

Abstract
Objectives
Carbon monoxide (CO) is a colorless and odorless gas that is an important cause of poisoning mortality and morbidity in the United States. At this time there is no reliable method to predict severity of poisoning or clinical prognosis following CO exposure. Whole blood cells such as peripheral blood mononuclear cells (PBMCs) and platelets have been explored for their potential to act as sensitive biomarkers for mitochondrial dysfunction.
Design
The objective of this study was to measure mitochondrial respiration using intact cells obtained from patients exposed to CO as a potential biomarker for mitochondrial inhibition with results that can be obtained in a time frame useful for guiding clinical care. This was a prospective, observational pilot study performed from July 2015 to July 2016 at a single academic tertiary care center that is the location of the region’s only multi chamber hyperbaric.
Measurements
Clinical characteristics, patient demographics, mitochondrial respiration and outcomes were recorded.
Main results
There were 7 patients enrolled with a mean COHb level was (26.8+/−10) with a mean lactate of (1.1+/−0.4 mmol/L). All 7 CO exposures were related to heat generators used during winter months with two deaths. There was a positive correlation between maximal respiration and COHb levels with both a high maximal respiration and high spare respiratory capacity correlating with a high COHb level. There was a subset of PBMCs (n=4) that were analyzed for Complex IV (cytochrome c oxidase) activity.
Conclusion
In this pilot study, measurement can be performed in an appropriate timeline for clinical care with potential to serve as a prognostic marker. Further work is necessary to develop high-resolution respirometry as a clinical tool for assessing severity of illness and guiding therapy.
Six words for indexing: Mitochondrial bioenergetics, carbon monoxide, biomarkers, acute care, mitochondrial respiration, poisoning
Introduction
Carbon monoxide (CO) is a colorless and odorless gas that is an important cause of poisoning mortality and morbidity in the United States due to a variety of causes that include hemoglobin binding, lipid peroxidation and mitochondrial inhibition.1 Death from CO poisoning has been reported to be over 1,000 in some years with over 50,000 CO cases seen in emergency departments in the US annually, with over half requiring hospitalization.2 The most serious complication for survivors of consequential CO exposure is delayed neurologic or neurocognitive sequela which can occur up to 50% of patients with symptomatic CO poisoning.3,4 At this time there is no reliable method to predict severity of poisoning or clinical prognosis following CO exposure in the acute setting.
The most common diagnostic test for suspected CO exposure is a blood carboxyhemoglobin (COHb) level: a COHb level (>10%) may confirm CO exposure but can be seen in heavy smokers. It is well-established that COHb levels do not predict either the severity of illness or patient prognosis resulting from exposure.5,6 While the COHb level is easily and rapidly available at most institutions other biomarkers having been proposed to serve as better predictors of severity and prognosis in CO poisoning that include lactate and S100B but have yielded variable results.7,8 The presence of microparticles in blood is also currently being explored as a potential biomarker for CO poisoning.9 Many of these markers act as secondary measures of CO poisoning but do not directly measure the effects of CO. Using a marker of the direct effects of CO would be preferable. Therefore measurement of mitochondrial respiration may serve as an effective biomarker in CO poisoning since it is a known Complex IV (cytochrome c oxidase) inhibitor.
Advances in relevant instruments now allow the sensitive measurement of mitochondrial respiration of human blood cells in clinical disease that includes metabolic disorders such as diabetes and heart disease. The study of mitochondrial respiration has also extended to areas of acute care that include: sepsis, traumatic injuries, and toxicology.10 Whole blood cells such as peripheral blood mononuclear cells (PBMCs) and platelets have been explored for their potential to act as sensitive biomarkers for mitochondrial dysfunction in lieu of invasive and time consuming tissue biopsies.11,12
Measurement of mitochondrial respiration in cells isolated from human blood may offer a more sensitive assay for CO by directly measuring mitochondrial respiration. The objective of this study is two-fold: (1) To explore the logistics of measuring mitochondrial respiration that fits in an analytical timeframe that may be useful to identify clinical abnormalities or to help guide therapy. (2) To evaluate that the measurement of mitochondrial respiration using intact cells obtained from patients exposed to CO as a potential biomarker for mitochondrial inhibition to indicate disease severity. We hypothesize that mitochondrial oxygen consumption is decreased in patients with clinical significant CO poisoning and that cellular oxygen utilization better correlates with severity of CO poisoning better than do COHb levels.
