

Abstract
N′-Nitrosonornicotine (NNN) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) are carcinogenic tobacco-specific nitrosamines believed to play a vital role in the initiation of tobacco-related cancers. To exhibit their carcinogenicity, both NNN and NNK must be metabolically activated by cytochrome P450s, specifically P450 2A6 and P450 2A13, respectively. Prior research has focused on α-hydroxylation, which leads to the formation of several DNA adducts that have been identified and quantified in vivo. However, some studies indicate that P450s can retain substrates within their active sites and perform processive oxidation. For nitrosamines, this would oxidize the highly unstable α-hydroxynitrosamines to potentially more stable nitrosamides, which could also alkylate DNA. Thus, we hypothesized that both NNN and NNK are processively oxidized in vitro to nitrosamides by P450 2A6 and P450 2A13, respectively. To test this hypothesis, we synthesized the NNN- and NNK-derived nitrosamides, determined their half-lives at pH 7.4 and 37 °C, and monitored for nitrosamide formation in an in vitro P450 system with product analysis by LC-NSI+-HRMS/MS. Half-lives of the nitrosamides were determined by HPLC-UV and ranged from 7–35 min, which is more than 40 times longer than the corresponding α-hydroxynitrosamines. Incubation of NNN in the P450 2A6 system resulted in the formation of the nitrosamide, N'-nitrosonorcotinine (NNC) at low levels. Similarly, the nitrosamide 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanedione (CH2-oxo-NNK), was detected in low amounts in the incubation of NNK with the P450 2A13 system. The other possible NNK-derived nitrosamide, 4-(nitrosoformamido)-1-(3-pyridyl)-1-butanone (CH3-oxo-NNK), was not observed in the P450 2A13 reactions. CH2-oxo-NNK readily formed O6meGua in reactions with dGuo and calf thymus DNA. These results demonstrate that NNC and CH2-oxo-NNK are novel metabolites of NNN and NNK, respectively. Though low-forming, their increased stability may allow for mutagenic DNA damage in vivo. More broadly, this study provides the first account of a cytochrome-P450 mediated conversion of nitrosamines to nitrosamides which warrants further studies to determine how general this phenomenon is in nitrosamine metabolism.
TOC Graphic
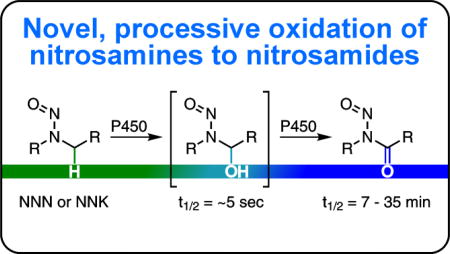
Introduction
Tobacco use is the leading preventable cause of cancer death in the United States resulting in an estimated 160,000 deaths annually,1 or 30% of all cancer deaths nationwide.2 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK, 1) and N′-nitrosonornicotine (NNN, 2) are tobacco-specific nitrosamines thought to play a critical role in tobacco carcinogenesis. Both compounds cause tumors in animal models and have been classified as carcinogenic to humans.1,3 To exhibit their carcinogenicity, NNK and NNN must first be metabolically activated by cytochrome P450s.4–6 In the case of NNK, P450 2A13-mediated hydroxylation at the methylene or methyl carbon adjacent to the nitrosamino group results in highly unstable α-hydroxy species 3 and 43,4 (Scheme 1A). Prior work indicates that α-hydroxynitrosamines have half-lives of ~5 sec7,8, decomposing to diazohydroxides such as 7 and 8,8 which lose H2O producing diazonium ions that either hydrolyze to urinary products 10 and 11 or react with DNA and proteins to form adducts4 (Scheme 1A). If left unrepaired, DNA adducts can cause mutations in critical oncogenes or tumor suppressor genes, initiating tumor development. Methyl adducts such as O6-methylguanine (O6meG) are especially tumorigenic in animal models.9 For NNN, α-hydroxylation at the 2′ or 5′ position by P450 2A6 initiates a similar cascade leading to a variety of DNA adducts and urinary products (Scheme 1A). Both NNK- and NNN-derived DNA adducts have been identified and quantified in several animal models and show promise as biomarkers for carcinogen activation in humans.10–14
Scheme 1.

(A) Established in vivo metabolism of NNK (1) and NNN (2) by P450 2A13- or P450 2A6-mediated oxidation, respectively. Oxidation results in unstable α-hydroxynitrosamines (3 – 6) which spontaneously decompose to diazohydroxides (7 – 9). These either hydrolyze to products excreted in the urine (10 – 12) or react with DNA to form adducts. (B) Proposed P450-mediated oxidation of NNK (1) and NNN (2) to nitrosamides (13 – 15) through retention of the α-hydroxynitrosamines 3, 4, and 6 within the P450 active site. If formed in vivo, we anticipate these species would also form adducts with DNA.
While the α-hydroxynitrosamine hypothesis outlined above accounts nicely for many of the metabolic products and DNA adducts produced in the metabolism of NNK, NNN, and other carcinogenic nitrosamines, there are also inconsistencies in this hypothesis.15,16 Important among these is the short lifetime of α-hydroxynitrosamines which raises questions about their ability to alkylate nuclear DNA after having been formed in the endoplasmic reticulum. A few studies have shown these intermediates exist long enough for glucuronidation, but measured levels were minor. In addition, it is unclear if this pathway mediates detoxification or intracellular transportation to the nucleus17–19. Elespuru et al and Guttenplan have explored the hypothesis that the α-hydroxynitrosamines are further oxidized to nitrosamides, which are also direct DNA alkylating agents, but this alternate hypothesis lacks compelling supportive data15,16. In the study reported here, we explore this hypothesis with respect to the metabolic activation of NNK and NNN. We hypothesize that the α-hydroxynitrosamines could be retained in the active site of P450 2A13 or P450 2A6 and further oxidized to the corresponding nitrosamides: 4-(methylnitrosamino)-1-(3-pyridyl)-1,4-butanedione (CH2-oxo-NNK, 13), 4-(nitrosoformamido)-1-(3-pyridyl)-1-butanone (CH3-oxo-NNK, 14) from NNK and N′-nitrosonorcotinine (NNC, 15) from NNN.
There is precedent for retention of substrates in P450s leading to processive oxidation. Metabolism of nicotine by P450 2A6 proceeds through a retained iminium ion or hemiaminal intermediate before releasing cotinine as the major metabolite.20 The Guengerich group has shown that P450 2E1 oxidizes ethanol directly to acetic acid with limited substrate dissociation.21 Likewise, they showed that formaldehyde and acetaldehyde formed in the metabolism of dimethylnitrosamine and diethylnitrosamine by P450 2A6 are directly oxidized to formic acid and acetic acid, respectively, without release of the intermediate aldehydes.22 They later proposed an alternate route for acid formation via a nitrosamide intermediate; however, it was undetectable.23
Nitrosamides are recognized as direct-acting carcinogens with common half-lives being on the scale of minutes.24 Extensive research shows that their stability and reactivity is dependent on temperature, steric and electronic factors, and solvent composition.25–29 In nucleophilic environments,30 the major products are the corresponding carboxylic acid derivatives and the diazonium species discussed earlier (Scheme 1). Thus, if released into a cell, nitrosamides should not only have better stability for traversing the hydrolytic environment of the cytosol, but also the ability to alkylate DNA. If nitrosamines are oxidized to nitrosamides, this could lead to the identification of new classes of DNA adducts and a better understanding of the mechanisms of nitrosamine carcinogenesis.
In the present study, we examine the hypothesis that NNK and NNN are metabolized to their corresponding nitrosamides. We synthesized these three nitrosamides and evaluated their half-lives and major degradation products in vitro. We describe our finding that CH2-oxo-NNK and NNC are minor metabolites of NNK and NNN in in vitro assays with human cytochrome P450s. Further evaluation of CH2-oxo-NNK demonstrated that it methylates dGuo in DNA and is thus potentially mutagenic. Together, this work provides the first account of a nitrosamine being converted metabolically to a nitrosamide and furthers our understanding of the metabolism of NNK and NNN.
Experimental Procedures
Caution
NNN and NNK are carcinogenic in animal models and are IARC Group 1 carcinogens. All nitrosamides are presumed to be carcinogens based on their structure and reactivity. Handle these in a well-ventilated fume hood with personal protective equipment and extreme care.
Chemicals and Enzymes
NNK, NNN, 4-oxo-4-(3-pyridyl)-butanol (keto alcohol, 11), 5-(3-pyridyl)-2-hydroxytetrahydrofuran (lactol, 12), O6-methylguanine (O6meGua), and [CD3]O6meGua were synthesized as previously described.31–34 5-(3-Pyridyl)-2-pyrrolidinone (norcotinine, 28) was obtained from AKos GmbH (Steinen, Germany). P450 2A6 Baculosomes, regeneration system and reaction buffer were available as a Vivid CYP450 Screening Kit from Life Technologies (Carlsbad, CA). Purified P450 2A13 and P450 reductase were a generous gift from Dr. Sharon Murphy (University of Minnesota). All other chemicals and solvents used were obtained from either Sigma Aldrich (Milwaukee, WI) or Thermo Scientific (Waltham, MA) in reagent grade and used without further purification.
