

Abstract
Epigenetic modifications affect gene expression without changes in the actual DNA sequence. Two most important mechanisms include DNA methylation and histone tail modifications (especially acetylation and methylation). Epigenetic modulation is a part of normal physiologic development; its dysregulation is an important mechanism of pathogenesis of some cancers including acute myeloid leukemia (AML). Despite significant progress in understanding the pathogenesis of AML, therapeutic options remain quite limited. Technological advances have facilitated understanding of aberrant DNA methylation and histone methylation/acetylation as key elements in the development of AML and uncovered several recurrent mutations in genes important for epigenetic regulation. However, much remains to be learned about how to exploit this knowledge for epigenetic therapeutic targeting. Currently, no epigenetic therapy is approved for the treatment of AML although two DNA methyltransferase inhibitors (azacitidine and decitabine) are commonly used in clinical practice. Among the other epigenetic modifiers undergoing research in AML, the histone deacetylase inhibitors are the most studied. Other promising drugs such as inhibitors of histone methylation (e.g. EZH2 and DOT1L inhibitors), inhibitors of histone demethylases (e.g. LSD1 inhibitors), inhibitors of bromodomain-containing epigenetic “reader” BET proteins, and inhibitors of mutant isocitrate dehydrogenases are at early stages of clinical evaluation.
Keywords: epigenetics, acute myeloid leukemia, DNA methylation, histones
INTRODUCTION
In AML, primitive malignant clonal hematopoietic cells proliferate and accumulate in bone marrow, peripheral blood and occasionally other tissues1. AML accounts for 90% of all acute leukemia in adults, and 15–20% in children2. Despite extensive clinical research of many agents and exponential progress in dissecting the underlying molecular mechanisms of pathogenesis, and in contrast to most other hematologic malignancies where we have seen the approval of multiple novel agents, the backbone of induction chemotherapy for AML remains the same anthracycline/cytarabine combination regimen (3+7) developed in the 1970s3.
The etiologies for AML have not been fully elucidated. Risk factors include previous exposure to chemotherapy and radiation. Myelodysplastic syndromes (MDS) and myeloproliferative neoplasms (MPN) can also progress to AML. Along with many genetic alterations resulting in activation of oncogenes or inactivation of tumor suppressor genes (TSG)4, epigenetic changes have been shown to play an important role in carcinogenesis. Epigenetic modification refers to a stable, mitotically perpetuated regulatory mechanism of gene expression that is not associated with a change in the actual DNA sequence5. Main mechanisms of epigenetic modifications include DNA methylation, histone tail modifications such as acetylation and methylation6, 7 and changes in the small regulatory non-coding anti-sense RNAs (i.e. microRNA) which can result in transcriptional or posttranscriptional gene silencing or activation8. However, the epigenetic dysregulation that contributes to leukemogenesis is not fully understood9. A better understanding of epigenetic dysregulation will shed light on the mechanism of leukemogenesis, provide prognostic information, and elucidate potential therapeutic targets. In this review, we overview the current understanding of the epigenetic dysregulation in AML, discuss the available epigenetic therapies, and forecast the future directions of research in this area. Due to space limitations, we will focus on DNA methylation and histone modifications and their therapeutic targeting while inhibitory RNAs and the use of antisense oligonucleotides will not be discussed.
DNA METHYLATION IN AML
DNA methylation involves the addition of a methyl group to cytosine or adenine residues in DNA resulting in impaired gene transcription and translation. As such, DNA methylation provides an epigenetic method for controlling gene expression. This process is an essential component of normal development and cell differentiation. Certain cancer cells have altered patterns of DNA methylation via the global change of DNA methylation, or hypermethylation in specific regions of the genome10. While global hypomethylation is associated with chromosomal instability in vitro and may play a role in carcinogenesis11, it is believed that aberrant DNA hypermethylation contributes to carcinogenesis by silencing TSG.
In contrast to normal hematopoietic cells, hypermethylation of TSG promoters is found at a high frequency in AML12. However, only a few of the frequently methylated genes in AML have known tumor suppressor function13. In addition, promoter hypermethylation in AMLin vitro does not always silence critical genes, especially TSGin vivo13, 14. Therefore, only some of the genes which are hypermethylated in promoters are clearly significant to the neoplastic process. Regardless of the role of hypermethylation of TSG in the pathogenesis of AML, frequent gene methylation is generally associated with poor prognosis in AML13 and clinical outcomes could be predicted based on DNA methylation cluster, including the groups without specific mutation14.
MUTATIONS IN GENES WHICH INFLUENCE DNA METHYLATION
DNMT mutations in AML
DNA methylation is induced by a group of epigenetic “writer” enzymes called DNA methyltransferases (DNMTs) that add a methyl group to the cytosine residues, mostly within CpG islands, leading to epigenetic silencing of target genes15, 16. Of the 3 described DNMTs, DNMT1 mostly maintains existing methylation during replication of the DNA while DNMT3A and DNMT3B on the other hand are generally responsible for de novo methylation of previously unmethylated DNA17.
The frequent occurrence of recurrent mutations in enzymes associated with DNA methylation in AML cells suggests that aberrant epigenetic modulation of genome plays an important role in leukemogenesis. DNMT3A is one of the most commonly mutated genes in AML (4% to 22% of adult AML patients and up to 36% of cytogenetically normal [CN] AML)17. DNMT3A mutations are associated with monocytic subtype, older age, and concurrent mutations including FLT3, NPM1, andisocitrate dehydrogenase (IDH)1 and218, 19. Interestingly, AML patients with mutations in DNMT3A (along with NPM1 or fusions in MLL1) appear to benefit from dose intensification of daunorubicin (90mg/m2) during induction chemotherapy20.
TET2
TET (Ten-Eleven Translocation) 1~3 genes encode for epigenetic “eraser” enzymes that induce hydroxymethylation by catalyzing the conversion of 5-methylcytosine to 5-hydroxymethylcytosine, which is followed by passive demethylation16, 21. Mutations in TET2 have been detected in 7–23% of AML22, 23. Recent study suggests that TET2 mutations occur more frequently in CN-AML and are associated with older age, higher white blood cell counts, and lower platelet counts16, 23. In addition, TET2 and IDH mutations are mutually exclusive, which supports the role of aberrant hydroxymethylation in leukemogenesis as IDH gain of function mutations produce the oncometabolite 2-hydroxyglutarate (2-HG) which inhibits TET2 catalytic activity (see next section)16, 24. Experimental models suggest that TET2 mutations result in a dysregulation of hematopoietic cell-renewal control mechanisms and facilitate the acquisition of additional somatic mutations24–26. Nonetheless, the precise mechanism by which TET2 mutations drive leukemogenesis are still to be defined.
