

Abstract
Background
Single-inhaler fluticasone furoate/umeclidinium/vilanterol (FF/UMEC/VI) 100/62.5/25 μg has been shown to improve lung function and health status, and reduce exacerbations, versus budesonide/formoterol in patients with chronic obstructive pulmonary disease (COPD). We evaluated the non-inferiority of single-inhaler FF/UMEC/VI versus FF/VI + UMEC using two inhalers.
Methods
Eligible patients with COPD (aged ≥40 years; ≥1 moderate/severe exacerbation in the 12 months before screening) were randomized (1:1; stratified by the number of long-acting bronchodilators [0, 1 or 2] per day during run-in) to receive 24-week FF/UMEC/VI 100/62.5/25 μg and placebo or FF/VI 100/25 μg + UMEC 62.5 μg; all treatments/placebo were delivered using the ELLIPTA inhaler once-daily in the morning. Primary endpoint: change from baseline in trough forced expiratory volume in 1 s (FEV1) at Week 24. The non-inferiority margin for the lower 95% confidence limit was set at − 50 mL.
Results
A total of 1055 patients (844 [80%] of whom were enrolled on combination maintenance therapy) were randomized to receive FF/UMEC/VI (n = 527) or FF/VI + UMEC (n = 528). Mean change from baseline in trough FEV1 at Week 24 was 113 mL (95% CI 91, 135) for FF/UMEC/VI and 95 mL (95% CI 72, 117) for FF/VI + UMEC; the between-treatment difference of 18 mL (95% CI -13, 50) confirmed FF/UMEC/VI’s was considered non-inferior to FF/VI + UMEC. At Week 24, the proportion of responders based on St George’s Respiratory Questionnaire Total score was 50% (FF/UMEC/VI) and 51% (FF/VI + UMEC); the proportion of responders based on the Transitional Dyspnea Index focal score was similar (56% both groups). A similar proportion of patients experienced a moderate/severe exacerbation in the FF/UMEC/VI (24%) and FF/VI + UMEC (27%) groups; the hazard ratio for time to first moderate/severe exacerbation with FF/UMEC/VI versus FF/VI + UMEC was 0.87 (95% CI 0.68, 1.12). The incidence of adverse events was comparable in both groups (48%); the incidence of serious adverse events was 10% (FF/UMEC/VI) and 11% (FF/VI + UMEC).
Conclusions
Single-inhaler triple therapy (FF/UMEC/VI) is non-inferior to two inhalers (FF/VI + UMEC) on trough FEV1 change from baseline at 24 weeks. Results were similar on all other measures of efficacy, health-related quality of life, and safety.
Trial registration
GSK study CTT200812; ClinicalTrials.gov NCT02729051 (submitted 31 March 2016).
Keywords: COPD, Exacerbations, FEV1, Lung function, Fluticasone furoate/umeclidinium/vilanterol, Randomized controlled trial, Single-inhaler triple therapy
Background
The Global Initiative for Chronic Obstructive Lung Disease (GOLD) strategy document recommends escalating to combination triple therapy with a long-acting β2-agonist (LABA), a long-acting muscarinic antagonist (LAMA) and an inhaled corticosteroid (ICS) for patients with advanced chronic obstructive pulmonary disease (COPD) and persistent symptoms (GOLD Group D) who experience further symptoms or exacerbations on dual LABA/LAMA or LABA/ICS therapy [1]. Although triple therapy for COPD using multiple inhalers is common in current clinical practice [2, 3], the comparative benefits of COPD treatment regimens using single or multiple inhalers are not well understood.
Triple therapy with a LAMA plus ICS/LABA administered using multiple inhalers has been shown to improve forced expiratory volume in 1 s (FEV1) and health status, and reduce exacerbations and rescue medication use, in patients with COPD compared with ICS plus LABA dual therapy or LAMA monotherapy [4–9]. Several recent large randomized controlled trials have also assessed the efficacy and safety of triple ICS/LABA/LAMA therapy using a single fixed-dose combination inhaler for patients with COPD at increased exacerbation risk [10–12].
The FULFIL study demonstrated improvements in trough FEV1, health status, and reductions in moderate/severe exacerbation rate, with once-daily, single-inhaler fluticasone furoate/umeclidinium/vilanterol (FF/UMEC/VI) versus twice-daily budesonide/formoterol (FOR) [10]. Similarly, the TRILOGY study showed improvements in lung function and exacerbation frequency with a twice-daily, single-inhaler ICS/LABA/LAMA combination of beclomethasone dipropionate (BDP)/FOR/glycopyrronium bromide (GB) compared with BDP/FOR alone [11]. Furthermore, results from the TRINITY study confirmed that the twice-daily, single-inhaler BDP/FOR/GB combination was non-inferior to twice-daily BDP/FOR plus tiotropium using multiple inhalers on change from baseline in pre-dose FEV1 [12].
Given that single-inhaler triple therapy is soon expected to be widely available, the current study was specifically designed to demonstrate the non-inferiority of the only currently available once-daily, single-inhaler triple therapy (FF/UMEC/VI) to an alternative once-daily triple therapy regimen using two inhalers (FF/VI + UMEC) on trough FEV1 after 24 weeks of treatment. To our knowledge, this is the first study to specifically evaluate the same individual component molecules administered using either a single inhaler or multiple inhalers.
