

Abstract
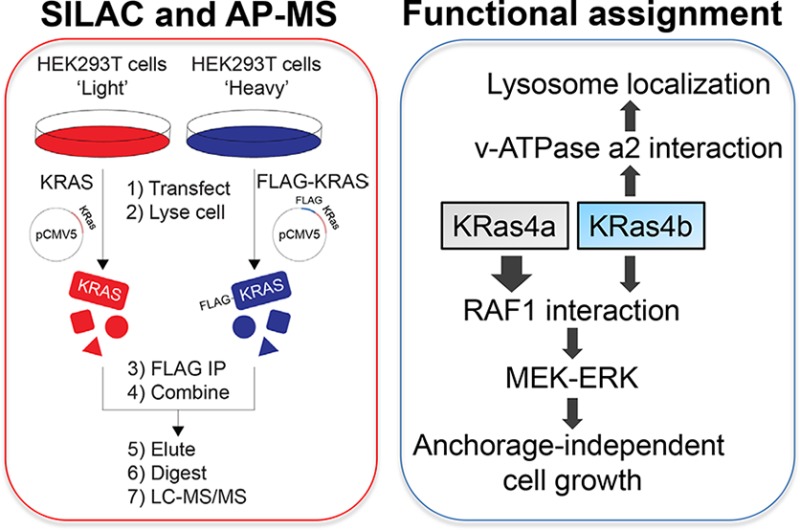
The KRAS gene encodes two isoforms, KRas4a and KRas4b. Differences in the signaling functions of the two KRas proteins are poorly understood. Here we report the comparative and nucleotide-dependent interactomes of KRas4a and KRas4b. Many previously unknown interacting proteins were identified, with some interacting with both isoforms while others prefer only one. For example, v-ATPase a2 and eIF2Bδ interact with only KRas4b. Consistent with the v-ATPase interaction, KRas4b has a significant lysosomal localization. Comparing WT and constitutively active G12D mutant KRas, we examined differences in the effector proteins of the KRas4a and KRas4b. Interestingly, KRas4a binds RAF1 stronger than KRas4b. Correspondingly, KRas4a can better promote ERK phosphorylation and anchorage-independent growth than KRas4b. The interactome data represent a useful resource to understand the differences between KRas4a and KRas4b and to discover new function or regulation for them. A similar proteomic approach would be useful for studying numerous other small GTPases.
Short abstract
The comparative and nucleotide-dependent interactomes of KRas4a and KRas4b identify many previously unknown and isoform selective interacting proteins of KRas4a and KRas4b.
Introduction
The Ras superfamily of small GTPases consists of more than 150 members and plays important roles in numerous biological processes such as signal transduction, membrane trafficking, nuclear export/import, and cytoskeletal dynamics.1 Among all these members, four Ras proteins (HRas, NRas, KRas4a, and KRas4b) encoded by three RAS genes (HRAS, NRAS, and KRAS) attract broad interest because their deregulation is frequently found in various human cancers.2 It is well established that mutant Ras proteins are cancer drivers.3 Ras proteins are active in their GTP-bound state and inactive in their GDP-bound state. Guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs) regulate the GTP and GDP exchange on Ras proteins.4 GEFs facilitate the formation of GTP-bound Ras, while GAPs activate Ras intrinsic GTP hydrolysis and promote the formation of GDP-bound Ras. GTP loading on Ras proteins induces a conformational change in their switch I region, which allows the recruitment of effector proteins, turning on Ras signaling.5 The four Ras proteins share a high sequence identity in their conserved domain (residues 1–165), which includes nucleotide and effector protein binding regions. The Ras proteins diverge in their C-terminal hypervariable region (HVR, residues 166–188 or −189), which plays important roles in membrane targeting, protein–protein interaction, and signal transduction.6 Different Ras proteins share many effector proteins, such as RAF1, RalGDS, PI3K, and PLCε,5 and were originally thought to have similar biological functions. However, accumulating evidence has shown that different Ras proteins exhibit different signaling and biological functions. For example, HRas, NRas, and KRas exhibit different leukemogenic potentials in mice.7 KRas but not HRas can translocate from the plasma membrane (PM) to Golgi complex and early/recycling endosomes in a Ca2+/calmodulin dependent manner.8 The different functions of Ras proteins suggest that they may recruit different proteins that determine the signaling outputs, or bind to the same effector protein with different affinities which could also lead to different signaling outputs. Even KRas4a and KRas4b, two alternatively spliced products from KRAS gene, were shown to have different subcellular localizations and biological functions.9,10 However, the molecular basis for the different signaling functions of various Ras proteins, especially the two KRas isoforms, is poorly understood.
The interactome of a protein of interest can provide important functional clues for that protein, thereby facilitating the discoveries of new functions for the protein. This is especially important for proteins whose functions mainly rely on recruiting other proteins, such as the Ras proteins. Currently, the most well-established protein interactome database is BioPlex 2.0, which includes interactome data for almost half of the human proteome.11 However, for each single protein, only the most abundant and high-confident interacting proteins are present in the database. For example, we searched KRas interacting proteins in BioPlex 2.0 and only found RIN1 and BRAF (http://bioplex.hms.harvard.edu), two known Ras interacting proteins.12,13 This is far fewer than the number of known KRas interacting proteins. Moreover, many protein–protein interactions are protein state dependent. For example, Ras–effector interactions are GTP-dependent. Knowing the nucleotide-dependent interactome is thus important for understanding protein functions. However, such nucleotide-dependent protein–protein interaction information is not available in the reported interactome database. In this study, we report the nucleotide-dependent interaction map of the two KRas isoforms, KRas4a and KRas4b, acquired using stable isotope labeling with amino acids in cell culture (SILAC) and affinity-purification mass spectrometry (AP-MS). The use of SILAC makes the interactome analysis more reliable and quantitative, which enables us to identify finer differences between the interactomes of KRas4a and KRas4b. These interactions not only can explain some of the functional differences between KRas4a and KRas4b but also can facilitate the discovery of new signaling functions of the two KRas isoforms.
