

Abstract
A collection of new approaches to building and training neural networks, collectively referred to as deep learning, are attracting attention in theoretical chemistry. Several groups aim to replace computationally expensive ab initio quantum mechanics calculations with learned estimators. This raises questions about the representability of complex quantum chemical systems with neural networks. Can local-variable models efficiently approximate nonlocal quantum chemical features? Here, we find that convolutional architectures, those that only aggregate information locally, cannot efficiently represent aromaticity and conjugation in large systems. They cannot represent long-range nonlocality known to be important in quantum chemistry. This study uses aromatic and conjugated systems computed from molecule graphs, though reproducing quantum simulations is the ultimate goal. This task, by definition, is both computable and known to be important to chemistry. The failure of convolutional architectures on this focused task calls into question their use in modeling quantum mechanics. To remedy this heretofore unrecognized deficiency, we introduce a new architecture that propagates information back and forth in waves of nonlinear computation. This architecture is still a local-variable model, and it is both computationally and representationally efficient, processing molecules in sublinear time with far fewer parameters than convolutional networks. Wave-like propagation models aromatic and conjugated systems with high accuracy, and even models the impact of small structural changes on large molecules. This new architecture demonstrates that some nonlocal features of quantum chemistry can be efficiently represented in local variable models.
Short abstract
A deep learning architecture that propagates information in waves, back and forth across a molecule, efficiently represents aromatic and conjugated systems, a task on which convolutional networks fail.
Introduction
A surge of interest in deep learning has encouraged its application to a range of problems in theoretical chemistry and chemical biology. Deep learning is a collection of new techniques that have substantially increased the power, flexibility, and applicability of neural networks.1−4 There is currently a resurgence in chemistry research groups using neural networks to predict the properties of small molecules.5−11 The best deep learning models consistently outperform other machine learning approaches in modeling drug metabolism,5,7,12,13 electrophilic and nucleophilic reactivity,8,14 chemical reactions,15 logP,16 and pKa.17
This track record raises hope that deep learning might approximate solutions to the Schrödinger equation, thereby efficiently estimating atomic and molecular properties. Large data sets of quantum chemical calculations have been compiled to enable researchers to test machine learning methods.18 To date, machine learning techniques have been used to estimate atomization energies,19−22 bond energies,23 molecular orbital energies,24 ground state Hamiltonians,25 and ab initio molecular dynamics,26 among others. For small molecules with fewer than 10 heavy atoms, deep learning models can approximate experimental observations with accuracy comparable to density functional theory and other methods.27,28 Encouraged by this success, several groups aim to replace computationally expensive ab initio calculations with accurate approximations in molecular simulations.
This hope is curtailed severely if nonlocal features, like aromaticity, cannot be efficiently represented by deep learning models. Fully connected networks might represent nonlocal features, but they are not computationally or representationally efficient.25 Likewise, graph-walking architectures proposed thus far are also inefficient, and require collapsing rings into pseudoatoms.15 Consequently, most effort has focused on different types of convolutional networks, which are efficient because they locally aggregate information in molecular graphs14,29 (Figures 1A and 5A–D). Convolutional networks, however, do not efficiently propagate long-range information. This raises a fundamental question: are the nonlocal features of chemistry representable in efficient, local-variable models?
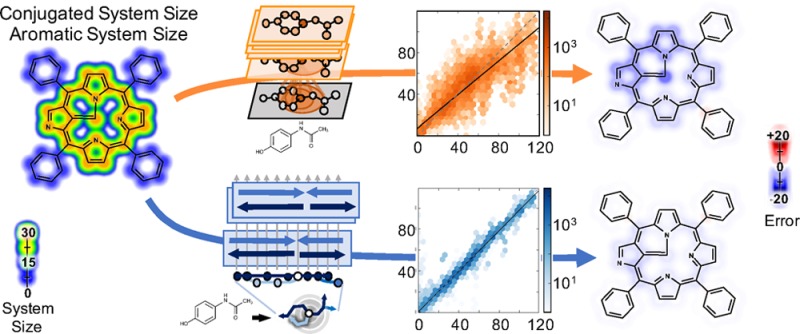
Figure 1.

Wave-like propagation of information more accurately represents aromatic and conjugated system size. (A) Four architectures were tested, including two convolutional models, the neighborhood model (red), which aggregates features over nearby atoms, and the weave model (orange), which repeatedly aggregates information over a local neighborhood of atoms and bonds; the WAVE model (blue) propagates waves of nonlinear computation forward and backward across a molecule; and a hybrid N-WAVE (purple) architecture combines the neighborhood and WAVE models. (B, C) Models of all four architectures were trained to label each atom with the size of the aromatic and conjugated systems of which it was a part. Both the WAVE and N-WAVE models outperform neighborhood and weave models. Dashed green lines indicate 95% confidence intervals of the interalgorithmic agreement on aromatic system size between RDKit and OpenBabel.
Figure 5.

Current state of the art neighborhood and weave deep learning architectures aggregate over local neighborhoods, while the WAVE architecture efficiently propagates information across a molecule using a breadth-first-search. (A, B) The neighborhood convolution architecture sums atom descriptors over increasing neighborhood depth. These neighborhood sums are then used as input to a fully connected neural network. (C, D) The undirected graph or weave architecture also aggregates over local neighborhoods. However, instead of a simple summation, aggregation is learnable and performed over pairs of atoms in a local neighborhood. Furthermore, multiple aggregation steps are performed. (E) In contrast, the WAVE model propagates information across a molecule in waves using a breadth-first search with a recurrent neural network. For the example acetaminophen, colored arrows indicate separate paths of information propagation. (F) The WAVE architecture uses gated recurrent units (GRU) to combine information from multiple input bonds and compute an output. This output then flows along bonds to the next atom in the BFS, and also flows up to the next layer. (G) The GRU uses a carefully constructed mix gate to combine information from multiple input bonds. The mix gate computes both linear-weighted sums and softmax-weighted sums of input bond data. In addition, tree and cross edges enter at separate ports, with separate weight computations.