Methods and Material
Study Design
This was a prospective, observational pilot study performed from July 2015 to July 2016 at a single academic tertiary care center that is the location of the region’s only multichamber hyperbaric chamber managed by the Division of Hyperbaric Medicine. All patients 18 years and older who presented to the Emergency Department with suspected CO exposure based on history or a positive COHb level were screened for eligibility. This study was approved by the institutional review board and informed consent was obtained from the patient or an appropriate surrogate.
Clinical Measurements
Clinical characteristics, patient demographics, and outcomes were recorded by academic research associates blinded to the results of the study measurements. All patients had COHb level and lactate levels performed as part of routine care for suspected CO poisoning. Other clinical data recorded include abnormal physical exam findings, all laboratory results ordered as part of clinical care, electrocardiograms, any treatments received that include normobaric oxygen and if hyperbaric oxygen treatment was received. Enrolled patients were followed up to discharge from the hospital to determine survival and any neurocognitive defects in the acute phase.
Outcomes and Statistics
The primary outcome of this study was the association between COHb and corrected PBMC mitochondrial respiration to severity of CO poisoning defined as end-organ injury or death. To account for multiple statistical testing (7 tests) on same data, an adjusted p–value of .007 using a Bonferroni correction was considered statistically significant and data was fitted to a linear model. All analyses were performed using SAS statistical software (Version 9.4, SAS Institute, Cary NC).
Blood collection and cell isolation
A single non-tourniqueted venous blood sample was collected from patients with CO exposure after informed consent. Peripheral blood mononuclear cells (PBMCs) were isolated from patient blood for the measurement of mitochondrial respiration. PBMCs consisting of lymphocytes and monocytes were isolated from citrated whole blood by density gradient centrifugation in the following manner: Blood samples were diluted 1:1 using a balanced salt solution (anhydrous d-glucose 5.5mM, CaCl2 5mM, MgCl2 0.98mM, KCl 5.4mM, Tris 145mM, and NaCl 140mM with pH adjusted to 7.6) and layered on top of Ficoll-Paque PLUS (density 1.077g/mL; Amersham Biosciences, Piscataway, NJ). The sample was centrifuged at 400g for 40 minutes at 20°C. The layer containing the PBMCs at the interface was gently aspirated and centrifuged again at 1,800g for 5 minutes to remove any platelets.11 The PBMC pellet was then resuspended in Hank balanced salt solution (pH 7.40) containing 5.5mM glucose, 1mM pyruvate, and 10mM 4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid. Cell counts were performed using trypan blue exclusion (Countess II, Life Technologies, Grand Island, NY) with more than 95% viability.
Measurement of mitochondrial respiration: Intact PBMCs were placed in a 2-mL chamber at a final concentration of 2–3×106 cells/mL with a goal concentration of 4–6×106 cells per chamber. Measurement of oxygen consumption was performed at 37°C in a high-resolution oxygraph (Oxygraph-2k OROBOROS Instruments, Innsbruck, Austria) corrected for cell number. Oxygen flux (in pmol O2/s/106 cells), which is directly proportional to oxygen consumption, was recorded continuously using DatLab software 6 (Oroboros Instruments). Figure 1 demonstrates the sequence of compounds used to assess mitochondrial oxygen utilization and a typical oxygraph tracing. In addition, 20 ul of succinate (final concentration of 10 mM) was administrated between the injections of rotenone and antimycin to check for any disruption of the cell’s outer membrane. Succinate is a Complex II substrate and does not permeate intact cellular membranes. Any elevation in mitochondrial respiration upon injection of succinate after Complex I blockade with rotenone indicates injured/dead cells.
Figure 1.
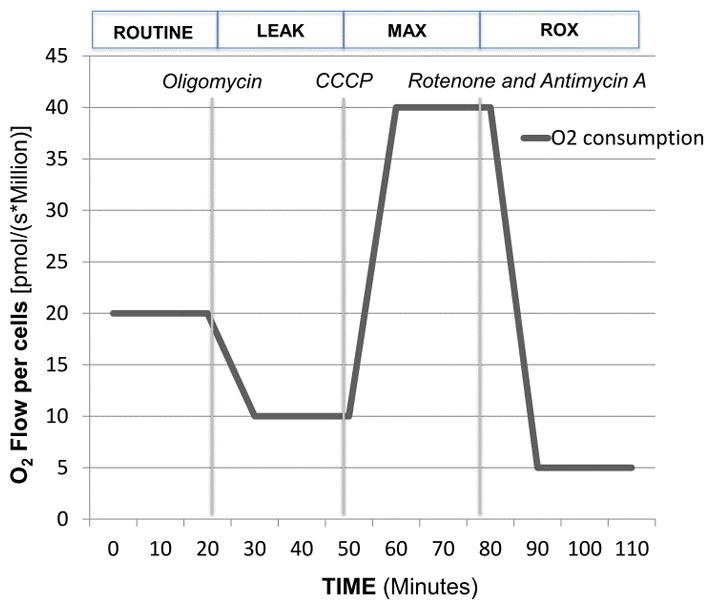
After routine oxygen consumption was recorded for 10 minutes, the following sequential injections of select compounds were carried out adhering to a SUIT (substrate-uncoupler-inhibitor-titration) protocol that provides a wealth of information on the key parameters of mitochondrial respiration for intact cells. The following terms are important in the interpretation of mitochondrial respiration:10
Routine respiration (ROUTINE): It is the oxygen consumption due to the combination of ATP production and proton leak. This represents energy demand under steady state conditions. Changes in routine respiration can be from a change in leak and/or ATP-linked respiration.