General Synthetic Procedures
NMR spectra were recorded on a Bruker 500 MHz spectrometer. Chemical shifts are reported as parts per million (ppm). Residual solvent peaks were used as an internal reference for 1H-NMR (7.26 ppm CDCl3; 2.50 ppm D6-DMSO) and 13C-NMR (77.2 ppm CDCl3; 39.5 ppm D6-DMSO). Peak splitting used the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, dt = doublet of triplets, ddd = doublet of doublet of doublets, bs = broad singlet, and m = multiplet. High resolution mass spectrometry (HRMS) for selected compounds was performed on an LTQ Orbitrap Velos (Thermo Scientific, Carlsbad, CA) and reported as m/z. Thin-layer chromatography (TLC) utilized Polygram pre-coated silica gel TLC plate (40 × 80 mm, 0.2 mm thick) with 254 nm fluorescent indicator. TLC plates were visualized with permanganate stain when necessary, otherwise UV lamp irradiation sufficed. Flash chromatography was performed on SiliCycle 60 (70–150) mesh silica gel. Reactions were performed under an atmosphere of N2 unless specified otherwise.
Methyl 4-oxo-4-(3-pyridyl)-1-butanoate (18)
Sodium cyanide (0.104 g, 2.12 mmol) was suspended in anhydrous N,N-dimethylformamide (DMF, 10 mL) and brought to 35 °C. 3-Pyridinecarboxaldehyde (2.27 g, 21.2 mmol, 2 mL) was added dropwise to the suspension. After 10 min of stirring, the resulting red solution was treated dropwise with methyl acrylate (1.90 g, 22.1 mmol, 2 mL). Over 4 h, the solution became increasingly yellow and slightly viscous. The reaction was quenched with acetic acid (100 µL). The resulting yellow solution was diluted in CH2Cl2 and washed with H2O and brine. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo to a crude, yellow solid. Purification by column chromatography (50 to 100% EtOAc in hexanes) yielded pure product as a white, crystalline solid (3.02 g, 73.8%). 1H-NMR (500 MHz; CDCl3): δ 9.21 (dd, J = 2.2, 0.8 Hz, 1H, 2-Py), 8.79 (dd, J = 4.8, 1.7 Hz, 1H, 6-Py), 8.25 (ddd, J = 8.0, 2.2, 1.8 Hz, 1H, 4-Py), 7.43 (ddd, J = 8.0, 4.8, 0.9 Hz, 1H, 5-Py), 3.72 (s, 3H, CH3), 3.33 (t, J = 6.5 Hz, 2H, COCH2CH2), 2.80 (t, J = 6.5 Hz, 2H, COCH2CH2).; 13C-NMR (126 MHz; CDCl3): δ 197.1 (CO), 173.2 (CO2CH3), 153.8 (2-Py), 149.8 (5-Py), 135.5 (4-Py), 131.9 (3-Py), 123.8 (5-Py), 52.1 (CH3), 33.8 (COCH2CH2CO2), 27.9 (COCH2CH2CO2) ppm.
4-Oxo-4-(3-pyridyl)-butanoic acid (19)
Compound 18 (393 mg, 2.03 mmol) was dissolved in 1N NaOH (4 mL) and stirred for 3 h at room temperature. The solution went from colorless to yellow. The pH was adjusted to ~5–6 with an equal volume of 1N HCl and a precipitate formed. The solid was filtered and dried under vacuum to give a white, crystalline solid (251 mg, 69%).
1H-NMR (500 MHz; DMSO-d6): δ 12.18 (s, 1H, COOH), 9.14 (d, J = 2.1 Hz, 1H, 2-Py), 8.80 (dd, J = 4.8, 1.6 Hz, 1H, 6-Py), 8.31 (dd, J = 8.0, 1.8 Hz, 1H, 4-Py), 7.57 (dd, J = 8.0, 4.8 Hz, 1H, 5-Py), 3.30 (t, J = 6.3 Hz, 2H, COCH2), 2.60 (t, J = 6.3 Hz, 2H, CH2COOH). 13C-NMR (126 MHz; DMSO): δ 198.1 (CO), 173.7 (COOH), 153.5 (2-Py), 149.1 (6-Py), 135.4 (4-Py), 131.7 (3-Py), 123.9 (5-Py), 33.4 (COCH2), 27.7 (CH2COOH) ppm.
Methyl 4-oxo-4-(3-pyridyl)-butanamide (20a)
A solution of 19 (43.57 mg, 0.243 mmol), methylamine hydrochloride (25.41 mg, 0.376 mmol), and N-hydroxysuccinimide (NHS, 45.2mg, 0.393 mmol) in anhydrous dimethylsulfoxide (DMSO, 3 mL) was treated with N,N-diisopropylethylamine (DIPEA, 75.7 mg, 0.586 mmol, 102 µL) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC, 196 mg, 1.03 mmol). The reaction was stirred for 22 h at room temperature before diluting with EtOAc. The mixture was washed with H2O and brine, dried over MgSO4, filtered, and concentrated in vacuo to yield a crude, pink oil. Purification by column chromatography (5:100 MeOH/CH2Cl2) yielded pure product as an off-white solid (32.9 mg, 70%).
1H-NMR (500 MHz; DMSO-d6): δ 9.13 (d, J = 1.2 Hz, 1H, 2-Py), 8.79 (dd, J = 4.7, 1.3 Hz, 1H, 6-Py), 8.30 (d, J = 7.9 Hz, 1H, 4-Py), 7.84 (s, 1H, NH), 7.57 (dd, J = 7.9, 4.7 Hz, 1H, 5-Py), 3.27 (t, J = 6.6 Hz, 2H, COCH2), 2.57 (d, J = 4.6 Hz, 3H, CH2CONH), 2.48 (t, J = 6.6 Hz, 2H, NHCH3). 13C NMR (126 MHz; DMSO): δ 198.6 (CO), 171.3 (CONH), 153.3 (6-Py), 149.1 (2-Py), 135.4 (4-Py), 131.8 (3-Py), 123.9 (5-Py), 33.7 (COCH2), 29.0 (CH2CONH), 25.5 (NHCH3) ppm.
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanedione (CH2-oxo-NNK, 13)
A solution of 20a (31.4mg, 0.163 mmol) in a 5:1 mixture of acetic anhydride and acetic acid (6 mL) was brought to 0 °C. To this was added NaNO2 (30.7 mg, 0.445 mmol) all at once. After 4 h, the mixture was poured onto ice-cold H2O. The aqueous mixture was extracted with CH2Cl2. The pooled organics were dried over MgSO4, filtered, and concentrated in vacuo to yield a crude yellow oil. Purification by column chromatography on silica gel (100% EtOAc) yielded pure product as a bright, yellow oil (28.8 mg, 65%). 1H-NMR (500 MHz; CDCl3): δ 9.27 (s, 1H, 2-Py), 8.83 (d, J = 4.1 Hz, 1H, 6-Py), 8.31 (d, J = 7.9 Hz, 1H, 4-Py), 7.48 (dd, J = 7.8, 4.9 Hz, 1H, 5-Py), 3.67 (t, J = 6.0 Hz, 2H, COCH2), 3.55 (t, J = 6.0 Hz, 2H, CH2CON), 3.14 (s, 3H, CH3). 13C NMR (126 MHz; CDCl3): δ 196.7 (CO), 175.9 (CONNO), 153.5 (6-Py), 149.4 (2-Py), 135.6 (4-Py), 131.9 (3-Py), 123.8 (5-Py), 33.0 (CH2CONNO), 28.7 (COCH2), 25.9 (NCH3) ppm. HRMS Calc: 222.08732, Found: 222.08719
CH2-oxo-NNK from 5′-Hydroxycotinine
5'-Hydroxycotinine (54.23 mg, 0.282 mmol) in CH2Cl2 (25 mL) was brought to 0 °C and treated with p-toluenesulfonic acid (111 mg, 0.584 mmol). After 5 min of stirring, NaNO2 (151.2 mg, 2.2 mmol) was added. Stirring was continued for 3 h before pouring onto ice-cold H2O. The organic layer was separated and the aqueous extracted with CH2Cl2. The pooled organics were dried over MgSO4, filtered, and concentrated in vacuo to give a crude orange oil. The product was purified by HPLC using a 150 × 4.6 mm, 5 µm, Kinetix HILIC column (Phenomenex) with 1:1 hexanes/CHCl3 and isopropanol as mobile phases. The gradient was 5% to 20% isopropanol over 10 min at 1 mL/min. The product eluted at ~4 min. The purified product was a bright, yellow oil (4.98 mg, 8%).
