

Atherosclerosis is an underlying cause of various cardiovascular diseases including coronary artery disease, chronic kidney disease, abdominal aortic aneurysm development, stroke and heart failure.1, 2 This progressive vascular disease is characterized by subendothelial lipoprotein retention, immune cell infiltration, a maladaptive inflammatory response and vascular smooth muscle (VSMC)-mediated fibrous cap formation.1 As atherosclerotic lesions progress, cellular and tissue dysfunction give way to cell death and fibrous cap thinning, leading to thrombotic events and plaque rupture.3
Mitochondrial function is a critical regulator of cellular homeostasis due to its role in ATP production, ion transport, reactive oxygen species generation, and apoptotic signaling.4 Given the evidence for cellar dysfunction and the metabolic underpinning of atherosclerosis, mitochondrial dysfunction is largely hypothesized to contribute to lesion development and progression.5 Indeed, recent work has pointed to a role for mitochondrial reactive oxygen species (mitoROS) and mitochondrial DNA (mtDNA) damage in animal models of atherosclerosis.6, 7 In particular, mtDNA damage has been found to precede lesion development, and genetic inhibition of mouse mitochondrial polymerase-γ exonuclease activity results in increased mtDNA damage, decreased respiratory function and augmented lesion development and necrosis.8 In addition, human studies have shown a correlation between mtDNA damage and lesion development/progression;8-10 however, the mechanism through which mtDNA damage exacerbates lesion development has remained a mystery. Specifically, mtDNA damage is correlated with mitochondrial respiration deficiencies, but the role of mitochondrial respiration in plaque development and progression remains unclear.
In this issue of Arteriosclerosis Thrombosis and Vascular Biology, Yu & Reinhold et al.11 delve into this topic. They show that human plaque samples exhibit reduced mtDNA copy number and mitochondrial respiration which are associated with increased mitophagy in plaque-derived VSMC samples. Further, aortic samples from ApoE−/− mice fed a high-fat diet for 14 weeks show mtDNA damage and decreased mitochondrial respiration, and overexpression of the mtDNA helicase Twinkle, a regulator of mtDNA copy number and maintenance, rescues this phenotype. In mouse-derived VSMCs, Twinkle overexpression reduces mtDNA damage but does not affect mtDNA copy number. Conversely, macrophages overexpressing Twinkle exhibit increased mtDNA copy number without changes in mtDNA damage. In both cases, possibly through protection of, or increased mitochondrial-derived electron transport chain subunit transcription and translation, Twinkle overexpression enhances respiration. Twinkle-mediated increases in respiration promote thickening of the fibrous cap and a decrease in necrotic core formation. Changes in necrosis were found to be primarily regulated by macrophages, whereas increased VSMC proliferation and reduced apoptosis contributed to fibrous cap thickening. Thus, Yu & Reinhold et al. provide a link, using human and mouse samples, between mtDNA damage-induced mitochondrial respiratory dysfunction and the progression of atherosclerosis and plaque instability. These novel findings also raise new and interesting questions concerning the critical role of mitochondria in atherosclerosis.
First, what is the role of mitoROS? There has been contentious debate in recent years regarding the role of mitoROS in the development of atherosclerosis and whether it contributes to, and/or is a consequence of, mitochondrial dysfunction. mitoROS has been shown to promote mtDNA oxidative damage,12 and recent evidence indicates mitoROS plays a significant role in lesion development.7, 13 However, mtDNA damage has also been shown to promote atherosclerosis independent of mitoROS.8 In the current study, Twinkle overexpression in mice had no effect on high-fat diet induced mitoROS production. It could be hypothesized that an increase in respiration would result in elevated mitoROS production unless mitochondrial antioxidant systems were also elevated; however, this avenue was unexplored. There also appears to be a tissue- and disease-dependent role for Twinkle in mitoROS regulation given that overexpression of Twinkle has been shown to reduce mitoROS.14 Future investigation should aim to tease out the precise role of mitoROS in lesion development and its potential contribution to changes in mtDNA damage and mitochondrial dysfunction.
In addition to the role of mitoROS, how do changes in mitochondrial respiration influence plaque stability? Increased respiration resulted in decreased VSMC apoptosis and an increase in proliferation. While proliferation may be dependent upon ATP production, how does respiration influence mitochondrial-dependent apoptotic signaling? It would also be interesting to observe if enhanced mitochondrial respiration influences mitochondrial membrane potential and ion transport and their role in plaque stability. Furthermore, recent evidence indicates inflammatory macrophages experience metabolic reprogramming resulting in increased glycolysis-derived ATP and a decrease in oxygen consumption.15 Given the role of Twinkle-overexpressing macrophages in necrotic core formation,11 future investigation should explore how enhanced respiration affects inflammatory signaling as well as inflammation resolution mechanisms such as efferocytosis. In addition, how does enhanced respiration affect mitophagy and mitochondrial biogenesis and vice versa? The authors noted that VSMCs derived from human plaque samples exhibited increased mitophagy without subsequent increases in mitochondrial biogenesis which may help to explain a decrease in mtDNA copy number in human plaques. However, how Twinkle-induced respiratory increases affect mitophagy and/or mitochondrial biogenesis is unknown and warrants future investigation. Delineating the contribution of mtDNA damage and mtDNA copy number to respiratory deficiencies and the resultant phenotype is also warranted. Twinkle overexpression elicits differing responses in VSMCs and macrophages (decreased mtDNA damage vs increased mtDNA copy number). While it is likely that mtDNA damage and mtDNA copy number both contribute to the observed phenotype, their specific roles remain unclear.
