Hemelsoet K. Vibrational Fingerprint of the Absorption Properties of UiO-Type MOF Materials. Theor. Chem. Acc. 2016, 135, 102. 10.1007/s00214-016-1842-8. [DOI] [Google Scholar]
- De Moor B. A.; Ghysels A.; Reyniers M. F.; Van Speybroeck V.; Waroquier M.; Marin G. B. Normal Mode Analysis in Zeolites: Toward an Efficient Calculation of Adsorption Entropies. J. Chem. Theory Comput. 2011, 7, 1090–1101. 10.1021/ct1005505. [DOI] [PubMed] [Google Scholar]
- Kreevoy M. M.; Truhlar D. G. In Investigation of Rates and Mechanisms of Reactions, 4th ed.; Bernasconi C. F., Ed.; Techniques of Chemistry, 4th ed. Vol. 6, Part 1; John Wiley and Sons: New York, 1986; pp 13−95. [Google Scholar]
- Truhlar D. G.; Garrett B. C. Variational Transition-State Theory. Acc. Chem. Res. 1980, 13, 440–448. 10.1021/ar50156a002. [DOI] [Google Scholar]
- Fernandez-Ramos A.; Ellingson B. A.; Garrett B. C.; Truhlar D. G. Variational Transition State Theory with Multidimensional Tunneling. Rev. Comp. Chem. 2007, 23, 125–232. 10.1002/9780470116449.ch3. [DOI] [Google Scholar]
- Spivey J. J.; Krishna K. S.; Kumar C. S. S. R.; Dooley K. M.; Flake J. C.; Haber L. H.; Xu Y.; Janik M. J.; Sinnott S. B.; Cheng Y.-T.; Liang T.; Sholl D. S.; Manz T. A.; Diebold U.; Parkinson G. S.; Bruce D. A.; de Jongh P. Synthesis, Characterization, and Computation of Catalysts at the Center for Atomic-Level Catalyst Design. J. Phys. Chem. C 2014, 118, 20043–20069. 10.1021/jp502556u. [DOI] [Google Scholar]
- Morris W.; Volosskiy B.; Demir S.; Gándara F.; McGrier P. L.; Furukawa H.; Cascio D.; Stoddart F. J.; Yaghi O. M. Synthesis, Structure, and Metalation of Two New Highly Porous Zirconium Metal–Organic Frameworks. Inorg. Chem. 2012, 51, 6443–6445. 10.1021/ic300825s. [DOI] [PubMed] [Google Scholar]
- Feng D.; Gu Z. Y.; Li J. R.; Jiang H. L.; Wei Z.; Zhou H. C. Zirconium-Metalloporphyrin PCN-222: Mesoporous Metal–Organic Frameworks with Ultrahigh Stability as Biomimetic Catalysts. Angew. Chem., Int. Ed. 2012, 51, 10307–10310. 10.1002/anie.201204475. [DOI] [PubMed] [Google Scholar]
- Troya D. Reaction Mechanism of Nerve-Agent Decomposition with Zr-Based Metal Organic Frameworks. J. Phys. Chem. C 2016, 120, 29312–29323. 10.1021/acs.jpcc.6b10530. [DOI] [Google Scholar]
- Taddei M. When Defects Turn into Virtues: The Curious Case of Zirconium-Based Metal-Organic Rrameworks. Coord. Chem. Rev. 2017, 343, 1–24. 10.1016/j.ccr.2017.04.010. [DOI] [Google Scholar]
- Trickett C. A.; Gagnon K. J.; Lee S.; Gándara F.; Bürgi H. B.; Yaghi O. M. Definitive Molecular Level Characterization of Defects in UiO-66 Crystals. Angew. Chem., Int. Ed. 2015, 54, 11162–11167. 10.1002/anie.201505461. [DOI] [PubMed] [Google Scholar]
- Yuan S.; Zou L.; Li H.; Chen Y.-P.; Qin J.; Zhang Q.; Lu W.; Hall M. B.; Zhou H.-C. Flexible Zirconium Metal-Organic Frameworks as Bioinspired Switchable Catalysts. Angew. Chem., Int. Ed. 2016, 55, 10776–10780. 10.1002/anie.201604313. [DOI] [PubMed] [Google Scholar]