Local-variable models of quantum mechanics have a long history, of both proposals and proofs of nonexistence.30,31 Recently, hydrodynamics experiments have reawakened interest in these models by showing that many nonlocal features of quantum mechanics can arise in classical systems from waves, which are clearly governed exclusively by local-variables and interactions.31 In these experiments, bouncing oil droplets interact with the waves they create on a vibrating bath. The droplets’ dynamics exhibit several nonlocal features, with correspondence to pilot-wave theory interpretations of quantum mechanics. Though not a perfect quantum analogue, these systems exhibit nonlocal behavior like double-slit diffraction, suggesting that local-variable models might explain more of quantum mechanics than first appreciated. With this in view, we aimed to test whether commonly used definitions of aromatic and conjugated systems are representable in a local-variable model.
Propagating Information in Waves
We hypothesize that aromatic and conjugated systems are representable with local-variables when information is propagated in waves, back and forth, across a molecule. Information is propagated locally, but can travel across the entire molecule in a single pass.
This hypothesis is motivated by two lines of reasoning. The first line of reasoning is inspired by chemical informatics. Efficient algorithms to compute both aromaticity and conjugated system size are usually implemented as depth-first searches and complex nonlinear rules.32,33 Though speculative, we imagine it is possible to reformulate these algorithms as multiple passes along a breadth-first search, visiting atoms in a wave-like order to compute local interactions. A breadth-first search is a way of efficiently bookkeeping local interactions across arbitrarily complex euclidian graphs. If our intuition is correct, this would prove an efficient way of representing chemicals to capture long-range interactions. The second line of reasoning is inspired by the pilot-wave interpretation of quantum mechanics.31,34 Nonlocal waves can arise in local-variable models as local interactions propagate across a system, and these waves can describe a great deal of quantum mechanics; this guides our intuition that a local variable model of quantum chemistry could propagate information nonlocally. Neither chemical informatics algorithms nor the mathematical details of pilot-wave theory are directly encoded in the algorithm. These lines of reasoning are offered merely to motivate our intuition that information propagated in waves of local interactions might give rise to the nonlocal behavior required to model quantum mechanics.
Guided by this intution, we constructed two architectures (WAVE and N-WAVE) to pass information in waves of local interactions across a molecule’s graph (Figures 1A and 5E–G). Local variables are maintained at each atom, and a nonlinear computation updates them in each pass using information from local interactions on one side of the wavefront. In this way, this architecture is a local-variable model, just like pilot-wave models of quantum mechanics. The precise details of the interactions and local variables are not prespecified, and these details are learned from data during a training phase.
Results and Discussion
We focused on aromatic and conjugated systems, both of which arise from nonlocal interactions across a molecule. The training and validation sets included both small and large molecules, and several adversarial examples (see Data and Methods). The modeling task is to annotate each atom and bond with the size of its aromatic or conjugated system, which in turn is determined by nonlocal features. This is a challenging task that requires architectures to represent nonlocality; small changes to a molecule can dramatically alter the system size at distant sites. Architectures must be capable, at minimum, of representing Hückel’s rule35 and the aromaticity algorithms in chemical informatics software.36,37 This is a key test case, because architectures that cannot represent aromaticity are not suitable for modeling quantum chemistry.
Though there is ongoing controversy over the precise definition of aromaticity,38 conjugated system size is more reliably defined and equally important to quantum chemistry. Both aromatic and conjugated systems are characterized by electron delocalization across an array of aligned pi-bonds. This delocalization determines several properties of molecules, most noticeably by imparting some with brilliant colors.41,42 It is delocalization that enables some molecules to transfer charge or interact with light with precisely tuned energetics. For this reason, large aromatic and conjugated systems are important in both chemistry and biology.36−38,43,44 For example, metalloporphyrins, nicknamed the pigments of life, contain large aromatic systems that are necessary for photosynthesis, oxygen transport, and electron transfer in several enzymes.45 Effective models of quantum chemistry should be able to represent aromaticity in complex chemicals like these.
Aromaticity is a real phenomenon, but its definition in quantum mechanics and analytical chemistry is not settled, especially for many boundary cases and rings with non-carbon atoms.38 For this reason, all models of aromaticity are subject to intractable debate. We define both aromatic and conjugated system sizes using the RDKit39 software package, which defines aromaticity using a graph-based algorithm. Error in this definition was estimated by comparing to the OpenBabel40 aromaticity model, which is also commonly used but is less documented than RDKit. Using an established and documented algorithm ensures reproducibility of our results while also quantifying confidence intervals on the training targets. Using a quantum mechanics definition of aromaticity would introduce additional problems without reducing this ambiguity. Simulation dependent definitions are sensitive to initial conditions and are difficult to exactly reproduce, while still remaining subject to the same ambiguity in defining aromaticity. Ultimately, prediction of quantum chemical properties as determined by experiment or simulation is most important, but at minimum architectures used in quantum chemistry should be able to reproduce aromaticity and conjugation calculated deterministically from molecular graphs. Defining aromaticity by a well-documented, graph-based algorithm establishes a straightforward and reproducible diagnostic of the limits of deep learning models. Ambiguity notwithstanding, architectures incapable of modeling aromaticity computed with a graph-based algorithm are not expected to effectively model quantum mechanics, which will include similar long-range behavior. As we will see, convolutional networks fail this test, but wave-like propagation succeeds.
Representing Aromatic and Conjugated Systems
We assessed wave-like propagation by comparing it to convolutional architectures. Controlling for differences unrelated to the information propagation model, each architecture was evaluated within a common framework. The same input features, output architecture, and training regimen were used in all cases, with the tested architecture inserted between the standardized input and output layers (Figure S1).
After training, the accuracy of each model was assessed. The two convolutional models, neighborhood convolution and weave, did not closely fit the data (Figure 1B,C). The output of the neighborhood convolution was nonlinearly related to the true values and exhibited high error on both aromatic (7.34 root-mean-square error (RMSE)) and conjugated systems (23.1 RMSE). The weave model was more effective, but still exhibited high error that increased with molecule size. In contrast, the two wave-like models represented the data with much better accuracy on both aromatic (4.36 vs 3.68 and 3.25 RMSE) and conjugated system size (13.3 vs 3.87 and 3.44 RMSE). The output of wave-like models is linear, and the error is constant with molecule size. Moreover, the error of the best wave-like model, N-WAVE, was within the interalgorithm error of two commonly used aromaticity detection algorithms (RSME 3.58, Figure S2).
