

Abstract
Bryostatin 1 (henceforth bryostatin) is in clinical trials for the treatment of Alzheimer’s disease and for HIV/AIDS eradication. It is also a preclinical lead for cancer immunotherapy and other therapeutic indications. Yet nothing is known about the conformation of bryostatin bound to its protein kinase C (PKC) target in a membrane microenvironment. As a result, efforts to design more efficacious, better tolerated, or more synthetically accessible ligands have been limited to structures that do not include PKC or membrane effects known to influence PKC–ligand binding. This problem extends more generally to many membrane-associated proteins in the human proteome. Here, we use rotational-echo double-resonance (REDOR) solid-state NMR to determine the conformations of PKC modulators bound to the PKCδ-C1b domain in the presence of phospholipid vesicles. The conformationally limited PKC modulator phorbol diacetate (PDAc) is used as an initial test substrate. While unanticipated partitioning of PDAc between an immobilized protein-bound state and a mobile state in the phospholipid assembly was observed, a single conformation in the bound state was identified. In striking contrast, a bryostatin analogue (bryolog) was found to exist exclusively in a protein-bound state, but adopts a distribution of conformations as defined by three independent distance measurements. The detection of multiple PKCδ-C1b-bound bryolog conformers in a functionally relevant phospholipid complex reveals the inherent dynamic nature of cellular systems that is not captured with single-conformation static structures. These results indicate that binding, selectivity, and function of PKC modulators, as well as the design of new modulators, are best addressed using a dynamic multistate model, an analysis potentially applicable to other membrane-associated proteins.
Short abstract
REDOR NMR detects multiple conformations for a bryostatin analogue complexed with a PKCδ-C1b domain in functionally relevant phospholipid vesicles, thereby establishing a platform to evaluate other ligands and effects on PKC function.
Introduction
While membrane proteins comprise a significant percentage of the human proteome,1 the determination of the structures of ligands and drug candidates bound to these proteins in a membrane microenvironment is an ongoing and formidable challenge.2 Furthermore, membrane-associated proteins (MAPs) interact with phospholipid bilayers in a dynamic fashion and introduce additional complexity for relevant X-ray structure determinations as crystal packing forces could disrupt or distort the MAP–ligand–membrane complex.3 The large size of a MAP–ligand–membrane complex with restricted mobility also poses a challenge to solution-state NMR analysis.3 Representative of MAPs, protein kinase C (PKC) isoforms4,5 play a central role in cellular signal transduction through the phosphorylation of a diverse array of proteins involved in cell growth, differentiation, proliferation, and apoptosis.6,7 More generally, PKC isoforms8−12 and mutated PKCs13,14 have been implicated in numerous therapeutic indications including HIV/AIDS eradication, Alzheimer’s disease, cancer immunotherapy, cardiovascular disease, pain, and stroke, among others. Yet nothing is known about the bound conformations of endogenous (e.g., diacylglycerols) or exogenous (e.g., phorbol esters, prostratin, bryostatins, daphnane diterpenoid orthoesters, ingenanes, aplysiatoxin, and teleocidins) PKC modulatory ligands in a membrane environment. Such information is critical for understanding how these agents function at a molecular level and more importantly how one might design superior, more accessible, and better tolerated analogues.
Bryostatin15−17 is a marine-derived PKC modulator now in clinical trials for Alzheimer’s disease and HIV/AIDS eradication. Its original GMP (good manufacturing practice) supply is nearly depleted.18 Efforts to design superior, better tolerated, and more synthetically accessible ligands19 have progressed20−23 but rely on computer-guided ligand comparisons24−26 and models27−29 that lack experimental information on the structure(s) of the PKC-bound ligand in a membrane microenvironment. Here, we use REDOR solid-state NMR30−35 to determine the conformations of PKC modulators bound to the 51-residue PKCδ-C1b regulatory domain with phosphatidylserine (PS) multilamellar vesicles as the membrane surrogate. Using the conformationally limited phorbol diacetate (PDAc, Figure 1) as an initial test ligand, we find unanticipated partitioning of PDAc between a mobile lipid-associated state and an immobilized protein-bound state, but identify a single conformation in the bound state. In striking contrast, the bryostatin analogue, bryolog 1, adopts multiple bound conformations. The molecular distribution revealed by REDOR NMR is supported by molecular dynamics simulations and demonstrates a departure from prior studies which have sought out and presented only a single, lowest-energy, PKC-bound conformation. This finding represents a fundamentally new way to think about PKC ligand binding and calls attention to the importance of multistage contributions to the binding and function of membrane-bound protein–ligand complexes. The work indicates the need to consider molecular flexibility when designing and developing new ligands to modulate PKC function. For example, PDAc is rigid and is a tumor promoter, whereas the more flexible bryostatin exhibits therapeutic potential in cancer therapy. Furthermore, we present an experimental platform to address outstanding questions involving the binding of other bryologs and other MAP ligands.
Figure 1.