Methods
Study design
This was a phase III, 24-week, randomized, double-blind, parallel group, multicenter non-inferiority study (GSK study CTT200812; ClinicalTrials.gov identifier NCT02729051) that assessed the efficacy of once-daily FF/UMEC/VI 100 μg/62.5 μg/25 μg using a single ELLIPTA inhaler versus once-daily FF/VI 100 μg/25 μg plus UMEC 62.5 μg using two ELLIPTA inhalers (Fig. 1). Prior to the beginning of the 24-week treatment period, there was a 2-week run-in period, during which patients continued their existing COPD medications. At randomization following the run-in period, all existing COPD medications were discontinued and patients started their assigned study treatment, with short-acting albuterol/salbutamol provided as rescue medication throughout the study. Study clinic visits occurred at pre-screening (visit 0), screening (visit 1), randomization (Week 0, visit 2), Week 4 (visit 3), Week 12 (visit 4), and Week 24 (visit 5). A safety follow-up telephone contact or clinic visit was conducted a week after study completion, or in the event of an early withdrawal.
Fig. 1.
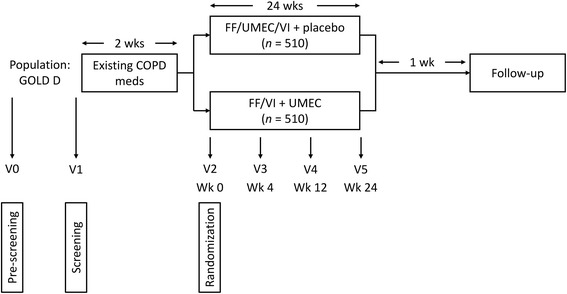
Study design
Patients
Patients aged ≥40 years with COPD and who were current/former smokers with a ≥ 10-pack-year smoking history were eligible for enrollment. Other key inclusion criteria included: COPD Assessment Test™ (CAT) score ≥ 10 [13, 14]; a post-albuterol/salbutamol FEV1/forced vital capacity ratio < 0.70; and a post-bronchodilator FEV1 < 50% of predicted and ≥1 moderate/severe exacerbation in the previous 12 months or a post-bronchodilator FEV1 ≥ 50% to < 80% of predicted and ≥2 moderate exacerbations or ≥1 severe exacerbation requiring hospitalization in the previous 12 months.
Exclusion criteria included: a current diagnosis of asthma (patients with a prior history of asthma were eligible if they had a current diagnosis of COPD that was the primary cause of their respiratory symptoms); α1-antitrypsin deficiency; active tuberculosis; other respiratory disorders that were the primary cause of respiratory symptoms; lung resection surgery in the previous 12 months; risk factors for pneumonia (including immunosuppression and neurological disorders affecting control of the upper airway [e.g. Parkinson’s disease or myasthenia gravis]; pneumonia and/or moderate/severe exacerbation that had not resolved at least 14 days prior to screening; respiratory infections; abnormal findings on chest X-ray; clinically significant comorbidities; unstable liver or cardiac disease; and cancer. Patients with a high risk for pneumonia (e.g. very low body mass index, severely malnourished, or very low FEV1) were only to be included at the discretion of the investigator.
All patients provided written, informed consent prior to enrollment.
Treatments
Eligible patients were randomized (1:1) to receive 24 weeks of FF/UMEC/VI 100 μg/62.5 μg/25 μg in a single inhaler and placebo (second inhaler) or FF/VI 100 μg/25 μg and UMEC 62.5 μg, in separate inhalers; all treatments/placebo were delivered using the ELLIPTA inhaler once daily in the morning. Randomization was stratified by the number of long-acting bronchodilators (0, 1, or 2) per day during the run-in.
Study assessments
The primary efficacy endpoint was change from baseline in trough FEV1 at Week 24. Secondary efficacy endpoints included: proportion of responders based on the St George’s Respiratory Questionnaire (SGRQ) Total score at Week 24; change from baseline in SGRQ Total score at Week 24; proportion of responders based on Transitional Dyspnea Index (TDI) focal score at Week 24; TDI focal score at Week 24; and time to first moderate/severe exacerbation. Safety endpoints included the incidence of adverse events (AEs), serious AEs (SAEs), and AEs of special interest (AESIs).
Spirometry was performed at screening and pre-dose at each scheduled study visit during the treatment period using standardized equipment according to American Thoracic Society–European Respiratory Society guidelines [15]. The SGRQ for COPD patients [16] was completed by patients at randomization and Weeks 12 and 24. The Baseline Dyspnea Index was measured at randomization and the TDI was measured at Weeks 12 and 24; these assessments were completed electronically by patients using self-administered computerized versions. Potential COPD exacerbations were identified based on patient-reported symptoms in an eDiary and confirmed by follow-up with the investigator. Exacerbations were defined as worsening of COPD symptoms that were mild (self-managed by the patient; corticosteroids or antibiotics were not required), moderate (required oral/systemic corticosteroids and/or antibiotics), or severe (required hospitalization). The investigators and their study staff were responsible for detecting, documenting, and reporting AEs at each study visit. The CAT was completed at screening using the eDiary, before any other assessments, to assess eligibility.
Statistical analyses
Sample size calculations used a one-sided 2.5% significance level and an estimate of residual standard deviation (SD) for trough FEV1 at Week 24 of 220 mL (the SD estimate was based on previous phase III studies in patients with COPD). A study with 816 evaluable patients for the primary analysis would have 90% power to determine non-inferiority of FF/UMEC/VI versus FF/VI + UMEC based on trough FEV1 at Week 24, when the margin of non-inferiority is 50 mL and the true mean treatment difference is assumed to be 0 mL. It was estimated that ~ 20% of patients who were randomized would either discontinue study treatment or be excluded from the modified per-protocol (mPP) population at Week 24, and so ~ 1020 patients were planned for randomization.