Results and Discussion
Identifying KRas4a and KRas4b Interacting Proteins in HEK293T Cells by SILAC and AP-MS
We utilized SILAC and AP-MS to construct the interactome network of wild-type (WT) and constitutively active Gly12Asp mutant (G12D) of KRas4a and KRas4b. It has been shown that most WT KRas is in the GDP-bound state in cells (∼93%).14 In contrast, mutation of KRas Gly12 to any other amino acids except proline blocks GAP arginine finger assisted GTP hydrolysis, leading to most of the G12D mutant KRas bound to GTP.15 We chose the G12D mutant for interactome study because KRas G12D is the most abundant mutation in many cancers, such as pancreatic ductal adenocarcinoma (PDAC) and colon and rectal carcinoma (CRC).2 Comparing the interactome of KRas4a/b WT and G12D mutant may reveal KRas4a/b nucleotide-dependent interacting proteins.
We used SILAC to rule out nonspecific proteins and false positives, which are common problems for AP-MS based interactome studies. The experimental design is shown in Figure 1a. Taking the identification of KRas4a WT interacting proteins as an example, we transiently transfected tag-free KRas4a WT into “light” HEK293T cells and FLAG-tagged KRas4a WT into “heavy” HEK293T cells at similar expression levels (Figure S1). Then we carried out FLAG immunoprecipitation (IP) and combined the FLAG resin from light and heavy samples. After elution and trypsin digestion, the proteins were identified and quantified by MS. In the SILAC results, we picked proteins with a heavy/light (H/L) ratio >1.5 and with at least two unique peptides as potential KRas4a WT interacting proteins. To enhance data reliability and reduce false positive hits, we also performed reverse SILAC experiments in parallel, in which the “heavy” and “light” samples were swapped. In the reverse SILAC, proteins with H/L ratio <0.67 and with at least two unique peptides would be potential interacting proteins. We plotted KRas4a interacting proteins (with ≥2 unique peptides identified) against their heavy/light ratios in both forward and reverse SILAC for easy visualization of the result (Figure 1b). Over 85% of proteins from the original SILAC results were ruled out, leaving a list of high-confident interacting proteins (Table S1).
Figure 1.

Identifying KRas4a and KRas4b interacting proteins in HEK293T cells by SILAC and AP-MS. (a) Scheme showing identification of KRas4a/b interacting proteins in HEK293T cells by SILAC and AP-MS. (b) Plotting KRas4a/b interacting proteins (with ≥2 unique peptides) against their heavy/light ratios. (c) Nonspecific versus confident interacting proteins of KRas4a/b identified in HEK293T cells. (d) Heat map showing the heavy/light ratios of KRas4a/b interacting proteins in HEK293T cells. (e) iBAQ values (from HEK293T cells) showing the abundance distribution of KRas4a/b interacting proteins. (f) Heavy/light ratio and peptide number (within parentheses) of known KRas4a/b interacting proteins. (g) Venn diagrams showing the numbers of shared and unique interacting proteins of KRas4a/b in HEK293T cells. (h) Biological process analysis of KRas4a/b interacting proteins. Categories were assigned based on DAVID analysis UP_KEYWORDS.
Similar SILAC experiments and analysis were done for KRas4a G12D, KRas4b WT, and KRas4b G12D. Comparable numbers of interacting proteins were identified for each KRas protein (KRas4a WT, 103 proteins; KRas4a G12D, 96 proteins; KRas4b WT, 94 proteins; KRas4b G12D, 110 proteins. Figure 1c,d). The KRas4a/b interacting proteins spanned a broad range in abundance across the HEK293T proteome16 (Figure 1e). Many known KRas interacting proteins were identified (Figure 1f), suggesting that the interactome data set was reliable.
For each KRas isoform, more than half of the interacting proteins were shared between WT and G12D (Figure 1g), suggesting that GTP/GDP binding on KRas affected some but not the majority of interacting proteins. One example of the shared interacting proteins is FNTB (protein farnesyltransferase subunit beta, Figure 1f), which is involved in posttranslational modification of Ras proteins. Among the interacting proteins that were not shared between WT and G12D, we found many known KRas4a/b effector proteins that only interacted with G12D, such as ARAF and BRAF (Figure 1f). This suggested that comparing the WT and G12D interactome could help identify new KRas4a/b effector proteins.
Comparison of different KRas isoforms (KRas4a versus KRas4b, or KRas4a G12D versus KRas4b G12D) suggested that more than half of interacting proteins were isoform specific (Figure 1g). For each KRas isoform, we combined the WT and G12D interactome and analyzed the biological processes that the interacting proteins are involved in. We found many shared biological processes such as nucleotide-binding and alternative splicing (Figure 1h). Some biological processes showed KRas isoform specificity. For example, KRas4a-specific interacting proteins are involved in mitosis, DNA damage, and ion transport, while KRas4b-specific interacting proteins are involved in neurodegeneration, mRNA transport, lipid metabolism, and protein biosynthesis (Figure 1h).
Biochemical Validation of K-Ras Isoform Specific Interacting Proteins
Among the proteins that only interacted with one of the KRas isoforms, we chose two proteins for validation, v-ATPase a2 and the δ subunit of eukaryotic initiation factor 2B (eIF2Bδ), which only interacted with KRas4b but not KRas4a based on the interactome data (Figure 2a and 3a). When we transfected FLAG-tagged KRas4a and KRas4b into HEK293T cells, immunoprecipitation of FLAG-tagged KRas4b, but not KRas4a, pulled out endogenous v-ATPase a2 (Figure 2b), confirming the interactome data.
Figure 2.