Proton Leak (LEAK): The remaining mitochondrial respiration after the injection of a Complex V inhibitor, oligomycin (final concentration of 2.5 uM), is due to proton leak. A degree of proton leak is expected under normal mitochondrial respiration. Significant proton leak can be an indication of mitochondrial damage from direct injury.
Electron Transport System Maximal Respiration (ETS or MAX): The addition of a mitochondrial uncoupler such as dinitrophenol (DNP) or Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (CCCP) stimulates maximal respiration by mimicking a physiological energy demand leading to a increase in oxygen consumption. The final concentration of CCP ranged from 0.5–5 uM with maximal respiration obtained usually obtained with a final concentration of 1–2 uM.
Residual oxygen consumption (ROX): The addition of mitochondrial inhibitors such as the combination of rotenone (final concentration of 0.5 uM) and antimycin A (final concentration of 2.5 uM) will completely shut down the electron transport chain. The remaining oxygen consumption is due to non-mitochondrial respiration such as oxidases and other cellular enzymes. All respiration data is corrected for ROX.
ATP-linked production: The difference between ROUTINE and LEAK is oxygen consumption related to ATP production. The decrease in oxygen consumption with the injection of oligomycin represents the oxygen consumption utilized for ATP production and often referred to as ATP-linked respiration.
Spare respiratory capacity (SRC): The difference between maximal respiration and routine respiration represents the cell’s SRC. The SRC is thought to indicate the ability of the cell to respond to the energetic demand and thus a measure of a cell’s fitness. A decrease in the SRC may limit the cell’s ability to handle a stress response resulting in mitochondrial dysfunction.
In a subset of patients (n=4) we performed a comprehensive SUIT protocol as depicted in Figure 2. We used frozen patient samples stored less than a month containing PBMCs obtained in the same manner as intact PBMCs and digitonin was used to permeabilized PBMCs. We permeabilized PBMCs stored at −80°C to better examine the function of individual complexes with particular focus on Complex IV. For permeabilization we used digitonin (8 ug/106 cells), with the sequence of titrations found in Figure 2.
Figure 2.
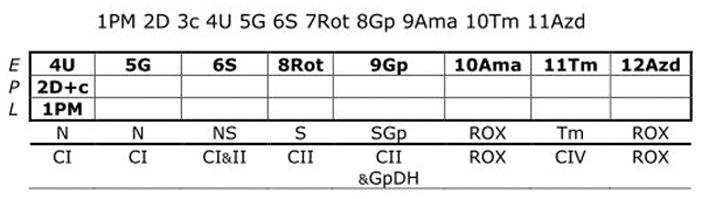
This SUIT protocol allows for the examination of Complex I and II-linked activity as well as Complex IV. Correction for chemical background was done with DatLab for Complex IV activity obtained with ascorbate and N,N,N′,N′-tetramethyl-p-phenylenediamine dihydrochloride (TMPD). In short, once proper permeabilization of cells was achieved with digitonin, cytochrome c was used as a quality check to make sure over-permeabilization did not occur and the sequence of injections given as seen in Figure 2. Key parameters in respiration was obtained for each injection with Complex IV activity recorded. Further details of cell permeabilization in intact cells and muscle fibers can be found here.13
The time from ED presentation to blood collection for mitochondrial analysis was less than 1 hour. All samples were processed and mitochondrial respiration was obtained in less than 2 hours with the actual measurement of mitochondrial respiration taking less than 30-minutes in most cases after chamber calibration.