MUTATIONS IN GENES THAT IMPACT BOTH DNA METHYLATION AND HISTONE POSTTRANSLATIONAL MODIFICATIONS
IDH1 and 2
IDH 1 and 2 are a group of NADP+ dependent enzymes which catalyze the conversion of isocitrate to α-ketoglutarate (α-KG) in the Krebs cycle and are thought to be involved in the prevention of oxidative damage within the cell16, 24, 27. IDH mutations are found in 15~30% of de novo AML and secondary AML24, 28. These neomorphic mutations confer a novel activity to the enzymes catalyzing the conversion of α-KG to its D stereoisomer 2-HG24, 27–29. Indeed, high2-HGlevels are detectable in AML patients who have IDH mutations, and serum levels of 2-HG have been shown to have a diagnostic utility, to function as a biomarker for monitoring of disease activity and therapeutic response, and to potentially predict clinical outcomes27–29. The mechanisms by which mutatingIDH 1 and 2 drive pathogenesis of AML are under active investigation but it appears that the functional loss of TET2 activity by the depleted level of the TET2 cofactor α-KG and the inhibitory effects of 2-HG on TET2 function play important roles16, 24. IDH mutations also contribute to leukemogenesis in TET2-independent manners16. The oncometabolite 2-HG can also inhibit several other key epigenetic modifiers involved in histone and DNA methylation and demethylation leading to aberrant DNA and histone methylation and epigenetic remodeling30. The activity of histone demethylases are also dependent on α-KG and the epigenetic functional impact of inhibition of histone demethylase by low levels of α-KG may contribute to leukemogenesis by blocking differentiation30, 31. In addition, it is thought that high levels of α-HG may increase the production of reactive oxygen species and subsequent DNA damage31. The prognostic significance of IDH mutations remains debated.
MUTATIONS IN GENES WHICH AFFECT HISTONE POSTTRANSLATIONAL MODIFICATIONS
ASXL-1
Somatic nonsense, missense, frameshift and point mutations of the additional sex combs-like gene (ASXL-1), one of the Polycomb group (PcG) of proteins are found in 3 to 30% AML9, 16, 32. ASXL-1 mutations were initially detected in MDS and chronic myelomonocytic leukemia (CMML) but were subsequently identified in AML32–37. These mutations are more prevalent in older AML patients (vs. younger patients) and those with therapy-related AML (vs.de novo AML)32–34. ASXL-1 is important for PRC2-mediated trimethylation of histone 3 lysine 27 (H3K27); an epigenetic modification with a transcriptionally repressive effect. Inactivating mutations of ASXL-1 therefore lead to H3K27me3level reductions at critical sites and potentially promote leukemogensis by de-repressing important leukemogenic oncogenes16, 34. Mutations inASXL-1 are also reported to be associated with interruption of ubiquitin removal from specific histone lysine residues35 and promoting HOX gene expression34 which are important in leukemic transformation. However, the mechanisms underlying leukemogenesis in patients with ASXL-1 mutations remain to be full identified. The prognostic implications of ASXL-1 mutations in AML have been elusive due to several conflicting data. Still, it is generally accepted that ASXL-1 mutation is an adverse prognostic indicator in intermediate-risk AML32, 36, 37.
EPIGENETIC THERAPY
DNMT Inhibitors
5-Azacytidine (azacitidine) and its deoxy analogue 5-aza-2’-deoxycytidine (decitabine) are the two most extensively studied DNMT inhibitors and are approved for clinical use in hematologic malignancies (specifically MDS) in the USA. These medications were first invented in 1960s, but were clinically abandoned due to excessive toxicity and narrow therapeutic window38. Subsequently it was realized that dose-response curve of azacitidine is U-shape suggesting that a much lower dose of the drug than initially used could be clinically effective and safe39.
Despite being recognized for several decades and being commercially available for a decade, the mechanisms of action of both approved DNMT inhibitors remain to be fully elucidated. Azacitidine is metabolized to decitabine and the tri-phosphorylated product (i.e. decitabine triphosphate) is incorporated into DNA and subsequently binds to DNMT covalently at the C-6 position40, 41. The incorporation of active metabolites results in cytotoxicitic effect at high concentrations while at lower concentrations the predominant effect appears to be depletion of DNMTs with subsequent loss of DNA methylation following DNA replication and therapeutic epigenetic modulation40–44. In contrast to decitabine which has no direct effects on RNA, most of the phosphorylated azacitidine is actually incorporated into the RNA thereby interfering with protein synthesis40, 43. In addition, these drugs appear to have immune-mediated effects45, 46. The degree to which the beneficial clinical effects of DNMT inhibitors are dependent on any of these (or other) mechanisms and whether differential molecular mechanisms of azacitidine versus decitabine (e.g. direct impact on protein synthesis) affect the final clinical effects are yet to be established. Recently, the use of decitabine with 75 to 90% dose reduction in patients with MDS with a more frequent administration (1 to 3 days/week instead of 5 days every 28 days schedule) showed that DNMT depletion effect was predominant with minimial direct cytotoxic effect via conventional apoptotic pathways44. These data suggest that lower dose of decitabine (or possibly azacitidine) might be effective in MDS or AML with mutation of key apoptotic genes including TP53 and that different mechanisms of DNMT inhibitors depending on doses, administration schedule and their clinical applicability should be further investigated44.
Table 1 summarizes the results of some of the important clinical trials of DNMT inhibitors in AML patients. A subgroup analysis of the AZA-001, a trial that compared azacitidine to conventional care regimens (CCR) in patients with higher-risk MDS, in patients with 20–30% bone marrow blasts who are currently classified as having AML according to the World Health Organization (WHO) classification demonstrated a significantly better median overall survival (OS, 24.5 vs. 16.0 months, respectively, hazard ratio=0.47; 95% CI, 0.28 to 0.79; p=0.005) and probability of OS at 2-year OS rate (50% vs 16%, respectively, p=0.001)47. Another subgroup analysis of patients with 20–30% bone marrow blasts (reclassified by WHO criteria) in the Cancer and Leukemia Group B (CALGB) randomized trial that led to FDA approval of azacitidine in MDS demonstrated a significant survival advantage of azacitidine as well48. Subsequent two phase II trials of decitabine in AML demonstrated similar response rates (25% and 26%, respectively)49, 50. A phase III randomized study of first-line decitabine therapy in elderly patients with AML (>65 years old at time of diagnosis) with intermediate or poor cytogenetics showed that while decitabine significantly increased CR rate compared to best supportive care (BSC) or low-dose cytarabine (17.8% vs. 7.8%, odds ratio, 2.5; 95% CI, 1.4 to 4.8; p=0.001), the difference in median OS was not statistically significant between the two groups (7.7 months vs 5.0 months, p=0.108)51.
Table 1.