KRas4b interacts with v-ATPase a2 through its C-terminal HVR. (a) Heavy/light ratio of v-ATPase a2 in KRas4a/b SILAC and the primary mass spectra of one v-ATPase a2 peptide (residues 638–650) in forward and reverse KRas4b SILAC. (b) Immunoprecipitation of FLAG-tagged KRas4b, but not KRas4a, pulled out endogenous v-ATPase a2 in HEK293T cells. (c) Immunoprecipitation of FLAG-tagged HRas(1–164)-KRas4b (165–188) (labeled as FLAG-HRas+K4b), but not FLAG-tagged HRas(1–164)-KRas4a (165–189) (labeled FLAG-HRas+K4a), pulled out endogenous v-ATPase a2 as FLAG-tagged KRas4b did. (d) Immunoprecipitation of both FLAG-tagged KRas4b G12D and S17N pulled out similar levels of endogenous v-ATPase a2. RAF1 was used as a positive control, which only interacted with KRas4b G12D but not S17N. (e) Confocal images showing the colocalization of GFP-KRas4a WT or GFP-KRas4b WT with LAMP1 in HEK293T cells. Quantification of colocalization was shown on the left using Pearson’s coefficient (n = 10 for each sample). Statistical evaluation was done using a two-way ANOVA. Scale bar: 5 μm.
Figure 3.

KRas4b interacts with eIF2Bδ through its C-terminal HVR. (a) Heavy/light ratio of eIF2Bδ in KRas4a/b SILAC and the primary mass spectra of one eIF2Bδ peptide (residues 210–225) in forward and reverse KRas4b SILAC. (b) Immunoprecipitation of FLAG-tagged KRas4b, but not KRas4a, pulled out endogenous eIF2Bδ in HEK293T cells. (c) Immunoprecipitation of FLAG-tagged HRas(1–164)-KRas4b (165–188), but not HRas(1–164)-KRas4a (165–189), pulled out endogenous eIF2Bδ as FLAG-tagged KRas4b did. (d) Immunoprecipitation of FLAG-tagged KRas4b WT, G12D and S17N pulled out similar levels of endogenous eIF2Bδ. (e) Heavy/light ratios of eIF2Bα, eIF2Bβ, eIF2Bγ, and eIF2Bε in KRas4a/b SILAC.
Since KRas4a and KRas4b are only different at the C-terminal HVR, we hypothesized that for any proteins that show KRas4a or KRas4b specificity, the C-terminal HVR should contribute to the specificity. We also found that FLAG-tagged HRas did not interact with endogenous v-ATPase a2 (Figure 2c). Therefore, to confirm whether the KRas4b C-terminal HVR contributed to the interaction with v-ATPase a2, we made FLAG-tagged HRas-KRas4a/b chimeric constructs by adding the KRas4a/b C-terminal HVR (residues 165–189 on KRas4a and residues 165–188 on KRas4b) to the HRas conserved domain (residues 1–164) (Figure 2c). In HEK293T cells, immunoprecipitation of FLAG-tagged HRas(1–164)-KRas4b (165–188), but not FLAG-tagged HRas(1–164)-KRas4a (165–189), pulled out endogenous v-ATPase a2 to similar levels as FLAG-tagged KRas4b did (Figure 2c), suggesting that the KRas4b C-terminal HVR accounted for the specific binding to v-ATPase a2.
Both the WT and G12D mutant of KRas4b interacted with v-ATPase a2 based on the interactome data, suggesting that v-ATPase a2 is unlikely a KRas4b effector protein. We further confirmed this by examining the interactions between v-ATPase a2 and KRas4a G12D (constitutively active, always binds to effector protein) or S17N (dominant negative, cannot bind to effector protein).17 KRas4b G12D and S17N showed comparable v-ATPase a2 interactions (Figure 2d), suggesting that v-ATPase a2 is not a KRas4b effector protein.
v-ATPase is known to localize on the lysosome.18,19 The specific interaction between KRas4b and v-ATPase a2 suggests that KRas4b may have more lysosome localization than KRas4a. Using confocal microscopy, we performed colocalization analysis of KRas4a and KRas4b with the lysosome marker LAMP1. The result suggested that KRas4b had significantly higher colocalization with LAMP1 than KRas4a (Figure 2e). Thus, the higher v-ATPase a2 interaction of KRas4b is consistent with the higher lysosomal localization.
Similarly, we confirmed biochemically that eIF2Bδ only interacted with KRas4b but not KRas4a in a nucleotide-independent and the C-terminal HVR-dependent fashion (Figure 3b–d). eIF2B is a heterotrimeric G protein that consists of five subunits (eIF2Bα, eIF2Bβ, eIF2Bγ, eIF2Bδ, and eIF2Bε).20 All the other four eIF2B subunits were identified as KRas4b specific interacting proteins (Figure 3e), suggesting that KRas4b may use its C-terminal HVR to form a complex with eIF2B.
KRas4a Has More RAF1 Interaction than KRas4b in Cells
We next asked whether we could utilize the interactome data to identify the KRas4a/b interacting proteins that could contribute to the different biological functions of KRas4a and -4b. We found that RAF1, a well characterized Ras effector protein, had a higher peptide number and a higher protein score in the KRas4a WT interactome than in the KRas4b WT interactome (Figure 1f, peptide number, 13 vs 5; protein score, 42.7 vs 16.6), suggesting that KRas4a had more RAF1 interaction than KRas4b. Moreover, in the reverse SILAC, RAF1 was identified as an interacting protein for KRas4a WT but not for KRas4b WT, which was likely due to a lower binding affinity between RAF1 and KRas4b WT. Although both KRas4a G12D and KRas4b G12D pulled out similar levels of RAF1 (Figure 1f), we reasoned that the high percentage of GTP-loading on overexpressed KRas4a/b G12D may saturate RAF1 binding and obscure the difference in binding.