Determination of the bound conformation and accounting of PKCδ-C1b-bound PDAc. (A) Phorbol diacetate is depicted as an overlay of two energetically accessible conformers, highlighting the in silico calculated distance between the C12-OAc carbonyl and the internuclear average of the C13-OAc deuterons. (B) 13C cross-polarization magic-angle-spinning (CPMAS) spectrum of the [13C,2H3]PDAc–PKCδ-C1b–phosoholipid vesicle complex, acquired as an 8 Tr (rotor period) echo with 20,000 scans and MAS at 7143 Hz. By spin counting, there are 1.2 μmol of PDAc in the NMR sample. (C) C{D} REDOR spectra were obtained at 11 evolution times, and the REDOR dephasing is best fit to an internuclear distance of 5.1 ± 0.2 Å (reduced χ2 value of 1.85), corresponding to a 35 ± 4 Hz best fit for the dipolar coupling. Error bars are based on the signal-to-noise ratio of each spectrum. Approximately 140,000 scans of S0 and S were obtained for the [13C,2H3]PDAc complex at the early evolution times of 4.48 and 6.72 ms where the REDOR differences were small (13 days total). 60–90,000 scans were collected for later evolution times (requiring an additional 30 days of acquisition). Experimentally corrected S0 spectra corresponding to the 13C label in PDAc without contributions from natural abundance carbons were obtained by subtracting the corresponding full-echo spectrum of [2H3]PDAc (12 days acquisition) from the [13C,2H3]PDAc spectrum (normalized to the 172 ppm carbonyl peak intensity) for each evolution time (as shown in Figure S1). (D) C{D} REDOR spectra resulting from 160 Tr (22.40 ms) of evolution for the doubly labeled [13C,2H3]PDAc complex (left) and a control complex in which the PDAc was labeled only with 2H (right). 40% dephasing is observed for the PDAc carbonyl (left), and there is no dephasing for other natural abundance carbons (right). (E) 2H Hahn echo spectra of the [13C,2H3]PDAc–PKC–phospholipid vesicle complex. 2H Hahn echoes were obtained with a long (8 s, red) and a short (0.5 s, blue) recycle delay. The 0.5 s delay 2H spectrum is right-shifted for clarity and is the result of 100,000 scans, and the 8 s delay 2H spectrum is the result of 11,000 scans, both with MAS at 7143 Hz.
Results and Discussion
Experimental measurements of long-range intraligand dipolar couplings report directly on distances and, thus, the ligand structure or conformational ensemble of ligand structures in a macromolecular assembly.32−34 We introduced a design strategy involving the synthesis of bryolog 1, selectively installing 13C, 19F, and 2H isotopic labels across the molecule (Figure 2A) to enable the measurement of long-range intraligand distances of the membrane-associated PKCδ-C1b-bound bryolog using REDOR NMR.36 The feasibility and sensitivity of the REDOR-based approach were first evaluated with a more conformationally limited PKC substrate, PDAc, designed and synthesized to report on one 13C–2H distance of approximately 5 Å. As a phorbol diester, PDAc belongs to one of the most studied classes of biologically active molecules.37In silico analysis of PDAc revealed that the C12 and C13 acyloxy carbonyls are 5.0 ± 0.5 Å away from the α-position of their counterpart, regardless of the conformation of the C12 acetoxy group (Figure 1A). A 13C label was installed at the C12-OAc carbonyl position, and 2H was incorporated as a CD3 group on the C13-OAc, where the REDOR-determined distance would extend from the C12 carbonyl to the average position of the three deuterons (Figure 1B). Labeled ligands were complexed with a 51-residue peptide corresponding to the mouse PKCδ-C1b domain,38 allowing comparison of previous structural studies39−50 using this isoform domain, which recapitulates the ligand binding capabilities of the full-length protein.51 The folding and function of PKC and its C1 domain are sensitive to its environment, and in the case of membrane-associated PKC, we have shown that the C1 domain does not fold properly for binding in water alone but only in association with a PS vesicle.51 Thus, complexes were assembled in association with PS vesicles as the membrane surrogate, using a 1:1:50:50 molar ratio of ligand:PKCδ-C1b:phosphatidylserine:trehalose in Tris buffer. This peptide-phospholipid composition yielded a Kd value of 1.5 nM for the high affinity ligand phorbol 12,13-dibutyrate (PDBu), comparable to affinity observed when using a ratio of ∼4300:1 that is employed in standard radioactivity-based binding assays52 (0.36–0.52 nM; see Table S1).
Figure 2.
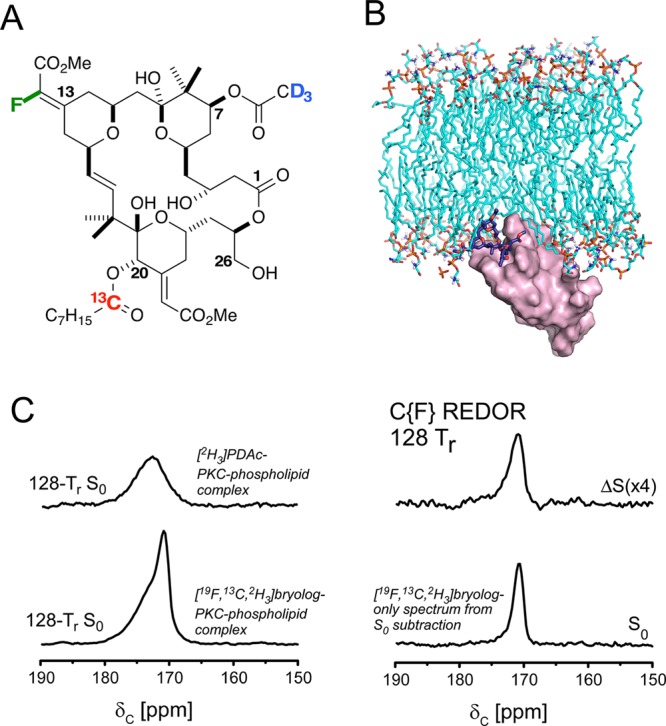
Isotope-labeling strategy and representative REDOR NMR spectra of the PKCδ-C1b-bound bryolog 1. (A) Chemical structure and locations of isotopic labels in bryolog 1. (B) Molecular view of PKCδ-C1b-bound bryolog 1 in association with phospholipids. (C) C{F} REDOR spectra of [19F,13C,2H3]bryolog–PKCδ-C1b–phospholipid complex and [2H3]PDAc–PKCδ-C1b–phospholipid complex at 128 Tr (17.92 ms). The bryolog full-echo S0 (bottom left spectrum) contains contributions from 13C label in bryolog 1 and the natural abundance 13C from PKCδ-C1b and phospholipids. The bryolog-only S0 (bottom right spectrum) is obtained by subtracting the PKCδ-C1b and phospholipids natural abundance 13C (using the natural abundance PDAc full-echo S0, top left spectrum) from the bryolog full-echo S0.