Selected phase II and III clinical trials of DNMT inhibitors in AML
| Study | Design | Participants | N | Responses | Median Overall survival (OS) |
Ref |
|---|---|---|---|---|---|---|
| Azacitidine(AZ A-001) |
Randomized phase III |
Reclassification of AML based on WHO criteria, Bone marrow (BM) blast 20~30% |
55 | 18% CR | 24.5ms (Aza) vs 16 ms (CCR) (p=0.005) |
47 |
| Azacitidine(CAL BG 8421, 8921, 9221) |
Randomized phase III |
Reclassification of AML based on WHO criteria, BM blast 20~30% |
103 | 35~48% (CR/PR/HI) |
19.3 ms (Aza) vs 12.9 ms (observation) (only from CALGB 9221) (p value not indicated) |
48 |
| Decitabine(DA CO-017) |
Single arm phase II |
AML with age>60, intermediate/poor risk (de novo or secondary) |
55 | 25% (CR/PR) | 7.7 ms | 49 |
| Decitabine (00332/AMLSG 14-09) |
Single arm phase II |
AML with age>60, intermediate/poor risk (de novo or secondary) |
277 | 26% (CR/PR) | 5.5 ms | 50 |
| Decitabine(DA CO-016) |
Randomized phase III |
AML with age>65, intermediate/poor risk (de novo or secondary) |
242 | 17.8% (CR+CRi) (vs. 7.8% treatment choice, p=0.001) |
7.7 ms (decitabine) vs 5.0 ms (treatment choice) (p=0.0373) |
51 |
Pts: patients
CR (complete remission): the disappearance of all signs and symptoms related to the disease, a bone marrow with 5% or fewer blasts and peripheral blood count with an absolute neutrophil count of 109/L or more and platelet count of 100 × 109/L or more.
CRp: CR except for a platelet count increase by 50% to more than 30 × 109/L but less than 100 × 109/L.
PR (Partial remission): A cellular marrow aspirate with 5% to 25% blasts, with a platelet count greater than 100×109/L and WBC less than 1.5×109/L
Bone marrow (BM) response: bone marrow blast of 5% or less but without meeting the peripheral blood count criteria for CR or CRp.
Hematologic improvement (HI): HI-E: a hemoglobin increase by at least 15g/L or transfusion independence, HI-P: an absolute increase of platelet counts from less than 20 to more than 20 × 109/L and by at least 100% or if more than 20 × 109/L, by an absolute increase of at least 30 × 109/L, HI-N: a granulocyte increase by at least 100% and by an absolute increase of at least 0.5 × 109/L.
CCR: conventional care regimen
N: number of patients
Aza: azacitidine
HDAC Inhibitors
Histones play a role in forming nucleosomes and chromatins with DNA in eukaryotic cells52. Acetylation and deacetylation in histone tails by histone acetyltransferases (HAT) and histone deacetylases (HDAC) is one of the important epigenetic mechanisms of control of gene transcription40. Several clinical trials have been conducted with histone deacetylase (HDAC) inhibitors in patients with MDS and AML (Table 2). In general, single agent HDAC inhibitor therapy has been associated with low response rates, usually in the range of 10–20%52. Based on in vitro studies, revealing HDAC inhibitors and DNMT inhibitors act synergistically to induce re-expression of silenced genes in cancer cells53, clinical trials have been conducted to use HDAC inhibitors in combination with other agents, such as DNMT inhibitors and conventional chemotherapeutic agents52.
Table 2.
Selected phase I and II clinical trials of HDAC inhibitors in AML
| Study | Design | Participants | N | Responses | Median OS | Ref |
|---|---|---|---|---|---|---|
| Vorinostat | Single arm phase I | Relapsed or refractory leukemia, and MDS |
41 (31 of AML) | 17 % (2CR, 2CRi, 3 HI) | Not reported | 56 |
| Vorinostat (NCT00305773) |
Two-stage, randomized phase II |
Relapsed AML or untreated AML with age >65, antecedent MDS, poor cytogenetics |
37 (15 pts in arm A, 22 pts in arm B, arm A and arm B have different administration schedule) |
0% (arm A), 4.5% (CR) (arm B) |
105 days (arm A), 153 days (arm B) |
57 |
| Romidepsin (Depsipeptide) |
Two cohort, single arm phase II |
Second relapsed AML <60 years, first relapsed AML >60 years, previously untreated AML>60 years who are not candidates for or who refuse conventional chemotherapy |
20 (Cohort A: absence of chromosomal aberrations, Cohort B: presence of chromosomal aberrations (t(8;21), inv(16), t(15;17) |
Cohort A: 0%, Cohort B: no objective responses by criteria, yet 75% >50% decrease in BM blasts, and HI in 3 out of 7 patients. |
Not reported | 58 |
| VPA monotherapy or in combination with ATRA |
Single arm phase II | AML/MDS, AML/MPN, de novo AML |
75 (32 of AML) | 16% (3% CR, 13% HI) | Not reported | 59 |
| MGCD0103 (Mocetinostat) |
Single arm phase I | Relapsed or refractory AML, untreated AML with age >60 |
29 (22 of AML) | 2 complete BM response (blasts<5%) |
Not reported | 63 |
| MS-275 (Entinostat) |
Single arm phase I | Relapsed AML or untreated AML with age >65, antecedent MDS, poor risk features (complex karyotype, antecedent hematologic disease) |
38 | No CR/PR. 12 pts in BM response, decreased transfusion requirement, ANC improvement etc. |
Not reported | 64 |
| LBH589 (Panobinostat) |
Single arm phase I | Relapsed or refractory AML | 15 (13 of AML) | 8 out of 11 pts with peripheral blasts showed transient reduction. |
Not reported | 65 |
Pts: patients
CR (complete remission): the disappearance of all signs and symptoms related to the disease, a bone marrow with 5% or fewer blasts and peripheral blood count with an absolute neutrophil count of 109/L or more and platelet count of 100 × 109/L or more.
CRp: CR except for a platelet count increase by 50% to more than 30 × 109/L but less than 100 × 109/L.
PR (Partial remission): A cellular marrow aspirate with 5% to 25% blasts, with a platelet count greater than 100×109/L and WBC less than 1.5×109/L
Bone marrow (BM) response: bone marrow blast of 5% or less but without meeting the peripheral blood count criteria for CR or CRp.
Hematologic improvement (HI): HI-E: a hemoglobin increase by at least 15g/L or transfusion independence, HI-P: an absolute increase of platelet counts from less than 20 to more than 20 × 109/L and by at least 100% or if more than 20 × 109/L, by an absolute increase of at least 30 × 109/L, HI-N: a granulocyte increase by at least 100% and by an absolute increase of at least 0.5 × 109/L.
CCR: conventional care regimen
N: number of patients
Aza: azacitidine
Panobinostat in an HDAC inhibitor under investigation especially for core-binding factor (CBF)-AML. CBF-AML and APL are characterized by chimeric proteins that aberrantly recruit HDAC to target gene promoters leading to differentiation block and blast proliferation54. In a murine model of t(8;21) AML, panobinostat resulted in a significant anti-leukemic effect and triggered terminal myeloid differentiation suggesting the need for further studies of HDAC inhibitors in this setting55.
Vorinostat (Suberoylanilide hydroxamic acid, SAHA), another potent HDAC inhibitor containing a hydroxamic acid moiety, binds to the zinc-containing pocket in the catalytic site of HDAC 1, 2, 3 and 6, causing their reversible inhibition. In a phase I trial of oral vorinostat for hematologic malignancies, responses were observed in leukemia including AML (7 of 41 patients,17%), including 2 CR, 2 CRs with incomplete hematologic recovery, and 3 with hematologic improvement56. In another study, 37 patients with AML were treated with vorinostat in a phase II trial, but only one patient had a hematologic improvement57.