The intent-to-treat (ITT) population included all randomized patients, except those randomized in error. The mPP population included all patients in the ITT population who did not have a protocol deviation affecting efficacy. Data following a severe/moderate COPD exacerbation or pneumonia were excluded from the analysis due to the potential impact of the event or the medications used to treat it. Patients with partial protocol deviations considered to impact efficacy were included in the mPP population but had their data excluded from analyses from the time of deviation onwards.
The primary efficacy endpoint was analyzed for the mPP population using a mixed model repeated measures (MMRM) analysis, including trough FEV1 at Weeks 4, 12, and 24. The model included covariates of stratum (number of long-acting bronchodilators per day during the run-in [0/1 or 2]), baseline FEV1, visit, center group, treatment, visit by baseline, and visit by treatment interaction. The non-inferiority margin was set at 50 mL (half the minimal clinically important difference [MCID] for trough FEV1 in COPD [17]. If the lower bound of the two-sided 95% confidence interval (CI) around the FF/UMEC/VI versus FF/VI + UMEC treatment difference was above − 50 mL, then FF/UMEC/VI was considered non-inferior to FF/VI + UMEC. The MMRM analysis was repeated for the ITT population. All other endpoints were analyzed for the ITT population only.
The proportion of responders based on the SGRQ Total score at Weeks 12 and 24 was analyzed using a generalized linear mixed model, including covariates of baseline SGRQ Total score, treatment group, number of long-acting bronchodilators per day during the run-in (0/1 or 2), geographic region, visit, visit by baseline interaction, and visit by treatment interaction. The number and proportion of responders and non-responders for each treatment at Weeks 12 and 24 was calculated and an odds ratio (OR) for the comparison between FF/UMEC/VI and FF/VI + UMEC with associated 95% CI was provided. The proportion of responders based on the TDI focal score was analyzed in the same way for proportion of responders based on SGRQ Total score. Change from baseline in SGRQ Total score and change from baseline in TDI total score at Weeks 12 and 24 were analyzed separately as described for the primary analysis.
A Cox proportional hazards model was used to compare the time to first moderate/severe COPD exacerbation during 24 weeks of treatment with either FF/UMEC/VI or FF/VI + UMEC. This model used covariates of treatment group, gender, exacerbation history (0, 1, or 2 moderate/severe exacerbations within 12 months of screening), smoking status at screening, number of long-acting bronchodilators per day during the run-in (0/1 or ≥2), geographic region, and baseline percent predicted FEV1. A Kaplan–Meier analysis was performed to produce a figure showing Kaplan–Meier survivor functions of the proportion of patients with a first moderate/severe exacerbation over time for each treatment group.
The number and proportion of patients experiencing at least one AE of any type, AEs within each body system, and AEs within each Medical Dictionary for Regulatory Activities preferred term were recorded for each treatment group. Separate summaries were provided for all AEs, drug-related AEs, fatal AEs, non-fatal SAEs, AESIs, and AEs leading to withdrawal. SAEs and deaths were documented in case-narrative format.
Results
Patients
A total of 1311 patients were enrolled, of whom 1055 were randomized to receive study treatment (ITT population; FF/UMEC/VI, n = 527; FF/VI + UMEC, n = 528); 956 patients were included in the mPP population (FF/UMEC/VI, n = 478; FF/VI + UMEC, n = 478). In the ITT population, 94% of patients completed the study in each treatment group. Patient demographics and clinical characteristics in the ITT population were generally well balanced between the treatment groups, with no significant differences in terms of disease severity, GOLD grade or exacerbation history at baseline (Table 1); current and past medical conditions and the incidence of cardiovascular risk factors at baseline were also similar between the two treatment arms. Patient characteristics in the mPP population were similar (not shown).
Table 1.