Results
We enrolled 7 patients in this preliminary study for detailed measurement of mitochondrial respiration. At enrollment, the mean COHb level was (26.8+/−10) and the mean lactate was (1.1+/−0.4 mmol/L). All of the CO exposures were related to heat generators used during winter months. Patient demographics and clinical characteristics of the entire cohort are summarized include the following: Of these patients 57% (n=4) were men and 43% (n=3) were women. The mean age was 52.2 +/− 13.4 years. On presentation to the emergency department 57% of patients (n=4) had no complaints, 57% (n=4) had headache, 28.5% (n=2) had syncope, 14.2% (n=1) had chest pain or shortness of breath, and 28.5% (n=2) had abdominal pain. EKG abnormalities were noted in 57% (n=4) of patients with sinus tachycardia being the most common finding. In regards to treatment, 100% (n=7) of patients received normobaric oxygen and 71.4% (n=5) of patients underwent hyperbaric oxygen treatment. As an outcome index, two of the patients died. Both had presented to the emergency department related to faulty heat generators with confirmed CO exposure (COHb less than 10%).
The first fatality was found unresponsive at home by a family member with an alarming CO detector. The family member called EMS who measured the CO level in the air above 150 ppm and the patient was brought into the ED with agonal respirations with loss of pulses where resuscitation was initiated. The patient expired during resuscitation with the sample being drawn in addition to other labs at the start of the resuscitation. The family member was found to have a COHb of 19 with a mild headache. The second fatality was also the result of a faulty heat generator used during a power outage in the winter. The patient was found unresponsive while sleeping on the ground level next to a heat generator by their significant other who was sleeping upstairs. The patient was brought in as a cardiac arrest without successful resuscitation. The family member was found to have a COHb of 26 and underwent HBO treatment.
The PBMC cell count in each O2K chamber after PBMC isolation ranged between 3–4×106 PBMCs/chamber. One titration of oligomycin was required to inhibit Complex V resulting in leak and on average 2–3 titrations of CCCP were required to obtain maximal respiration consistent with prior studies involving intact PBMCs. Figure 3a and 3b illustrate that there is a positive correlation between maximal respiration and COHb levels with both a high MAX and high SRC correlating with a high COHb level. There was no correlation between COHb levels and with the following parameters of mitochondrial respiration: ROUTINE, LEAK, ROX, and ATP-linked respiration.
Figure 3.
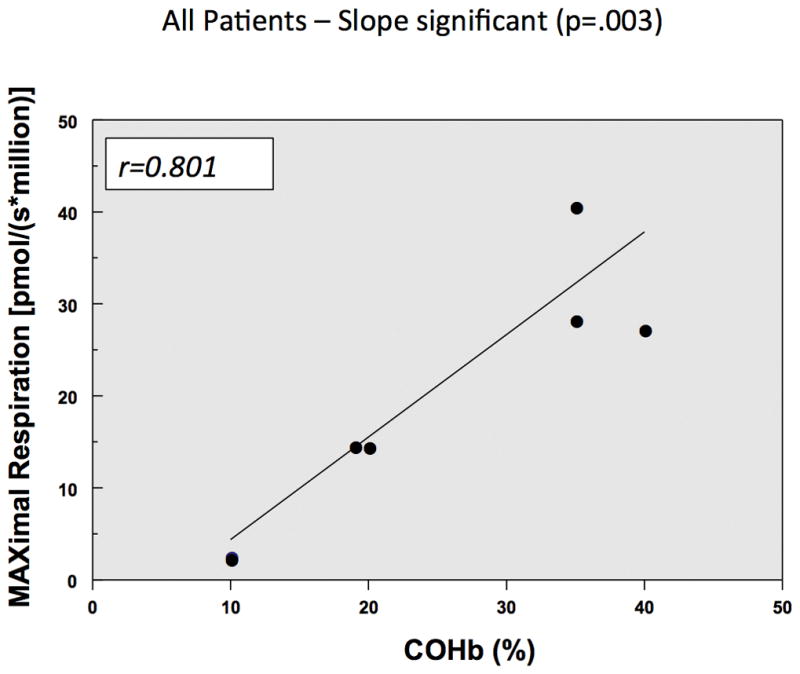
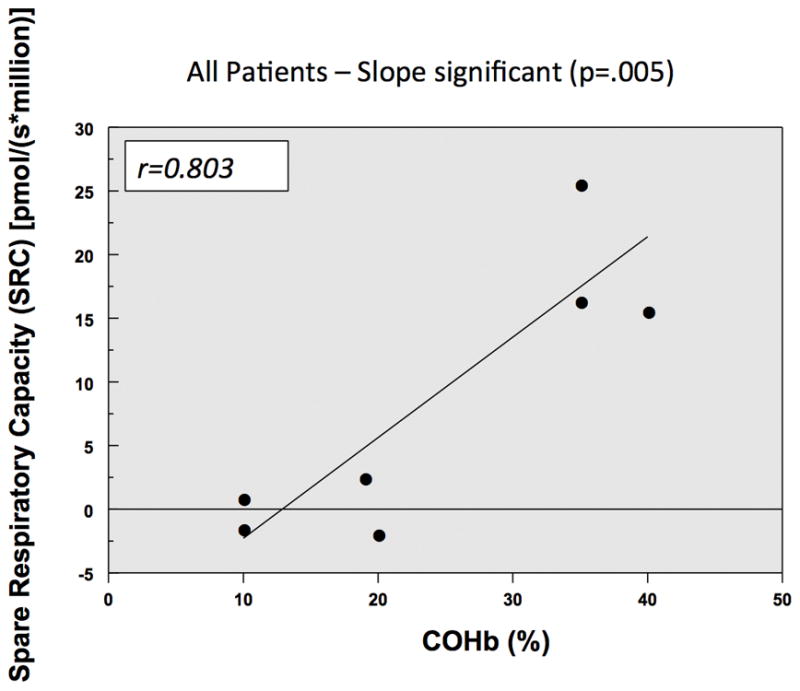
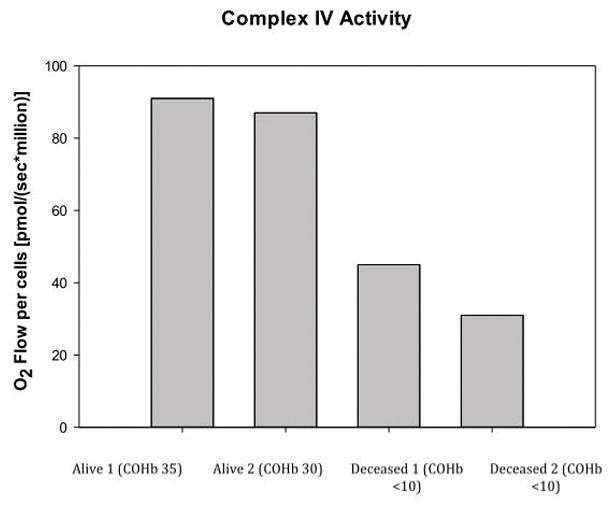