These results demonstrate a critical and heretofore unrecognized limitation of convolutional networks; they cannot represent long-range interactions known to be important to chemical properties. Having demonstrated this failure point, it should guide future work in deep learning architectures. To this end, these results also support the hypothesis that wave-like propagation of information can better represent properties requiring propagation of long-range information.
The N-WAVE and weave models were selected for further study, because they achieved the best performances in their classes, wave-like and convolutional, respectively.
Modeling Long-Range Effects
Wave-like models also more accurately represent changes in aromatic and conjugated system sizes due to modifications at distant locations. We generated adversarial examples by converting double bonds to single bonds. Converted bonds were chosen to disrupt the aromatic or conjugated system (Figure 2A,D). For these adversarial examples, the convolutional model estimates aromatic and conjugated system sizes with poor accuracy (RMSE of 3.18 and 11.8, respectively, Figure 2B,E). In contrast, the wave-like model’s estimate is more accurate for both aromatic and conjugated system sizes (RMSE of 2.48 and 4.08, respectively).
Figure 2.

Wave-like propagation correctly models adversarial examples. (A, D) Adversarial aromatic and conjugated systems were generated from PubChem molecules by changing a double bond to a single bond. (B, E) Wave-like models more accurately estimate the size of aromatic and conjugated systems than convolutional models in these adversarial examples. (C, F) Critically, wave-like models more accurately estimate the change in aromatic and conjugated system sizes between adversarial molecules and their unmodified parents. Dashed green lines indicate 95% confidence intervals of the interalgorithmic agreement on aromatic system size between RDKit and OpenBabel.
Critically, wave-like models more accurately estimate the change in aromatic and conjugated system size of the adversarial molecules when compared to their unmodified parent. The convolutional model does not accurately model the change in aromatic and conjugated system sizes between a molecule and its modified counterparts (RMSE of 3.31 and 10.5, respectively, Figure 2C,F). In contrast, the wave-like model estimates these pairwise changes much more accurately (RMSE of 2.50 and 3.75, respectively).
Consistency on Individual Systems
Our hypothesis was further supported by comparing the consistency of estimates within individual aromatic and conjugated systems. Effective models compute the same estimate for all atoms in the same system (Figure 3). The wave-like estimate was more consistent than convolutional models (within system average standard deviation of 0.56 vs 0.83). As a typical example, the convolutional model overestimated aromatic system size for atoms near the center of a large polycyclic hydrocarbon system and underestimated size for atoms near the edge. In contrast, this type of error is not visible in wave-like estimates.
Figure 3.

The error from wave-like models is better behaved than convolutional models. Three molecules were painted with their aromatic system size (top) alongside the error of convolutional (middle) and wave-like (bottom) models. These examples include (left) a large polycyclic compound, (center) a porphyrin, and (right) an adversarial porphyrin, with a key double bond (circled in black) switched to a single bond to disrupt the aromatic macrocycle. The errors are higher for the convolutional model than the wave-like model. (far right) Moreover, the convolutional model often does not produce the same prediction across large systems, causing a larger variance in predictions that increases with aromatic system size.
Likewise, the wave-like model accurately identified cases where aromatic status is defined by long-range interactions. Changing a double bond to a single bond in porphyrin derivatives obliterates the large aromatic system (Figure 3). Convolutional models overestimate the size of the aromatic system for atoms far from the modified bond, but the wave-like model estimates were more accurate.
We further quantified the long-range effect visible in the porphyrin case using the full set of adversarial examples. We quantified the accuracy of aromatic status predictions on atoms more than five bonds away from the modified bond. This test evaluates the long-range propagation of information about subtle changes in a molecule. On these cases, the wave-like model is much more accurate than convolutional models (91.6% vs 85.8% area under the receiver operator curve, respectively, Table S1).
Representational Efficiency
We hypothesize that wave-like propagation efficiently represents aromatic and conjugated system size. In most modeling tasks, there is a trade-off between model error and complexity, which is quantified by the number of parameters in a model. All else being equal, models with more parameters will fit the data better. Simple models should be preferred, unless a more complex model improves generalization accuracy.
Comparing representational efficiency, therefore, requires measuring the performance of a wide range of models of each architectural class, to determine which architecture has the best trade-off between complexity and error. The architecture with the best trade-off most efficiently represents the task. Put another way, representational efficiency can be assessed by controlling either for complexity or for accuracy. Controlling for complexity, models using efficient architectures will perform better with the same number of parameters as those using less efficient architectures. Controlling for accuracy, models using more efficient architectures will have fewer parameters than those with equivalent performance but less efficient architectures.
We compared the trade-off between model complexity as defined by the number of tunable model parameters and accuracy with hyperparameter sweeps on the structural parameters of each architecture (Table S2). Each combination of hyperparameters was used to train and evaluate a model. In total, we trained and tested 567 convolutional models and 324 wave-like models (Figure 4). We found that wave-like models were more accurate with fewer parameters than convolutional models.
Figure 4.

Wave-like propagation is more representationally efficient than convolution. A large search was conducted over structural parameters of the convolutional (orange) and wave-like (purple) models (Table S2). In total, 567 weave architectures and 324 wave-like architectures were tested. (A, D) Most wave-like architectures with more than one forward–backward pass exhibited better validation accuracy than the best convolutional models. Furthermore, wave-like models require far fewer parameters. (B, E) Depth is a critical determinant of convolutional model accuracy, with increasing depth exhibiting diminishing returns on validation accuracy. Wave-like models with depth greater than one have nearly equivalent performance. (C, F) In contrast, the number of local variables (defined by the width of the feature vector at each atom and bond) is critical for wave-like model accuracy, but does not improve convolutional models.
Patterns of performance in the best models of each architecture support this hypothesis. The best wave-like models had lower test error and lower parameter counts than the best convolutional models (140,024 parameters vs 453,099 parameters, aromatic system size RMSE 2.34 vs 2.88, conjugated system size RMSE 2.88 vs 7.61). Furthermore, wave-like models outperformed convolutional models 2 orders of magnitude more complex (e.g., 6,704 parameters vs 453,099 parameters, conjugated system size RMSE 6.04 vs 7.61). Similar patterns of performance are observed globally. Most wave-like models with more than one pass outperformed the best convolutional model (232 of 243 models). These experiments provide strong evidence that wave-like propagation is more representationally efficient than local aggregation.