We first validated KRas4a/b and RAF1 interaction in HEK293T cells. Immunoprecipitation of FLAG-tagged KRas4a WT pulled out more endogenous RAF1 than FLAG-tagged KRas4b WT did (Figure 4a). We also included FLAG-tagged HRas and NRas in our co-IP experiment and found that KRas4a had the highest RAF1 interaction among the four Ras proteins (Figure 4a). FLAG-tagged KRas4a G12D and KRas4b G12D had comparable RAF1 interactions (Figure 4a), which is consistent with the SILAC result. To reduce the effect of saturated RAF1 binding on KRas4a/b G12D, we lowered KRas4a/b G12D expression levels by stably expressing them in NIH 3T3 cells. NIH 3T3 cells were also a better and widely used cell model for studying Ras related cancer biology than HEK293T cells.21 We selected cells that expressed FLAG-tagged Ras proteins at levels similar to that of the corresponding endogenous Ras (Figure 4b) to reduce the artifact caused by too much protein overexpression. Immunoprecipitation of KRas4a G12D pulled out more RAF1 than KRas4b G12D and HRas G12D did (Figure 4c). The other two members of the RAF family, ARAF and BRAF, showed comparable interactions with KRas4a G12D and KRas4b G12D (Figure 4c), suggesting that the higher binding affinity to KRas4a was specific to RAF1.
Figure 4.

KRas4a has more RAF1 interaction than KRas4b in cells. (a) Detection of interactions between endogenous RAF1 and FLAG-tagged WT and G12D mutants of HRas, NRas, KRas4a, and KRas4b in HEK293T cells. (b) FLAG-tagged KRas4a/b G12D and endogenous Ras expression levels in NIH 3T3 cells. FLAG-tagged KRas4a/b had higher molecular weight than endogenous Ras and ran higher on the gel. (c) Immunoprecipitation of FLAG-tagged KRas4a G12D pulled out more endogenous RAF1, but not ARAF and BRAF, than FLAG-tagged HRas G12D and KRas4b G12D did in NIH 3T3 cells. (d) Immunoprecipitation of FLAG-tagged HRas(1–164)-KRas4a(165–189) pulled out more endogenous RAF1 than FLAG-tagged HRas(1–164)-KRas4b(165–188) did in HEK293T cells. (e) p-ERK (Thr202, Tyr204) and ERK levels in NIH 3T3 cells expressing pCDH vector, FLAG-tagged HRas G12D, KRas4a G12D, or KRas4b G12D. (f) FLAG-tagged HRas G12D, KRas4a G12D, and KRas4b G12D increased the phosphorylation levels of several key proteins (p-Akt Thr308, p-Akt Ser473, p-S6K Thr389, and p-4E-BP1 Thr37,46) in the PI3K-Akt-mTOR pathway to similar extents in NIH 3T3 cells. (g) Normal 2D cell proliferation of NIH 3T3 cells expressing pCDH vector, FLAG-tagged HRas G12D, KRas4a G12D, or KRas4b G12D. Statistical evaluation was examined using an unpaired two-tailed Student t test. Error bars represent SD in three biological replicates. (h) Anchorage-independent soft agar assay showing that KRas4a G12D expressing NIH 3T3 cells had higher colony number than HRas G12D or KRas4b G12D expressing NIH 3T3 cells. Statistical evaluation was examined using an unpaired two-tailed Student t test. Error bars represent SD in three biological replicates. ***P < 0.001. (i) Knocking down RAF1 by two different shRNAs dramatically decreased KRas4a G12D and KRas4b G12D induced colony formation in soft agar assay. Top figure shows the Western blot of endogenous RAF1 in empty vector, KRas4a G12D, or KRas4b G12D expressing NIH 3T3 cells and the quantification of bands on the Western blot membrane. Statistical evaluation was examined using an unpaired two-tailed Student t test. Error bars represent SD in three biological replicates. ***P < 0.001. (j) Scheme showing that in NIH 3T3 cells, increased KRas4a-RAF1 interaction may contribute to increased anchorage-independent cell growth.
To test whether the difference in RAF1 binding affinity was due to the C-terminal HVRs, we transfected FLAG-tagged HRas(1–164)-KRas4a(165–189) and HRas(1–164)-KRas4b(165–188) into HEK293T cells. Immunoprecipitation of HRas(1–164)-KRas4a(165–189) pulled out more endogenous RAF1 than HRas(1–164)-KRas4b(165–188) did (Figure 4d). In contrast, ARAF showed similar bindings to these chimeric proteins (Figure 4d). This result suggested that, besides the switch I region of Ras protein, KRas4a C-terminal HVR also contributed to RAF1 binding.
We next tested whether the stronger interaction with RAF1 has any functional significance. We measured the phosphorylation level of ERK, a well-established RAF kinase downstream protein.22 At similar expression levels, KRas4a G12D expressing cells had higher ERK phosphorylation than KRas4b G12D and HRas G12D expressing cells (Figure 4e), suggesting that the increased KRas4a-RAF1 interaction led to increased RAF-MEK-ERK signaling as expected.
The PI3K-Akt-mTOR pathway is another well-known Ras effector pathway.23 To make sure that the differential effect of KRas4a G12D and KRas4b G12D was not due to a minor difference in expression levels, we also examined the activation of the PI3K-Akt-mTOR pathway by KRas4a G12D and KRas4b G12D. All three Ras proteins increased p-Akt (Thr308), p-Akt (Ser473), p-S6K (Thr389), and p-4EBP1 (Thr37,46) to similar extents (Figure 4f), suggesting that the differential effect of KRas4a and KRas4b on RAF-MEK-ERK is unique and attributable to the differential RAF1 binding affinity.