Romidepsin (depsipeptide) is a cyclic tetrapeptide that is a potent HDAC inhibitor in vitro. In a multicenter phase II trial, romidepsin was administrated to 20 patients with refractory or relapsed AML58. Two patients had disappearance of bone marrow blasts with concomitant recovery of near-normal hematopoiesis following one or two cycles of therapy, and three additional achieved a >50% reduction in bone marrow blasts58.
Valproic acid (VPA) is a short-chain fatty acid oral antiepileptic agent that has been shown to inhibit HDAC activity at low levels. In a phase II study of single agent VPA or in combination with all-trans retinoic acid (ATRA), 75 patients with MDS or AML (n=32) were treated with VPA, including nine patients who also received ATRA from the start of treatment59. Hematologic improvement was observed in 24% of patients (30% in MDS, 16% in AML). Other studies using VPA either as a single agent or in combination with ATRA as therapy of poor-risk AML and MDS have also reported minimal activity60–62. Therefore, it seems that the activity of single-agent VPA in AML and MDS is limited.
Mocetinostat, entinostat and panobinostat have been studied mostly in phase I studies63–65. In summary, single-agent HDAC inhibitor therapy results in modest responses in AML at the best. In order to improve outcomes, studies have been conducted using HDAC inhibitors in combination with other agents such as DNMT inhibitors and conventional chemotherapeutic agents (See the next section).
Combinations of DNMT inhibitors and HDAC inhibitors
Elucidation of multiple interacting mechanisms of gene silencing has led to combining drugs that affect multiple epigenetic pathways (Table 3). A number of clinical trials have tested the combination of DNMT inhibitors with HDAC inhibitors (i.e. VPA, vorinostat, entinostat and mocetinostat etc.) in both MDS and AML. Since Gore et al. have developed a well-tolerated combination schedule of azacitidine and HDAC inhibitor which induces promoter methylation reversal and global histone acetylation66, multiple studies demonstrated that the combination of DNMT inhibitors with HDAC inhibitors confers a synergistic effect but most of these studies are in phase I or II63, 67–69. DNMT inhibitors have been combined with non-epigenetic agents including gemtuzumab ozogamicin70, and sorafenib71. Combination of DNMT inhibitors with other agents such as erythropoietin72, romiplostim73, arsenic74, and lenalidomide75 were investigated mostly in MDS. The combination of bortezomib with decitabine seems to be interesting as it showed clinical activity in a limited number of older patients with AML76.
Table 3.
Selected clinical trials of combination epigenetic therapy with DNMT inhibitors and HDAC inhibitors in AML
| Study | Design | Participants | N | Responses | Median OS | Ref |
|---|---|---|---|---|---|---|
| Azacitidine/ phenylbutyate |
Single arm II | MDS, AML/MDS, Relapsed AML or untreated AML with age >65, antecedent MDS, poor cytogenetics |
29 (18 of AML) |
38% (4 CR, 1PR, 6HI) |
Not reported | 66 |
| Azacitidine/ VPA/ATRA |
Single arm I/II |
Relapsed or refractory AML, high-risk MDS, untreated AML with age >60, who refused or were not candidates for front-line chemotherapy |
53 (49 of AML) |
42% (12 CR, 3 CRp, 7 BM response) |
With a median follow up of 21 wks, the median survival in patients achieving CR or CRp has not been reached |
67 |
| Decitabine/ VPA |
Single arm I/II |
Relapsed or refractory AML, high-risk MDS, untreated AML with age >60, who refused or were not candidates for front-line chemotherapy |
54 (48 of AML) |
22% (10 CR, 2 CRp) | 15.3 ms in responders vs. 4.9 ms in non- responders (p value not reported) |
68 |
| Decitabine/ VPA |
Single arm I | Relapsed AML or untreated AML with age >60, who were not candidates for intensive chemotherapy |
25 | 44% (4 CR) | Not reported | 69 |
| Gemtuzumab ozogamicin/ vorinostat/ azacitidine |
Single arm I/II |
Primary refractory or relapsed AML (age>50) | 43 | 41.9% (10 CR, 8 CRi) |
7.4 ms in responders vs. 3.1 ms in non- responders (p=0.0023) |
70 |
| Azacitidine/ sorafenib |
Single arm I/II |
Refractory or relapsed AML | 43 | 46% (6 CR, 10 CRi, 1 PR) |
7.8 ms in responders vs. 6.0 ms in non- responders (p=0.01) |
71 |
| Bortezomib/ decitabine |
Single arm I | Relapsed or refractory AML or previously untreated AML with age >65 |
19 | 36% (7 CR+CRi) | Not reported | 76 |
| Vorinostat/ idarubicin/ cytarabine |
Single arm II | Previously untreated AML or higher-risk MDS with age 15 to 65 years |
75 (52 of AML) |
85% (57 CR, 7 CRi) | 82 wks | 77 |
| Decitabine/ VPA |
Randomized phaseII |
Previously untreated AML or higher-risk MDS with age > 65 years | 149 (62 of AML) |
ORR: 51% in decitabine alone vs 35% in decitabine/VPA (p=0.208), CR: 33% in decitabine alone vs 9% in decitabine/VPA (p=0.729) |
9.6 ms in decitabine alone vs. 7.9 ms in decitabine/VPA (p=0.729) |
78 |
| Azacitidine/ Entinostat (E1905) |
Randomized phase II |
MDS, CMML, AML with MDS related change (excluding therapy related MDS or AML) |
149 (52 of AML) |
44~46% (not analyzed in AML subgroup) |
18 ms in aza alone vs 13 ms in aza/entinostat |
79 |
Pts: patients
CR (complete remission): the disappearance of all signs and symptoms related to the disease, a bone marrow with 5% or fewer blasts and peripheral blood count with an absolute neutrophil count of 109/L or more and platelet count of 100 × 109/L or more.
CRp: CR except for a platelet count increase by 50% to more than 30 × 109/L but less than 100 × 109/L.
PR (Partial remission): A cellular marrow aspirate with 5% to 25% blasts, with a platelet count greater than 100×109/L and WBC less than 1.5×109/L
Bone marrow (BM) response: bone marrow blast of 5% or less but without meeting the peripheral blood count criteria for CR or CRp.
Hematologic improvement (HI): HI-E: a hemoglobin increase by at least 15g/L or transfusion independence, HI-P: an absolute increase of platelet counts from less than 20 to more than 20 × 109/L and by at least 100% or if more than 20 × 109/L, by an absolute increase of at least 30 × 109/L, HI-N: a granulocyte increase by at least 100% and by an absolute increase of at least 0.5 × 109/L.