The 13C spectrum of the peptide–ligand–phospholipid complex includes natural abundance 13C contributions from the PKCδ-C1b and phosphatidylserine in addition to the labeled 13C carbonyl contribution from the [13C,2H3]PDAc, fortunately shifted upfield of carbonyl peaks associated with the peptide and phospholipids (Figure 1B). The spectrum of a comparative sample of protein complexed with [2H3]PDAc confirmed the [13C,2H3]PDAc 13C chemical shift assignment (Figure S1). A full C{D} REDOR dephasing curve plotted as the percent dephasing (ΔS/S0) for multiple evolution times was obtained, with 42% dephasing observed after 24.64 ms of dipolar evolution (Figure 1C). No dephasing was observed for the [2H3]PDAc complex (Figure 1D), confirming that the dephasing in the [13C,2H3]PDAc complex arises only from the PDAc 13C–2H dipolar coupling. The experimental REDOR curve fit a distance of 5.1 ± 0.2 Å, matching the in silico prediction. However, the REDOR dephasing exhibited a plateau of 42% and did not reach the maximum theoretical C{D} REDOR dephasing of 73% (see Supporting Information), indicating that some 13C–2H dipolar couplings were not detected. REDOR experiments on [13C,2H3]zinc acetate yielded the theoretically expected maximum dephasing of 74%.53
We hypothesized that the reduced plateau could be attributed to mobile PDAc that was phospholipid-associated but not peptide-bound. Anisotropic motion faster than the strength of the ∼35 Hz 13C–2H dipolar coupling would interfere with REDOR dephasing, but still allow 13C to be observed in the REDOR full-echo reference (S0) spectrum. In this way, the experimental determination of the REDOR ΔS/S0 plateau established a total accounting only of the rigid ligand. A 2H Hahn echo spectrum independently served as a reporter of PDAc motion and revealed that a significant fraction of the PDAc was mobile, with a prominent center band (red) corresponding to motional averaging of the 2H quadrupolar anisotropy tensor (Figure 1E). The 2H Hahn echo spectrum acquired with a shorter recycle delay of 0.5 s, instead of 8 s, exhibited further diminution of the broad sideband pattern corresponding to the rigid component (with a longer T1) relative to the mobile 2H center band. Comparison of the integrated peak intensities of the two 2H Hahn echo spectra indicated that the rigid peptide-bound fraction is 55%, where the spectrum collected with the 8 s recycle delay (in red) has all PDAc contributions and the spectrum resulting from the shorter recycle delay (blue) reports only on mobile PDAc (Figure 1E). The REDOR plateau would be 40% if 55% of the PDAc was rigid and peptide-bound (55% of the maximum theoretical dephasing of 73%), which is in close agreement with the experimentally observed plateau of 42%. Thus, based on the C{D} REDOR NMR and the independent evaluation of the amount of mobile ligand by observing 2H, we conclude that 55–57% of the PDAc is bound to PKC. The remaining 43–45% is mobile. The specific distance of 5.1 Å between the PDAc C12-OAc 13C and the C13-OAc CD3 indicates that PDAc adopts a conformation in which a C9-OH is hydrogen bonded to a C13-OAc when bound to PKCδ-C1b in phospholipid vesicles. This intramolecular hydrogen bond is known to be central to function,54 as phorbol (C12-OH, C13-OAc) disrupts this interaction in favor of a C12-OH to C13-OAc hydrogen bond and does not achieve nanomolar binding affinity to PKC.
REDOR NMR and molecular dynamics were next employed to define the conformation of bryolog 1 bound to PKCδ-C1b in phospholipid vesicles. The positioning of labels in this analogue was guided by computational analysis and intended to enable differentiation of a bound conformer from a range of conformational possibilities. Bryolog 1 exhibited a binding affinity of approximately 1 nM to PKCδ, statistically indistinguishable from bryostatin itself.36 The three possible heteronuclear distance measurements were made between 13C, 2H, and 19F using REDOR NMR. In each case, the analytical REDOR results provided unambiguous evidence of distance distributions55 (Figure S2) and were interpreted with the aid of molecular dynamics simulations.