As the most common oncogene in human cancer, KRAS plays crucial roles in tumorigenesis and tumor growth.24 However, which KRas isoform plays more important roles remains a matter of debate.9 The difference in RAF1 binding affinity thus prompted us to examine whether KRas4a and KRas4b would exhibit different transforming abilities. We employed two classical and widely used assays, the anchorage-independent soft agar assay and the normal 2D cell proliferation assay, to evaluate KRas4a and KRas4b transformed NIH 3T3 cells. Both assays reflect integrated phenotypes that are contributed by different molecular signaling events, but anchorage-independent cell growth is generally thought to be mainly regulated by RAF-MEK-ERK,25−27 and the PI3K-Akt-mTOR pathway more contributes to the normal 2D cell proliferation.28 Based on the above observation that KRas4a G12D and KRas4b G12D affect the RAF-MEK-ERK pathway differentially, but the PI3K-Akt-mTOR pathway similarly, we predicted that KRas4a G12D transformed cells and KRas4b G12D transformed cells should have similar normal 2D cell proliferation but different anchorage-independent growth. These predictions were indeed supported by the experimental observations: KRas4a G12D transformed and KRas4b G12D transformed NIH 3T3 cells showed similar proliferation rates under normal 2D cell culture (Figure 4g), but the KRas4a G12D transformed NIH 3T3 cells had significantly higher colony numbers than KRas4b G12D or HRas G12D transformed cells on soft agar anchorage-independent growth assay (Figure 4h).
To further confirm that RAF1-MEK-ERK signaling cascade plays a key role in KRas4a/b G12D induced anchorage-independent cell growth, we knocked down RAF1 by 68% and 59% with two different shRNAs, respectively, in KRas4a G12D and KRas4b G12D transformed NIH 3T3 cells (Figure 4i) and performed soft agar assay. As shown in Figure 4i, knocking down RAF1 dramatically blocked colony formation in both KRas4a G12D and KRas4b G12D transformed 3T3 cells. The colony numbers decreased 93% and 84% respectively with two different shRNAs in KRas4a G12D transformed cells, and 97% and 86% respectively in KRas4b G12D transformed cells. This result suggests that RAF1 plays an essential role in KRas4a/b G12D induced anchorage-independent cell growth. Considering that KRas4a has higher RAF1 interaction than KRas4b does (Figure 4a and 4c), the higher KRas4a transforming ability can be attributed to the increased RAF1–KRas4a interaction (Figure 4j).
Using a quantitative proteomic approach, here we examined the nucleotide-dependent interactomes of two splice variants of KRas, KRas4a and KRas4b. In addition to identifying known interacting proteins, we identified many previously unknown KRas4a and KRas4b interacting proteins. Some proteins interact with only one KRas isoform, such as v-ATPase a2 (Figure 2, interacts with KRas4b) and eIF2B (Figure 3, interacts with KRas4b). v-ATPase is known to localize on the surface of the lysosome membrane.18,19 Correspondingly, KRas4b has more lysosome localization than KRas4a (Figure 2e), suggesting that KRas4a and -4b have different intracellular localizations, which may contribute to different signaling functions of KRas4a and -4b. Although KRas4b is reported to mainly localize on the plasma membrane, our study suggests that KRas4b can localize on the lysosome via its interaction with v-ATPase. Another KRas4b interacting protein, eIF2B, is the GEF of eIF2α and plays pivotal roles in canonical translation initiation.29 Our data suggests that KRas4b may regulate protein translation initiation by interacting with eIF2B. Although the functional significance of these newly discovered interactions awaits further investigation, these previously unknown interacting proteins that show isoform specificity may uncover new functions or new regulatory mechanisms of KRas4a and KRas4b.
The quantitative information that is available in our experimental approach also allows us to identify proteins that preferentially bind to the active GTP-bound form of KRas4a and KRas4b. Such proteins are more likely to be effector proteins of KRas. Some of these proteins interact with both KRas isoforms, but at different levels. RAF1 has more KRas4a interaction than KRas4b, which is due to the contribution of KRas4a C-terminal HVR to the interaction (Figure 4d). Interestingly, KRas4a and KRas4b have similar interactions with ARAF or BRAF (Figure 4c). The stronger interaction between KRas4a and RAF1 led to stronger RAF-MEK-ERK pathway activation, while the PI3K-Akt-mTOR pathway activation is similar for KRas4a and KRas4b. Consistent with these signaling properties, we found that KRas4a and KRas4b increased normal 2D cell proliferation similarly, but KRas4a increased anchorage-independent cell growth better than KRas4b did. Our interactome data suggest that the increased anchorage-independent colony formation in KRas4a transformed NIH 3T3 cells is likely attributed to increased KRas4a–RAF1 interaction and RAF1-MEK-ERK signaling cascade.
RAS is the most frequently mutated oncogene in human cancer.2 Constitutively active mutants of RAS are found in 60–90% of pancreatic cancer, 36% of colorectal cancer, and 19% of lung cancer.30,31 Among all RAS-driven cancers, KRAS is the most frequently mutated RAS (86%) (COSMIC database). Most early studies have focused on KRas4b in KRas-driven cancers since KRas4b was found to be more abundant.32 However, accumulating evidence suggests that the KRas4a isoform is widely expressed in different human cancers and also plays important roles in tumorigenesis.9,33−35 Therefore, an important question is whether there is any difference in the transforming ability of KRas4a and KRas4b and what the underlying molecular mechanism is. Our interactome study described here thus has provided important insights into this question.
The Ras superfamily of small GTPases consists of more than 150 members. They work similarly to KRas, activating different pathways by recruiting different effector proteins when bound to GTP. Therefore, the comparative nucleotide-dependent interactome study described here for KRas could be similarly used to study the specificities of other small GTPases and discover previously unknown functions for them. We thus believe that this is a powerful approach that can be used to gain useful biological insights for numerous cell signaling pathways.