- Klet R. C.; Liu Y.; Wang T. C.; Hupp J. T.; Farha O. K. Evaluation of Brønsted Acidity and Proton Topology in Zr-and Hf-Based Metal–Organic Frameworks Using Potentiometric Acid–Base Titration. J. Mater. Chem. A 2016, 4, 1479–1485. 10.1039/C5TA07687K. [DOI] [Google Scholar]
- Islamoglu T.; Atilgan A.; Moon S. Y.; Peterson G. W.; DeCoste J. B.; Hall M.; Hupp J. T.; Farha O. K. Cerium (IV) vs Zirconium (IV) Based Metal–Organic Frameworks for Detoxification of a Nerve Agent. Chem. Mater. 2017, 29, 2672–2675. 10.1021/acs.chemmater.6b04835. [DOI] [Google Scholar]
- Valenzano L.; Civalleri B.; Chavan S.; Bordiga S.; Nilsen M. H.; Jakobsen S.; Lillerud K. P.; Lamberti C. Disclosing the Complex Structure of UiO-66 Metal Organic Framework: A Synergic Combination of Experiment and Theory. Chem. Mater. 2011, 23, 1700–1718. 10.1021/cm1022882. [DOI] [Google Scholar]
- Chavan S.; Vitillo J. G.; Gianolio D.; Zavorotynska O.; Civalleri B.; Jakobsen S.; Nilsen M. H.; Valenzano L.; Lamberti C.; Lillerud K. P.; Bordiga S. H2 storage in Isostructural UiO-67 and UiO-66 MOFs. Phys. Chem. Chem. Phys. 2012, 14, 1614–1626. 10.1039/C1CP23434J. [DOI] [PubMed] [Google Scholar]
- Vandichel M.; Hajek J.; Ghysels A.; De Vos A.; Waroquier M.; Van Speybroeck V. Water Coordination and Dehydration Processes in Defective UiO-66 Type Metal Organic Frameworks. CrystEngComm 2016, 18, 7056–7069. 10.1039/C6CE01027J. [DOI] [Google Scholar]
- Platero-Prats A. E.; Mavrandonakis A.; Gallington L. C.; Liu Y.; Hupp J. T.; Farha O. K.; Cramer C. J.; Chapman K. W. Structural Transitions of the Metal-Oxide Nodes within Metal–Organic Frameworks: On the Local Structures of NU-1000 and UiO-66. J. Am. Chem. Soc. 2016, 138, 4178–4185. 10.1021/jacs.6b00069. [DOI] [PubMed] [Google Scholar]
- Guan S.-H.; Zhang X.-J.; Liu Z.-P. Energy Landscape of Zirconia Phase Transitions. J. Am. Chem. Soc. 2015, 137, 8010–8013. 10.1021/jacs.5b04528. [DOI] [PubMed] [Google Scholar]
- Yang D.; Bernales V.; Islamoglu T.; Farha O. K.; Hupp J. T.; Cramer C. J.; Gagliardi L.; Gates B. C. Tuning the Surface Chemistry of Metal Organic Framework Nodes: Proton Topology of the Metal-oxide-like Zr6 nodes of UiO-66 and NU-1000. J. Am. Chem. Soc. 2016, 138, 15189–15196. 10.1021/jacs.6b08273. [DOI] [PubMed] [Google Scholar]
- Vandichel M.; Hajek J.; Vermoortele F.; Waroquier M.; De Vos D. E.; Van Speybroeck V. Active Site Engineering in UiO-66 Type Metal–Organic Frameworks by Intentional Creation of Defects: A Theoretical Rationalization. CrystEngComm 2015, 17, 395–406. 10.1039/C4CE01672F. [DOI] [Google Scholar]
- Vermoortele F.; Bueken B.; Le Bars G.; Van de Voorde B.; Vandichel M.; Houthoofd K.; Vimont A.; Daturi M.; Waroquier M.; Van Speybroeck V.; Kirschhock C.; De Vos D. E. Synthesis Modulation as a Tool to Increase the Catalytic Activity of Metal–Organic Frameworks: The Unique Case of UiO-66(Zr). J. Am. Chem. Soc. 2013, 135, 11465–11468. 10.1021/ja405078u. [DOI] [PubMed] [Google Scholar]