CCR: conventional care regimen
N: number of patients
Aza: azacitidine
A phase II study suggested the combination of vorinostat with idarubicin and cytarabine is viable in AML, but phase III study will be necessary to confirm these results77. A recently published randomized phase II trial in 149 patients with high-risk MDS and AML showed that the addition of VPA to decitabine did not improve CR rates, ORR, or survival in comparison to decitabine alone78. The E1905 study in which 149 patients with MDS (97 patients) and AML with trilineage dysplasia (52 patients) were randomized to azacitidine or a combination of azacitidine with the HDAC inhibitor, entinostat did not only show lack of significant differences in responses and survival between the two groups, but also observed lower degree of demethylation with the combination versus azacitidine monotherapy suggesting pharmacodynamic antagonism79. These findings suggest that the choice of the individual agents, their target specificity, the administration schedules, and the particular doses are all likely to significantly impact the outcome of combination studies. Further studies will be needed to determine whether there is a synergistic or additive clinical effects to combining DNMT inhibitors with HDAC inhibitors.
Novel DNMT inhibitors
The high frequency of somatic alterations in epigenetic modifiers in AML patients, combined with the clinical importance of DNMT inhibitors in AML, has led to a great interest in the development of novel epigenetic therapies. Next generation DNMT inhibitors have been developed. For example, SGI-110 is a novel DNMT inhibitor that was designed to enhance the efficacy of decitabine by combining it with deoxyguanosine which induces resistance to degradation by cytidine deaminase therefore increasing the bioavailability of the drug52, 80. A phase II of SGI-110 in patients with relapsed and refractory AML or elderly treatment-naïve AML show that overall remission rate in treatment naïve elderly AML was 53%; a rate which is comparable to that using conventional DNMT inhibitors81.
Novel epigenetic modifiers
Several classes of novel epigenetic modifiers are in early phase clinical trials (Figure 1). In addition to methylation of DNA, methylation of histones is another important form of epigenetic modulation that controls critical cellular functions and whose aberrant regulation has been shown to contribute to leukemogenesis especially in mixed lineage leukemia (MLL)-fusion leukemias (which account for approximately 10% of adult AML). The H3K27 methyltransferase Enhancer of Zeste Homolog 2 (EZH2), the main component in the polycomb repressive complex 2 (PRC2) which tri-methylates H3K27 to H3K27me3 and recruits PRC1 to promoters of target genes, and the H3K79 histone methyltransferase Disruptor of Telomere Silencing 1-like (DOT1L), which di-methylates H3K79 to H3K79me2, both contribute to important cellular functions including maintenance of stemness of cells82. Mutations of EZH2 have been observed in various hematologic malignancies including MLL-rearranged leukemias and Inhibition of EZH2 using selective inhibitors such as 3-deazaneplanocin A (DZNep) has shown promising efficacy in preclinical studies in MLL-fusion leukemias82, 83. Importantly, in murine models of MLL-rearranged leukemias not only did DZNep suppress leukemia proliferation, but it also reduced leukemia-initiating cells [LIC] frequency through up-regulation of p1682. Double inhibition of EZH2 and EZH1 with an oral selective inhibitor, UNC1999, prolonged survival in a MLL-rearranged murine model further suggesting that inhibition of EZH2 (and EZH1) is a promising therapeutic intervention in MLL-rearranged leukemias84. Furthermore, combined epigenetic therapy using a combination of an EZH2 inhibitor and a HDAC inhibitor (DZNep and panobinostat) and even a triple combination (DZNep, the HDAC inhibitor trichostatin-A and the DNMT inhibitor decitabine) was effective and synergistic in preclinical studies of AML suggesting that triple targeting of the 3 epigenetic mechanisms that silence TSG merits further evaluation in AML85, 86.
Figure 1.
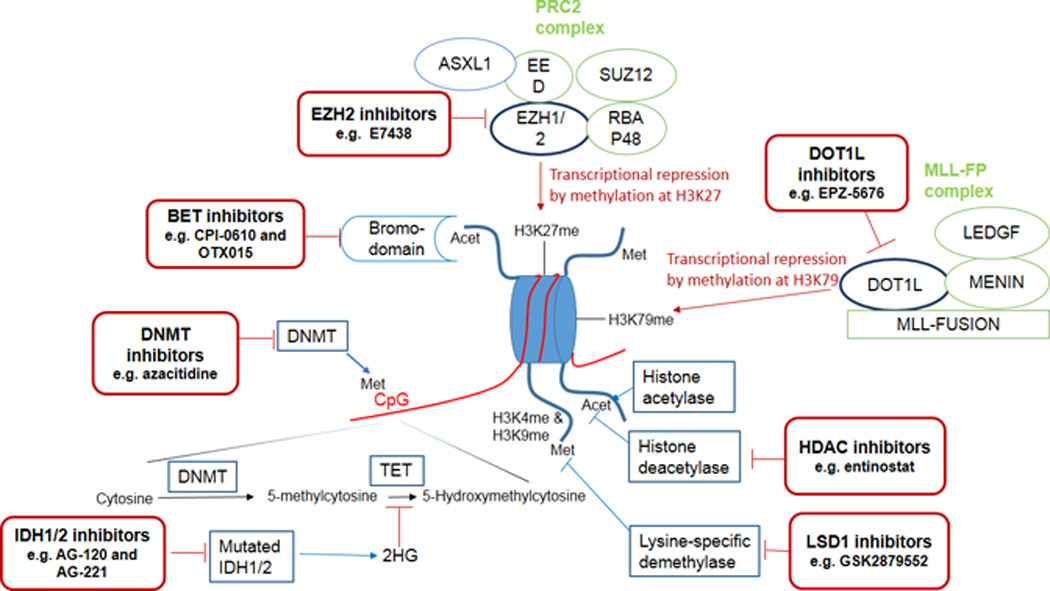
Schematic view of the mechanism of epigenetic modifications and targeting sites for treatment
Misguiding of DOT1L with subsequent aberrant H3K79 methylation and transcriptional up-regulation of HoxA and Meis1 genes are important for the initiation and maintenance of MLL-fusion leukemias while the loss of DOT1L function selectively reduces expression of the MLL-fusion driven transcriptional programs; therefore identifying DOT1L as a rational therapeutic target in MLL-rearranged leukemias87, 88. Indeed, a potent and selective aminonucleoside inhibitor of DOT1L, EPZ-5676, caused complete and sustained regression of the tumor in a rat xenograft model of MLL-fusion leukemia89 and is currently undergoing phase 1 clinical trials in relapsed/refractory adult and pediatric MLL-rearranged leukemias (Clinicaltrials.gov identifier: NCT02141828 and NCT01684150). Studies in MLL-rearranged leukemia cell lines showed synergistic effects when EPZ-5676 was combined with DNMT inhibitors, cytarabine or daunorubicin, providing a rationale for clinical evaluation of these combinations90. Furthermore, inhibition of DOT1L with another small molecular inhibitor, SYC-522, was shown to increase chemo-sensitivity of MLL-rearranged leukemia by preventing DNA damage response91. Recent data suggest activity of DOT1L inhibitors might extend to other subtypes of AML aside from MLL-rearranged leukemias. For example, EPZ004777, a DOT1L inhibitor revealedin vitro responses in primary AML cells with IDH1 or IDH2 mutations, therefore warranting further evaluation of these agents in other subtypes of AML92.