Patient demographics and clinical characteristics (ITT population)
| Characteristic | FF/UMEC/VI 100/62.5/25 μg | FF/VI 100/25 μg + UMEC 62.5 μg | Total |
|---|---|---|---|
| (N = 527) | (N = 528) | (N = 1055) | |
| Age (years), mean (SD) | 66.7 (8.5) | 65.9 (8.8) | 66.3 (8.6) |
| Female, n (%) | 136 (26) | 134 (25) | 270 (26) |
| Current smoker at screening, n (%) | 209 (40) | 192 (36) | 401 (38) |
| Smoking pack-years, mean (SD) | 43.4 (23.9) | 44.2 (25.2) | 43.8 (24.6) |
| Current cardiovascular risk factors, n (%) | 379 (72) | 367 (70) | 746 (71) |
| Number of exacerbations in previous 12 months, n (%)a | |||
| 1 moderate/severe | 236 (45) | 227 (43) | 463 (44) |
| ≥ 2 moderate/severe | 291 (55) | 301 (57) | 592 (56) |
| ≥ 2 moderate or ≥1 severe | 352 (67) | 360 (68) | 712 (67) |
| History of pneumonia, n (%) | 86 (16) | 100 (19) | 186 (18) |
| Screening lung function, mean (SD) | n = 515 | n = 512 | n = 1027 |
| Post-bronchodilator FEV1, mL | 1247 (465) | 1297 (471) | 1272 (469) |
| Post-bronchodilator FVC, mL | 2879 (885) | 2896 (849) | 2887 (867) |
| Post-bronchodilator FEV1/FVC ratio | 0.440 (0.116) | 0.455 (0.119) | 0.447 (0.118) |
| Post-bronchodilator percent predicted FEV1 | 44.5 (14.5) | 45.5 (14.1) | 45.0 (14.3) |
| Percent reversibility | 9.02 (11.22) | 8.87 (10.15)b | 8.95 (10.69) |
| Number of long-acting bronchodilators per day during the run-in, n (%) | |||
| 0/1 | 225 (43) | 226 (43) | 451 (43) |
| 2 | 302 (57) | 302 (57) | 604 (57) |
| Concomitant COPD medications taken at screening, n (%) | |||
| Single-inhaler maintenance bronchodilator | 40 (8) | 42 (8) | 82 (8) |
| LAMA | 32 (6) | 35 (7) | 67 (6) |
| LABA | 8 (2) | 7 (1) | 15 (1) |
| Combination therapy | 448 (85) | 443 (84) | 891 (84) |
| ICS + LABA+LAMA | 198 (38) | 193 (37) | 391 (37) |
| ICS + LABA | 144 (27) | 137 (26) | 281 (27) |
| LABA+LAMA | 62 (12) | 76 (14) | 138 (13) |
| ICS + LABA+LAMA+ xanthine | 29 (6) | 25 (5) | 54 (5) |
| ICS + LAMA | 7 (1) | 9 (2) | 16 (2) |
| LABA+LAMA+xanthine | 8 (2) | 3 (< 1) | 11 (1) |
| COPD severity at screening | |||
| GOLD grade, n (%) | n = 515 | n = 512 | n = 1027 |
| 1 (mild) | 0 | 1 (< 1) | 1 (< 1) |
| 2 (moderate) | 174 (34) | 189 (37) | 363 (35) |
| 3 (severe) | 251 (49) | 253 (49) | 504 (49) |
| 4 (very severe) | 90 (17) | 69 (13) | 159 (15) |
| Reversible, n (%)c | n = 515 | n = 511 | n = 1026 |
| Yes | 73 (14) | 74 (14) | 147 (14) |
| GOLD grade/exacerbation history, n (%)a | n = 514 | n = 512 | n = 1026 |
| Grade 1/2 with ≥2 moderate or ≥1 severe | 173 (34) | 190 (37) | 363 (35) |
| Grade 3/4 with < 2 moderate and no severe | 171 (33) | 164 (32) | 335 (33) |
| Grade 3/4 with ≥2 moderate or ≥1 severe | 170 (33) | 158 (31) | 328 (32) |
| CAT score, mean (SD) | 19.6 (5.8) | 20.1 (6.1) | 19.9 (6.0) |
CAT COPD Assessment Test™, COPD chronic obstructive pulmonary disease, FEV1 forced expiratory volume in 1 s, FF fluticasone furoate, FVC forced vital capacity, GOLD Global Initiative for Chronic Obstructive Lung Disease, ICS inhaled corticosteroid, ITT intent-to-treat, LABA long-acting β2-agonist, LAMA long-acting muscarinic antagonist, SD standard deviation, UMEC umeclidinium, VI vilanterol
aModerate exacerbations were defined as exacerbations requiring oral/systemic corticosteroids and/or antibiotics (not involving hospitalization). Severe exacerbations were defined as exacerbations that required in-patient hospitalization
bFF/VI + UMEC, n = 511
cReversible was an increase in FEV1 of ≥12% and ≥200 mL following administration of salbutamol. Not reversible was an increase in FEV1 of < 200 or ≥200 mL increase that is < 12% of the pre-salbutamol FEV1
Efficacy
In the mPP population, the mean change from baseline in trough FEV1 at Week 24 was 113 mL (95% CI 91, 135) for FF/UMEC/VI and 95 mL (95% CI 72, 117) for FF/VI + UMEC; the between-treatment difference was 18 mL (95% CI -13, 50) (Fig. 2). As the lower bound of the 95% CI for the comparison was above the predefined non-inferiority margin (− 50 mL), FF/UMEC/VI was considered non-inferior to FF/VI + UMEC. Comparable findings were observed in the ITT population: the mean change from baseline in trough FEV1 at Week 24 was 107 mL (95% CI 87, 126) for FF/UMEC/VI and 81 mL (95% CI 61, 100) for FF/VI + UMEC; the between-treatment difference was 26 mL (95% CI -2, 53).
Fig. 2.