N′-Nitrosonorcotinine (NNC, 15)
A solution of norcotinine (31.4 mg, 0.163 mmol) in a 5:1 mixture of acetic anhydride and acetic acid (6 mL) was brought to 0 °C. To this was added NaNO2 (30.7 mg, 0.445 mmol) all at once. After 4 h, the mixture was poured onto ice-cold H2O. The aqueous mixture was extracted with CH2Cl2. The pooled organics were dried over MgSO4, filtered, and concentrated in vacuo to yield a crude yellow oil. Purification by column chromatography (100% EtOAc) yielded pure product as a bright, yellow oil (92%). 1H-NMR (500 MHz; CDCl3): δ 8.56 (d, J = 4.3 Hz, 1H, 6-Py), 8.46 (d, J = 1.8 Hz, 1H, 2-Py), 7.36 (d, J = 8.0 Hz, 1H, 4-Py), 7.30-7.28 (m, 1H, 5-Py), 5.29 (dd, J = 9.1, 3.1 Hz, 1H, NCH), 2.97 (dt, J = 18.6, 9.4 Hz, 1H, COCH2), 2.87 (ddd, J = 18.5, 9.4, 4.0 Hz, 1H, COCH2'), 2.66 (dq, J = 13.4, 9.4 Hz, 1H, CHCH2), 2.15-2.09 (m, 2H, CHCH2') ppm; 13C-NMR (126 MHz; CDCl3): δ 172.7 (CO), 149.4 (6-Py), 147.1 (2-Py), 134.1 (3-Py), 132.7 (4-Py), 123.8 (5-Py), 55.7 (NCH), 26.2 (COCH2), 22.2 (CHCH2) ppm. HRMS Calc: 192.07675, Found: 192.07670
2-(3-Pyridyl)-1,3-dithiane (21)
A solution of 3-pyridylcarboxaldehyde (114 mg, 1.06 mmol, 100 µL) and 1,3-propanedithiol (162 mg, 1.50 mmol, 150 µL) in anhydrous tetrahydrofuran (THF, 5 mL) was treated with BF3•Et2O (173 mg, 1.22 mmol 300 µL) dropwise at room temperature. The mixture was then heated to reflux at 80 °C and stirred for 24 h before quenching with sat'd NaHCO3 solution. The aqueous phase was extracted several times with CH2Cl2. The pooled organic layers were washed with brine, dried over MgSO4, filtered, and evaporated in vacuo to give crude yellow crystals. Purification by column chromatography (hexanes/EtOAc 1:1) yielded the product as a fine, white powder (199.4 mg, >95%).
1H-NMR (500 MHz; CDCl3): δ 8.72 (d, J = 2.0 Hz, 1H, 2-Py), 8.57 (dd, J = 4.9, 1.5 Hz, 1H, 6-Py), 7.85 (dt, J = 7.9, 2.0 Hz, 1H, 4-Py), 7.31 (dd, J = 7.9, 4.9 Hz, 1H, 5-Py), 5.21 (s, 1H, S2CH), 3.10 (ddd, J = 14.6, 12.3, 2.4 Hz, 2H, SCH2CH2-ax), 2.96 (dt, J = 14.0, 3.8 Hz, 2H, SCH2CH2 -eq), 2.22 (dtt, J = 14.2, 4.6, 2.4 Hz, 1H, SCH2CH2 -ax), 2.02-1.93 (m, 1H, SCH2CH2 -eq) ppm; 13C NMR (126 MHz; CDCl3): δ 150.1 6-Py, 149.5 (2-Py), 135.7 (4-Py), 135.4 (3-Py), 124.0 (5-Py), 48.8 (S2CH), 32.3 (SCH2CH2), 25.2 (SCH2CH2) ppm.
tert-Butyl 3-(2-(3-pyridyl)-1,3-dithianyl)-1-propylcarbamate (22)
A solution of 21 (222 mg, 1.13 mmol) and tetramethylethylenediamine (TMEDA, 131.8 mg, 1.13 mmol, 170 µL) in anhydrous THF (6 mL) was cooled to −78 °C and treated with n-BuLi in hexanes dropwise (1.28 mmol, 800 µL). The resulting dark red solution was stirred at −78 °C for 30 min before dropwise addition of 27 in THF (360 mg, 1.26 mmol, 3 mL). The mixture was stirred at −78 °C for 2 h before allowing the bath to come to room temperature. After 14 h of stirring, the reaction was quenched with H2O. The aqueous phase was extracted several times with EtOAc. The pooled organics were dried over MgSO4, filtered, and concentrated in vacuo to give a crude, yellow oil. Purification by column chromatography (hexanes/EtOAc 1:1) yielded the product as yellow crystals (290 mg, 72.5%).
1H-NMR (500 MHz; CDCl3): δ 9.13 (d, J = 2.1 Hz, 1H, 2-Py), 8.52 (dd, J = 4.7, 1.6 Hz, 1H, 6-Py), 8.20 (ddd, J= 8.1, 2.4, 1.6 Hz, 1H, 4-Py), 7.32 (ddd, J = 8.1, 4.7, 0.6 Hz, 1H, 5-Py), 4.41 (bs, 1H, NH), 3.03-3.00 (m, 2H, NHCH2), 2.72-2.61 (m, 4H, SCH2CH2), 2.03-2.00 (m, 2H, CCH2), 1.99-1.91 (m, 2H, SCH2CH2), 1.48-1.42 (m, 2H, CH2CH2CH2), 1.40 (s, 9H, C(CH3)3) ppm; 13C NMR (126 MHz; CDCl3): δ 155.9 (NHCO), 150.8 (2-Py), 148.4 (6-Py), 137.6 (3-Py), 136.8 (4-Py), 123.4 (5-Py), 79.4 (C(CH3)3), 56.5 (SCS), 42.5 (CCH2), 40.4 (NHCH2), 28.5 (CH3), 27.6 (SCH2CH2), 25.1 (SCH2CH2), 24.7 (CH2CH2CH2) ppm.
3-(2-(3-Pyridyl)-1,3-dithianyl)-1-propylformamide (23)
A solution of 22 (87.6 mg, 0.247 mmol) in CH2Cl2 (3 mL) was treated with trifluoroacetic acid (TFA, 1.49 g, 13.1 mmol, 1 mL), which resulted in gas evolution. After 3 h, the solvent was evaporated in vacuo to remove excess TFA. The resulting oil was reconstituted in CH2Cl2 and washed with sat'd. NaHCO3 solution and brine. The organic layer was dried over MgSO4, filtered, and concentrated in vacuo to give a yellow oil. This was dissolved in MeOH (5 mL) and treated with triethylamine (29.04 mg, 0.288 mmol, 40 µL) and methyl formate (95.7 mg, 1.60 mmol, 110 µL). The flask was sealed and heated to 55 °C with stirring. After 4 h, the mixture was concentrated in vacuo to yield a yellow oil. 1H-NMR indicated the product was a 9:1 mixture of cis- and trans-formamide isomers. Purity was sufficient to carry forward without column purification.
1H-NMR (500 MHz; CDCl3): δ 9.09 (d, J = 2.5 Hz, trans-2-Py), 9.08 (d, J = 2.1 Hz, 1H, cis-2-Py), 8.49 (dd, J = 4.8, 1.6 Hz, trans-6-Py), 8.48 (dd, J = 4.7, 1.5 Hz, 1H, cis-6-Py), 8.19-8.16 (m, 1H, 4-Py), 8.07 (d, J = 1.1 Hz, 1H, cis-NHCHO), 7.92 (d, J = 11.9 Hz, trans-NHCHO), 7.32-7.29 (m, 1H, 5-Py), 5.94 (s, 1H, cis-NHCHO), 5.84 (s, trans-NHCHO), 3.18 (q, J = 6.7 Hz, 2H, cis-CH2NH), 3.10 (q, J = 6.8 Hz, trans-CH2NH), 2.63 (m, 4H, SCH2), 2.02-1.99 (m, 2H, CCH2), 1.98-1.85 (m, 2H, SCH2CH2), 1.52-1.46 (m, 2H, CH2CH2NH). 13C-NMR (126 MHz; CDCl3): δ 164.4 (trans-CHO), 161.2 (cis-CHO), 150.5 (2-Py), 148.37 (trans-6-Py), 148.25 (cis-6-Py), 137.45 (cis-3-Py), 137.35 (trans-3-Py), 136.73 (cis-4-Py), 136.67 (trans-4-Py), 123.33 (trans-5-Py), 123.29 (cis-5-Py), 56.28 (cis-SCS), 56.17 (trans-SCS), 42.3 (cis-CCH2), 42.0 (trans-CCH2), 41.4 (trans-NHCH2), 37.6 (cis-NHCH2), 27.5 (SCH2), 25.7 (trans-CH2CH2CH2), 24.85 (cis-SCH2CH2), 24.79 (trans-SCH2CH2), 24.1 (cis-CH2CH2CH2) ppm.