The bromodomain and extra terminal (BET) family of proteins include a number of Bromodomain-containing “reader” proteins (BRD [BRD2, BRD3, BRD4, and BRDT]) which use their acetyl-lysine recognition motifs (bromodomains) to read the post-translational acetylated motifs of histones and influence transcription of target genes93, 94. Dysregulation of BET adaptors has been shown to contribute to leukemogenesis across a variety of AML subtypes driven by different mutations95. Dislocation of BRD3 or BRD4 in AML (for instance, dissociation of BRD4 from mutant NPM1) enhances the expression of oncogenes, such as c-Myc, Bcl-2 and CDK6, by recruiting super elongation complexes96. Inhibition of BRD4 using small hairpin inhibitory RNAs (shRNAs) or small molecule inhibitors (e.g. JQ1 and I-BET151) induces robust in vivo and in vitro anti-leukemic effects via down-regulation of oncogenes, suggesting this approach could be a potentially effective therapeutic strategy in some subtypes of AML94, 95, 97–99. Preclinical studies of the selective small molecule BRD4 inhibitors JQ1 and I-BET151 showed terminal myeloid differentiation and elimination of leukemia stem cells in MLL-rearranged AML, synergistic activity with HDAC inhibitors and anthracyclines to enhance p53-mediated apoptosis in DNMT3A/NPM1-mutated leukemic cell lines, and activity against NPM1-mutated AML and JAK2V617F-driven neoplasms94, 95, 98, 99. BET inhibitors (e.g. CPI-0610 and OTX015) have entered phase 1 clinical trials for refractory acute leukemias and MDS (NCT02158858 and NCT01713582). Lysine-specific demethylase1 (LSD1) is an epigenetic eraser that removes methyl groups from H3K4me1/2 and H3K9me1/2 in a location- and context-specific fashion acting as a transcriptional repressor or activator100. Data suggest that LSD1 is involved in leukemogenesis and maintenance of leukemic stem cells especially in MLL-rearranged leukemias101. Preclinical data suggest that inhibitors of LSD1 have activity in AML102 and some are already in phase 1 trials (e.g.NCT02177812 evaluating the LSD1 inhibitor GSK2879552).
Finally, inhibitors of mutant IDH1 have also demonstrated preclinical evidence of efficacy in IDH1-mutated AML103. There are ongoing phase 1 clinical trials of mutant IDH1inhibitors (e.g. AG-120 in NCT02074839) and mutant IDH2 inhibitors (e.g. AG-221 in NCT01915498) in advanced hematologic malignancies with these respective IDH mutations. In addition, given that IDH-mutated AML are addicted to glutamine as the main source of α-KG104, 105, selective glutamine depletion is being explored clinically as a potential therapeutic approach in IDH-mutated AML using Erwinase (Asparaginase Erwinia Chrysanthemi; NCT02283190).
CONCLUSIONS
Recent DNA sequencing and other techniques have facilitated understanding of the role of epigenetics in leukemogenesis, especially in AML. Aberrant DNA methylation (hypermethylation in promoters of TSG and hypomethylation in promoters of oncogenes) and aberrant histone acetylation and methylation are widely seen in AML. Mutations in genes important in the regulation of the epigenome, such as TET2, DNMT3A, ASXL-1, IDH1/2, and EZH2, have also provided insights into the process of leukemogenesis in AML and suggested opportunities for therapeutic targeting. These mutations also contribute to prognostication and risk stratification in AML.
Currently, DNMT inhibitors which are approved for treatment of AMDS are widely used as an off-label treatment option for AML patients who are not candidates for conventional intensive chemotherapy. HDAC inhibitors are among the better-studied epigenetic modifiers but are yet to improve outcomes in AML. Several other promising epigenetic modifiers are undergoing early phase clinical research in AML including inhibitors of histone methylation (e.g. EZH2 and DOT1L inhibitors), inhibitors of histone demethylases (e.g. LSD1 inhibitors), inhibitors of bromodomain-containing epigenetic “reader” BET proteins, and inhibitors of mutant IDH. In addition, combination-based approaches of different epigenetic modifiers are under active investigation. While epigenetic therapy for AML is still in its infancy, it is fair to say the future is likely to see a significant increase in the use of these agents as single agents, in combinations with each other, or with conventional chemotherapy.
Acknowledgments
SDG receives research funding from and consults for Celgene Corp.
Footnotes
Conflicts of interest: AMZ and TKK have no relevant disclosures or conflicts to declare.
REFERENCES
- 1.Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet. 2013;381:484–495. doi: 10.1016/S0140-6736(12)61727-9. [DOI] [PubMed] [Google Scholar]
- 2.O'Donnell MR, Abboud CN, Altman J, Appelbaum FR, Arber DA, Attar E, et al. Acute myeloid leukemia. Journal of the National Comprehensive Cancer Network : JNCCN. 2012;10:984–1021. doi: 10.6004/jnccn.2012.0103. [DOI] [PubMed] [Google Scholar]
- 3.Yates JW, Wallace HJ, Jr, Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer chemotherapy reports Part 1. 1973;57:485–488. [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 5.Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nature reviews Cancer. 2012;12:599–612. doi: 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nature reviews Genetics. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 11.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 12.Melki JR, Vincent PC, Clark SJ. Cancer-specific region of hypermethylation identified within the HIC1 putative tumour suppressor gene in acute myeloid leukaemia. Leukemia. 1999;13:877–883. doi: 10.1038/sj.leu.2401401. [DOI] [PubMed] [Google Scholar]
- 13.Toyota M, Kopecky KJ, Toyota MO, Jair KW, Willman CL, Issa JP. Methylation profiling in acute myeloid leukemia. Blood. 2001;97:2823–2829. doi: 10.1182/blood.v97.9.2823. [DOI] [PubMed] [Google Scholar]
- 14.Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. The New England journal of medicine. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 16.Conway O'Brien E, Prideaux S, Chevassut T. The epigenetic landscape of acute myeloid leukemia. Advances in hematology. 2014;2014:103175. doi: 10.1155/2014/103175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcucci G, Metzeler KH, Schwind S, Becker H, Maharry K, Mrozek K, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:742–750. doi: 10.1200/JCO.2011.39.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. The New England journal of medicine. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thol F, Damm F, Ludeking A, Winschel C, Wagner K, Morgan M, et al. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:2889–2896. doi: 10.1200/JCO.2011.35.4894. [DOI] [PubMed] [Google Scholar]
- 20.Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. The New England journal of medicine. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdel-Wahab O, Mullally A, Hedvat C, Garcia-Manero G, Patel J, Wadleigh M, et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114:144–147. doi: 10.1182/blood-2009-03-210039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metzeler KH, Maharry K, Radmacher MD, Mrozek K, Margeson D, Becker H, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: a Cancer and Leukemia Group B study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weissmann S, Alpermann T, Grossmann V, Kowarsch A, Nadarajah N, Eder C, et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia. 2012;26:934–942. doi: 10.1038/leu.2011.326. [DOI] [PubMed] [Google Scholar]