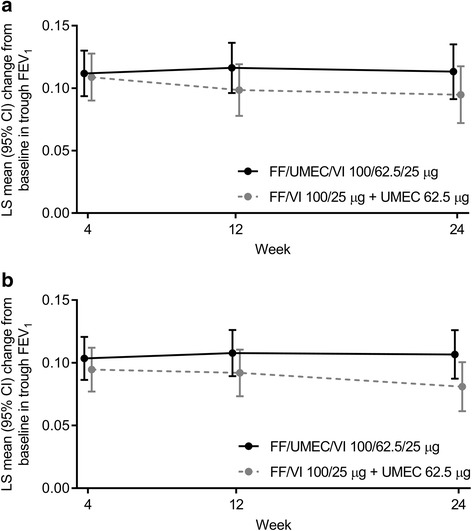
Mean change from baseline in trough FEV1 over 24 weeks in a: the mPP population and b: the ITT population
In the ITT population, the proportion of responders based on the SGRQ Total score at Week 24 was similar in the FF/UMEC/VI group (50%) and FF/VI + UMEC group (51%); the OR of response versus non-response for FF/UMEC/VI versus FF/VI + UMEC was 0.92 (95% CI 0.71, 1.20). The mean change from baseline in SGRQ Total score at Week 24 was − 5.8 (95% CI -7.0, − 4.7) for FF/UMEC/VI and − 4.9 (95% CI -6.1, − 3.8) for FF/VI + UMEC; the between-treatment difference was − 0.9 (95% CI -2.5, 0.7) (Fig. 3). The proportion of responders based on TDI focal score at Week 24 was the same for FF/UMEC/VI and FF/VI + UMEC (56% in each group; OR of response versus non-response for FF/UMEC/VI versus FF/VI + UMEC was 0.95 [95% CI 0.72, 1.25]). The mean TDI focal score at Week 24 was 2.0 (95% CI 1.8, 2.3) for FF/UMEC/VI and 1.9 (95% CI 1.6, 2.1) for FF/VI + UMEC; the between-treatment difference was 0.1 (95% CI -0.2, 0.5).
Fig. 3.
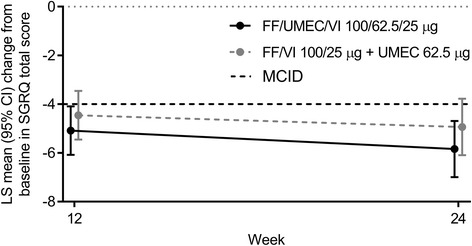
Mean change from baseline in SGRQ Total score over 24 weeks (ITT population)
A similar proportion of patients (ITT population) experienced a moderate/severe exacerbation in the FF/UMEC/VI group (24%) and FF/VI + UMEC group (27%) (Table 2). The hazard ratio for time to first on-treatment moderate/severe exacerbation with FF/UMEC/VI versus FF/VI + UMEC was 0.87 (95% CI 0.68, 1.12) (Fig. 4).
Table 2.
Summary of on-treatment COPD exacerbations (ITT population)
| F/UMEC/VI 100/62.5/25 μg | FF/VI 100/25 μg + UMEC 62.5 μg | |
|---|---|---|
| (N = 527) | (N = 528) | |
| Patients with a mild, moderate or severe exacerbation, n (%) | 134 (25) | 145 (27) |
| Mild | 8 (2) | 5 (< 1) |
| Moderate | 111 (21) | 118 (22) |
| Severe | 22 (4) | 31 (6) |
| Moderate/Severe | 129 (24) | 142 (27) |
| Number of moderate/severe exacerbations, n (%) | ||
| 0 | 398 (76) | 386 (73) |
| 1 | 105 (20) | 111 (21) |
| ≥ 2 | 24 (5) | 31 (6) |
COPD chronic obstructive pulmonary disease, FF fluticasone furoate, ITT intent-to-treat, SD standard deviation, UMEC umeclidinium, VI vilanterol
Fig. 4.
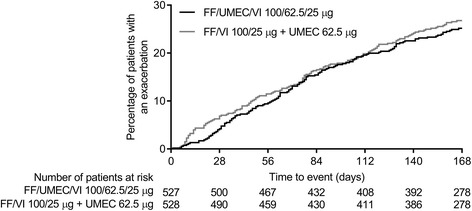
Time to first on-treatment moderate/severe exacerbation over the 24-week treatment period (ITT population)
Safety
The proportion of patients who experienced at least one AE was comparable between both treatment groups (48%); the proportion of patients who had at least one SAE was 10% in the FF/UMEC/VI group and 11% in the FF/VI + UMEC group (Table 3). The most frequent AEs were viral upper respiratory tract infection (FF/UMEC/VI, 11%; FF/VI + UMEC, 10%), headache (6% in each group), and COPD (FF/UMEC/VI, 4%; FF/VI + UMEC, 6%). The incidence of AESIs was similar between the treatment groups, including pneumonia (FF/UMEC/VI, 3%; FF/VI + UMEC, 4%) and cardiovascular events (FF/UMEC/VI, 6%; FF/VI + UMEC, 5%) (Table 3).
Table 3.