All in all, the work performed by Yu & Reinhold et al. establishes a direct relationship between mtDNA damage, mitochondrial respiratory deficiencies, and lesion progression in atherosclerosis. These novel findings encourage continued investigation into the precise role of mitochondrial function in the development of atherosclerosis and cardiovascular disease.
Figure.
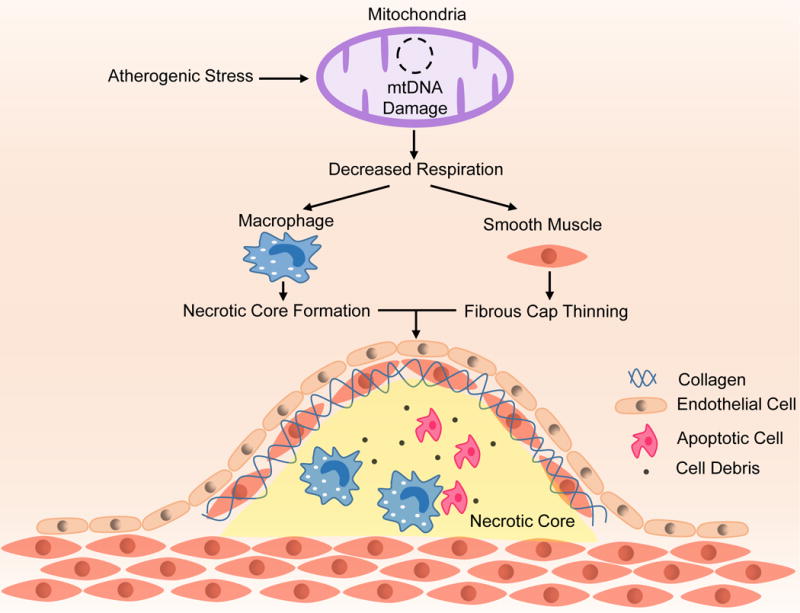
Atherogenic stimuli cause mtDNA damage leading to a decline in mitochondrial respiration. Decreased mitochondrial respiration leads to thinning of the fibrous cap through VSMC dysfunction and apoptosis and increased necrotic core formation as a result of macrophage activation. Increasing mitochondrial respiration attenuates these effects.
Acknowledgments
Sources of Funding
SJF is supported by N.I.H. 5T32HL007745-24. This work was supported by NIH P01 HL095070.
Footnotes
Disclosures
None.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Robertson A-KL, Söderberg-Nauclér C. Inflammation and atherosclerosis. Annual Review of Pathology: Mechanisms of Disease. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 3.Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis. Update and Therapeutic Implications. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 4.Nunnari J, Suomalainen A. Mitochondria: In sickness and in health. Cell. 148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circulation Research. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 6.Yu EPK, Bennett MR. Mitochondrial DNA damage and atherosclerosis. Trends in Endocrinology & Metabolism. 25:481–487. doi: 10.1016/j.tem.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017 doi: 10.1161/ATVBAHA.117.309580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu E, Calvert PA, Mercer JR, et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128:702–712. doi: 10.1161/CIRCULATIONAHA.113.002271. [DOI] [PubMed] [Google Scholar]
- 9.Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat Res. 1992;275:169–180. doi: 10.1016/0921-8734(92)90021-g. [DOI] [PubMed] [Google Scholar]
- 10.Fetterman JL, Holbrook M, Westbrook DG, Brown JA, Feeley KP, Bretón-Romero R, Linder EA, Berk BD, Weisbrod RM, Widlansky ME, Gokce N, Ballinger SW, Hamburg NM. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovascular Diabetology. 2016;15:53. doi: 10.1186/s12933-016-0372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu EPK, Reinhold J, Yu H, Starks L, Uryga AK, Foote K, Finigan A, Figg N, Pung Y-F, Logan A, Murphy MP, Bennett M. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017 doi: 10.1161/ATVBAHA.117.310042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mikhed Y, Daiber A, Steven S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. International Journal of Molecular Sciences. 2015;16:15918–15953. doi: 10.3390/ijms160715918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nf-κb-mediated inflammation in macrophages. Circulation research. 2014;114:421–433. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeda M, Ide T, Fujino T, Arai S, Saku K, Kakino T, Tyynismaa H, Yamasaki T, Yamada K-I, Kang D, Suomalainen A, Sunagawa K. Overexpression of tfam or twinkle increases mtdna copy number and facilitates cardioprotection associated with limited mitochondrial oxidative stress. PLoS ONE. 2015;10:e0119687. doi: 10.1371/journal.pone.0119687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mills EL, Kelly B, Logan A, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 167:457–470.e413. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]