Methods
Reagents
ERK (#4696), p-ERK Thr202, Tyr204 (#4370), Akt (#4691), p-Akt Thr308 (#13038), p-Akr Ser473 (#4060), S6K (#9202), p-S6K Thr389 (#9234), 4E-BP1 (#9644), and p-4E-BP1 Thr37,46 (#2855) antibodies were purchased from Cell Signaling Technology. ARAF (sc-408), BRAF (sc-166), RAF1 (sc-227), and β-actin (sc-4777) antibodies were purchased Santa Cruz Biotechnology. eIF2Bδ (11332-1-AP) antibody was purchased from Proteintech. v-ATPase a2 (GTX111275) antibody was purchased from GeneTex. Anti-FLAG affinity gels (#A2220) and FLAG antibody (#A8592) were purchased from Sigma. Mouse RAF1 shRNAs (#1, TRCN0000012628, TRCN #2, TRCN0000312820) were purchased from Sigma. Protease inhibitor cocktail, puromycin, crystal violet, [13C6,15N2]-l-lysine, [13C6,15N4]-l-arginine, l-lysine, and l-arginine were purchased from Sigma. FuGENE 6 transfection reagent and sequencing grade modified trypsin were purchased from Promega. MEM nonessential amino acids and ECL plus Western blotting detection reagent were purchased from ThermoFisher. Sep-Pak C18 cartridge was purchased from Waters. LAMP1-RFP (Addgene plasmid #1817) was obtained from Walther Mothes.36
Cell Culture
Human embryonic kidney (HEK) 293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM) medium (ThermoFisher) with 10% heat inactivated FBS (ThermoFisher). Mouse embryonic fibroblast NIH 3T3 cells were cultured in DMEM medium with 15% heat inactivated FBS and MEM nonessential amino acids. All the cell lines had been tested for mycoplasma contamination and showed no mycoplasma contamination.
Cloning, Transfection, and Transduction
Human HRAS, NRAS, KRAS4A, and KRAS4B were inserted into pCMV5 and pCDH-CMV-MCS-EF1-Puro vectors with N-terminal FLAG tag. Human KRAS4A and KRas4B were inserted into pEGFP C1 vector with N-terminal GFP tag. All mutants were generated by QuikChange site-directed mutagenesis. All transient transfections were performed using FuGENE 6 transfection reagent according to the manufacturer’s protocol. HRas G12D, KRas4a G12D, KRas4b G12D, and RAF1 shRNA lentiviruses were generated by cotransfection of HRas/KRas4a/KRas4b G12D in pCDH vector or RAF1 shRNA in pLKO.1 vector, pCMV-dR8.2, and pMD2.G into HEK293T cells. To obtain the HRas/KRas4a/KRas4b G12D stably overexpressed NIH 3T3 cells, cells were treated with 2 mg/mL of puromycin 48 h after lentivirus infection.
Co-Immunoprecipitation
Cells were collected and lysed in 1% NP40 lysis buffer (1% NP40, 25 mM Tris-HCl pH 7.4, 150 mM NaCl, and 10% glycerol) with protease inhibitor cocktail (1:100 dilution) on ice for 30 min. After centrifuging at 15000g for 10 min, the supernatant (total lysates) was collected for FLAG immunoprecipitation following the manufacturer’s protocol. The affinity gel was washed three times with NP40 washing buffer (0.2% NP40, 25 mM Tris-HCl pH 7.4, and 150 mM NaCl). To detect the interacting proteins, the affinity gel was heated at 95 °C for 10 min in 2× protein loading buffer, followed by Western blot analysis.
Western Blot
Western blot analysis was performed following previously published methods.37 The proteins of interest were detected and visualized using a Typhoon FLA 7000 scanner (GE Healthcare).
SILAC and Nano LC–MS/MS Analysis
“Heavy” HEK293T cells were cultured in DMEM with [13C6,15N2]-l-lysine, [13C6,15N4]-l-arginine, and 10% dialyzed FBS for 5 generations. “Light” HEK293T cells were cultured in DMEM with normal l-lysine, l-arginine, and 10% dialyzed FBS for 5 generations. After transient transfection of desired plasmids, “heavy” and “light” cells were lysed in 1% NP40 lysis buffer separately, according to the protocol described above. Protein input of 8 mg for each “heavy” and “light” total lysate was subjected for FLAG immunoprecipitation separately. After washing the affinity gel three times with NP40 washing buffer, “heavy” and “light” samples were mixed and washed two more times with NP40 washing buffer. To elute FLAG-tagged protein with its interacting proteins, the affinity gel was heated at 95 °C for 10 min in 1% SDS elution buffer (1% SDS, 25 mM Tris-HCl pH 7.4, and 150 mM NaCl), followed by methanol/chloroform protein precipitation. The protein pellets were denatured in 6 M urea, 10 mM DTT, and 50 mM Tris-HCl pH 8.0 at room temperature for 1 h. The proteins were alkylated by incubating with 40 mM iodoacetamide at room temperature for 1 h. DTT was then added to stop alkylation at room temperature for 1 h. After diluting the protein sample 7 times with 50 mM Tris-HCl pH 8.0 and 1 mM CaCl2, 1 μg of trypsin was added and incubated with the protein at 37 °C for 18 h. Trifluoroacetic acid (0.1% in water) was added to quench the trypsin digestion, followed by desalting using a Sep-Pak C18 cartridge. The lyophilized peptide powders were collected for LC–MS/MS analysis (LTQ-Orbitrap Elite mass spectrometer coupled with nanoLC). The lyophilized peptide powders were dissolved in 2% acetonitrile (ACN) with 0.5% formic acid (FA). The reconstituted peptides were injected into an Acclaim PepMap nano Viper C18 trap column (5 μm, 100 μm × 2 cm, Thermo Dionex) and separated in a C18 RP nano column (5 μm, 75 μm × 50 cm, Magic C18, Bruker). The flow rate was set as 0.3 μL/min. The gradient was set as follows: 5–38% ACN with 0.1% FA (0–120 min), 38–95% ACN with 0.1% FA (120–127 min), 95% ACN with 0.1% FA (127–135 min). Positive ion mode was used in an LTQ-Orbitrap Elite mass spectrometer (spray voltage 1.6 kV, source temperature 275 °C). The precursor ions scan from m/z 375 to 1800 at resolution 120,000 using an FT mass analyzer. Collision-induced dissociation (CID) was used for the MS/MS scan at resolution 15,000 on the 10 most intensive peaks, isolation width was set as 2.0 m/z, and normalized collision energy was set as 35%. Xcalibur 2.2 operation software was used for collecting the data. The MS data was further processed using Sequest HT in Proteome Discoverer 1.4.1.14 (PD 1.4, Thermo Scientific).