Overview of safety findings (ITT population)
| Category, n (%) | FF/UMEC/VI 100/62.5/25 μg | FF/VI 100/25 μg + UMEC 62.5 μg |
|---|---|---|
| (N = 527) | (N = 528) | |
| On-treatment AEs | 255 (48) | 253 (48) |
| On-treatment drug-related AEs | 27 (5) | 19 (4) |
| On-treatment SAEs | 52 (10) | 57 (11) |
| On-treatment non-fatal SAEs | 50 (9) | 54 (10) |
| On-treatment fatal SAEs | 4 (< 1) | 4 (< 1) |
| On-treatment AESIsa | ||
| Adrenal suppression | 1 (< 1) | 0 |
| Anticholinergic syndrome | 12 (2) | 5 (< 1) |
| Cardiovascular events | 30 (6) | 28 (5) |
| Cardiac arrhythmia | 6 (1) | 8 (2) |
| Cardiac failure | 7 (1) | 7 (1) |
| Ischemic heart disease | 6 (1) | 3 (< 1) |
| Hypertension | 10 (2) | 13 (2) |
| CNS hemorrhage/cerebrovascular conditions | 3 (< 1) | 1 (< 1) |
| Decreased bone mineral density and associated fractures | 5 (< 1) | 6 (1) |
| Hyperglycemia/new-onset diabetes mellitus | 7 (1) | 6 (1) |
| Hypersensitivity | 7 (1) | 9 (2) |
| LRTI excluding pneumonia | 16 (3) | 11 (2) |
| Local steroid effects | 12 (2) | 14 (3) |
| Ocular effects | 4 (< 1) | 5 (< 1) |
| Pneumonia | 14 (3) | 21 (4) |
| Tremor | 1 (< 1) | 0 |
AE adverse event, AESI adverse event of special interest, CNS central nervous system, FF fluticasone furoate, ITT intent-to-treat, LRTI lower respiratory tract infection, SAE serious adverse event, SD standard deviation, UMEC umeclidinium, VI vilanterol
aNo events were reported for the asthma/bronchospasm, effects on potassium, gastrointestinal obstruction, or urinary retention AESI groups
Discussion
This study aimed to evaluate an important clinical outcome in patients with COPD following treatment with the same three individual component molecules administered using either a single inhaler or two inhalers. While this appears to be scientifically self-evident, this was the first study to specifically demonstrate non-inferiority of a single-inhaler triple pharmacologic regimen to the same treatments delivered using multiple inhalers.
This study showed that single-inhaler FF/UMEC/VI 100 μg/62.5 μg/25 μg is non-inferior to FF/VI 100 μg/25 μg plus UMEC 62.5 μg using two inhalers based on change from baseline in trough FEV1 at Week 24. The non-inferiority margin for the lower 95% confidence limit was set at − 50 mL, which is half the MCID for trough FEV1 in COPD [17]. The proportions of responders based on SGRQ Total score and TDI focal score, and the time to first on-treatment moderate/severe exacerbation, were similar between the treatment groups. As expected, the incidence of AEs, SAEs, and AESIs was comparable for FF/UMEC/VI and FF/VI + UMEC; the incidence of pneumonia AESI was 3% and 4% for FF/UMEC/VI and FF/VI + UMEC, respectively. These results demonstrate that the efficacy and safety of single-inhaler FF/UMEC/VI are similar to the same treatments delivered using two inhalers in patients with COPD. Our findings are supported by previous research, which showed that the systemic exposure to FF, UMEC, and VI following administration as a single-inhaler combination is similar to that observed with the dual therapies FF/VI and UMEC/VI [18].
The mean change from baseline in trough FEV1 at Week 24 with FF/VI/UMEC in the ITT population in the current study (107 mL) is lower than the change from baseline reported with FF/UMEC/VI in the ITT population in the FULFIL study at 24 weeks (142 mL) [10]. This likely reflects the fact that 80% of the enrolled population in the current study used dual, triple, or quadruple combination therapies at baseline. Nevertheless, this reduced level of improvement in lung function was associated with a clinically meaningful improvement from baseline in SGRQ Total score at Week 24 of − 5.8 units, which is consistent with the − 6.6-unit change reported with FF/UMEC/VI in the FULFIL study [10]. It should also be noted that the mean improvement from baseline in trough FEV1 at Week 24 with FF/VI/UMEC in the ITT population in the current study was 26 mL (95% CI -2, 53) greater than that observed with FF/VI + UMEC using two inhalers. This indicates some potential for greater consistency in the bronchodilator effect for the single-inhaler combination across all patients, compared with the two-inhaler regimen.
Our findings are also in line with a previous 52-week study comparing twice-daily, single-inhaler BDP/FOR/GB triple therapy with twice-daily BDP/FOR plus tiotropium using multiple inhalers, which showed non-inferiority for the single-inhaler formulation based on change from baseline in pre-dose FEV1 (between-treatment difference: − 3 mL [95% CI -33, 27]) [12]. The incidence of AEs and SAEs with FF/UMEC/VI observed in this study (48% and 10%, respectively) was higher than the incidences observed with FF/UMEC/VI in the FULFIL trial (39% and 5%, respectively). The current study was designed to recruit a population with higher disease burden and exacerbation risk, compared to the population recruited to the FULFIL trial [10], which likely accounts for the differences seen in adverse event reporting between the two studies. These differences are therefore not considered to be clinically relevant. The incidence of pneumonia AESI in the FF/UMEC/VI group in the current study was relatively low (3%) despite 16% of patients enrolled in this group having a history of pneumonia.
A once-daily, single-inhaler treatment regimen offers a simplified dosing option that may provide a number of benefits to patients with COPD, such as reducing the number of obligatory co-pays in markets where patients are required to subsidize the cost of their medicines. A single-inhaler regimen may also help to ensure that the prescribed combination is delivered consistently and reduce the risk of inhaler errors [19–21]. This may improve patient adherence and outcomes, and reduce associated healthcare costs. However, in the current study, patients in both groups received their assigned study treatment using double-dummy blind inhalers, and so we were unable to directly assess the impact on adherence of a simplified single-inhaler triple therapy regimen versus the same treatments delivered using two inhalers. Nevertheless, as numerical treatment differences in favor of FF/VI/UMEC versus UMEC+FF/VI were consistently observed in the ITT population for both mean change from baseline in FEV1 and SGRQ total score, it is possible that a more pragmatic, real-world efficacy study might demonstrate such efficacy benefits.