4-(Formamido)-1-(3-pyridyl)-1-butanone (24)
N-Chlorosuccinimide (NCS, 101.8 mg, 0.762 mmol) and AgNO3 (168.5 mg, 0.992 mmol) were suspended in 1:1 MeCN/H2O (1 mL) and cooled to 0 °C. To this was added a solution of 23 in MeCN (0.247 mmol, 1.5 mL), which resulted in immediate precipitate formation. After 30 min, the reaction was quenched with sat. Na2SO3, sat'd NaHCO3, and brine solutions in succession (1 mL each). The mixture was filtered and extracted with CH2ClS. The pooled organics were dried over MgSO4, filtered, and concentrated in vacuo to give a crude solid. Purification by column chromatography on silica gel (6% MeOH in CHCl3) yielded pure product as a white solid (20.5 mg, 43% over three steps). 1H-NMR (500 MHz; CDCl3): δ 9.15 (dd, J = 2.2, 0.7 Hz, 1H, 2-Py), 8.78 (dd, J = 5.5, 1.7 Hz, 1H, trans-6-Py), 8.77 (dd, J = 4.8, 1.7 Hz, 1H, cis-6-Py), 8.22 (dt, J = 8.0, 2.1 Hz, 1H, trans-4-Py), 8.21 (dt, J = 8.0, 2.0 Hz, 1H, cis-4-Py), 8.18 (s, 1H, cis-CHO), 8.06 (d, J = 11.9 Hz, trans-CHO), 7.42 (m, 1H, 5-Py), 5.96 (s, 1H, NHCHO), 3.41 (q, J = 6.6 Hz, 2H, cis-CH2NH), 3.36 (q, J = 6.8 Hz, trans-CH2NH), 3.08 (t, J = 6.9 Hz, 2H, cis-COCH2), 3.07 (t, J = 6.8 Hz, trans-COCH2), 2.01 (quintet, J = 6.9 Hz, 2H, cis-CH2CH2CH2), 2.01 (quintet, J = 6.9 Hz, trans-CH2CH2CH2); 13C-NMR (126 MHz; CDCl3): δ 198.6 (CO), 161.5 (CHO), 153.8 (6-Py), 149.7 (2-Py), 135.5 (4-Py), 132.0 (3-Py), 123.8 (5-Py), 37.8 (CH2NH), 36.3 (COCH2), 23.5 (CH2CH2CH2) ppm.
tert-Butyl 3-hydroxypropyl-1-carbamate (26)
A solution of 3-amino-1-propanol (4.94 g, 65.8 mmol, 25) and triethylamine (7.26 g, 71.7 mmol, 10 mL) in CH2Cl2 (175 mL) was treated with Boc anhydride (16.15 g, 74.0 mmol,17 mL) dropwise, which resulted in vigorous gas evolution. Once bubbling ceased, the reaction was quenched with sat'd NH4Cl. The organic layer was collected and the aqueous layer was further extracted with CH2Cl2. The pooled organics were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. NMR indicated the product was sufficiently pure to be brought directly to the next step.
1H-NMR (500 MHz; CDCl3): δ 4.84 (bs, 1H, OH), 3.67 (q, J = 5.5 Hz, 2H, HOCH2), 3.30 (d, J = 5.7 Hz, 2H, CH2NH), 3.07 (bs, 1H, NH), 1.68 (quintet, J = 5.7 Hz, 2H, CH2CH2CH2), 1.46 (s, 9H, t-Bu) ppm. 13C NMR (126 MHz; CDCl3): δ 157.2 (CONH2), 79.6 (C(CH3)3, 59.2 (HOCH2), 36.9 (CH2NH), 32.9 (CH2CH2CH2), 28.4 (t-Bu) ppm.
tert-Butyl 3-iodopropyl-1-carbamate (27)
Imidazole (Im., 5.42 g, 79.7 mmol) and triphenylphospine (20.72 g, 79.0 mmol) were dissolved in CH2Cl2 (250 mL) and brought to 0 °C. To this was added I2 (20.50 g, 80.8 mmol) scoopwise, resulting in a dark orange solution. After 20 min of stirring, 26 in CH2Cl2 (11.5 g, 65.7 mmol, 50 mL) was added and the solution was allowed to come to room temperature. After 22 h of stirring, the mixture was filtered over Celite and washed with 5% Na2S2O3. The organics were dried over MgSO4, filtered, and concentrated in vacuo to give a yellow solid. Purification by column chromatography (hexanes/EtOAc 4:1) gave pure product as a light yellow solid (83.2% over 2 steps).1H-NMR (500 MHz; CDCl3): δ 4.63 (bs, 1H, NH), 3.19 (m, 4H, NHCH2/ICH2), 2.00 (quintet, J = 6.6 Hz, 2H, CH2CH2CH2), 1.44 (s, 9H, t-Bu) ppm. 13C NMR (126 MHz; CDCl3): δ 155.9 (CONH), 79.4 (C(CH3)3), 41.0 (CH2NH), 33.4 (CH2), 28.4 (C(CH3)3), 3.1 (ICH2) ppm.
4-(Nitrosoformamido)-1-(3-pyridyl)-1-butanone (CH3-oxo-NNK, 14)
A solution of 24 (31.4mg, 0.163 mmol) in a 5:1 mixture of acetic anhydride and acetic acid (6 mL) was brought to 0 °C. To this was added NaNO2 (30.7mg, 0.445 mmol) all at once. After 4 h, the mixture was poured onto ice-cold H2O. The aqueous mixture was extracted with CH2Cl2. The pooled organics were dried over MgSO4, filtered, and concentrated in vacuo to yield a crude yellow oil. Purification by column chromatography (100% EtOAc) yielded pure product as a bright, yellow oil (28.8mg, 80%). 1H-NMR (500 MHz; CDCl3): δ 10.00 (s, 1H, CHO), 9.13 (d, J = 1.1 Hz, 1H, 2-Py)), 8.80 (dd, J = 4.8, 1.1 Hz, 1H, 6-Py), 8.24 (dd, J = 8.0, 1.6 Hz, 1H, 4-Py), 7.47 (dd, J = 8.0, 4.9 Hz, 1H, 5-Py), 4.11 (q, J = 7.1 Hz, 1H, NCH2), 2.92 (t, J = 6.9 Hz, 2H, COCH2), 1.91 (quintet, J = 6.9 Hz, 2H, CH2CH2CH2) ppm. 13-C NMR (126 MHz; CDCl3): δ 196.8 (CO), 168.5 (CHO), 153.1 (6-Py), 149.0 (2-Py), 136.0 (4-Py), 132.2 (3-Py), 124.1 (5-Py), 37.6 (NCH2), 35.8 (COCH2), 20.9 (CH2CH2CH2) ppm; HRMS Calc: 222.08732, Found: 222.08719
Determination of t1/2 of Nitrosamides
HPLC-UV analysis was performed using a 250×4.6 mm Gemini-NX C18 column (Phenomenex, Torrance, CA) with the following solvent gradients for the analyses indicated below: (1) isocratic for 5 min at 4% B followed sequentially by a linear gradient to 12% B over 15 min, a 10 min hold, a linear gradient to 30% B over 10 min, and a final linear gradient to 40% B over 2 min; (2) isocratic for 5 min at 12% B followed sequentially by a linear gradient to 30% B over 10 min, a linear gradient to 40% B over 15 min, and a final linear gradient to 70% B over 2 min. In both systems, solvent A was 15 mM NH4OAc and solvent B was methanol.
For t1/2 determination, an aliquot of NNC (180 nmol) in CH2Cl2 was dried and reconstituted in 30 µL of 0.5X P450 2A6 Reaction Buffer (Life Technologies) and incubated for 0 to 30 min at 37 °C. After the desired incubation time, 5 µL of sample was analyzed by HPLC using gradient 1. A254 was monitored and the peaks for NNC and its decomposition products were integrated. NNC eluted at 36.0 min. Peak area for NNC was fit to a single-order exponential plot while using the 0 min incubation peak area as a normalizing factor. The analysis was similarly performed for CH2-oxo-NNK and CH3-oxo-NNK, except HPLC gradient 2 was used. CH2-oxo-NNK and CH3-oxo-NNK eluted at 33.0 and 29.5 min, respectively. Decomposition products were identified by retention time comparisons and co-injection with synthetic standards.
In vitro detection of CH2-oxo-NNK using P450 2A13
Incubations with P450 2A13 were performed as previously reported.35 Briefly, purified P450 2A13 and cytochrome P450 reductase were reconstituted with dilauroylphosphatidylcholine (DLPC, Sigma Aldrich) for 45 min on ice before diluting with Tris buffer to give a final concentration of 1 µM P450 2A13, 2 µM P450 reductase, 0.1 µg/µL DLPC, and 50 mM Tris, pH = 7.4. To initiate the reaction, an aliquot of this (containing 5 pmol P450) was added to a Tris-buffered solution of NNK (4 µM) and NADPH (0.2 mM). Final reaction volumes were always 100 µL. The mixture was brought to 37 °C for 1–60 min before quenching with 10 µL of both Ba(OH)2 and ZnSO4. After centrifuging the sample at 8000 g for 4 min, the supernatant was collected and 2 µL were immediately analyzed by liquid chromatography-positive nanoelectrospray-ionization high-resolution tandem mass spectrometry (LC-NSI+-HRMS/MS) with an LTQ Orbitrap Velos (Thermo Scientific, Carlsbad, CA). LC employed a hand-packed, Luna C18 (5 µm), 100 mm × 75 µm, 15 µm orifice capillary column with a multi-step gradient. Initially, 5% B at 1 µL/min from 0–5 min was used to load the sample. Afterwards, the flow rate was dropped to 0.3 µL/min and a linear gradient was started from 5% to 20% B over 4 min, followed by a ramp to 55% B over 10 min, and re-equilibration, where solvent A was 5 mM NH4OAc and solvent B was acetonitrile. CH2-oxo-NNK and CH3-oxo-NNK were monitored by both full scan and MS2 fragmentation. Full scan was performed at a resolution of 60,000 and the accurate parent mass of both nitrosamides (m/z = 222.08719) was extracted at a mass tolerance of 5 ppm. For MS2 fragmentation, parent ions were isolated (2.0 amu isolation width) and fragmented by collision-induced dissociation (CID) with a collision energy of 25 eV, resolution of 15,000, and scan time of 30 ms. Accurate product ion masses from characteristic transitions for CH2-oxo-NNK (m/z 222 → m/z 180.06542, -H3CNNO +OH), CH3-oxo-NNK (m/z 222 → m/z 106.02852), NNK (m/z 208 → m/z 178.11002), and keto alcohol 11 (m/z 166 → m/z 148.07564) were extracted at a mass tolerance of 5 ppm.