- 24.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fathi AT, Sadrzadeh H, Borger DR, Ballen KK, Amrein PC, Attar EC, et al. Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood. 2012;120:4649–4652. doi: 10.1182/blood-2012-06-438267. [DOI] [PubMed] [Google Scholar]
- 28.Janin M, Mylonas E, Saada V, Micol JB, Renneville A, Quivoron C, et al. Serum 2-hydroxyglutarate production in IDH1- and IDH2-mutated de novo acute myeloid leukemia: a study by the Acute Leukemia French Association group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:297–305. doi: 10.1200/JCO.2013.50.2047. [DOI] [PubMed] [Google Scholar]
- 29.DiNardo CD, Propert KJ, Loren AW, Paietta E, Sun Z, Levine RL, et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood. 2013;121:4917–4924. doi: 10.1182/blood-2013-03-493197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Human pathology. 2012;43:1541–1551. doi: 10.1016/j.humpath.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Pratcorona M, Abbas S, Sanders MA, Koenders JE, Kavelaars FG, Erpelinck-Verschueren CA, et al. Acquired mutations in ASXL1 in acute myeloid leukemia: prevalence and prognostic value. Haematologica. 2012;97:388–392. doi: 10.3324/haematol.2011.051532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schnittger S, Eder C, Jeromin S, Alpermann T, Fasan A, Grossmann V, et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia. 2013;27:82–91. doi: 10.1038/leu.2012.262. [DOI] [PubMed] [Google Scholar]
- 34.Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer cell. 2012;22:180–193. doi: 10.1016/j.ccr.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. Journal of hematology & oncology. 2012;5:12. doi: 10.1186/1756-8722-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrozek K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118:6920–6929. doi: 10.1182/blood-2011-08-368225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y, Zhu YM, Fan X, Shi JY, Wang QR, Yan XJ, et al. Gene mutation patterns and their prognostic impact in a cohort of 1185 patients with acute myeloid leukemia. Blood. 2011;118:5593–5603. doi: 10.1182/blood-2011-03-343988. [DOI] [PubMed] [Google Scholar]
- 38.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 39.Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, et al. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2'-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–1640. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 40.Oki Y, Issa JP. Epigenetic mechanisms in AML - a target for therapy. Cancer treatment and research. 2010;145:19–40. doi: 10.1007/978-0-387-69259-3_2. [DOI] [PubMed] [Google Scholar]
- 41.Jackson-Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R. Mutagenicity of 5-aza-2'-deoxycytidine is mediated by the mammalian DNA methyltransferase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:4681–4685. doi: 10.1073/pnas.94.9.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nature biotechnology. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin T, Jelinek J, Si J, Shu J, Issa JP. Mechanisms of resistance to 5-aza-2'-deoxycytidine in human cancer cell lines. Blood. 2009;113:659–667. doi: 10.1182/blood-2008-02-140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saunthararajah Y, Sekeres M, Advani A, Mahfouz R, Durkin L, Radivoyevitch T, et al. Evaluation of noncytotoxic DNMT1-depleting therapy in patients with myelodysplastic syndromes. The Journal of clinical investigation. 2015 doi: 10.1172/JCI78789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hambach L, Ling KW, Pool J, Aghai Z, Blokland E, Tanke HJ, et al. Hypomethylating drugs convert HA-1-negative solid tumors into targets for stem cell-based immunotherapy. Blood. 2009;113:2715–2722. doi: 10.1182/blood-2008-05-158956. [DOI] [PubMed] [Google Scholar]
- 46.Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, et al. De novo induction of a cancer/testis antigen by 5-aza-2'-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer research. 2006;66:1105–1113. doi: 10.1158/0008-5472.CAN-05-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:562–569. doi: 10.1200/JCO.2009.23.8329. [DOI] [PubMed] [Google Scholar]
- 48.Silverman LR, McKenzie DR, Peterson BL, Holland JF, Backstrom JT, Beach CL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 49.Cashen AF, Schiller GJ, O'Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:556–561. doi: 10.1200/JCO.2009.23.9178. [DOI] [PubMed] [Google Scholar]
- 50.Lubbert M, Ruter BH, Claus R, Schmoor C, Schmid M, Germing U, et al. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica. 2012;97:393–401. doi: 10.3324/haematol.2011.048231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2670–2677. doi: 10.1200/JCO.2011.38.9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Navada SC, Steinmann J, Lubbert M, Silverman LR. Clinical development of demethylating agents in hematology. The Journal of clinical investigation. 2014;124:40–46. doi: 10.1172/JCI69739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nature genetics. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 54.Yang G, Khalaf W, van de Locht L, Jansen JH, van der Reijden BA, Muller-Tidow C, et al. Epigenetic regulation of tumor suppressors in t(8:21)-containing AML. Annals of hematology. 2004;83:329–330. doi: 10.1007/s00277-003-0841-8. [DOI] [PubMed] [Google Scholar]
- 55.Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood. 2014;123:1341–1352. doi: 10.1182/blood-2013-03-488114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 57.Schaefer EW, Loaiza-Bonilla A, Juckett M, DiPersio JF, Roy V, Slack J, et al. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica. 2009;94:1375–1382. doi: 10.3324/haematol.2009.009217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Odenike OM, Alkan S, Sher D, Godwin JE, Huo D, Brandt SJ, et al. Histone deacetylase inhibitor romidepsin has differential activity in core binding factor acute myeloid leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008;14:7095–7101. doi: 10.1158/1078-0432.CCR-08-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuendgen A, Knipp S, Fox F, Strupp C, Hildebrandt B, Steidl C, et al. Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Annals of hematology. 2005;84(Suppl 1):61–66. doi: 10.1007/s00277-005-0026-8. [DOI] [PubMed] [Google Scholar]
- 60.Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, et al. The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer. 2006;106:112–119. doi: 10.1002/cncr.21552. [DOI] [PubMed] [Google Scholar]
- 61.Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, et al. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104:2717–2725. doi: 10.1002/cncr.21589. [DOI] [PubMed] [Google Scholar]
- 62.Raffoux E, Chaibi P, Dombret H, Degos L. Valproic acid and all-trans retinoic acid for the treatment of elderly patients with acute myeloid leukemia. Haematologica. 2005;90:986–988. [PubMed] [Google Scholar]