Confocal Imaging
Cells were seeded in 35 mm glass bottom dishes (MatTek) and cotransfected with GFP-KRas4a/b and LAMP1-RFP. After 24 h, cells were rinsed with PBS twice and fixed with 4% paraformaldehyde (in PBS) for 15 min. The fixed cells were washed twice with PBS and imaged with Zeiss LSM880 inverted confocal microscopy.
Quantitative Analyses of Colocalization
KRas4a/b-LAMP1 colocalization was quantitatively analyzed using Fiji software. Image background was first subtracted, and then the cell was selected and quantified for both GFP and RFP channels. Pearson’s correlation coefficient was calculated using the Fiji plug-in Coloc2 program.38
Normal 2D Cell Proliferation Assay
NIH 3T3 cells stably expressing pCDH-HRas G12D, KRas4a G12D, or KRas4b G12D were seeded into a 12-well plate (200 cells/well). The medium was changed every 48 h. After 9 days of culture, the cells were washed with PBS and fixed with ice-cold methanol for 10 min. After removing the methanol, the cells were stained with crystal violet staining solution (0.2% in 2% ethanol) for 5 min. Then the cells were rinsed with water to remove extra crystal violet. The absorption of crystal violet was measured at 550 nm after the stained cells were solubilized with 0.5% SDS in 50% ethanol.
Anchorage-Independent Soft Agar Assay
1.5 mL of 0.6% base low melting point agarose was added into a 6-well plate. After the agarose was solidified, 5.0 × 103 of NIH 3T3 cells stably expressing pCDH, HRas G12D, KRas4a G12D, or KRas4b G12D were mixed with 0.3% low melting point agarose and plated on top of the 0.6% base agarose layer. 150 μL of normal culture medium was added on top of the 0.3% low melting point agarose. The medium was changed every 48 h. After 14 days of culture, colonies were stained with crystal violet staining solution (0.1% in 25% methanol) for 30 min. Then the cells were rinsed with 50% methanol to remove extra crystal violet. To observe the effect of RAF1 knockdown on KRas4a G12D and KRas4b G12D induced anchorage-independent cell growth, NIH 3T3 cells stably expressing KRas4a G12D or KRas4b G12D were treated with lentiviruses carrying luciferase shRNA (shCtrl), RAF1 shRNA#1, or RAF1 shRNA#2 for 48 h before seeding into the 6-well plate. The soft agar assay was perfomed with the same method described above.
Statistical Analysis
Quantitative data were expressed as mean ± SD (standard deviation, represented by error bar). Differences were examined by two-tailed Student’s t test (for cell proliferation assay and soft agar assay) or two-way ANOVA (for confocal imaging).
Acknowledgments
This work is supported in part by a grant (1R01GM121540-01A1) from NIH. H.J. is a Howard Hughes Medical Institute International Student Research Fellow. We thank Prof. Maurine Linder at Cornell University for providing HRas, NRas, and KRas4b plasmids as the cloning template. We thank Dr. Sheng Zhang and Dr. Ievgen Motorykin at the Proteomic and MS Facility of Cornell University for help with the SILAC experiments. The Orbitrap Fusion mass spectrometer is supported by the NIH SIG 1S10 OD017992-01 grant. We thank Cornell University Biotechnology Resource Center (BRC) Imaging Facility for the support on the confocal microscopy, which is supported in part by NIH S10RR025502.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00440.
Author Contributions
X.Z. and H.L. designed the research, analyzed the results, and wrote the manuscript; X.Z. performed all the biochemical and cellular studies except those noted below; J.C. performed RAF1 knockdown experiment and soft agar assay; S.P.M. validated some KRas interacting proteins and cloned HRas and NRas constructs; H.J. performed soft agar assay.
The authors declare no competing financial interest.
Supplementary Material
References
- Wennerberg K.; Rossman K. L.; Der C. J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118 (5), 843. 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- Cox A. D.; Fesik S. W.; Kimmelman A. C.; Luo J.; Der C. J. Drugging the undruggable RAS: Mission possible?. Nat. Rev. Drug Discovery 2014, 13 (11), 828. 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub A. E.; Weinberg R. A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9 (7), 517. 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigil D.; Cherfils J.; Rossman K. L.; Der C. J. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy?. Nat. Rev. Cancer 2010, 10 (12), 842. 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Jang H.; Muratcioglu S.; Gursoy A.; Keskin O.; Nussinov R.; Zhang J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116 (11), 6607. 10.1021/acs.chemrev.5b00542. [DOI] [PubMed] [Google Scholar]
- Ahearn I. M.; Haigis K.; Bar-Sagi D.; Philips M. R. Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13 (1), 39. 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh C.; Subrahmanyam R.; Ren R. Oncogenic NRAS, KRAS, and HRAS exhibit different leukemogenic potentials in mice. Cancer Res. 2007, 67 (15), 7139. 10.1158/0008-5472.CAN-07-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivaz M.; Meyer T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J. Cell Biol. 2005, 170 (3), 429. 10.1083/jcb.200409157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai F. D.; Lopes M. S.; Zhou M.; Court H.; Ponce O.; Fiordalisi J. J.; Gierut J. J.; Cox A. D.; Haigis K. M.; Philips M. R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (3), 779. 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalonga P.; Lopez-Alcala C.; Bosch M.; Chiloeches A.; Rocamora N.; Gil J.; Marais R.; Marshall C. J.; Bachs O.; Agell N. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell. Biol. 2001, 21 (21), 7345. 10.1128/MCB.21.21.7345-7354.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin E. L.; Bruckner R. J.; Paulo J. A.; Cannon J. R.; Ting L.; Baltier K.; Colby G.; Gebreab F.; Gygi M. P.; Parzen H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545 (7655), 505. 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Bliss J. M.; Wang Y.; Colicelli J. RIN1 is an ABL tyrosine kinase activator and a regulator of epithelial-cell adhesion and migration. Curr. Biol. 2005, 15 (9), 815. 10.1016/j.cub.2005.03.049. [DOI] [PubMed] [Google Scholar]