The 13C chemical shift of the bryolog carbonyl label is upfield and partially resolved from the PKCδ-C1b and phospholipid carbonyl peaks, as evident in the C{F} REDOR S0 spectrum at 128 Tr (17.92 ms) (Figure 2C, bottom left spectrum). Thus, a direct spectral subtraction was performed for each REDOR measurement using spectra obtained identically from the [2H3]PDAc–PKCδ-C1b–phospholipid complex, lacking 13C enrichment (Figure 2C, top left spectrum). This yielded a REDOR S013C spectrum wherein the only carbonyl contributions arose from the bryolog 13C label (Figure 2C, bottom right spectrum). C{F} REDOR spectra were obtained for six REDOR evolution times for the [19F,13C,2H3]bryolog–PKCδ-C1b–phospholipid complex and the [2H3]PDAc–PKCδ-C1b–phospholipid complex, requiring a total signal acquisition time of 42 and 45 days, respectively. The C{F} REDOR results yielded a dephasing curve that could only be attributed to a distribution of distances (Figure 3A, top panel, and Figure S2).
Figure 3.

Identification and quantification of distance distributions for PKCδ-C1b-bound bryolog 1 by REDOR NMR and molecular dynamics simulations. (A) C{F} REDOR, (B) C{D} REDOR, and (C) D{F} REDOR dephasing data with fits by distribution of distances (top panels) and corresponding distributions of the C–F, C–D, and D–F distances of bryolog 1 as a function of the free energy of the PKCδ-C1b–bryolog–phospholipid complex as calculated by molecular dynamics simulations.
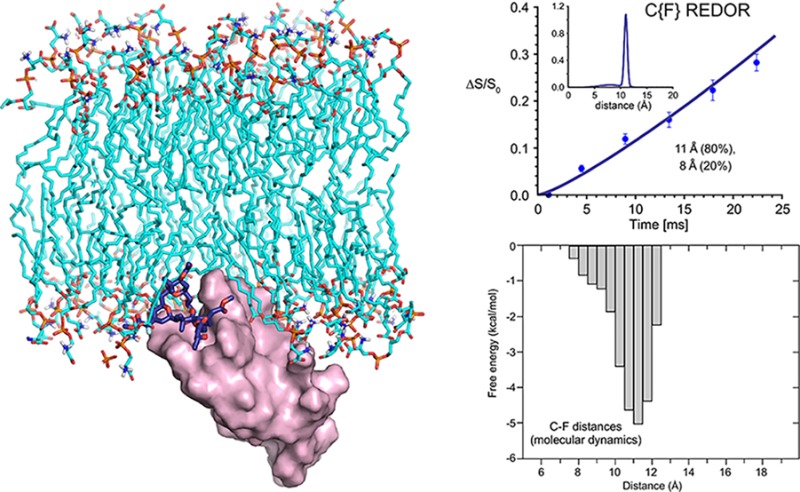
Long time scale (∼500 μs aggregate simulation time), unbiased molecular dynamics simulations of the PKCδ-C1b–phospholipid–bryolog complex29 were employed to examine low-energy conformations and identify distances between the 13C-, 2H-, and 19F-labeled sites. For the 13C- and 19F-labeled sites, the low-energy conformations (Figure 3A, bottom panel) mapped to a minor distribution with the 13C–19F distances centered around 8 Å and a major distribution with the 13C–19F distances centered around 11 Å (Figure 3A, bottom panel). These 13C–19F distance distributions were used as guidance to fit the experimental REDOR data points. A good fit was obtained (Figure 3A, top panel, solid line) with distance distributions (Figure 3A, top panel, inset) very similar to those of the MD simulations (Figure 3A, bottom panel). The ultimate dephasing should reach a plateau of approximately 1.0, accounting for all the 13C–19F internuclear pairs. This was supported by a 2H Hahn echo spectrum, indicating that almost all bryolog 1 was PKCδ-C1b bound (Figure S3). Thus, the REDOR NMR results confirmed the presence of the multiple low-energy conformers found by molecular dynamics, with a distribution of distances centered around two distances, a minor population (20%) at ∼8 Å and a major population (80%) at 11 Å (Figure 3A).
C{D} REDOR results were also indicative of long-range contacts (Figure 3B). Molecular dynamics suggested that bryolog 1 bound to PKCδ-C1b would contain two major distributions of 13C–2H distances centered near 10 and 16 Å. Practically, a distance of 16 Å would be too far to measure experimentally with REDOR, with less than 1% theoretical dephasing even after a REDOR evolution time of 22.40 ms. Yet, the presence of such a conformation was supported by the REDOR data which matched the simulated dephasing expected for the presence of a distribution wherein 55% of the C–D distances are centered at the closer distance of 10 Å and 45% centered at the longer 16 Å distance (Figure 3B, top panel). The distribution of distances centered at 10 Å dominated the dephasing, as expected, wherein the C{D} REDOR ΔS/S0 plateau was expected to reach 41% (∼55% of the theoretical 74%), accounting for the observed population of shorter 13C–2H distances. The remaining 45% of the 13C–2H distances are too distant to be measured with REDOR and were predicted to be centered at 16 Å by the molecular dynamics simulations. These C{D} REDOR results required a total acquisition time of 53 days.
The molecular dynamics simulations predicted a narrow distribution of 2H–19F distances centered at 13 Å (Figure 3C, bottom panel). The simulated REDOR curve for this single distribution centered at 13 Å reached a maximum dephasing of only 2.4% after 22.40 ms. However, results from D{F} REDOR revealed the presence of strong dephasing at early evolution times, e.g., 6.5% dephasing after just 4.48 ms. Thus, there is a shorter than anticipated 2H–19F distance that dominated the observed dephasing. The dephasing (acquired with an acquisition time of 46 days) could be fit to a distribution in which 22% of 2H–19F distances are centered at 6 Å (Figure 3C, top panel). A 6 Å intramolecular proximity is not likely based on the molecular dynamics free energy calculations and the unfavorable ligand distortion that would be required. This suggests the presence of an intermolecular distance associated with an arrangement in which the 2H of one bryolog is proximate to the 19F of a neighboring bryolog. Furthermore, the individual spinning sidebands exhibited different levels of dephasing, indicating that the observed D{F} REDOR dephasing arose from a 2H–19F pair with orientational preferences (Figure 4B).