- Bristow J. K.; Svane K. L.; Tiana D.; Skelton J. M.; Gale J. D.; Walsh A. Free Energy of Ligand Removal in the Metal–Organic Framework UiO-66. J. Phys. Chem. C 2016, 120, 9276–9281. 10.1021/acs.jpcc.6b01659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serna P.; Gates B. C. Molecular Metal Catalysts on Supports: Organometallic Chemistry Meets Surface Science. Acc. Chem. Res. 2014, 47, 2612–2620. 10.1021/ar500170k. [DOI] [PubMed] [Google Scholar]
- Copéret C.; Chabanas M.; Petroff Saint-Arroman R.; Basset J.-M. Homogeneous and Heterogeneous Catalysis: Bridging the Gap through Surface Organometallic Chemistry. Angew. Chem., Int. Ed. 2003, 42, 156–181. 10.1002/anie.200390072. [DOI] [PubMed] [Google Scholar]
- Copéret C.; Comas-Vives A.; Conley M. P.; Estes D. P.; Fedorov A.; Mougel V.; Nagae H.; Núñez-Zarur F.; Zhizhko P. V. Surface Organometallic and Coordination Chemistry toward Single-Site Heterogeneous Catalysts: Strategies, Methods, Structures, and Activities. Chem. Rev. 2016, 116, 323–421. 10.1021/acs.chemrev.5b00373. [DOI] [PubMed] [Google Scholar]
- Manna K.; Ji P.; Greene F. X.; Lin W. Metal–Organic Framework Nodes Support Single-Site Magnesium–Alkyl Catalysts for Hydroboration and Hydroamination Reactions. J. Am. Chem. Soc. 2016, 138, 7488–7491. 10.1021/jacs.6b03689. [DOI] [PubMed] [Google Scholar]
- Kim I. S.; Borycz J.; Platero-Prats A. E.; Tussupbayev S.; Wang T. C.; Farha O. K.; Hupp J. T.; Gagliardi L.; Chapman K. W.; Cramer C. J.; Martinson A. B. F. Targeted Single-Site MOF Node Modification: Trivalent Metal Loading via Atomic Layer Deposition. Chem. Mater. 2015, 27, 4772–4778. 10.1021/acs.chemmater.5b01560. [DOI] [Google Scholar]
- Rimoldi M.; Gallington L. C.; Chapman K. W.; MacRenaris K.; Hupp J. T.; Farha O. K. Catalytically Active Silicon Oxide Nanoclusters Stabilized in a Metal-Organic Framework. Chem. - Eur. J. 2017, 23, 8532–8536. 10.1002/chem.201701902. [DOI] [PubMed] [Google Scholar]
- Nguyen H. G. T.; Schweitzer N. M.; Chang C.-Y.; Drake T. L.; So M. C.; Stair P. C.; Farha O. K.; Hupp J. T.; Nguyen S. T. Vanadium-Node-Functionalized UiO-66: A Thermally Stable MOF-Supported Catalyst for the Gas-Phase Oxidative Dehydrogenation of Cyclohexene. ACS Catal. 2014, 4, 2496–2500. 10.1021/cs5001448. [DOI] [Google Scholar]
- Manna K.; Ji P.; Lin Z.; Greene F. X.; Urban A.; Thacker N. C.; Lin W. Chemoselective Single-Site Earth-Abundant Metal Catalysts at Metal-Organic Framework Nodes. Nat. Commun. 2016, 7, 12610. 10.1038/ncomms12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyzavi M. H.; Vermeulen N. A.; Howarth A. J.; Tussupbayev S.; League A. B.; Schweitzer N. M.; Gallagher J. R.; Platero-Prats A. E.; Hafezi N.; Sarjeant A. A.; Miller J. T.; Chapman K. W.; Stoddart J. F.; Cramer C. J.; Hupp J. T.; Farha O. K. A Hafnium-Based Metal–Organic Framework as a Nature-Inspired Tandem Reaction Catalyst. J. Am. Chem. Soc. 2015, 137, 13624–13631. 10.1021/jacs.5b08440. [DOI] [PubMed] [Google Scholar]