In vitro detection of NNC using P450 2A6
Incubations with P450 2A6 were performed as described by the manufacturer with modifications.36 After thawing the P450 2A6 Baculosomes and Vivid-NADPH-Regeneration System (Life Technologies) on ice, aliquots were combined and diluted 1:10 and 1:50, respectively, with 0.5X Vivid Reaction Buffer (Life Technologies). For each incubation, an aliquot of the combined-enzyme system (containing 5 pmol P450) was added to a 0.5X Reaction-Buffered solution of NNN (4 µM) and this new mixture was pre-incubated for 2 min at 37 °C. To initiate the reaction, an aliquot of NADP+ (containing 3 nmol) was added. Final reaction volumes were 100 µL. The incubation and work-up were as described earlier for NNK-P450 2A13 incubations. NNC detection was performed by adapting the NNK-P450 2A13 LC-NSI+-HRMS/MS method described above to the accurate parent mass of NNC (m/z 192.07670) in full scan. Likewise, the MS2 analysis was used to monitor for the accurate product ion masses from characteristic transitions of NNC (m/z 192 → m/z 134.04739, 162.07874), NNN (m/z 178 → m/z 148.09941), and lactol 12 (m/z 166 → m/z 148.07571).
in vitro Methylation of dGuo by CH2-oxo-NNK
A solution of CH2-oxo-NNK in CH2Cl2 was dried under a stream of N2 and reconstituted in a phosphate-buffered solution of dGuo (4.34 mM dGuo, 25 mM NaHPO4, pH = 7.4) so that the molar ratio of CH2–oxo-NNK to dGuo was 1:1. This was brought to 37 °C and incubated for 18 h. To assess methylation, 200 fmol of [CD3]O6meGua was added as internal standard. Samples were brought up to 1 mL with 0.1N HCl and incubated at 90 °C for 30 min. After cooling on ice, the samples were neutralized with 1.0N NaOH and purified by solid-phase extraction (Strata-X polymeric reversed phase, 30 mg, Phenomenex, Torrance, CA). Before sample addition, the cartridge was activated using 1 mL each of MeOH and H2O. After sample addition, the cartridge was washed with 1 mL of both H2O and 10% MeOH. The sample was eluted and collected with 1 mL of MeOH. The collected fraction was evaporated to dryness in a Speedvac. The residue was reconstituted in 30 µL of H2O and analyzed by LC-MS/MS.37
We used a well-established liquid chromatography-positive electrospray ionization-tandem mass spectrometry (LC-ESI+-MS/MS) method. A 0.5 × 150 mm Zorbax SB-C18, 5 µm column (Agilent, Santa Clara, CA) was eluted with a multi-step gradient and flow rate of 10 µL/min. After a linear gradient from 5% to 10% B over 10 min, the eluant was brought to 40% B over 5 min, followed by a wash at 90% B and re-equilibration, where solvent A was 15mM NH4OAc and solvent B was methanol. MS was performed on a TSQ Vantage triple quadrupole mass analyzer (Thermo Scientific). The SRM transitions were m/z 166.1 → m/z 149.1 and m/z 166.1 → m/z 124.1 for O6meGua and m/z 169.1 → m/z 152.1 for [CD3]O6meGua using a collision energy of 30 eV and a 0.2 amu scan width.
in vitro Methylation of calf thymus DNA by CH2-oxo-NNK
A solution of CH2-oxo-NNK was dried under a stream of N2 and reconstituted in a phosphate-buffered solution of calf thymus DNA so that the ratio was 3 nmol CH2-oxo-NNK :1 µg DNA. This was brought to 37 °C and incubated for 18 h. The aqueous sample was extracted twice with equal volumes of CHCl3:isoamyl alcohol (24:1). The DNA was precipitated by addition of an equal volume of isopropanol and gentle shaking. Isolated DNA was washed with 500 µL of 70% EtOH and 100% EtOH, and dried under N2. To assess methylation, isolated DNA was dissolved in 100 µL of sodium phosphate buffer (25 mM, pH = 7.4) and 200 fmol of [CD3]O6meGua was added as internal standard. The samples were then processed and analyzed as described above for dGuo methylation.
Results
Synthesis of Nitrosamides
Retrosynthetic analysis identified 3-pyridinecarboxaldehyde (17) as a common precursor for both NNK-derived nitrosamides (Scheme 2). The synthesis of CH2-oxo-NNK started with the formation of keto ester 18 by using the Stetter reaction38 to couple aldehyde 17 with methyl acrylate, followed by hydrolysis of 18 to keto acid 19. This method is a convenient alternative to the more commonly used routes to this compound.39,40 Keto acid 19 was coupled to methylamine using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDAC) and N-hydroxysuccinimide (NHS) in dimethylsulfoxide (DMSO), resulting in the open-chain and lactam conformers of 20 in a 2:1 ratio, respectively. Nitrosation of the open-chain conformer produced CH2-oxo-NNK (13) as a single rotamer.41 Nitrosation of the lactam conformer also produced CH2-oxo-NNK; however, new conditions using a strong acid catalyst were required. These conditions also degraded the product which limited the isolatable yield (<10%). Ultimately, only the open-chain route was synthetically useful for CH2-oxo-NNK production.
Scheme 2.

Synthesis of Nitrosamides (A) Methyl Acrylate, NaCN, DMF, 40 °C, 4h; (B) NaOH, H2O, RT, 3h; (C) EDAC, NHS, MeNH2•HCl, DMSO, RT, 22h; (D) NaNO2, Ac2O:HOAc, 0 °C, 4h; (E) HS(CH2)3SH, BF3•OEt2, THF, 80 °C, 24h; (F) (i) n-BuLi, TMEDA, THF, −78 °C, 1h; (ii) 27, THF, −78 °C to RT, 16h; (G) 25% TFA, CH2Cl2, RT, 3h; (H) HCO2Me, Et3N, MeOH, 55 °C, 4h; (I) AgNO3, NCS, MeCN:H2O (1:1), 0 °C, 30 min; (J) Boc2O, Et3N, CH2Cl2, RT, 30 min; (K) I2, PPh3, Im., CH2Cl2, 0 °C-RT, 22h.
Synthesis of CH3-oxo-NNK (Scheme 2) started with protection of 17 with 1,3-propanedithiol in nearly quantitative yield to give 21. This was coupled to 27 by classic umpolung chemistry42 to give 22 in excellent yield. Compound 27 was prepared in two steps from 25 on a multi-gram scale by first Boc-protecting the amine and then converting the alcohol to an iodide using a modified Appel reaction.43 After Boc removal from 22 and N-formylation to achieve 23, the dithiane group was oxidatively removed44 to produce 24 in 43% yield over 3 steps. To complete the synthesis, 24 was nitrosated to give CH3-oxo-NNK (14) in excellent yield.
NNC (15) was prepared by nitrosation of norcotinine (28) in 92% yield. The three nitrosamides were stored in CH2Cl2 at 4 °C. They were stable for at least three months under these conditions. Attempts to store these compounds neat or in H2O-miscible solvents (MeOH, MeCN, acetone, etc.) resulted in decomposition.
Stability of Nitrosamides
The stabilities of the nitrosamides were determined at pH 7.4, 37 °C, in buffers to be used in our P450 assays. Reactions were followed by HPLC and major products were identified (Figure S1, Supporting Information). Decay curves for each nitrosamide are shown in Figure 1. The half-lives of CH2-oxo-NNK and CH3-oxo-NNK were 35.5 min and 6.7 min, respectively. The half-life of NNC under these conditions was 12.3 min. The major product in each case was that expected by nitrosamide hydrolysis, namely keto acid 19 from CH2-oxo-NNK, keto alcohol 11 from CH3-oxo-NNK, and hydroxy acid 29 from NNC (Scheme 3).
Figure 1.

Stability of CH2-oxo-NNK (blue diamonds), CH3-oxo-NNK (green triangles), and NNC (red squares) in reaction buffer at 37 °C. The half-lives were determined to be 35.5, 6.7, and 12.3 min, respectively, by HPLC-UV. Nitrosamide peak areas were normalized to the 0-min peak area and fit to a first-order exponential. Relative amounts of each nitrosamide were determined at each time point in triplicate with error bars denoting the standard deviation.
Scheme 3.

(A) Mechanism of hydrolysis of nitrosamides. Hydrolysis results in a carboxylic acid and an alcohol via a transient diazohydroxide that decomposes to a diazonium ion. (B) The hypothesized decomposition products of CH2-oxo-NNK, CH3-oxo-NNK, and NNC in assay buffer (pH = 7.4) at 37 °C.