Figure 4.

Structures of the PKCδ-C1b-bound bryolog 1 and plausible PKCδ-C1b dimers by REDOR NMR and molecular dynamics simulations. (A) Two representative bryolog conformers from the molecular dynamics simulations. (B) D{F} REDOR spectra obtained for the evolution time of 13.44 ms reported in Figure 3C. Preferential dephasing of the spinning sidebands indicates orientational preferences for the 2H–19F internuclear vector. (C) An intermolecular arrangement of two PKC-bound bryolog complexes that maintains a short 5–7 Å 2H–19F intermolecular distance and reveals the presence of plausible protein–protein interactions in a possible dimeric complex.
We examined whether such a short interligand 2H–19F distance was compatible with an arrangement in which two bryolog-bound PKCδ-C1b complexes are proximate. With only the restraint of a 5–7 Å 2H–19F intermolecular proximity, a pairing of two PKC peptides emerged with regions of complementarity between the two peptides, as illustrated in Figure 4C, specifically including interactions between H16 and K45 of peptide 1 and E44 of peptide 2, and interactions between D15 of peptide 1 and K26 and H40 of peptide 2. There are also no intermolecular 13C–2H or 13C–19F distances in the modeled dimer that would have been in the detection range of the REDOR measurements. Thus, the REDOR data are compatible with a model in which some bryolog–PKCδ-C1b complexes can dimerize through contacts consistent with specific protein–protein interactions. This represents a new type of protein–protein interaction for PKC and could potentially contribute to the multiple functions of PKC modulators.
Conclusions
REDOR NMR and molecular dynamics simulations on the three-component PKCδ-C1b–ligand–phospholipid system independently support the presence of a distribution of PKC-bound bryolog conformations. In contrast to the bryolog, PDAc exhibits partitioning between a mobile lipid-associated state and a conformationally static PKCδ-C1b-bound state (Figure 1C and Figure S11 of ref (29)). We believe that these molecular differences ultimately contribute to their functional differences, wherein bryostatin is a promising candidate for various therapeutic indications and the phorbol esters are tumor promoters. The presence of a distribution of conformations for the bryolog could have contributed to previous failed efforts to obtain crystalline bryostatin–PKC complexes, and the relatively low resolution (4.0 Å) of the only full-length PKC crystal structure.43 Molecular dynamics simulations of protein folding and macromolecular interactions have been transformative in generating in silico atomistic views of the possible pathways and conformational sampling involved in molecular recognition. Yet, drug design, biological assays, and drug development strategies are often based on single static structures such as the single-snapshot total structures obtained from X-ray crystallography. In contrast, REDOR NMR captured the conformational plasticity of the bryostatin–PKCδ-C1b–phospholipid system. These REDOR studies suggest that PKC modulators, and possibly other enzyme modulators (for example, Factor Xa bound ligands56), exist in multiple bound states that contribute collectively to affinity but individually or collectively to function. This changes approaches to analogue design from a focus on static single-state structures to multistate dynamic systems accessible through long-range molecular dynamics simulations and REDOR experiments that integrate microenvironment contributions.
Flexibility in the ligand structure and binding could be important to the functional consequences of bryostatin binding and PKC function, in contrast to PDAc and phorbol esters as tumor promoters. Our work now provides a platform to evaluate the binding modes of other analogues and to establish possible correlations for the presence of ligand binding distributions with effects on PKC-associated function. We believe that the evaluation of selectivity and function of candidate PKC modulators, as well as the design of new modulators, should be addressed in terms of quantitative structure activity relationships using a dynamic multistate model, a recommendation also applicable to other membrane-associated proteins. Finally, the direct detection of a short 2H–19F distance was identified only through the REDOR dephasing, and we hypothesize that this could reflect the presence of PKC protein–protein interactions where two bryologs come into proximity, as illustrated in Figure 4C.
PKC and its C1 domain have many physiological states, and we have established fundamental atomic-level distances of biologically active ligands in the membrane-associated structure of the PKCδ-C1b domain. Biorelevant atomic-level detail for bryostatin, phorbol esters, and other PKC ligands is invaluable for the design of more PKC isoform selective ligands and therapeutic leads and, more generally, for understanding the functional role of PKC in the membrane microenvironment. By extending the reach of structural analysis from single-state static systems to multistate systems of higher order complexity, this work presents new opportunities to investigate how protein–ligand structures are influenced by membrane composition and type of bound modulator, and how these connect to function.
Acknowledgments
We acknowledge Professor Kazuhiro Irie for generously providing synthetic PKCδ-C1b peptide for preliminary binding assays.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00475.
Materials and methods, Tables S1 and S2, and Figures S1–S5 (PDF)
Author Contributions
‡ H.Y., D.S., and S.M.R. contributed equally to this work.
This research was supported by grants from the National Institutes of Health to P.A.W. (CA031845 and AI124743), J.S. (EB002058), and L.C. (GM117278). A.D.A., A.B.B., and B.A.L. were supported through the Stanford Center for Molecular Analysis and Design Postdoctoral Fellowship Program. D.S. was supported by the Amgen Graduate Fellowship.