Critical Determinants of Representational Power
The parameter sweep also tests what components of an architecture are critical determinants of representational efficiency. The convolutional architecture requires increasing depth to propagate information across a molecule, but this only slowly improves accuracy with diminishing returns while substantially increasing the number of tunable parameters (Figure 4B,E). The performance of convolutional models improves only slightly between depth 30 and 50 (mean RMSE of 9.07 vs 8.63), but depth 50 models have nearly twice as many parameters (mean parameter count of 172,171 vs 286,731). In contrast, increasing the number of local variables did not improve convolutional model estimates (Figure 4C,F).
In wave-like propagation, the most important determinant of performance was depth. At least two forward–backward passes were required for accurate models. However, additional passes did not improve performance substantially. After depth, the number of local variables most strongly influenced model accuracy (average conjugated system size RMSE of 8.39, 6.46, and 5.89 for widths of 10, 20, and 30, Figure 4F). This suggests that performance is primarily influenced by the wave-like information propagation and by the number of local variables.
The WAVE model is defined by wave-like propagation of information, but it encodes local interactions with recurrent units making use of several components. Most components are standard in deep learning, but they also include a new component, a mix gate, which mixes information from multiple inputs together. The relative importance of each component of the model was assessed by studying the performance of the model as each component was removed or altered, one at a time (Table S3). The largest increases in error are observed when using only one forward–backward pass and when removing the mix gate. Other components only subtly affect error. We conclude that these two features of the WAVE model, along with the number of local variables, are critical determinants of its representational power. This supports the hypothesis that wave-like propagation is a critical determinant of efficient representation of nonlocal features.
Computational Complexity of Wave-like Propagation
Wave-like
propagation is sublinear in computational complexity, improving substantially
on the O(N3) complexity
of ab initio calculations and fully connected networks.
Most organic molecules are 1D or 2D, but condensed phase simulations
are 3D. Moreover, parallelism is easily exploitable with the current
generation of computational hardware. Consequently, the computational
complexity of an efficient parallel implementation of wave-like propagation
is proportional to a system’s width, or O(N), 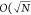 ),
and
),
and 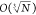 ), respectively, for 1D, 2D, and
3D systems
(see Supporting Information). In contrast,
the weave and neighborhood convolutions have constant computational
complexity, but do not propagate information across entire systems.
With sublinear complexity on large systems, wave-like propagation
may be a practically applied to modeling nonlocal properties in much
larger systems, including perhaps macromolecules and condensed phase
simulations.
), respectively, for 1D, 2D, and
3D systems
(see Supporting Information). In contrast,
the weave and neighborhood convolutions have constant computational
complexity, but do not propagate information across entire systems.
With sublinear complexity on large systems, wave-like propagation
may be a practically applied to modeling nonlocal properties in much
larger systems, including perhaps macromolecules and condensed phase
simulations.
Conclusion
It may be surprising that convolutional neural networks struggle to represent aromatic and conjugated systems. This finding initially appears to contradict several universal approximation proofs.46−48 These proofs, however, only apply to fully connected networks, with infinite training data, and with arbitrarily large numbers of hidden units. Under these conditions, neural networks can approximate simple quantum systems to arbitrary accuracy.25 These proofs do not extend to convolutional networks, which are not fully connected networks. Demonstrating the limitations of convolutional networks in chemistry is a foundational finding for the field.
It may be equally surprising that wave-like propagation efficiently represents nonlocal features with local variables. As demonstrated by hydrodynamic analogues of quantum mechanics,31 surprising nonlocal behavior can emerge from local interactions. Hydrodynamics models, however, are not perfect analogues of quantum mechanics, and they may never scale to large many-body problems. Our findings, nonetheless, empirically add to the argument by demonstrating that important nonlocal features in more complex chemical systems can, in principle, emerge from local variable models when information is propagated in waves.
While this study focused on aromatic and conjugated system size computed using graph-based algorithms, the findings here have immediate practical relevance in ongoing efforts to model quantum chemistry with deep learning. Convolutional networks are not efficient models of nonlocal properties, but other efficient models are possible and might be preferred. It remains an open question how much of quantum chemistry is representable with local-variable models, but we may soon find out as deep learning is extended to quantum mechanical simulations of large molecules. As efforts to model quantum chemistry with deep learning progress, they could become more than engineering efforts solving a purely practical problem. They might also empirically test whether quantum chemistry is fully representable in local-variable models.
Data and Methods
Large Aromatic and Conjugated Systems from PubChem
Diverse training and test sets with many types and sizes of aromatic and conjugated systems were extracted from the PubChem database.49 First, a random sample of 200,000 compounds was collected (Figure S3A,B). Most compounds in PubChem were small (fewer than 50 heavy atoms), with small conjugated systems (fewer than 25 atoms) and small aromatic systems (fewer than 10 atoms). To ensure representation of larger, more difficult molecules, we selected a stratified subsample of training and test sets from the full range of aromatic and conjugated system sizes (Figure S3C–F). Only molecules with fewer than 200 atoms were included. Aromatic system sizes in the final data set ranged from 0 to 80 heavy atoms, while conjugated system sizes ranged between 0 and 165 heavy atoms. The final data set included 2,678 molecules with 139,138 heavy atoms for the training set and 471 molecules with 24,240 heavy atoms for the test set.
Adversarial Modifications to Aromatic and Conjugated Systems
To study the efficiency of information propagation in deep learning chemistry architectures, we added adversarial examples to both the training and test sets. For each PubChem compound in the data sets, between two and four adversarial examples were constructed. In each example, a single double bond from a kekulized form of the molecule was chosen and then changed to a single bond. Bonds were chosen so that the change in aromatic or conjugated system sizes was large. For aromatic system sizes, two adversarial examples were constructed with bond modifications leading to (1) the maximum change in aromatic system size and (2) the median change in aromatic system size (Figure S4A). The RMSE difference in aromatic system sizes between the normal and adversarial examples was 6.61 and 7.29 for the training and test sets, respectively (Figure S4B,C). For conjugated system sizes, the same strategy was used to generate two additional adversarial examples (Figure S5A). The RMSE difference in conjugated system sizes between normal and adversarial examples was 20.1 and 19.3 for the training and test sets, respectively (Figure S5B,C). Including adversarial examples, the final training set contained 11,123 molecules with 605,000 heavy atoms, and the final test set contained 1,990 molecules with 105,726 atoms.