- Peters A. W.; Li Z.; Farha O. K.; Hupp J. T. Atomically Precise Growth of Catalytically Active Cobalt Sulfide on Flat Surfaces and within a Metal−Organic Framework via Atomic Layer Deposition. ACS Nano 2015, 9, 8484–8490. 10.1021/acsnano.5b03429. [DOI] [PubMed] [Google Scholar]
- Li Z.; Peters A. W.; Bernales V.; Ortuño M. A.; Schweitzer N. M.; DeStefano M. R.; Gallington L. C.; Platero-Prats A. E.; Chapman K. W.; Cramer C. J.; Gagliardi L.; Hupp J. T.; Farha O. K. Metal–Organic Framework Supported Cobalt Catalysts for the Oxidative Dehydrogenation of Propane at Low Temperature. ACS Cent. Sci. 2017, 3, 31–38. 10.1021/acscentsci.6b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji P.; Manna K.; Lin Z.; Feng X.; Urban A.; Song Y.; Lin W. Single-Site Cobalt Catalysts at New Zr12(μ3-O)8(μ3-OH)8(μ2-OH)6 Metal–Organic Framework Nodes for Highly Active Hydrogenation of Nitroarenes, Nitriles, and Isocyanides. J. Am. Chem. Soc. 2017, 139, 7004–7011. 10.1021/jacs.7b02394. [DOI] [PubMed] [Google Scholar]
- Li Z.; Schweitzer N. M.; League A. B.; Bernales V.; Peters A. W.; Getsoian A. B.; Wang T. C.; Miller J. T.; Vjunov A.; Fulton J. L.; Lercher J. A.; Cramer C.; Gagliardi L.; Hupp J. T.; Hupp J. T.; Farha O. K. Sintering-Resistant Single-Site Nickel Catalyst Supported by Metal–Organic Framework. J. Am. Chem. Soc. 2016, 138, 1977–1982. 10.1021/jacs.5b12515. [DOI] [PubMed] [Google Scholar]
- Platero-Prats A. E.; Li Z.; Gallington L. C.; Peters A. W.; Hupp J. T.; Farha O. K.; Chapman K. W. Addressing the Characterisation Challenge to Understand Catalysis in MOFs: The Case of Nanoscale Cu Supported in NU-1000. Faraday Discuss. 2017, 201, 337–350. 10.1039/C7FD00110J. [DOI] [PubMed] [Google Scholar]
- Gallington L. C.; Kim I. S.; Liu W. G.; Yakovenko A. A.; Platero-Prats A. E.; Li Z.; Wang T. C.; Hupp J. T.; Farha O. K.; Truhlar D. G.; Chapman K. W. Regioselective Atomic Layer Deposition in Metal–Organic Frameworks Directed by Dispersion Interactions. J. Am. Chem. Soc. 2016, 138, 13513–13516. 10.1021/jacs.6b08711. [DOI] [PubMed] [Google Scholar]
- Klet R. C.; Tussupbayev S.; Borycz J.; Gallagher J. R.; Stalzer M. M.; Miller J. T.; Gagliardi L.; Hupp J. T.; Marks T. J.; Cramer C. J.; et al. Single-Site Organozirconium Catalyst Embedded in a Metal–Organic Framework. J. Am. Chem. Soc. 2015, 137, 15680–15683. 10.1021/jacs.5b11350. [DOI] [PubMed] [Google Scholar]
- Ahn S.; Thornburg N. E.; Li Z.; Wang T. C.; Gallington L. C.; Chapman K. W.; Notestein J. M.; Hupp J. T.; Farha O. K. Stable Metal–Organic Framework-Supported Niobium Catalysts. Inorg. Chem. 2016, 55, 11954–11961. 10.1021/acs.inorgchem.6b02103. [DOI] [PubMed] [Google Scholar]
- Noh H.; Cui Y.; Peters A. W.; Pahls D. R.; Ortuño M. A.; Vermeulen N. A.; Cramer C. J.; Gagliardi L.; Hupp J. T.; Farha O. K. An Exceptionally Stable Metal–Organic Framework Supported Molybdenum (VI) Oxide Catalyst for Cyclohexene Epoxidation. J. Am. Chem. Soc. 2016, 138, 14720–14726. 10.1021/jacs.6b08898. [DOI] [PubMed] [Google Scholar]