The authors declare no competing financial interest.
Supplementary Material
References
- Wallin E.; von Heijne G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cournia Z.; Allen T. W.; Andricioaei I.; Antonny B.; Baum D.; Brannigan G.; Buchete N. V.; Deckman J. T.; Delemotte L.; del Val C.; Friedman R.; Gkeka P.; Hege H. C.; Henin J.; Kasimova M. A.; Kolocouris A.; Klein M. L.; Khalid S.; Lemieux M. J.; Lindow N.; Roy M.; Selent J.; Tarek M.; Tofoleanu F.; Vanni S.; Urban S.; Wales D. J.; Smith J. C.; Bondar A. N. Membrane protein structure, function, and dynamics: a perspective from experiments and theory. J. Membr. Biol. 2015, 248, 611–640. 10.1007/s00232-015-9802-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. K.; Zhang Z. F.; Tang X. Q.; Li J. P.; Glaubitz C.; Yang J. Conformation and topology of diacylglycerol kinase in E. coli membranes revealed by solid-state NMR spectroscopy. Angew. Chem., Int. Ed. 2014, 53, 5624–5628. 10.1002/anie.201311203. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Kishimoto A.; Takai Y.; Nishizuka Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium-dependent protease from rat brain. J. Biol. Chem. 1977, 252, 7610–7616. [PubMed] [Google Scholar]
- Nakamura S.; Yamamura H. Yasutomi Nishizuka: father of protein kinase C. J. Biochem. 2010, 148, 125–130. 10.1093/jb/mvq066. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumor promotion. Nature 1984, 308, 693–698. 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- Newton A. C. Protein kinase C: poised to signal. Am. J. Physiol. Endocrinol. Metabol. 2010, 298, E395–E402. 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochly-Rosen D.; Das K.; Grimes K. V. Protein kinase C, an elusive therapeutic target?. Nat. Rev. Drug Discovery 2012, 11, 937–957. 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu-Zhang A. X.; Newton A. C. Protein kinase C pharmacology: refining the toolbox. Biochem. J. 2013, 452, 195–209. 10.1042/BJ20130220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert B. J.; Niu A.; Ramani R.; Marshall G. R.; Wender P. A.; Williams R. M.; Ratner L.; Barnes A. B.; Kyei G. B. Combinations of isoform-targeted histone deacetylase inhibitors and bryostatin analogues display remarkable potency to activate latent HIV without global T-cell activation. Sci. Rep. 2017, 7, 7456. 10.1038/s41598-017-07814-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden M. D.; Loy B. A.; Wu X.; Ramirez C. M.; Schrier A. J.; Murray D.; Shimizu A.; Ryckbosch S. M.; Near K. E.; Chun T.-W.; Wender P. A.; Zack J. A. In vivo activation of latent HIV with a synthetic bryostatin analog effects both latent cell“ kick” and“ kill” in strategy for virus eradication. PLoS Pathog. 2017, 13, e1006575. 10.1371/journal.ppat.1006575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biberacher V.; Decker T.; Oelsner M.; Wagner M.; Bogner C.; Schmidt B.; Kreitman R. J.; Peschel C.; Pastan I.; Meyer zum Buschenfelde C.; Ringshausen I. The cytotoxicity of anti-CD22 immunotoxin is enhanced by bryostatin 1 in B-cell lymphomas through CD22 upregulation and PKC-βII depletion. Haematologica 2012, 97, 771–779. 10.3324/haematol.2011.049155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso S. I.; Callender J. A.; Hooli B.; Antal C. E.; Mullin K.; Sherman M. A.; Lesne S. E.; Leitges M.; Newton A. C.; Tanzi R. E.; Malinow R. Gain-of-function mutations in protein kinase Cα (PKCα) may promote synaptic defects in Alzheimer’s disease. Sci. Signaling 2016, 9, ra47. 10.1126/scisignal.aaf6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton A. C.; Brognard J. Reversing the paradigm: protein kinase C as a tumor suppressor. Trends Pharmacol. Sci. 2017, 38, 438–447. 10.1016/j.tips.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit G. R.; Day J. F.; Hartwell J. L.; Wood H. B. Antineoplastic components of marine animals. Nature 1970, 227, 962–963. 10.1038/227962a0. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Herald C. L.; Doubek D. L.; Herald D. L.; Arnold E.; Clardy J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. 10.1021/ja00388a092. [DOI] [Google Scholar]
- Wender P. A.; Hardman C. T.; Ho S.; Jeffreys M. S.; Quiroz R. V.; Ryckbosch S. M.; Shimizu A. J.; Sloane J. L.; Stevens M. C.; Maclaren J. K. Scalable synthesis of bryostatin 1 and analogs, adjuvant leads against latent HIV. Science 2017, 358, 218–223. 10.1126/science.aan7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaufelberger D. E.; Koleck M. P.; Beutler J. A.; Vatakis A. M.; Alvarado A. B.; Andrews P.; Marzo L. V.; Muschik G. M.; Roach J.; Ross J. T.; Lebherz W. B.; Reeves M. P.; Eberwein R. M.; Rodgers L. L.; Testerman R. P.; Snader K. M.; Forenza S. The large-scale isolation of bryostatin 1 from Bugula neritina following current good manufacturing practices. J. Nat. Prod. 1991, 54, 1265–1270. 10.1021/np50077a004. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Quiroz R. V.; Stevens M. C. Function through synthesis-informed design. Acc. Chem. Res. 2015, 48, 752–760. 10.1021/acs.accounts.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; De Brabander J.; Harran P. G.; Jimenez J. M.; Koehler M. F. T.; Lippa B.; Park C. M.; Shiozaki M. Synthesis of the first members of a new class of biologically active bryostatin analogues. J. Am. Chem. Soc. 1998, 120, 4534–4535. 10.1021/ja9727631. [DOI] [Google Scholar]