Common Atom-Level Input Descriptors
All models used the same minimal set of numerical descriptors which were computed across the heavy atoms in each molecule. These descriptors included 11 binary indicators denoting the element of the atom, the number of covalently bonded hydrogens, and the formal charge of the atom. Atom indicators included boron, carbon, nitrogen, oxygen, fluorine, phosphorus, sulfur, chlorine, arsenic, bromine, and iodine.
Common Atom-Level and Bond-Level Targets
All models were trained to predict the same set of targets at the atom and bond level. For each atom, 11 atom-level targets were calculated: (1) membership in any ring, (2) membership in an aromatic ring, (3–8) membership in a ring of size three, four, five, six, seven, or eight heavy atoms, (9) the number of atoms in the largest ring of which this atom is a member, (10) the number of atoms in the aromatic system of which this atom is a member, and (11) the number of atoms in the conjugated system of which this atom is a member. Aromatic and conjugated systems were defined as the set of all atoms reachable by walking from atom to atom on aromatic or conjugated bonds, respectively. Bond aromaticity was included as a bond-level target. RDKit39 was used to label aromatic and conjugated bonds. OpenBabel40 was used to determine ring sizes and membership.
Common Input and Output Architecture
To evaluate each architecture’s ability to propagate information across a molecule, a common architecture was used at the first input layer and final output (Figure S1A). At the input, a single fully connected layer with rectified linear activation scales the atom-level input size to the input size of the test network. The test network produces a vector of atom features for each atom in the input data. Two separate networks use these features to predict atom or bond targets. The atom network consists of a single hidden layer of ten units, eight output units with sigmoid activations, and three output units with rectified linear activations. The bond network consists of an atom feature conversion layer, a single hidden layer of ten units, and one output unit with a sigmoid activation. Atom features are converted to bond features by application of an order invariant transformation. Specifically, features for each atom in a bond are processed by a fully connected layer with rectified linear activation, and the output for both atoms is summed to produce the bond-level features.
Common Training Protocol
All models were trained in the Tensorflow50 framework using the same protocol, using cross-entropy loss51 on the binary targets and normalized sum of squared differences loss on the integer targets (Figure S1B). All models were trained with 15,000 iterations of mini-batch gradient descent using the Adam optimizer52 and batches of 100 molecules. An initial learning rate of 10–3 was used, followed by a decay to 10–4 after 7,500 iterations.
Neighborhood Convolution (NC or XenoSite) Architecture
The NC architecture is used by several groups, including ours, to predict drug metabolism and other atom and bond level properties. The NC linearly sums descriptors of neighboring atoms up to a depth of five bonds (d = 5) once for each neighborhood depth (Figure 5A,B). The resulting feature vector has a width d times the number of input descriptors. The feature vector is input to a neural network. We used two hidden layers of size 40 and 20, sequentially, to compute an output feature vector for each atom of dimension 20. The performance of NC depends on passing quality descriptors known to be important in chemistry, like aromaticity and ring membership. In this study, however, only minimal descriptors are used. It is not expected that NC will perform well in these tests, and it serves as a baseline method against which to compare other architectures. Hyperparameters for this model include the neighborhood depth d over which to aggregate descriptors, the number of hidden layers, the sizes of those hidden layers, and the number of output features. The baseline model had 1,104 parameters.
Weave Architecture
Weave is a state-of-the art undirected graph model that is used by DeepChem29,53 to predict the biochemical properties of small molecules. The weave architecture aggregates information locally over pairs of atoms in a neighborhood using multiple layers (Figure 5C,D). Weave is fully described in the literature,29 but a brief description is included here with default parameters noted. We used 15 weave layers. Each layer nonlinearly aggregates atoms, using a fully connected rectified linear layer, to produce a new set of pair outputs. Then, it does the same over pairs of atoms to produce a set of atom outputs. Atom output vectors were dimension 30, and bond output vectors were dimension 10. At each step, pairs of atoms up to a depth of two are aggregated. After aggregation, batch normalization was applied to both the atom and pair outputs at each layer. The exact implementation details are explained in the references. There are six hyperparameters: the number of layers, the bond and atom width, the bond and atom hidden layer width, and the aggregation depth. The baseline model had 71,164 parameters. These hyperparameters were chosen for the baseline model to bring the number of parameters onto the same order as the baseline WAVE architecture (see next section).
Multi-Pass Breadth-First (WAVE) Architecture
This architecture propagates information across a molecule back and forth in waves (Figure 5E). Atoms and bonds are processed in order as determined by a breadth-first search. Information is passed along bonds. At each atom, information from previously visited atoms is used to update the local variables (the state vector), which are then passed on to the next set of neighboring atoms. The backward pass reverses the order in which atoms are visited, reversing the flow of information. In each pass, the neural network updates the current state of the atom based on all the information passed to it from neighboring atoms.
The breadth-first search is initiated at an atom selected near the center of the molecule. From here, layers of atoms that are one bond away, two bonds away, and so on are identified. Atom states are updated layer by layer, in order, passing on information along bonds from one layer to the next. In the backward pass, layers are updated in the opposite order and the information flow is reversed. The bonds between layers are labeled “tree edges”, and bonds between atoms in the same layer are labeled “cross edges”.54 For tree edges, one atom of the edge is updated first, and then its updated state is sent to the next atom. For cross edges, each atom in the cross edge receives the non-updated state of the other atom; consequently, all atoms in a layer can be processed simultaneously. In the backward pass, the information transfer and order of updates is reversed. In this way, information propagates in waves of back and forth across the molecule, from the central atom outward, then back again.
A gated recurrent unit (GRU)55—a repeated neural network—integrates information from bonds and updates the atom’s local state (Figure 5F). This unit is mathematically detailed in the next section, but a narrative description is included here. The weights for this network are identical for all atoms in the molecule, but different for each pass of the algorithm. A mix gate integrates information from multiple bonds. It calculates two sums of the inputs along each tree-edge bond, a softmax sum and a weighted sum (Figure 5G). Weighting for each sum is computed independently for each component of the vector by single layer network. Cross edge inputs enter in their own port with their own set of mixing weights different from tree-edge inputs. The complexity of the mix gate is necessary for the network to treat tree and cross edges differently, while learning dimension and context aggregations that can range from weighted averaging, to weighted sums, to the weighted maximum or minimum. Next, the mixed input is concatenated with the atom’s local state and fed forward into the update and output gates. The rest of the unit is a standard GRU architecture. The output gate computes a preliminary output vector. This output vector is mixed with the local variables using a component-wise, weighted sum tuned by the update gate. The update gate is a single fully connected layer with a sigmoid activation, which tunes how much the output vector should update the local variables. The updated memory vector then becomes the final output of the GRU, which replaces the current state and is handed forward to update additional atom states.