In vitro cytochrome P450-catalyzed metabolism of NNK to CH2-oxo-NNK
With synthetic nitrosamide standards in hand and an understanding of their stability, we designed an assay to detect their formation by P450-mediated metabolism. P450 2A13 is the most efficient enzyme for α-hydroxylation of NNK and was chosen for this part of the study.45 The NNK concentration was set at 4 µM, the Km for production of hydroxylated products. Samples were analyzed immediately after work-up to minimize nitrosamide decomposition. We used an LTQ High-Resolution Orbitrap Velos MS system to enhance sensitivity and minimize background noise. We monitored for the accurate masses of the most abundant product ions of CH2-oxo-NNK and CH3-oxo-NNK resulting from MS2 fragmentation, and also extracted their accurate parent masses in full scan mode. Similar monitoring was performed for NNK and keto alcohol 11 to ensure catalytic turnover as 11 is the most abundant product from methyl hydroxylation.4,45
When NNK was incubated with the relevant enzymes and cofactors, we detected a peak that matched the accurate parent ion mass, accurate product ion mass resulting from MS2 of [M+H]+ = 222, and the retention time of synthetic CH2-oxo-NNK (Figure 2). This peak was detectable as early as the 1-min time point, reached its maximum concentration at 5 min, tapered off by 10–30 min, and was nearly undetectable by 60 min (Figure 2C–F). We did not detect CH3-oxo-NNK at any time point.
Figure 2.

LC-NSI-HRMS chromatograms resulting from the NNK-P450 2A13 incubations. For all sections, the top chromatogram is the accurate parent mass extracted from full scan for CH2-oxo-NNK and CH3-oxo-NNK. The middle and bottom chromatogram is the accurate product ion masses extracted from MS2 fragmentation for CH2-oxo-NNK and CH3-oxo-NNK, respectively. Sections are as follows: (A) CH2-oxo-NNK standard, (B) CH3-oxo-NNK standard, and NNK-P450 2A13 incubations containing all relevant enzymes and cofactors with incubation times of (C) 1 min, (D) 5 min, (E) 10 min, and (F) 60 min. RT = retention time; MA = Mass Area.
The signal for NNK decreased 8-fold while the signal for keto alcohol 11 simultaneously increased 4-fold over the 60-min period (data not shown). This indicates that NNK metabolism was rapid over the assay time period. CH2-oxo-NNK was a minor metabolite as its signal was >4000-fold less than that of keto alcohol 11. No metabolites were observed in control incubations lacking enzyme or cofactors (data not shown).
When identical incubations containing CH2-oxo-NNK (10 nM) were performed, the peak area was 10-fold higher than in unspiked samples, indicating that CH2-oxo-NNK was recoverable under our conditions. Based on this, we estimate that CH2-oxo-NNK was produced at concentrations less than 1 nM in our incubations.
In vitro cytochrome P450-catalyzed metabolism of NNN to NNC
The NNN incubations were performed in essentially the same way as those with NNK except that P450 2A6 was used instead of P450 2A13 as it is the most efficient enzyme for NNN metabolism.46 Catalytic turnover was assessed by measuring lactol 12, because it is the major product from 5′-hydroxylation of NNN (Scheme 1A).47 We detected NNC as early as the 1-min time point (Figure 3). The peak matched the synthetic standard with respect to the accurate parent ion mass in full scan, the accurate mass of the most abundant product ions in the MS2 of [M + H]+ = 192, and retention time.
Figure 3.

LC-NSI-HRMS chromatograms resulting from the NNN-P450 2A6 incubations. For all sections, the top chromatogram is the accurate parent mass extracted from full scan for NNC. The middle and bottom chromatograms are two accurate product ion masses extracted from MS2 fragmentation for NNC. Sections are as follows: (A) NNC standard, and NNC-P450 2A6 incubations containing all relevant enzymes and cofactors with incubation times of (B) 1 min, (C) 5 min, and (D) 10 min. RT = retention time; MA = Mass Area.
The NNC signal was maximal at 5 min and was approximately 1000-fold lower in intensity than that of lactol 12 (Scheme 1A). The concentration of NNC at the 5-min time point was estimated to be ~10 nM.
In vitro Methylation of dGuo and DNA by CH2-oxo-NNK
We tested the ability of CH2-oxo-NNK to methylate DNA by incubating it with a molar equivalent of dGuo and calf thymus DNA in phosphate buffer for 18 h. Levels of O6-meGua were 61.7 and 802 µmol/mg Gua for the dGuo and DNA reactions, respectively, as determined by LC-MS/MS analysis. (Figures S2, Supporting Information). Formation of 7-meGua was also noted, but it was not quantified.
Discussion
This study presents the first account of nitrosamines being directly converted to nitrosamides by P450 catalysis. This breaks new ground in our knowledge of nitrosamine metabolism and provides an impetus to determine if this phenomenon applies to all nitrosamines. Specifically, we found that CH2-oxo-NNK and NNC are novel metabolites of P450-mediated oxidation of NNK and NNN, respectively. We did not observe formation of CH3-oxo-NNK, perhaps due to its short half-life (6.7 min). These novel metabolites also provide a potentially new mechanism for NNK- and NNN-DNA adduct formation (Scheme 1B). It has long been known that the α-hydroxynitrosamine intermediates 3–6 (Scheme 1A) alkylate DNA, but their short lifetimes raise questions regarding their ability to traverse the hydrolytic cytosol. The detected nitrosamides had half-lives of 12–35 min, 100-fold more than those of the α-hydroxynitrosamines.7,8 Additionally, we showed that CH2-oxo-NNK methylates both dGuo and calf thymus DNA (Figure S2, Supplementary Information). Thus, in the case of NNK, it is plausible that CH2-oxo-NNK could be partially responsible for the methyl adducts previously thought to be formed purely by α-hydroxynitrosamine 3.
However, we note that both CH2-oxo-NNK and NNC are quite minor metabolites of NNK and NNN. It was estimated that CH2-oxo-NNK and NNC form at concentrations of ~1 nM and 10 nM, respectively, while keto alcohol 11 and lactol 12, the hydrolysis products of α-hydroxyNNK and α-hydroxyNNN, form at levels ~4000-fold higher. Because P450 2A13 and P450 2A6 are the most efficient enzymes for NNK and NNN oxidation and the formation of their known products keto alcohol 11 and lactol 12 (Scheme 1A) was rapid, it is unlikely that the low levels of CH2-oxo-NNK and NNC result from low catalytic turnover. Likewise, the positive controls indicate that analyte recovery was achievable under our incubation conditions. We noted that formation of both CH2-oxo-NNK and NNC started at 1 min, peaked at 5 min, and that both were nearly undetectable by 30 min. This may indicate that as metabolism proceeds, newly formed side products and P450-related reactive-oxygen species are eliminating the nitrosamides via secondary reactions at a rate faster than nitrosamide formation.
We were not able to quantify nitrosamide formation in these reactions. We initially attempted quantification by HPLC-radioflow techniques, but this approach was not sensitive enough (LOD = 400 fmol on column, data not shown). After nitrosamide detection was achieved by LC-NSI+-HRMS, we attempted to trap these products with N-acetyl-lysine and N-acetyl-cysteine. Though trapping was achieved with synthetic standards, this method was unsuccessful in our assay due to low trapping efficiency and low sample recovery after solid phase extraction, which resulted in no analyte detection even with accurate mass detection (data not shown). Therefore, we settled on estimating formation levels by comparing peak areas to those of spiked positive controls.
Despite being minor metabolites, the long half-lives and strong DNA-binding properties of the nitrosamides suggest potential biological relevance. However, determining whether it is more important to be low-forming and stable versus high-forming and unstable would require further study. Additionally, though CH2-oxo-NNK and NNC are formed to low extents, the nitrosamide pathway may be more efficient for other nitrosamines. For example, studies by Chowdhury et al noted considerable, processive conversion of dimethylnitrosamine and diethylnitrosamine to acid byproducts by P450 2A6.22 It was hypothesized that the α-hydroxynitrosamine intermediate decomposed within the active site and the resulting aldehyde was then oxidized to the acid. The nitrosamide hypothesis was also tested, but detection was unsuccessful. Given our results, it is plausible that nitrosamides were readily produced, but instability limited their detection.
Our synthesis of each nitrosamide proceeded essentially as expected, except for a few key findings. First, our method for keto acid 19 (Scheme 2), though not entirely novel,48 is considerably more convenient than previously reported routes.39,40 The two-step process involves milder conditions, gives reproducible yields, and simpler product purification; the latter step provides >99% pure product after only filtration. Next, the conversion of keto acid 19 to keto amide 20 (Scheme 2) was noteworthy because a previous study49 reported compound 20 to be in a ring-chain equilibrium heavily favoring the lactam (~6:1). In our hands, the compounds were readily separable on silica gel and showed no isomerization while stored neat at 2–8 °C. They were clearly distinct by NMR (Figure S3, Supporting Information). The open-chain product had two clean triplets integrating to 2H each while these signals collapsed into non-distinct multiplets integrating to 4H in the ring product. Additionally, the methyl resonance in the lactam is a singlet as opposed to a doublet in the open-chain product. In support of the results reported by Nguyen et al,49 when we performed this reaction in solvents other than DMSO, the lactam 5′-hydroxycotinine (20b, Scheme 2), predominated. Similarly, 20b was the major product when harsher amide coupling conditions were used, such as in situ acid chloride formation by oxalyl chloride or AlMe3-mediated amide formation50,51 from 18. It is apparent that DMSO and mild coupling conditions favor the open chain conformer.