Our results are specific to FF/UMEC/VI administered using the ELLIPTA inhaler, so similar clinical findings may not be observed when different molecules or inhaler devices are combined. Another study limitation was the lack of a control group receiving dual ICS/LABA or LAMA/LABA therapy, although a dual therapy control group was included in the FULFIL and forthcoming IMPACT trials [10, 22]. A potential study strength was that 80% of enrolled patients continued their existing dual, triple or quadruple combination therapies during the 2-week run-in period, and 67% continued to remain at high risk of exacerbations. This meant that the study population more closely resembled a real-world COPD population who may benefit most from the simplicity afforded by a once-daily, single-inhaler triple therapy regimen.
Conclusions
This study showed that single-inhaler FF/UMEC/VI 100 μg/62.5 μg/25 μg was non-inferior to FF/VI 100 μg/25 μg plus UMEC 62.5 μg based on change from baseline in trough FEV1 at Week 24 in patients with advanced COPD. Our findings confirm that single-inhaler triple therapy with FF/UMEC/VI offers similar efficacy, health-related quality of life, and safety benefits as the same triple therapy administered using two inhalers.
Acknowledgments
We would like to thank the patients and their families who participated in this study, and the study investigators and their staff. We also thank Helen Barnacle (senior scientist for this study), Niki Day (GSK Pharmacovigilance), and Erik Steinberg (GSK Data Quality Leader).
Medical writing support in the form of development of the draft outline and manuscript drafts in consultation with the authors, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, referencing and graphic services was provided by Thomas Burton, BMBS, of Gardiner-Caldwell Communications, Macclesfield, UK and was funded by GSK.
Funding
This study was funded by GSK (GSK study CTT200812; ClinicalTrials.gov identifier NCT02729051). ELLIPTA is owned by or licensed to the GSK group of companies.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AE
Adverse event
- AESI
Adverse event of special interest
- BDP
Beclomethasone dipropionate
- CAT
COPD Assessment Test™
- CI
Confidence interval
- CNS
Central nervous system
- COPD
Chronic obstructive pulmonary disease
- FEV1
Forced expiratory volume in 1 s
- FF
Fluticasone furoate
- FOR
Formoterol
- FVC
Forced vital capacity
- GB
Glycopyrronium bromide
- GOLD
Global initiative for chronic obstructive lung disease
- ICS
Inhaled corticosteroid
- ITT
Intent-to-treat
- LABA
Long-acting β2-agonist
- LAMA
Long-acting muscarinic antagonist
- LRTI
Lower respiratory tract infection
- MCID
Minimal clinically important difference
- MMRM
Mixed model repeated measures
- mPP
Modified per-protocol
- OR
Odds ratio
- SAE
Serious adverse event
- SD
Standard deviation
- SGRQ
St George’s respiratory questionnaire
- TDI
Transitional dyspnea index
- UMEC
Umeclidinium
- VI
Vilanterol
Authors’ contributions
Literature search: DAL, NB. Study design: DAL, NB, RB, CQZ. Data collection: PRB. Data analysis: DAL, NB, RB, ASI, CQZ. Data interpretation: all authors. Writing/reviewing of the manuscript: all authors. Final approval of the manuscript: all authors.
Ethics approval and consent to participate
The protocol was approved by applicable institutional review boards/ethics committees at each participating site, and was conducted in accordance with applicable regulatory requirements, ICH Good Clinical Practice guidelines, and the guiding principles of the Declaration of Helsinki. All patients provided written, informed consent prior to enrollment.
Consent for publication
Not applicable.
Competing interests
RB, NB, ASI, C-QZ, and DAL are employees of GSK and hold stocks/shares in the company. ASI is also an unpaid faculty member at McMaster University, Canada. PRB has no conflicts of interest to disclose.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Peter R. Bremner, Email: peter@lungs.com.au
Ruby Birk, Email: ruby.k.birk@gsk.com.
Noushin Brealey, Email: noushin.s.brealey@gsk.com.
Afisi S. Ismaila, Email: afisi.s.ismaila@gsk.com
Chang-Qing Zhu, Email: chang-qing.2.zhu@gsk.com.