There are four hyperparameters: the number of forward–backward passes, the memory and output width, the number of output network layers, and the number of mix network layers. Within a forward or backward pass, all GRUs share the same weights, and all GRUs across all passes share the same structure. Default parameters are noted here, which should be assumed unless stated otherwise. The default WAVE uses three forward–backward passes. The GRUs return vectors of dimension 25. The output gate uses a single fully connected layer with exponential linear activations to compute a new output. The mix gate uses a separate fully connected layer for each type of edge (cross or tree) and each weighting (softmax and softsign). The baseline model had 50,289 parameters.
The WAVE Model GRU
The schematic and narrative overview of the WAVE model’s GRU summarizes and depicts the information flow through a set of equations (Figure 5E–G). The GRU computes an updated state from the current state of the atom and the states of bond-connected atoms. This updated state is passed to neighboring atoms in the next layer, which have not been updated. Using column vectors, all its inputs are states of dimension d. This updated state s* is the weighted average of the mix gate output m and a computed output vector o,
| 1 |
Here, u is an update vector that ranges from zero to one, y is the preliminary output vector, and m is the output of the mix state, all of which are vectors of length d. The operator ⊗ is the element-wise multiply. The update and output vectors are computed as
| 2 |
| 3 |
where s is the current state of the atom and the subscripted W and b variables denote tunable matrices and vectors of appropriate dimension and size. The relu function is the rectified linear activation, and σ is the logistic activation, both of which are element-wise functions. Both m and s are vectors of d length, which are concatenated into a vector of length 2d; m is the mixed neighbor state, computed as
| 4 |
where 1 is a unity vector of dimension i that collapses its operand into a vector of size d, N* is the previously updated states from neighboring atoms, stacked as columns alongside each other in a d by i matrix, and i is the number of input states. A and B are computed weighting matrices, and N* is composed of both tree and cross edge states, as determined by the breadth-first search.
| 5 |
where T* and C are the column stacked tree and cross edge states, which are integrated using different sets of weights from each other. To enable parallel processing of all atoms a given depth from the central atom, we use the updated states for the tree edge connected atoms, and non-updated states from the cross edge connected atoms. The weight matrices A and B are shaped the same as N*. A is computed by
| 6 |
In this nonstandard notation, the braces are broadcast operators that replicate and horizontally stack the s, b, and w vectors to match the correct dimensions for concatenation or element-wise arithmetic. The softmax is defined in the usual way, ranges from zero to one, and is oriented so as to normalize each row to sum to one, normalizing each component of the state independently. The matrix B is computed with a similar formula by
| 7 |
where the softsign is the standard element-wise function that ranges from zero to one. In this way, the tree and cross edges are integrated into the same aggregate state m using different parameters. The combined use of both softmax and softsign enables a context dependent and nonlinear aggregation that can range from a weighted average, to weighted sum, to a minimum or maximum. The GRU computes its update from the updated states of atoms in the previous layer and non-updated atoms from the current layer, and then passes the newly updated state s* to atoms in the next layer to be updated.
Hybrid N-WAVE Architecture
In addition to the WAVE architecture, we experimented with hybrid architectures, which combined the neighborhood convolution and WAVE architectures. In this architecture, neighborhood convolution is applied to compute an atom representation which is then used as the input to multiple passes of WAVE.
Variants of the WAVE Architecture
In addition to the standard WAVE, we studied several variants of the gated recurrent unit architecture used to propagate information. Optional architectural components included (1) the read gate (a single fully connected layer with sigmoid activation, which selects among the output features of the mix gate for input to the unit); (2) the update gate; (3) the mix gate (replacing with an unweighted sum of input bond features); (4) use of the atom input features in the mix gate weight computation; (5) the mix gate’s softmax weighted sum; (6) the mix gate’s linear weighted sum; (7) use of the same recurrent unit for both forward and backward passes; and (8) a hybrid model with a neighborhood convolution as the first layer before the wave WAVE (N-WAVE). Furthermore, we studied three ways for the mix gate to process bonds labeled as cross edges by the breadth-first search: (1) the same weight computations are used for tree or cross edges indiscriminately; (2) weights on cross edges are computed by separate networks, or ports, from those used on tree edges; or (3) there are separate ports for tree and cross edges, treating cross edges as undirected, with cross edge memory fetched from the previous layer. The standard model treated cross edges with the latter strategy.
Hyperparameter Sweeps
We assessed validation accuracy for many values of each model’s hyperparameters. For each combination of hyperparameter values, three models were trained. Of these three, the model with the lowest error on the training set was chosen for evaluation on the validation set. A complete list of studied hyperparameters and tested values can be found in Table S2. In total, 567 weave model architectures and 324 N-WAVE model architectures were evaluated.
Acknowledgments
We are grateful to the developers of the open-source cheminformatics tools Open Babel and RDKit. Research reported in this publication was supported by the National Library of Medicine of the National Institutes of Health under Award Numbers R01LM012222 and R01LM012482 and by the National Institutes of Health under Award Number GM07200. The content is the sole responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially funded by NIH Grants 1S10RR022984-01A1 and 1S10OD018091-01. We also thank the Department of Immunology and Pathology at the Washington University School of Medicine, the Washington University Center for Biological Systems Engineering, and the Washington University Medical Scientist Training Program for their generous support of this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00405.
The authors declare no competing financial interest.