Our nitrosamides were each isolated exclusively as one rotamer. This contrasts to nitrosamines commonly occur as a mixture of both (E)- and (Z)-isomers. Past studies indicate that the (E)-conformer is electronically favored for most nitrosamides.24 Furthermore, rotation to the (Z)-conformer is commonly believed to be the rate-limiting step for nitrosamide decomposition by a pericyclic process24 and thus, may not be isolatable.
The order of compound stability was CH2-oxo-NNK > NNC > CH3-oxo-NNK. This ranking fits with known factors contributing to nitrosamide decomposition.24,26,41 In hydrolytic environments,30 nitrosamides with bulkier groups adjacent to the carbonyl group are more stable. This suggests that CH3-oxo-NNK should be the least stable, consistent with our observations. Likewise, bulky groups adjacent to the nitrogen decrease stability. This is consistent with CH2-oxo-NNK being most stable and NNC being relatively less stable. The decomposition products suggest that the mechanism is primarily hydrolysis. The products shown in Scheme 3B were all either the major or only identified product. However, for NNC and CH3-oxo-NNK, we did identify a lactone and ester as minor products, respectively (Figure S1, Supporting Information). These presumably result from the extensively studied 1,3-sigmatropic rearrangement mechanism.28 Though this rearrangement is highly favored when nitrosamides are heated in organic solvents, aqueous conditions seem to favor hydrolysis and are most relevant to the in vivo situation.
With data supporting the formation of CH2-oxo-NNK, we were interested in testing one of its possible modes of DNA damage: methylation. Methylation was expected since keto acid is the major product of CH2-oxo-NNK hydrolysis (Scheme 2). This implies that methane diazohydroxide, a known methylating agent, is also released. Our results clearly demonstrated the forrmation of O6meGua in these reactions, indicating that CH2-oxo-NNK methylates DNA. In addition to the methyl DNA adducts readily formed by CH2-oxo-NNK, both CH2-oxo-NNK and NNC could potentially generate a set of novel DNA adducts (Scheme 1B). Further studies are needed to establish the structures, level, and importance of possible adducts derived from the nitrosamide pathway.
In summary, we hypothesized that NNK and NNN are metabolized by P450 2A13 and P450 2A6, respectively, to their corresponding nitrosamides in vitro. We tested this by synthesizing CH2-oxo-NNK, CH3-oxo-NNK, and NNC and evaluating their stability at pH = 7.4 and 37 °C. They were quite stable relative to the corresponding α-hydroxynitrosamines. We then showed that CH2-oxo-NNK and NNC are novel, though minor, metabolites of NNK and NNN, respectively, in an in vitro P450 model. With the knowledge that CH2-oxo-NNK has a relatively long half-life and methylates DNA, it could potentially play a role in the mechanism of carcinogenesis by NNK. More broadly, this is the first direct evidence for the conversion of nitrosamines to nitrosamides by P450 catalysis and provides rationale for further studies to determine whether this is a general transformation in nitrosamine metabolism.
Supplementary Material
Acknowledgments
Funding Information:
This study was supported by grant no. CA-81301 from the U.S. National Cancer Institute
We thank Bob Carlson for editorial assistance, Dr. Peter Villalta and Xun Ming for mass spectrometry assistance in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, and Dr. Linda Von Weymarn and the laboratory of Dr. Sharon Murphy for allowing use of their facilities and providing enzymes and advice for the cytochrome P450 incubations. We would also like to thank Dr. Adam T. Zarth and Dr. Anna K. Michel for their valuable discussions and input. The Analytical Biochemistry Shared Resource is partially supported by National Cancer Institute Cancer Center Support Grant CA-77598.
Abbreviation List
- CH2-oxo-NNK
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanedione
- CH3-oxo-NNK
4-(Nitrosoformamido)-1-(3-pyridyl)-1-butanone
- DCM
Dichloromethane
- DIPEA
N,N'-diisopropylethylamine
- DMF
N,N-dimethylformamide
- DMSO
Dimethylsulfoxide
- EDAC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- 5-HC
5'-hydroxycotinine
- Im
imidazole
- Keto alcohol
4-oxo-4-(3-pyridyl)-butanol
- Lactol
5-(3-pyridyl)-2-hydroxytetrahydrofuran
- NCS
N'-chlorosuccinimide
- NHS
N'-hydroxysuccinimide
- NNC
N'-nitrosonorcotinine
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- NNN
N'-nitrosonornicotine
- O6meGua
O6-methylguanine
- [CD3]O6meGua
O6-([(D3)methyl])guanine
- TFA
Trifluoroacetic acid
- TMEDA
Tetramethylethylenediamine
Footnotes
Select chromatograms for nitrosamide stability and decomposition, LC-MS/MS chromatograms for dGuo and DNA methylation by CH2-oxo-NNK, and 1H-NMR spectra for compounds 20a and 20b. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.United States Department of Health and Human Services, Center for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress A Report of the Surgeon General. Atlanta: 2014. [Google Scholar]
- 2.American Cancer Society. Cancer Facts Fig. Atlanta: 2014. Cancer Facts & Figures 2014. [Google Scholar]
- 3.International Agency for Research on Cancer. Smokeless Tob. Some Tobacco-specific N-Nitrosamines. Lyon: 2007. Monographs on the Evaluation of Carcinogenic Risks to Humans - Volume 89. [Google Scholar]
- 4.Hecht SS. Biochemistry, Biology, and Carcinogenicity of Tobacco-Specific N-Nitrosamines. Chem. Res. Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 5.Hecht SS, Castonguay A, Chung FL, Hoffmann D. Carcinogenicity and metabolic activation of tobacco-specific nitrosamines: current status and future prospects. IARC Sci. Publ. 1984:763–778. [PubMed] [Google Scholar]
- 6.Hecht SS. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. - Fundam. Mol. Mech. Mutagen. 1999;424:127–142. doi: 10.1016/s0027-5107(99)00014-7. [DOI] [PubMed] [Google Scholar]
- 7.Revis C, Fishbein JC, Carolina N, April RV. Effects of structure on the reactivity of α-hydroxydialkylnitrosamines in aqueous. J. Am. Chem. Soc. 1996;1:7412–7413. [Google Scholar]
- 8.Moohizuki M, Anjo T, Okada M. Isolation and characterization of N-alkyl-N-(hydroxymethyl)nitrosamines from N-alkyl-N-(hydroperoxymethyl)nitrosamines by deoxygenation. Tetrahedron Lett. 1980;21:3693–3696. [Google Scholar]
- 9.Peterson LA, Hecht SS. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991;51:5557–5564. [PubMed] [Google Scholar]
- 10.Balbo S, Johnson CS, Kovi RC, James-Yi SA, O'Sullivan MG, Wang M, Le CT, Khariwala SS, Upadhyaya P, Hecht SS. Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis. 2014;35:2798–2806. doi: 10.1093/carcin/bgu204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hecht SS, Spratt TE, Trushin N. Evidence for 4-(3-pyridyl)-4-oxobutylation of DNA in F344 rats treated with the tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N'-nitrosonornicotine. Carcinogenesis. 1988;9:161–165. doi: 10.1093/carcin/9.1.161. [DOI] [PubMed] [Google Scholar]
- 12.Stepanov I, Muzic J, Le CT, Sebero E, Villalta P, Ma B, Jensen J, Hatsukami D, Hecht SS. Analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB)-releasing DNA adducts in human exfoliated oral mucosa cells by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol. 2013;26:37–45. doi: 10.1021/tx300282k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zarth AT, Upadhyaya P, Yang J, Hecht SS. DNA adduct formation from metabolic 5′-hydroxylation of the tobacco-specific carcinogen N ′-nitrosonornicotine in human enzyme systems and in rats. Chem. Res. Toxicol. 2016;29:380–389. doi: 10.1021/acs.chemrestox.5b00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao L, Balbo S, Wang M, Upadhyaya P, Khariwala SS, Villalta PW, Hecht S. Quantification of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)-or (S)-N'-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol. 2013;26:1526–1535. doi: 10.1021/tx400235x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guttenplan JB. Effects of cytosol on mutagenesis induced by N-nitrosodimethylamine, N-nitrosomethylurea and α-acetoxy-N-nitrosodimethylamine in different strains of Salmonella: evidence for different ultimate mutagens fromN-nitrosodimethylamine. Carcinogenesis. 1993;14:1013–1019. doi: 10.1093/carcin/14.5.1013. [DOI] [PubMed] [Google Scholar]