Figure 4 illustrate that in the two patients who died from CO poisoning had decreased Complex IV mitochondrial respiration when compared to two living patients exposed to CO who had higher CIV mitochondrial respiration with their initial CO levels.
Figure 4.
Discussion
This study demonstrates the application of the measurement of mitochondrial respiration as a potential sensitive assay for cellular dysfunction from CO poisoning. We found that patients with elevated CO levels, but no clinically discernible end-organ injury or mortality, showed a normal response in key parameters of mitochondrial respiration when compared to those patients who expired from CO poisoning. We also found that SRC, which represents the mitochondrial bioenergetic reserve, was significantly decreased in patients who expired from CO poisoning.14 In addition a more comprehensive SUIT carried out in permeabilized PBMCs for a subset of patients which demonstrated a decrease in Complex IV respiration in expired patients when compared to patients who survived. While this is a small sample size, it is of interest to note that the measured COHb level was not predictive of outcome or end-organ injury in CO poisoning. It does appear that the measurement of mitochondrial respiration may hold greater utility in this regard. Patients with high COHb levels maintained intact mitochondrial respiration, which most likely indicates that despite high COHb levels being found, CO poisoning had not yet affected organ tissue; hence normal mitochondrial respiration was found. The two patients who expired from CO poisoning had low COHb levels but also perturbed mitochondrial respiration. It is important to note that the two deaths were from faulty heat generators making a mixed exposure to other mitochondrial inhibitors less likely when compared to a fire. This observation does further support the concept that the COHb level serves more as a marker of exposure and is not predictive of clinical status or outcome. A larger sample size than we have studied in this preliminary work is needed to make any definitive conclusion comparing the measurement of mitochondrial respiration to COHb levels.
In this study the mean lactate levels were 1.1+/−0.4 mmol/L with no correlation with COHb or mitochondrial respiration. The initially lactate have also been examined to determine the relationship between the COHb level and clinical status of patients with CO exposure.15–16 The limitations for these studies is the retrospective nature of the studies as well as in many cases the CO exposure were related to fires so the possibility of other mitochondrial inhibitors such as cyanide cannot be excluded as cyanide is well known to cause increase in lactate. In this study only patients with a presumable pure CO exposure were captured due to faulty heat generators.