- 63.Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, et al. Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood. 2008;112:981–989. doi: 10.1182/blood-2007-10-115873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781–2790. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:4628–4635. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 66.Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer research. 2006;66:6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 67.Soriano AO, Yang H, Faderl S, Estrov Z, Giles F, Ravandi F, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 68.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, et al. Phase 1/2 study of the combination of 5-aza-2'-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, Devine H, et al. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:3884–3891. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- 70.Walter RB, Medeiros BC, Gardner KM, Orlowski KF, Gallegos L, Scott BL, et al. Gemtuzumab ozogamicin in combination with vorinostat and azacitidine in older patients with relapsed or refractory acute myeloid leukemia: a phase I/II study. Haematologica. 2014;99:54–59. doi: 10.3324/haematol.2013.096545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121:4655–4662. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fili C, Malagola M, Follo MY, Finelli C, Iacobucci I, Martinelli G, et al. Prospective phase II Study on 5-days azacitidine for treatment of symptomatic and/or erythropoietin unresponsive patients with low/INT-1-risk myelodysplastic syndromes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3297–3308. doi: 10.1158/1078-0432.CCR-12-3540. [DOI] [PubMed] [Google Scholar]
- 73.Kantarjian HM, Giles FJ, Greenberg PL, Paquette RL, Wang ES, Gabrilove JL, et al. Phase 2 study of romiplostim in patients with low- or intermediate-risk myelodysplastic syndrome receiving azacitidine therapy. Blood. 2010;116:3163–3170. doi: 10.1182/blood-2010-03-274753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Welch JS, Klco JM, Gao F, Procknow E, Uy GL, Stockerl-Goldstein KE, et al. Combination decitabine, arsenic trioxide, and ascorbic acid for the treatment of myelodysplastic syndrome and acute myeloid leukemia: a phase I study. American journal of hematology. 2011;86:796–800. doi: 10.1002/ajh.22092. [DOI] [PubMed] [Google Scholar]
- 75.Sekeres MA, Tiu RV, Komrokji R, Lancet J, Advani AS, Afable M, et al. Phase 2 study of the lenalidomide and azacitidine combination in patients with higher-risk myelodysplastic syndromes. Blood. 2012;120:4945–4951. doi: 10.1182/blood-2012-06-434639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012;119:6025–6031. doi: 10.1182/blood-2012-03-413898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garcia-Manero G, Tambaro FP, Bekele NB, Yang H, Ravandi F, Jabbour E, et al. Phase II trial of vorinostat with idarubicin and cytarabine for patients with newly diagnosed acute myelogenous leukemia or myelodysplastic syndrome. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:2204–2210. doi: 10.1200/JCO.2011.38.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Issa JP, Garcia-Manero G, Huang X, Cortes J, Ravandi F, Jabbour E, et al. Results of phase 2 randomized study of low-dose decitabine with or without valproic acid in patients with myelodysplastic syndrome and acute myelogenous leukemia. Cancer. 2014 doi: 10.1002/cncr.29085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prebet T, Sun Z, Figueroa ME, Ketterling R, Melnick A, Greenberg PL, et al. Prolonged administration of azacitidine with or without entinostat for myelodysplastic syndrome and acute myeloid leukemia with myelodysplasia-related changes: results of the US Leukemia Intergroup trial E1905. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:1242–1248. doi: 10.1200/JCO.2013.50.3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Griffiths EA, Choy G, Redkar S, Taverna P, Azab M, Karpf AR. SGI-110 DNA Methyltransferase Inhibitor Oncolytic. Drug Future. 2013;38:535–543. [PMC free article] [PubMed] [Google Scholar]
- 81.Kantarjian HM, Jabbour E, Yee K, Kropf P, O'Connell C, Stock W, et al. First Clinical Results Of a Randomized Phase 2 Study Of SGI-110, a Novel Subcutaneous (SQ) Hypomethylating Agent (HMA), In Adult Patients With Acute Myeloid Leukemia (AML) Blood. 2013;122 [Google Scholar]
- 82.Ueda K, Yoshimi A, Kagoya Y, Nishikawa S, Marquez VE, Nakagawa M, et al. Inhibition of histone methyltransferase EZH2 depletes leukemia stem cell of mixed lineage leukemia fusion leukemia through upregulation of p16. Cancer science. 2014;105:512–519. doi: 10.1111/cas.12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood. 2011;118:2830–2839. doi: 10.1182/blood-2010-07-294827. [DOI] [PubMed] [Google Scholar]
- 84.Xu B, On DM, Ma A, Parton T, Konze KD, Pattenden SG, et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2014 doi: 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Momparler RL, Cote S, Momparler LF, Idaghdour Y. Epigenetic therapy of acute myeloid leukemia using 5-aza-2'-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clinical epigenetics. 2014;6:19. doi: 10.1186/1868-7083-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–2743. doi: 10.1182/blood-2009-03-213496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117:6912–6922. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood. 2013;122:1017–1025. doi: 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klaus CR, Iwanowicz D, Johnston D, Campbell CA, Smith JJ, Moyer MP, et al. DOT1L inhibitor EPZ-5676 displays synergistic antiproliferative activity in combination with standard of care drugs and hypomethylating agents in MLL-rearranged leukemia cells. The Journal of pharmacology and experimental therapeutics. 2014;350:646–656. doi: 10.1124/jpet.114.214577. [DOI] [PubMed] [Google Scholar]
- 91.Liu W, Deng L, Song Y, Redell M. DOT1L inhibition sensitizes MLL-rearranged AML to chemotherapy. PloS one. 2014;9:e98270. doi: 10.1371/journal.pone.0098270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sarkaria SM, Christopher MJ, Klco JM, Ley TJ. Primary acute myeloid leukemia cells with IDH1 or IDH2 mutations respond to a DOT1L inhibitor in vitro. Leukemia. 2014 doi: 10.1038/leu.2014.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stewart HJ, Horne GA, Bastow S, Chevassut TJ. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer medicine. 2013;2:826–835. doi: 10.1002/cam4.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28:311–320. doi: 10.1038/leu.2013.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luo Z, Lin C, Shilatifard A. The super elongation complex (SEC) family in transcriptional control. Nature reviews Molecular cell biology. 2012;13:543–547. doi: 10.1038/nrm3417. [DOI] [PubMed] [Google Scholar]
- 97.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wyspianska BS, Bannister AJ, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto FJ, et al. BET protein inhibition shows efficacy against JAK2V617F-driven neoplasms. Leukemia. 2014;28:88–97. doi: 10.1038/leu.2013.234. [DOI] [PubMed] [Google Scholar]
- 100.Mould DP, McGonagle AE, Wiseman DH, Williams EL, Jordan AM. Reversible Inhibitors of LSD1 as Therapeutic Agents in Acute Myeloid Leukemia: Clinical Significance and Progress to Date. Medicinal research reviews. 2014 doi: 10.1002/med.21334. [DOI] [PubMed] [Google Scholar]
- 101.Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 102.Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nature medicine. 2012;18:605–611. doi: 10.1038/nm.2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chaturvedi A, Araujo Cruz MM, Jyotsana N, Sharma A, Yun H, Gorlich K, et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood. 2013;122:2877–2887. doi: 10.1182/blood-2013-03-491571. [DOI] [PubMed] [Google Scholar]
- 104.Emadi A, Jun SA, Tsukamoto T, Fathi AT, Minden MD, Dang CV. Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Experimental hematology. 2014;42:247–251. doi: 10.1016/j.exphem.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 105.Elhammali A, Ippolito JE, Collins L, Crowley J, Marasa J, Piwnica-Worms D. A high-throughput fluorimetric assay for 2-hydroxyglutarate identifies Zaprinast as a glutaminase inhibitor. Cancer discovery. 2014;4:828–839. doi: 10.1158/2159-8290.CD-13-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]