- 16.Elespuru RK, Saavedra JE, Kovatch RM, Lijinsky W. Examination of α-carbonyl derivatives of nitrosodimethylamine and ethylnitrosomethylamine as putative proximate carcinogens. Carcinogenesis. 1993;14:1189–1193. doi: 10.1093/carcin/14.6.1189. [DOI] [PubMed] [Google Scholar]
- 17.Murphy SE, Spina DA, Nunes MG, Pullo DA. Glucuronidation of 4-((hydroxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chem. Res. Toxicol. 1995;8:772–9. doi: 10.1021/tx00047a018. [DOI] [PubMed] [Google Scholar]
- 18.Wiench K, Frei E, Schroth P, Wiessler M. 1-c-glucuronidation of N-nitrosodiethylamine and N-nitrosomethyl-N-pentylamine in vivo and in primary hepatocytes from rats pretreated with inducers. Carcinogenesis. 1992;13:867–872. doi: 10.1093/carcin/13.5.867. [DOI] [PubMed] [Google Scholar]
- 19.Wiessler M, Rossnagel G. Alpha-Glucuronides of N-nitrosomethylbenzylamine. In: Bartsch H, O’Neill IK, Schulte-Hermann R, editors. The Relevance of N-Nitroso Compounds to Human Cancer, Exposure and Mechanism. IARC Scientific Reports 84; Lyon, France: 1987. pp. 170–172. [PubMed] [Google Scholar]
- 20.von Weymarn LB, Retzlaff C, Murphy SE. CYP2A6- and CYP2A13-catalyzed metabolism of the nicotine Δ5'(1')iminium ion. J. Pharmacol. Exp. Ther. 2012;343:307–315. doi: 10.1124/jpet.112.195255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell-parikh LC, Guengerich FP. Kinetics of cytochrome P450 2E1-catalyzed oxidation of ethanol to acetic acid via acetaldehyde. J. Biol. Chem. 1999;274:23833–23840. doi: 10.1074/jbc.274.34.23833. [DOI] [PubMed] [Google Scholar]
- 22.Chowdhury G, Calcutt MW, Peter Guengerich F. Oxidation of N-nitrosoalkylamines by human cytochrome p450 2A6: Sequential oxidation to aldehydes and carboxylic acids and analysis of reaction steps. J. Biol. Chem. 2010;285:8031–8044. doi: 10.1074/jbc.M109.088039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chowdhury G, Calcutt MW, Nagy LD, Guengerich FP. Oxidation of methyl and ethyl nitrosamines by cytochromes P450 2E1 and 2B1. Biochemistry. 2012;51:9995–10007. doi: 10.1021/bi301092c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chow YL. N-Nitrosamines. American Chemical Society; Washington DC: 1979. Chemistry of N-Nitrosamides and Related N-Nitrosamino Acids; pp. 13–37. [Google Scholar]
- 25.White EH. The Chemistry of the N-Alkyl-N-nitrosoamides. II. A New Method for the Deamination of Aliphatic Amines. J. Am. Chem. Soc. 1955;77:6011–6014. [Google Scholar]
- 26.White EH, Dolak LA. N-Nitroamides and N-nitrocarbamates. IV. Rates of decomposition. A case of steric acceleration. J. Am. Chem. Soc. 1966;88:3790–3795. [Google Scholar]
- 27.Darbeau RW, White EH, Song F, Darbeau NR, Chou J. A study of essentially free carbocations derived via diazonium and oxo diazonium ions in the liquid phase. J. Org. Chem. 1999;64:5966–5978. [Google Scholar]
- 28.White EH, Field KW, Hendrickson WH, Dzadzic P, Roswell DF, Paik S, Mullen PW. Inert-molecule-separated ion pairs. Stereochemical, I8O, and product studies. J. Am. Chem. Soc. 1992;114:8023–8031. [Google Scholar]
- 29.Darbeau RW, Pease RS, Gibble RE. A study of electronic effects on the kinetics of thermal deamination ofN-nitrosoamides. J. Org. Chem. 2001;66:5027–5032. doi: 10.1021/jo001741l. [DOI] [PubMed] [Google Scholar]
- 30.Moss RA. The solvolysis of alkyl diazotates. I. Partition between carbonium ions and diazoalkanes in aqueous base. J. Org. Chem. 1966;31:1082–1087. [Google Scholar]
- 31.Loh T-P, Zhou J-R, Li X-R, Sim K-Y. A novel reductive aminocyclization for the syntheses of chiral pyrrolidines: stereoselective syntheses of (S)-nornicotine and 2-(2'-pyrrolidyl)-pyridines. Tetrahedron Lett. 1999;40:7847–7850. [Google Scholar]
- 32.Loozen HJJ, Godefroi EF, Besters JSMM. A Novel and efficient route to 5-arylated gamma-Lactones. J. Org. Chem. 1975;40:892–894. [Google Scholar]
- 33.Amin S, Desai D, Hecht SS, Hoffmann D. Synthesis of tobacco-specific N-nitrosamines and their metabolites and results of related bioassays. Crit. Rev. Toxicol. 1996;26:139–47. doi: 10.3109/10408449609017927. [DOI] [PubMed] [Google Scholar]
- 34.Balsiger RW, Montgomery JA. Synthesis of potential anticancer agents. XXV. Preparation of 6-alkoxy-2-aminopurines. J. Org. Chem. 1960;25:1573–1575. [Google Scholar]
- 35.von Weymarn LB, Zhang QY, Ding X, Hollenberg PF. Effects of 8-methoxypsoralen on cytochrome P450 2A13. Carcinogenesis. 2005;26:621–629. doi: 10.1093/carcin/bgh348. [DOI] [PubMed] [Google Scholar]
- 36.Wong HL, Murphy SE, Wang M, Hecht SS. Comparative metabolism ofN-nitrosopiperidine and N-nitrosopyrrolidine by rat liver and esophageal microsomes and cytochrome P450 2A3. Carcinogenesis. 2003;24:291–300. doi: 10.1093/carcin/24.2.291. [DOI] [PubMed] [Google Scholar]
- 37.Upadhyaya P, Lindgren BR, Hecht S. Comparative Levels of O6 -Methylguanine, pyridyloxobutyl-, and pyridylhydroxybutyl-DNA adducts in lung and liver of rats treated chronically with the tobacco-specific carcinogen 4- (methylnitrosamino)-1- (3-pyridyl)-1-butanone. Drug Metab. Dispos. 2009;37:1147–1151. doi: 10.1124/dmd.109.027078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stetter H, Schreckenberg M. A new method for addition of aldehydes to activated double bonds. Angew. Chemie Int. Ed. English. 1973;12:81. [Google Scholar]
- 39.McKennis H, Turnbull LB, Wingfield HN, Dewey LJ. Metabolites of nicotine and a synthesis of nornicotine. J. Am. Chem. Soc. 1958;80:1634–1636. [Google Scholar]
- 40.Wingfield HN., Jr 4-(3-Pyridyl)-4-ketobutyric Acid. J. Org. Chem. 1959;24:872–873. [Google Scholar]
- 41.White EH. The chemistry of the N-alkyl-N-nitrosamides. I. Methods of preparation. J. Am. Chem. Soc. 1955;77:6008–6010. [Google Scholar]
- 42.Corey EJ, Seebach D. Carbanions of 1,3-Dithianes. Reagents for C-C bond formation by nucleophilic displacement and carbonyl addition. Chemie Int. Ed. 1965;4:1075–1077. [Google Scholar]
- 43.Appel R. Tertiary phosphane/tetrachloromethane, a versatile reagent for chlorination, dehydration, and P-N linkage. Angew. Chemie Int. Ed. English. 1975;14:801–811. [Google Scholar]
- 44.Corey EJ, Erickson BW. Oxidative hydrolysis of 1, 3-dithiane derivatives to carbonyl compounds using N-halosuccinimide reagents. J. Org. Chem. 1971;36:3553–3560. [Google Scholar]
- 45.Jalas JR, Hecht SS, Murphy SE. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), a tobacco-specific carcinogen. Chem. Res. Toxicol. 2005;18:95–110. doi: 10.1021/tx049847p. [DOI] [PubMed] [Google Scholar]
- 46.Patten CJ, Smith TJ, Friesen MJ, Tynes RE, Yang CS, Murphy SE. Evidence for cytochrome P450 2A6 and 3A4 as major catalysts forN'-nitrosonornicotine α-hydroxylation by human liver microsomes. Carcinogenesis. 1997;18:1623–1630. doi: 10.1093/carcin/18.8.1623. [DOI] [PubMed] [Google Scholar]
- 47.Wong HL, Murphy SE, Hecht SS. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines:N'-Nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem. Res. Toxicol. 2005;18:61–69. doi: 10.1021/tx0497696. [DOI] [PubMed] [Google Scholar]
- 48.Stetter H, SchreckenBerg M. A new method for the addition of aldehydes to activated double bonds III. Addtion of aromatic and heterocyclic aldehydes to a,b-unsaturated nitriles. Chem. Ber. 1974;107:210. [Google Scholar]
- 49.Nguyen T, Dagne E, Gruenke L, Bhargava H, Castagnoli NJ. The tautomeric structures of 5-hydroxycotinine, a secondary mammalian metabolite of nicotine. J. Org. Chem. 1981;46:758–760. [Google Scholar]
- 50.Basha A, Lipton M, Weinreb SM. A mild, general method for conversion of esters to amides. Tetrahedron Lett. 1977;18:4171–4172. [Google Scholar]
- 51.Lipton, Michael F, Basha, Anwer, Weinreb SM. Conversion of esters to amides with dimethylaluminum amides: N,N-dimethylcyclohexanecarboxamide. Org. Synth. 1979;59:49. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.