The use of circulating blood cells such as white blood cells or platelets potentially serves as an early warning or “canary in the coal mine” for mitochondrial dysfunction that may occur in conditions of metabolic stress in areas of acute care such as sepsis.17–19 Once isolation of PBMCs has occurred, the time to obtain key mitochondrial respiration parameters (Routine, LEAK, MAX, ROX) is typically less than an hour, which may have relevance for time-sensitive clinically problems. Prior to the development of sensitive instruments to measure mitochondrial oxygen consumption the most common approach has been to use isolated mitochondria from biopsies that is invasive, painful and time-consuming. The advantages of our described method are the minimally non-invasive method (phlebotomy), and the ability to perform multiple measurements, monitor response to treatment, and established clinical prognosis. Another important consideration with the use of the O2K is that it is not labor or time-intensive requiring minimal training and upkeep that can be used both in the acute phase and also during the hospital course.
As noted above, one of the mitochondrial respiration parameters that can be calculated is SRC, which represents the energetic reserve of the mitochondria. A decrease in SRC may indicate dysfunction of the mitochondria and the inability to meet an increase in bioenergetic demand that occur in a stress response from acute poisoning or sepsis.14 Our ex vivo study demonstrates decrease in both in the maximal respiration and SRC in blood cells exposed to cyanide.11 Studies in the bioenergetic function of immune cells in sepsis and traumatic injuries often show a decreased SRC. When a stress response requires an energetic demand beyond that of the cell’s SRC, cellular dysfunction can occur and this leads to organ dysfunction.17,18
One of the molecular mechanisms of CO poisoning is the inhibition of Complex IV similar to cyanide and hydrogen sulfide. We were able to retrospectively perform a more detailed respiration analysis on 4 frozen patient samples (2 deceased and 2 alive) to study the effect of CO on Complex IV activity with the parameters of intact respiration similar to what was found in frozen samples.23 It is well-known that the measurement of mitochondrial function is well preserved in properly frozen samples at −80°C.22,23 While only performed in a limited sample, there was a significant decrease in Complex IV activity in the expired patients compared to samples obtained from surviving patients having good outcomes. The sample size is too small to draw any definitive conclusion so this does warrant further exploration in future studies involving the measurement of Complex IV activity with CO poisoning. One of the considerations in performing a more detailed respiration analysis is the permeabilization of the cell with a detergent such as digitonin or saponin, as well as more extensive use of various substrates. Such experiments will be more time consuming and more technically challenging compared to performing the measurement of mitochondrial respiration in intact cells.20
Currently at this time the primary treatment for significant CO poisoning includes high-flow oxygen and hyperbaric oxygen (HBO). At this juncture the efficacy of HBO in CO poisoning is debatable with mixed results in the literature. Our preliminary data suggest an additional mechanism of CO poisoning that includes mitochondrial dysfunction which offers an alternative pathway for therapeutic intervention. There are currently treatments available directed towards the mitochondrial respiratory chain to enhance respiration.24,25 As further work in this area that explores mitochondrial dysfunction in CO poisoning develops this may lead to potential therapeutic options at this level.
Our study demonstrates that measuring key parameters in mitochondrial respiration may be a more sensitive measure of cellular function in CO poisoning when compared to a COHb level. The use of mitochondrial bioenergetics represents a potential new avenue for diagnostics and the ability to measure response to therapy for other poisonings that affect the mitochondria such as cyanide and hydrogen sulfide where the need for immediate clinical intervention, thus making the measurement of mitochondrial respiration feasible.
Limitations
One of the limitations of this study is the use of intact PBMCs to study mitochondrial respiration. In our study we used PBMCs that are a mixed population of both monocytes and lymphocytes. It is not clear which cell type is the ideal cell as there may be differences in the bioenergetic profile in various cell types.26 Another limitation is whether circulating cells reflect the mitochondrial function of organs affected by mitochondrial poisons such as carbon monoxide.27 Also as mitochondrial function was obtained in the acute setting, it is less clear the relationship between mitochondrial function and the development of delayed neurologic sequelae (DNS).
Another factor involving CO poisoning involves sources that may include multiple mitochondrial poisons. Significant exposure to smoke inhalation and fires can also result in substantial cyanide and hydrogen sulfide exposure. A previous study demonstrated that in vitro exposure of human cells to cyanide resulted in decreased mitochondrial respiration.11 It is likely that a mixed exposure to various mitochondrial poisons will make the interpretation of mitochondrial respiration difficult.28 In this study the 7 enrolled patients were exposed to faulty heat generators as their source of CO that makes exposure to other mitochondrial poisons less likely.