Supplementary Material
References
- LeCun Y.; Bengio Y.; Hinton G. Deep learning. Nature 2015, 521, 436–444. 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Schmidhuber J. Deep learning in neural networks: An overview. Neural Networks 2015, 61, 85–117. 10.1016/j.neunet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Baldi P.; Pollastri G. The Principled Design of Large-Scale Recursive Neural Network Architectures-DAG-RNNs and the Protein Structure Prediction Problem. J. Mach. Learn. Res. 2003, 4, 575–602. [Google Scholar]
- Gers F. A.; Schmidhuber J.; Cummins F. Learning to Forget: Continual Prediction with LSTM. Neural Computation 2000, 12, 2451–2471. 10.1162/089976600300015015. [DOI] [PubMed] [Google Scholar]
- Hughes T. B.; Miller G. P.; Swamidass S. J. Modeling Epoxidation of Drug-like Molecules with a Deep Machine Learning Network. ACS Cent. Sci. 2015, 1, 168–180. 10.1021/acscentsci.5b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlock M. K.; Hughes T. B.; Swamidass S. J. XenoSite server: A web-available site of metabolism prediction tool. Bioinformatics 2015, 31, 1136–1137. 10.1093/bioinformatics/btu761. [DOI] [PubMed] [Google Scholar]
- Zaretzki J. M.; Browning M. R.; Hughes T. B.; Swamidass S. J. Extending P450 site-of-metabolism models with region-resolution data. Bioinformatics 2015, 31, 1966–1973. 10.1093/bioinformatics/btv100. [DOI] [PubMed] [Google Scholar]
- Hughes T. B.; Miller G. P.; Swamidass S. J. Site of reactivity models predict molecular reactivity of diverse chemicals with glutathione. Chem. Res. Toxicol. 2015, 28, 797–809. 10.1021/acs.chemrestox.5b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman J. H.; Cothren S. D.; Park S. H.; Yun C. H.; Darsey J. A.; Miller G. P. Predicting CYP2C19 catalytic parameters for enantioselective oxidations using artificial neural networks and a chirality code. Bioorg. Med. Chem. 2013, 21, 3749–3759. 10.1016/j.bmc.2013.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh G. B.; Hodas N. O.; Vishnu A. Deep learning for computational chemistry. J. Comput. Chem. 2017, 38, 1291–1307. 10.1002/jcc.24764. [DOI] [PubMed] [Google Scholar]
- Ma J.; Sheridan R. P.; Liaw A.; Dahl G. E.; Svetnik V. Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model. 2015, 55, 263–274. 10.1021/ci500747n. [DOI] [PubMed] [Google Scholar]
- Dang N. L.; Hughes T. B.; Krishnamurthy V.; Swamidass S. J. A simple model predicts UGT-mediated metabolism. Bioinformatics 2016, 32, 3183–3189. 10.1093/bioinformatics/btw350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. B.; Swamidass S. J. Deep Learning to Predict the Formation of Quinone Species in Drug Metabolism. Chem. Res. Toxicol. 2017, 30, 642–656. 10.1021/acs.chemrestox.6b00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes T. B.; Dang N. L.; Miller G. P.; Swamidass S. J. Modeling Reactivity to Biological Macromolecules with a Deep Multitask Network. ACS Cent. Sci. 2016, 2, 529–537. 10.1021/acscentsci.6b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayala M. A.; Azencott C. E.-A.; Chen J. H.; Baldi P. Learning to Predict Chemical Reactions. J. Chem. Inf. Model. 2011, 51, 2209–2222. 10.1021/ci200207y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusci A.; Pollastri G.; Baldi P. Deep Architectures and Deep Learning in Chemoinformatics: The Prediction of Aqueous Solubility for Drug-Like Molecules. J. Chem. Inf. Model. 2013, 53, 1563–1575. 10.1021/ci400187y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butina D.; Segall M. D.; Frankcombe K. Predicting ADME properties in silico: methods and models. Drug Discovery Today 2002, 7, S83–S88. 10.1016/S1359-6446(02)02288-2. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; Von Lilienfeld O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 140022. 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütt K. T.; Arbabzadah F.; Chmiela S.; Müller K. R.; Tkatchenko A. Quantum-chemical insights from deep tensor neural networks. Nat. Commun. 2017, 8, 13890. 10.1038/ncomms13890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montavon G.; Hansen K.; Fazli S.; Rupp M.; Biegler F.; Ziehe A.; Tkatchenko A.; Lilienfeld A. V.; Müller K.-R.. Learning invariant representations of molecules for atomization energy prediction. In Advances in Neural Information Processing Systems; 2012; pp 440–448.
- Montavon G.; Rupp M.; Gobre V.; Vazquez-Mayagoitia A.; Hansen K.; Tkatchenko A.; Müller K.-R.; Von Lilienfeld O. A. Machine learning of molecular electronic properties in chemical compound space. New J. Phys. 2013, 15, 095003. 10.1088/1367-2630/15/9/095003. [DOI] [Google Scholar]
- Hansen K.; Montavon G.; Biegler F.; Fazli S.; Rupp M.; Scheffler M.; Von Lilienfeld O. A.; Tkatchenko A.; Müller K.-R. Assessment and validation of machine learning methods for predicting molecular atomization energies. J. Chem. Theory Comput. 2013, 9, 3404–3419. 10.1021/ct400195d. [DOI] [PubMed] [Google Scholar]
- Yao K.; Herr J. E.; Brown S. N.; Parkhill J. Intrinsic Bond Energies from a Bonds-in-Molecules Neural Network. J. Phys. Chem. Lett. 2017, 8, 2689–2694. 10.1021/acs.jpclett.7b01072. [DOI] [PubMed] [Google Scholar]
- Hansen K.; Biegler F.; Ramakrishnan R.; Pronobis W.; Von Lilienfeld O. A.; Müller K.-R.; Tkatchenko A. Machine learning predictions of molecular properties: Accurate many-body potentials and nonlocality in chemical space. J. Phys. Chem. Lett. 2015, 6, 2326–2331. 10.1021/acs.jpclett.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carleo G.; Troyer M. Solving the quantum many-body problem with artificial neural networks. Science 2017, 355, 602–606. 10.1126/science.aag2302. [DOI] [PubMed] [Google Scholar]