David A. Lipson, Phone: (+1) 610-270-7166, Email: david.a.lipson@gsk.com
References
- 1.Global Initiative for Chronic Obstructive Lung Disease . Global strategy for the diagnosis, management and prevention of COPD. 2017. [Google Scholar]
- 2.Wurst KE, Punekar YS, Shukla A. Treatment evolution after COPD diagnosis in the UK primary care setting. PLoS One. 2014;9:e105296. doi: 10.1371/journal.pone.0105296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simeone JC, Luthra R, Kaila S, Pan X, Bhagnani TD, Liu J, et al. Initiation of triple therapy maintenance treatment among patients with COPD in the US. Int J Chron Obstruct Pulmon Dis. 2017;12:73–83. doi: 10.2147/COPD.S122013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aaron SD, Vandemheen KL, Fergusson D, Maltais F, Bourbeau J, Goldstein R, et al. Tiotropium in combination with placebo, salmeterol, or fluticasone–salmeterol for treatment of chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med. 2007;146:545–555. doi: 10.7326/0003-4819-146-8-200704170-00152. [DOI] [PubMed] [Google Scholar]
- 5.Welte T, Miravitlles M, Hernandez P, Eriksson G, Peterson S, Polanowski T, et al. Efficacy and tolerability of budesonide/formoterol added to tiotropium in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180:741–750. doi: 10.1164/rccm.200904-0492OC. [DOI] [PubMed] [Google Scholar]
- 6.Hanania HA, Crater GD, Morris AN, Emmett AH, O’Dell DM, Niewoehner DE. Benefits of adding fluticasone propionate/salmeterol to tiotropium in moderate to severe COPD. Resp Med. 2012;106:91–101. doi: 10.1016/j.rmed.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 7.Jung KS, Park HY, Park SY, Kim SK, Kim YK, Shim JJ, et al. Korea chronic obstructive pulmonary disease study group. Comparison of tiotropium plus fluticasone propionate/salmeterol with tiotropium in COPD: a randomized controlled study. Resp Med. 2012;106:382–389. doi: 10.1016/j.rmed.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Siler TM, Kerwin E, Tombs L, Fahy WA, Naya I. Triple therapy of umeclidinium + inhaled corticosteroids/long-acting beta2 agonists for patients with COPD: pooled results of randomized placebo-controlled trials. Pulm Ther. 2016;2:43–58. doi: 10.1007/s41030-016-0012-4. [DOI] [Google Scholar]
- 9.Lee SD, Xie CM, Yunus F, Itoh Y, Ling X, Yu WC, et al. Efficacy and tolerability of budesonide/formoterol added to tiotropium compared with tiotropium alone in patients with severe or very severe COPD: a randomized, multicentre study in East Asia. Respirology. 2016;21:119–127. doi: 10.1111/resp.12646. [DOI] [PubMed] [Google Scholar]
- 10.Lipson DA, Barnacle H, Birk R, Brealey N, Locantore N, Lomas DA, et al. FULFIL trial: once-daily triple therapy for patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;196:438–446. doi: 10.1164/rccm.201703-0449OC. [DOI] [PubMed] [Google Scholar]
- 11.Singh D, Papi A, Corradi M, Pavlišová I, Montagna I, Francisco C, et al. Single inhaler triple therapy versus inhaled corticosteroid plus long-acting β2-agonist therapy for chronic obstructive pulmonary disease (TRILOGY): a double-blind, parallel group, randomised controlled trial. Lancet. 2016;388:963–973. doi: 10.1016/S0140-6736(16)31354-X. [DOI] [PubMed] [Google Scholar]
- 12.Vestbo J, Papi A, Corradi M, Blazhko V, Montagna I, Francisco C, et al. Single inhaler extrafine triple therapy versus long-acting muscarinic antagonist therapy for chronic obstructive pulmonary disease (TRINITY): a double-blind, parallel group, randomised controlled trial. Lancet. 2017;389:1919–1929. doi: 10.1016/S0140-6736(17)30188-5. [DOI] [PubMed] [Google Scholar]
- 13.Jones PW, Harding G, Berry P, Wiklund I, Chen WH, Kline LN. Development and first validation of the COPD assessment test. Eur Respir J. 2009;34:648–654. doi: 10.1183/09031936.00102509. [DOI] [PubMed] [Google Scholar]
- 14.Jones PW, Harding G, Wiklund I, Berry P, Tabberer M, Yu R, et al. Tests of the responsiveness of the COPD assessment test following acute exacerbation and pulmonary rehabilitation. Chest. 2012;142:134–140. doi: 10.1378/chest.11-0309. [DOI] [PubMed] [Google Scholar]
- 15.Miller MR, Hankinson J, Odencrantz J. Standardisation of spirometry. Eur Respir J. 2005;26:319–388. doi: 10.1183/09031936.05.00034805. [DOI] [PubMed] [Google Scholar]
- 16.Meguro M, Barley EA, Spencer S, Jones PW. Development and validation of an improved COPD-specific version of the St. George respiratory questionnaire. Chest. 2007;132:456–463. doi: 10.1378/chest.06-0702. [DOI] [PubMed] [Google Scholar]
- 17.Donahue JF. Minimal clinically important differences in COPD lung function. COPD. 2005;2:111–124. doi: 10.1081/COPD-200053377. [DOI] [PubMed] [Google Scholar]
- 18.Brealey N, Gupta A, Renaux J, Mehta R, Allen A, Henderson A. Pharmacokinetics of fluticasone furoate, umeclidinium, and vilanterol as a triple therapy in healthy volunteers. Int J Clin Pharmacol Ther. 2015;53:753–764. doi: 10.5414/CP202390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chrystyn H, van der Palen J, Sharma R, Barnes N, Delafont B, Mahajan A, et al. Device errors in asthma and COPD: systematic literature review and meta-analysis. NPJ Prim Care Respir Med. 2017;27:22. doi: 10.1038/s41533-017-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horsley MG, Bailie GR. Risk factors for inadequate use of pressurised aerosol inhalers. J Clin Pharmacol Ther. 1988;13:139–143. doi: 10.1111/j.1365-2710.1988.tb00170.x. [DOI] [PubMed] [Google Scholar]
- 21.De Blaquiere P, Christensen DB, Carter WB, Martin TR. Use and misuse of metered-dose inhalers by patients with chronic lung disease. A controlled, randomized trial of two instruction methods. Am Rev Respir Dis. 1989;140:910–916. doi: 10.1164/ajrccm/140.4.910. [DOI] [PubMed] [Google Scholar]
- 22.Pascoe SJ, Lipson DA, Locantore N, Barnacle H, Brealey N, Mohindra R, et al. A phase III randomised controlled trial of single-dose triple therapy in COPD: the IMPACT protocol. Eur Respir J. 2016;48:320–330. doi: 10.1183/13993003.02165-2015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.