Conclusions
In this pilot study, measurement can be performed in an appropriate timeline for clinical care with potential to serve as a prognostic marker. Further work is necessary to develop high-resolution respirometry as a clinical tool for assessing severity of illness and guiding therapy.
Acknowledgments
Funding for study:
K12 HL109009 from the National Heart, Lung, and Blood Institute (DJ)
UPENN Department of Emergency Medicine start-up funding (DJ)
N000141612100 from the Office of Naval Research (DME).
Footnotes
Institution where work was performed:
University of Pennsylvania Perelman School of Medicine, Department of Emergency Medicine
Contributor Information
David H. Jang, Assistant Professor, University of Pennsylvania Perelman School of Medicine, Department of Emergency Medicine, Division of Medical Toxicology and Critical Care Medicine. Role: Primary author; performed majority of experiment.
Matthew Kelly, Assistant Professor, University of Pennsylvania Perelman School of Medicine, Department of Emergency Medicine, Division of Hyperbaric Medicine. No conflicts, no disclosures. Role: Patient recruitment and review.
Kevin Hardy, Assistant Professor, University of Pennsylvania Perelman School of Medicine, Department of Emergency Medicine, Division of Undersea and Hyperbaric Medicine. No conflicts, no disclosures. Role: Patient recruitment and review.
David S. Lambert, Assistant Professor, University of Pennsylvania Perelman School of Medicine, Department of Emergency Medicine, Division of Undersea and Hyperbaric Medicine.
Frances S. Shofer, Adjunct Professor, University of Pennsylvania Perelman School of Medicine, Department of Emergency Medicine. Role: Performed statistical analysis and review of manuscript.
David M. Eckmann, Horatio C. Wood Professor of Anesthesiology and Critical Care, Professor of Bioengineering, Institute for Medicine and Engineering, Cardiovascular Institute and Institute for Translational Medicine and Therapeutics, University of Pennsylvania. Role: Senior author, provided critical guidance for manuscript and reviewed.
References
- 1.Iqbal S, Clower JH, King M, et al. National carbon monoxide poisoning surveillance framework and recent estimates. Public Health Rep. 2012 Sep-Oct;127(5):486–96. doi: 10.1177/003335491212700504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sircar K, Clower J, Shin MK, et al. Carbon monoxide poisoning deaths in the United States, 1999 to 2012. Am J Emerg Med. 2015 Sep;33(9):1140–5. doi: 10.1016/j.ajem.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pepe G, Castelli M, Nazerian P, et al. Delayed neuropsychological sequelae after carbon monoxide poisoning: predictive risk factors in the Emergency Department. A retrospective study. Scand J Trauma Resusc Emerg Med. 2011 Mar 17;19:16. doi: 10.1186/1757-7241-19-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thom SR, Taber RL, Mendiguren II, et al. Delayed neuropsychologic sequelae after carbon monoxide poisoning: prevention by treatment with hyperbaric oxygen. Ann Emerg Med. 1995 Apr;25(4):474–80. doi: 10.1016/s0196-0644(95)70261-x. [DOI] [PubMed] [Google Scholar]
- 5.Kaldirim U, Yolcu U, Arziman I, et al. The relationship between blood lactate, carboxy-hemoglobin and clinical status in CO poisoning. Eur Rev Med Pharmacol Sci. 2014 Oct;18(19):2777. [PubMed] [Google Scholar]
- 6.Hampson NB, Hauff NM. Carboxyhemoglobin levels in carbon monoxide poisoning: do they correlate with the clinical picture? Am J Emerg Med. 2008 Jul;26(6):665–9. doi: 10.1016/j.ajem.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Akdemir HU, Yardan T, Kati C, et al. The role of S100B protein, neuron-specific enolase, and glial fibrillary acidic protein in the evaluation of hypoxic brain injury in acute carbon monoxide poisoning. Hum Exp Toxicol. 2014 Nov;33(11):1113–20. doi: 10.1177/0960327114521049. [DOI] [PubMed] [Google Scholar]