- Chmiela S.; Tkatchenko A.; Sauceda H. E.; Poltavsky I.; Schütt K. T.; Müller K.-R. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 2017, 3, e1603015. 10.1126/sciadv.1603015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber F. A.; Hutchison L.; Huang B.; Gilmer J.; Schoenholz S. S.; Dahl G. E.; Vinyals O.; Kearnes S.; Riley P. F.; von Lilienfeld O. A. Prediction errors of molecular machine learning models lower than hybrid DFT error. J. Chem. Theory Comput. 2017, 13, 5255–5264. 10.1021/acs.jctc.7b00577. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; von Lilienfeld O. A. Big data meets quantum chemistry approximations: the Δ-machine learning approach. J. Chem. Theory Comput. 2015, 11, 2087–2096. 10.1021/acs.jctc.5b00099. [DOI] [PubMed] [Google Scholar]
- Kearnes S.; McCloskey K.; Berndl M.; Pande V.; Riley P. Molecular graph convolutions: moving beyond fingerprints. J. Comput.-Aided Mol. Des. 2016, 30, 595–608. 10.1007/s10822-016-9938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm D. A suggested interpretation of the quantum theory in terms of ”hidden” variables. I. Phys. Rev. 1952, 85, 166–179. 10.1103/PhysRev.85.166. [DOI] [Google Scholar]
- Bush J. W. M. The new wave of pilot-wave theory. Phys. Today 2015, 68, 47–53. 10.1063/PT.3.2882. [DOI] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck C.; Han Y.; Kuhn S.; Horlacher O.; Luttmann E.; Willighagen E. The Chemistry Development Kit (CDK): An Open-Source Java Library for Chemo- and Bioinformatics. J. Chem. Inf. Model. 2003, 43, 493–500. 10.1021/ci025584y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couder Y.; Protière S.; Fort E.; Boudaoud A. Dynamical phenomena: Walking and orbiting droplets. Nature 2005, 437, 208–208. 10.1038/437208a. [DOI] [PubMed] [Google Scholar]
- Randic M. Aromaticity and conjugation. J. Am. Chem. Soc. 1977, 99, 444–450. 10.1021/ja00444a022. [DOI] [Google Scholar]
- Kikuchi S. A History of the Structural Theory of Benzene - The Aromatic Sextet Rule and Huckel’s Rule. J. Chem. Educ. 1997, 74, 194. 10.1021/ed074p194. [DOI] [Google Scholar]
- Ajami D.; Oeckler O.; Simon A.; Herges R. Synthesis of a Möbius aromatic hydrocarbon. Nature 2003, 426, 819–821. 10.1038/nature02224. [DOI] [PubMed] [Google Scholar]
- Feixas F.; Matito E.; Poater J.; Solà M.; Solà M.; Mandado M.; Sodupe M.; Tobe Y.; Kakiuchi K.; Odaira Y.; Huck V. J. Quantifying aromaticity with electron delocalisation measures. Chem. Soc. Rev. 2015, 44, 6434–6451. 10.1039/C5CS00066A. [DOI] [PubMed] [Google Scholar]
- Landrum G.RDKit: Open-Source Cheminformatics Software. http://www.rdkit.org (accessed March 1st, 2017).
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindy H. Basics, Mechanisms and Properties in the Chemistry of Cyanine Dyes: A Review Paper. Mini-Rev. Org. Chem. 2012, 9, 352–360. 10.2174/157019312804699528. [DOI] [Google Scholar]
- Sperling W.; Rafferty C. N. Relationship between Absorption Spectrum and Molecular Conformations of 11-cis-Retinal. Nature 1969, 224, 591–594. 10.1038/224591a0. [DOI] [PubMed] [Google Scholar]
- Fukui K.; Yonezawa T.; Shingu H. A molecular orbital theory of reactivity in aromatic hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. 10.1063/1.1700523. [DOI] [Google Scholar]
- Firouzi R.; Sharifi Ardani S.; Palusiak M.; Fowler P. W.; McKenzie A. D.; Mauksch M.; Hommes N. J. R. v. E.; Alkorta I.; Elguero J.; Su S. J.; Windus T. L.; Dupuis M.; Montgomery J. A. Description of heteroaromaticity on the basis of π-electron density anisotropy. Phys. Chem. Chem. Phys. 2014, 16, 11538. 10.1039/c4cp01125b. [DOI] [PubMed] [Google Scholar]
- Chandra R.; Tiwari M.; Kaur P.; Sharma M.; Jain R.; Dass S. Metalloporphyrins-Applications and clinical significance. Indian J. Clin. Biochem. 2000, 15, 183–99. 10.1007/BF02867558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum E. K.; Li L. K. Approximation theory and feedforward networks. Neural Networks 1991, 4, 511–515. 10.1016/0893-6080(91)90047-9. [DOI] [Google Scholar]
- Hornik K.; Stinchcombe M.; White H. Multilayer feedforward networks are universal approximators. Neural Networks 1989, 2, 359–366. 10.1016/0893-6080(89)90020-8. [DOI] [Google Scholar]
- Leshno M.; Lin V. Y.; Pinkus A.; Schocken S. Multilayer feedforward networks with a nonpolynomial activation function can approximate any function. Neural Networks 1993, 6, 861–867. 10.1016/S0893-6080(05)80131-5. [DOI] [Google Scholar]
- Bolton E. E.; Wang Y.; Thiessen P. A.; Bryant S. H. PubChem: integrated platform of small molecules and biological activities. Annu. Rep. Comput. Chem. 2008, 4, 217–241. 10.1016/S1574-1400(08)00012-1. [DOI] [Google Scholar]
- Abadi M.; Barham P.; Chen J.; Chen Z.; Davis A.; Dean J.; Devin M.; Ghemawat S.; Irving G.; Isard M.. TensorFlow: A System for Large-Scale Machine Learning. OSDI; 2016; pp 265–283.
- Shore J.; Johnson R. Properties of cross-entropy minimization. IEEE Trans. Inf. Theory 1981, 27, 472–482. 10.1109/TIT.1981.1056373. [DOI] [Google Scholar]
- Kingma D.; Ba J. Adam: A method for stochastic optimization. arXiv 2014, 1412.6980. [Google Scholar]
- Wu Z.; Ramsundar B.; Feinberg E. N.; Gomes J.; Geniesse C.; Pappu A. S.; Leswing K.; Pande V. MoleculeNet: A Benchmark for Molecular Machine Learning. arXiv 2017, 1703.00564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberg J.; Tardos E.. Algorithm design; Pearson Education India: 2006. [Google Scholar]
- Cho K.; van Merrienboer B.; Bahdanau D.; Bengio Y.. On the Properties of Neural Machine Translation: Encoder–Decoder Approaches. Proceedings of SSST-8, Eighth Workshop on Syntax, Semantics and Structure in Statistical Translation; 2014; pp 103–111. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.