- Bernales V.; Yang D.; Yu J.; Gümüşlü G.; Cramer C. J.; Gates B. C.; Gagliardi L. Molecular Rhodium Complexes Supported on the Metal-Oxide-Like Nodes of Metal–Organic Frameworks and on Zeolite HY: Catalysts for Ethylene Hydrogenation and Dimerization. ACS Appl. Mater. Interfaces 2017, 9, 33511–33520. 10.1021/acsami.7b03858. [DOI] [PubMed] [Google Scholar]
- Yang D.; Odoh S. O.; Borycz J.; Wang T. C.; Farha O. K.; Hupp J. T.; Cramer C. J.; Gagliardi L.; Gates B. C. Tuning Zr6 Metal–Organic Framework (MOF) Nodes as Catalyst Supports: Site Densities and Electron-Donor Properties Influence Molecular Iridium Complexes as Ethylene Conversion Catalysts. ACS Catal. 2016, 6, 235–247. 10.1021/acscatal.5b02243. [DOI] [Google Scholar]
- Yang D.; Odoh S. O.; Wang T. C.; Farha O. K.; Hupp J. T.; Cramer C. J.; Gagliardi L.; Gates B. C. Metal–Organic Framework Nodes as Nearly Ideal Supports for Molecular Catalysts: NU-1000- and UiO-66-Supported Iridium Complexes. J. Am. Chem. Soc. 2015, 137, 7391–7396. 10.1021/jacs.5b02956. [DOI] [PubMed] [Google Scholar]
- Larabi C.; Quadrelli E. A. Titration of Zr3(μ-OH) Hydroxy Groups at the Cornerstones of Bulk MOF UiO-67, [Zr6O4(OH)4(biphenyldicarboxylate)6], and Their Reaction with [AuMe(PMe3)]. Eur. J. Inorg. Chem. 2012, 2012, 3014–3022. 10.1002/ejic.201200033. [DOI] [Google Scholar]
- Desai S. P.; Malonzo C. D.; Webber T.; Duan J.; Thompson A. B.; Tereniak S. J.; DeStefano M. R.; Buru C. T.; Li Z.; Penn R. L.; Farha O.; Hupp J. T.; Stein A.; Lu C. Assembly of Dicobalt and Cobalt-Aluminum Oxide Clusters on Metal-Organic Framework and Nanocast Silica Supports. Faraday Discuss. 2017, 201, 287–302. 10.1039/C7FD00055C. [DOI] [PubMed] [Google Scholar]
- Thompson A. B.; Pahls D. R.; Bernales V.; Gallington L. C.; Malonzo C. D.; Webber T.; Tereniak S. J.; Wang T. C.; Desai S. P.; Li Z.; Kim I. S.; Gagliardi L.; Penn R. L.; Chapman K. W.; Stein A.; Farha O. K.; Hupp J. T.; Martinson A. B. F.; Lu C. C. Installing Heterobimetallic Cobalt-Aluminum Single-Sites on a Metal Organic Framework Support. Chem. Mater. 2016, 28, 6753–6762. 10.1021/acs.chemmater.6b03244. [DOI] [Google Scholar]
- Yang D.; Momeni M. R.; Demir H.; Pahls D. R.; Rimoldi M.; Wang T. C.; Farha O. K.; Hupp J. T.; Cramer C. J.; Gates B. C.; Gagliardi L. Tuning the Properties of Metal Organic Framework Nodes as Supports of Single-Site Iridium Catalysts: Node Modification by Atomic Layer Deposition of Aluminium. Faraday Discuss. 2017, 201, 195–206. 10.1039/C7FD00031F. [DOI] [PubMed] [Google Scholar]
- Rimoldi M.; Nakamura A.; Vermeulen N. A.; Henkelis J. J.; Blackburn A. K.; Hupp J. T.; Stoddart J. F.; Farha O. K. A Metal–Organic Framework Immobilised Iridium Pincer Complex. Chem. Sci. 2016, 7, 4980–4984. 10.1039/C6SC01376G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon H. R.; Lim D. W.; Suh M. P. Fabrication of Metal Nanoparticles in Metal–Organic Frameworks. Chem. Soc. Rev. 2013, 42, 1807–1824. 10.1039/C2CS35320B. [DOI] [PubMed] [Google Scholar]