- 8.Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011 Aug;30(8):836–43. doi: 10.1177/0960327110384527. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Yang M, Kosterin P, et al. Carbon monoxide inhalation increases microparticles causing vascular and CNS dysfunction. Toxicol Appl Pharmacol. 2013 Dec 1;273(2):410–7. doi: 10.1016/j.taap.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang DH, Greenwood J, Spyres MB, Eckmann DM. Measurement of mitochondrial respiration and motility in acute care: sepsis, trauma and poisoning. Journal of Intensive Care Medicine. 2016 Jul 21; doi: 10.1177/0885066616658449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang DH, Shofer FS, Weiss SL, et al. Impairment of Mitochondrial Respiration Following Ex-vivo Cyanide Exposure in Peripheral Blood Mononuclear Cells. Clin Tox. 2016 Feb;5:1, 5. doi: 10.3109/15563650.2016.1139712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jang DH, Lampe J, Becker LB. The Potential Application of Mitochondrial Medicine in Toxicologic Poisoning. J Med Toxicol. 2015 Jun;11(2):201–7. doi: 10.1007/s13181-015-0478-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pesta D, Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol. 2012;810:25–58. doi: 10.1007/978-1-61779-382-0_3. [DOI] [PubMed] [Google Scholar]
- 14.Chacko BK, Kramer PA, Ravi S, et al. The Bioenergetic Health Index: a new concept in mitochondrial translational research. Clin Sci (Lond) 2014 Sep;127(6):367–73. doi: 10.1042/CS20140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moon JM, Shin MH, Chun BJ. The value of initial lactate in patients with carbon monoxide intoxication: in the emergency department. Hum Exp Toxicol. 2011 Aug;30(8):836–43. doi: 10.1177/0960327110384527. [DOI] [PubMed] [Google Scholar]
- 16.Cervellin G, Comelli I, Rastelli G, et al. Initial blood lactate correlates with carboxyhemoglobin and clinical severity in carbon monoxide poisoned patients. Clin Biochem. 2014 Dec;47(18):298–301. doi: 10.1016/j.clinbiochem.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 17.Villarroel JP, Guan Y, Werlin E, et al. Hemorrhagic shock and resuscitation are associated with peripheral blood mononuclear cell mitochondrial dysfunction and immunosuppression. J Trauma Acute Care Surg. 2013 Jul;75(1):24–31. doi: 10.1097/TA.0b013e3182988b1f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiss SL, Selak MA, Tuluc F, et al. Mitochondrial dysfunction in peripheral blood mononuclear cells in pediatric septic shock. Pediatr Crit Care Med. 2015 Jan;16(1):e4–e12. doi: 10.1097/PCC.0000000000000277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Japiassú AM, Santiago AP, d’Avila JC, et al. Bioenergetic failure of human peripheral blood monocytes in patients with septic shock is mediated by reduced F1Fo adenosine-5′-triphosphate synthase activity. Crit Care Med. 2011 May;39(5):1056–63. doi: 10.1097/CCM.0b013e31820eda5c. [DOI] [PubMed] [Google Scholar]
- 20.Pesta D, Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol. 2012;810:25–58. doi: 10.1007/978-1-61779-382-0_3. [DOI] [PubMed] [Google Scholar]
- 21.Soloviov M, Meuwly M. CO-dynamics in the active site of cytochrome c oxidase. Chem Phys. 2014 Apr 14;140(14):145101. doi: 10.1063/1.4870264. [DOI] [PubMed] [Google Scholar]
- 22.Keane KN, Calton EK, Cruzat VF, et al. The impact of cryopreservation on human peripheral blood leucocyte bioenergetics. Clin Sci (Lond) 2015 May 1;128(10):723–33. doi: 10.1042/CS20140725. [DOI] [PubMed] [Google Scholar]
- 23.Nazarpour R, Zabihi E, Alijanpour E, et al. Optimization of Human Peripheral Blood Mononuclear Cells (PBMCs) Cryopreservation. Int J Mol Cell Med. 2012 Spring;1(2):88–93. [PMC free article] [PubMed] [Google Scholar]
- 24.Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med. 2013 Feb;45(1):4–16. doi: 10.3109/07853890.2011.605389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ehinger JK, Piel S, Ford R, et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat Commun. 2016 Aug 9;7:12317. doi: 10.1038/ncomms12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chacko BK, Kramer PA, Ravi S, et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. 2013 Jun;93(6):690–700. doi: 10.1038/labinvest.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karamercan MA, Weiss SL, Villarroel JP, et al. Can peripheral blood mononuclear cells be used as a proxy for mitochondrial dysfunction in vital organs during hemorrhagic shock and resuscitation? Shock. 2013 Dec;40(6):476–84. doi: 10.1097/SHK.0000000000000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alarie Y. Toxicity of fire smoke. Crit Rev Toxicol. 2002 Jul;32(4):259–89. doi: 10.1080/20024091064246. [DOI] [PubMed] [Google Scholar]