- Wender P. A.; Baryza J. L.; Brenner S. E.; DeChristopher B. A.; Loy B. A.; Schrier A. J.; Verma V. A. Design, synthesis, and evaluation of potent bryostatin analogs that modulate PKC translocation selectivity. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6721–6726. 10.1073/pnas.1015270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChristopher B. A.; Loy B. A.; Marsden M. D.; Schrier A. J.; Zack J. A.; Wender P. A. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat. Chem. 2012, 4, 705–710. 10.1038/nchem.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Donnelly A. C.; Loy B. A.; Near K. E.; Staveness D.. Rethinking the role of natural products: function-oriented synthesis, bryostatin, and bryologs. In Natural Products in Medicinal Chemistry; Hanessian S., Ed.; Wiley-VCH: Weinheim, 2014; pp 473–544. [Google Scholar]
- Jeffrey A. M.; Liskamp R. M. J. Computer-assisted molecular modeling of tumor promoters: rationale for the activity of phorbol esters, teleocidin B, and aplysiatoxin. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 241–245. 10.1073/pnas.83.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Koehler K. F.; Sharkey N. A.; Dell'Aquila M. L.; Blumberg P. M. Analysis of the phorbol ester pharmacophore on protein kinase C as a guide to the rational design of new classes of analogs. Proc. Natl. Acad. Sci. U. S. A. 1986, 83, 4214–4218. 10.1073/pnas.83.12.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Cribbs C. M.; Koehler K. F.; Sharkey N. A.; Herald C. L.; Kamano Y.; Pettit G. R.; Blumberg P. M. Modeling of the bryostatins to the phorbol ester pharmacophore on protein kinase C. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 7197–7201. 10.1073/pnas.85.19.7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. N.; Ziemba B. P.; Falke J. J.; Voth G. A. Interactions of protein kinase C-α C1A and C1B domains with membranes: a combined computational and experimental study. J. Am. Chem. Soc. 2014, 136, 11757–11766. 10.1021/ja505369r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangsunan P.; Tateing S.; Hannongbua S.; Suree N. Structural insights into the interactions of phorbol ester and bryostatin complexed with protein kinase C: a comparative molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2016, 34, 1561–1575. 10.1080/07391102.2015.1084479. [DOI] [PubMed] [Google Scholar]
- Ryckbosch S. M.; Wender P. A.; Pande V. S. Molecular dynamics simulations reveal ligand-controlled positioning of a peripheral protein complex in membranes. Nat. Commun. 2017, 8, 6. 10.1038/s41467-016-0015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullion T.; Schaefer J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989, 81, 196–200. 10.1016/0022-2364(89)90280-1. [DOI] [PubMed] [Google Scholar]
- Gullion T.; Schaefer J. Detection of weak heteronuclear dipolar coupling by rotational-echo double-resonance nuclear magnetic resonance. Adv. Magn. Opt. Reson. 1989, 13, 57–83. 10.1016/B978-0-12-025513-9.50009-4. [DOI] [Google Scholar]
- Matsuoka S.; Inoue M. Application of REDOR NMR in natural product chemistry. Chem. Commun. 2009, 5664–5675. 10.1039/b910230b. [DOI] [PubMed] [Google Scholar]
- Toke O.; Cegelski L.. REDOR applications in biology: an overview. In Solid State NMR Studies of Biopolymers; McDermott A. E., Polenova T., Eds.; John Wiley & Sons: Chichester, 2010; pp 473–490. [Google Scholar]
- Cegelski L. REDOR NMR for drug discovery. Bioorg. Med. Chem. Lett. 2013, 23, 5767–5775. 10.1016/j.bmcl.2013.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller K. T.; Jarvie T. P.; Aurentz D. J.; Roberts B. W. The REDOR transform: direct calculation of internuclear couplings from dipolar-dephasing NMR data. Chem. Phys. Lett. 1995, 242, 535–542. 10.1016/0009-2614(95)00773-W. [DOI] [Google Scholar]
- Loy B. A.; Lesser A. B.; Staveness D.; Billingsley K. L.; Cegelski L.; Wender P. A. Toward a biorelevant structure of protein kinase C bound modulators: design, synthesis, and evaluation of labeled bryostatin analogues for analysis with rotational echo double resonance NMR spectroscopy. J. Am. Chem. Soc. 2015, 137, 3678–3685. 10.1021/jacs.5b00886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. B.; Wang X. Y.; Liu L. P.; Qin G. W.; Kang T. G. Tigliane diterpenoids from the Euphorbiaceae and Thymelaeaceae families. Chem. Rev. 2015, 115, 2975–3011. 10.1021/cr200397n. [DOI] [PubMed] [Google Scholar]
- Irie K.; Oie K.; Nakahara A.; Yanai Y.; Ohigashi H.; Wender P. A.; Fukuda H.; Konishi H.; Kikkawa U. Molecular basis for protein kinase C isozyme-selective binding: the synthesis, folding, and phorbol ester binding of the cysteine-rich domains of all protein kinase C isozymes. J. Am. Chem. Soc. 1998, 120, 9159–9167. 10.1021/ja981087f. [DOI] [Google Scholar]