- Rösler C.; Fischer R. A. Metal–Organic Frameworks as Hosts for Nanoparticles. CrystEngComm 2015, 17, 199–217. 10.1039/C4CE01251H. [DOI] [Google Scholar]
- Vilhelmsen L. B.; Sholl D. S. Thermodynamics of Pore Filling Metal Clusters in Metal Organic Frameworks: Pd in UiO-66. J. Phys. Chem. Lett. 2012, 3, 3702–3706. 10.1021/jz301806b. [DOI] [PubMed] [Google Scholar]
- Vilhelmsen L. B.; Walton K. S.; Sholl D. S. Structure and Mobility of Metal Clusters in MOFs: Au, Pd, and AuPd Clusters in MOF-74. J. Am. Chem. Soc. 2012, 134, 12807–12816. 10.1021/ja305004a. [DOI] [PubMed] [Google Scholar]
- Platero-Prats A. E.; League A. B.; Bernales V.; Ye J.; Gallington L. C.; Vjunov A.; Schweitzer N. M.; Li Z.; Zheng J.; Mehdi B. L.; et al. Bridging Zirconia Nodes within a Metal–Organic Framework via Catalytic Ni-Hydroxo Clusters to Form Heterobimetallic Nanowires. J. Am. Chem. Soc. 2017, 139, 10410–10418. 10.1021/jacs.7b04997. [DOI] [PubMed] [Google Scholar]
- Serna P.; Gates B. C. A Bifunctional Mechanism for Ethene Dimerization: Catalysis byRhodium Complexes on Zeolite HY in the Absence of Halides. Angew. Chem., Int. Ed. 2011, 50, 5528–5531. 10.1002/anie.201008086. [DOI] [PubMed] [Google Scholar]
- Markova V. K.; Vayssilov G. N.; Rösch N. Hydrogen Adsorption on Small Zeolite-Supported Rhodium Clusters. A Density Functional Study. J. Phys. Chem. C 2015, 119, 1121–1129. 10.1021/jp510842q. [DOI] [Google Scholar]
- Serna P.; Gates B. C. Zeolite-Supported Rhodium Complexes and Clusters: Switching Catalytic Selectivity by Controlling Structures of Essentially Molecular Species. J. Am. Chem. Soc. 2011, 133, 4714–4717. 10.1021/ja111749s. [DOI] [PubMed] [Google Scholar]
- Liang A. J.; Craciun R.; Chen M.; Kelly T. G.; Kletnieks P. W.; Haw J. F.; Dixon D. A.; Gates B. C. Zeolite-Supported Organorhodium Fragments: Essentially Molecular Surface Chemistry Elucidated with Spectroscopy and Theory. J. Am. Chem. Soc. 2009, 131, 8460–8473. 10.1021/ja900041n. [DOI] [PubMed] [Google Scholar]
- Ye J.; Gagliardi L.; Cramer C. J.; Truhlar D. G. Single Ni Atoms and Ni4 Clusters Have Similar Catalytic Activity for Ethylene Dimerization. J. Catal. 2017, 354, 278–286. 10.1016/j.jcat.2017.08.011. [DOI] [Google Scholar]
- Pahls D. R.; Ortuño M. A.; Winegar P. H.; Cramer C. J.; Gagliardi L. Computational Screening of Bimetal-Functionalized Zr6O8MOF Nodes for Methane C–H Bond Activation. Inorg. Chem. 2017, 56, 8739–8743. 10.1021/acs.inorgchem.7b01334. [DOI] [PubMed] [Google Scholar]
- Simons M. C.; Ortuño M. A.; Bernales V.; Cramer C. J.; Bhan A.; Gagliardi L.. C-H Bond Activation on Bimetallic Two-Atom Co-M Oxide Clusters Deposited on Zr-Based MOF Nodes: Effects of Doping at the Molecular Level. 2017, submitted for publication.
- Ortuño M. A.; Bernales V.; Gagliardi L.; Cramer C. J. Computational Study of First-Row Transition Metals Supported on MOF NU-1000 for Catalytic Acceptorless Alcohol Dehydrogenation. J. Phys. Chem. C 2016, 120, 24697–24705. 10.1021/acs.jpcc.6b06381. [DOI] [Google Scholar]