- Weber C. K.; Slupsky J. R.; Kalmes H. A.; Rapp U. R. Active Ras induces heterodimerization of cRaf and BRaf. Cancer Res. 2001, 61 (9), 3595. [PubMed] [Google Scholar]
- Bollag G.; Adler F.; elMasry N.; McCabe P. C.; Conner E. Jr.; Thompson P.; McCormick F.; Shannon K. Biochemical characterization of a novel KRAS insertion mutation from a human leukemia. J. Biol. Chem. 1996, 271 (51), 32491. 10.1074/jbc.271.51.32491. [DOI] [PubMed] [Google Scholar]
- Bos J. L.; Rehmann H.; Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 2007, 129 (5), 865. 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- Geiger T.; Wehner A.; Schaab C.; Cox J.; Mann M. Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 2012, 11 (3), M111.014050. 10.1074/mcp.M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar N.; Singh K.; Garcia-Diaz M. Structure of the dominant negative S17N mutant of Ras. Biochemistry 2010, 49 (9), 1970. 10.1021/bi9020742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi T.; Forgac M. The vacuolar (H+)-ATPases--nature’s most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 2002, 3 (2), 94. 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- Zoncu R.; Bar-Peled L.; Efeyan A.; Wang S.; Sancak Y.; Sabatini D. M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334 (6056), 678. 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi K.; Takahashi M.; Nishimoto M.; Hiyama T. B.; Higo T.; Umehara T.; Sakamoto K.; Ito T.; Yokoyama S. Crystal structure of eukaryotic translation initiation factor 2B. Nature 2016, 531 (7592), 122. 10.1038/nature16991. [DOI] [PubMed] [Google Scholar]
- Stacey D. W.; Kung H. F. Transformation of NIH 3T3 cells by microinjection of Ha-ras p21 protein. Nature 1984, 310 (5977), 508. 10.1038/310508a0. [DOI] [PubMed] [Google Scholar]
- Lavoie H.; Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16 (5), 281. 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]
- Rubio I.; Rodriguez-Viciana P.; Downward J.; Wetzker R. Interaction of Ras with phosphoinositide 3-kinase gamma. Biochem. J. 1997, 326 (3), 891. 10.1042/bj3260891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrem J. M.; Shokat K. M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discovery 2016, 15 (11), 771. 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- Dohn M. R.; Brown M. V.; Reynolds A. B. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 2009, 184 (3), 437. 10.1083/jcb.200807096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S.; Chang J. T.; Andrechek E. R.; Matsumura N.; Baba T.; Yao G.; Kim J. W.; Gatza M.; Murphy S.; Nevins J. R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28 (31), 2796. 10.1038/onc.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulku A. S.; Schafer R.; Der C. J. Essential role of Raf in Ras transformation and deregulation of matrix metalloproteinase expression in ovarian epithelial cells. Mol. Cancer Res. 2003, 1 (14), 1077. [PubMed] [Google Scholar]
- Fruman D. A.; Chiu H.; Hopkins B. D.; Bagrodia S.; Cantley L. C.; Abraham R. T. The PI3K Pathway in Human Disease. Cell 2017, 170 (4), 605. 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson R. J.; Hellen C. U.; Pestova T. V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11 (2), 113. 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior I. A.; Lewis P. D.; Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72 (10), 2457. 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D. P.; Hong T. S.; Bardeesy N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371 (22), 2140. 10.1056/NEJMc1412266. [DOI] [PubMed] [Google Scholar]
- Pells S.; Divjak M.; Romanowski P.; Impey H.; Hawkins N. J.; Clarke A. R.; Hooper M. L.; Williamson D. J. Developmentally-regulated expression of murine K-ras isoforms. Oncogene 1997, 15 (15), 1781. 10.1038/sj.onc.1201354. [DOI] [PubMed] [Google Scholar]
- To M. D.; Wong C. E.; Karnezis A. N.; Del Rosario R.; Di Lauro R.; Balmain A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat. Genet. 2008, 40 (10), 1240. 10.1038/ng.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patek C. E.; Arends M. J.; Wallace W. A.; Luo F.; Hagan S.; Brownstein D. G.; Rose L.; Devenney P. S.; Walker M.; Plowman S. J.; et al. Mutationally activated K-ras 4A and 4B both mediate lung carcinogenesis. Exp. Cell Res. 2008, 314 (5), 1105. 10.1016/j.yexcr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Plowman S. J.; Berry R. L.; Bader S. A.; Luo F.; Arends M. J.; Harrison D. J.; Hooper M. L.; Patek C. E. K-ras 4A and 4B are co-expressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J. Exp. Clin. Cancer Res. 2006, 25 (2), 259. [PubMed] [Google Scholar]
- Sherer N. M.; Lehmann M. J.; Jimenez-Soto L. F.; Ingmundson A.; Horner S. M.; Cicchetti G.; Allen P. G.; Pypaert M.; Cunningham J. M.; Mothes W. Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 2003, 4 (11), 785. 10.1034/j.1600-0854.2003.00135.x. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Khan S.; Jiang H.; Antonyak M. A.; Chen X.; Spiegelman N. A.; Shrimp J. H.; Cerione R. A.; Lin H. Identifying the functional contribution of the defatty-acylase activity of SIRT6. Nat. Chem. Biol. 2016, 12 (8), 614. 10.1038/nchembio.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler J.; Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander’s overlap coefficient. Cytometry, Part A 2010, 77A (8), 733. 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.