- Hommel U.; Zurini M.; Luyten M. Solution structure of a cysteine rich domain of rat protein kinase C. Nat. Struct. Mol. Biol. 1994, 1, 383–387. 10.1038/nsb0694-383. [DOI] [PubMed] [Google Scholar]
- Ichikawa S.; Hatanaka H.; Takeuchi Y.; Ohno S.; Inagaki F. Solution structure of cysteine-rich domain of protein kinase Cα. J. Biochem. 1995, 117, 566–574. 10.1093/oxfordjournals.jbchem.a124745. [DOI] [PubMed] [Google Scholar]
- Zhang G. G.; Kazanietz M. G.; Blumberg P. M.; Hurley J. H. Crystal structure of the Cys2 activator-binding domain of protein kinase Cδ in complex with phorbol ester. Cell 1995, 81, 917–924. 10.1016/0092-8674(95)90011-X. [DOI] [PubMed] [Google Scholar]
- Xu R. X.; Pawelczyk T.; Xia T. H.; Brown S. C. NMR structure of a protein kinase C-γ phorbol-binding domain and study of protein-lipid micelle interactions. Biochemistry 1997, 36, 10709–10717. 10.1021/bi970833a. [DOI] [PubMed] [Google Scholar]
- Leonard T. A.; Rozycki B.; Saidi L. F.; Hummer G.; Hurley J. H. Crystal structure and allosteric activation of protein kinase C βII. Cell 2011, 144, 55–66. 10.1016/j.cell.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. D.; Morgan B.; Massi F.; Igumenova T. I. Probing the determinants of diacylglycerol binding affinity in the C1b domain of protein kinase Cα. J. Mol. Biol. 2011, 408, 949–970. 10.1016/j.jmb.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugasundararaj S.; Das J.; Sandberg W. S.; Zhou X. J.; Wang D.; Messing R. O.; Bruzik K. S.; Stehle T.; Miller K. W. Structural and functional characterization of an anesthetic binding site in the second cysteine-rich domain of protein kinase Cδ. Biophys. J. 2012, 103, 2331–2340. 10.1016/j.bpj.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman G. M.; Shanker S.; Lewin N. E.; Kedei N.; Hill C. S.; Prasad B. V. V.; Blumberg P. M.; Das J. Identification of the activator-binding residues in the second cysteine-rich regulatory domain of protein kinase Cθ (PKCθ). Biochem. J. 2013, 451, 33–44. 10.1042/BJ20121307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J.; Rahman G. M. C1 domains: structure and ligand-binding properties. Chem. Rev. 2014, 114, 12108–12131. 10.1021/cr300481j. [DOI] [PubMed] [Google Scholar]
- Igumenova T. I. Dynamics and membrane interactions of protein kinase C. Biochemistry 2015, 54, 4953–4968. 10.1021/acs.biochem.5b00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucic I.; Truebestein L.; Leonard T. K. Novel features of DAG-activated PKC isozymes reveal a conserved 3-D architecture. J. Mol. Biol. 2016, 428, 121–141. 10.1016/j.jmb.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Stewart M. D.; Igumenova T. I. Toggling of diacylglycerol affinity correlates with conformational plasticity in C1 domains. Biochemistry 2017, 56, 2637–2640. 10.1021/acs.biochem.7b00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Irie K.; Miller B. L. Identification, activity, and structural studies of peptides incorporating the phorbol ester-binding domain of protein kinase C. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 239–243. 10.1073/pnas.92.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irie K.; Nakahara A.; Nakagawa Y.; Ohigashi H.; Shindo M.; Fukuda H.; Konishi H.; Kikkawa U.; Kashiwagi K.; Saito N. Establishment of a binding assay for protein kinase C isozymes using synthetic C1 peptides and development of new medicinal leads with protein kinase C isozyme and C1 domain selectivity. Pharmacol. Ther. 2002, 93, 271–281. 10.1016/S0163-7258(02)00196-1. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Kowalewski T.; Schaefer J. Local packing in glassy polycarbonate by carbon-deuterium REDOR NMR. Macromolecules 1993, 26, 1729–1733. 10.1021/ma00059a036. [DOI] [Google Scholar]
- Krauter G.; Von Der Lieth C.-W.; Schmidt R.; Hecker E. Structure/activity relationships of polyfunctional diterpenes of the tigliane type. A pharmacophore model for protein-kinase-C activators based on structure/activity studies and molecular modeling of the tumor promoters 12-O-tetradecanoylphorbol 13-acetate and 3-O-tetradecanoylingenol. Eur. J. Biochem. 1996, 242, 417–427. 10.1111/j.1432-1033.1996.0417r.x. [DOI] [PubMed] [Google Scholar]
- Yang H.Solid-state NMR study of the tertiary structure of the peptidoglycan of Enterococcus faecalis and the structures of phorbol diacetate and bryostatin bound to protein kinase Cδ C1b domain. Ph.D. Dissertation, Department of Chemistry, Washington University, St. Louis, MO, 2015. [Google Scholar]
- Studelska D. R.; McDowell L. M.; Adler M.; O’Connor R. D.; Mehta A. K.; Guilford W. J.; Dallas J. L.; Arnaiz D.; Light D. R.; Schaefer J. Conformation of a bound inhibitor of blood coagulant factor Xa. Biochemistry 2003, 42, 7942–7949. 10.1021/bi027369g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.