

SUMMARY
Rationally engineering thermostability in proteins would create enzymes and receptors that function under harsh industrial applications. Several sequence-based approaches can generate thermostable variants of mesophilic proteins. To gain insight into the mechanisms by which proteins become more stable, we use structural and dynamic analyses to compare two popular approaches, ancestral sequence reconstruction (ASR) and the consensus method, used to generate thermostable variants of Elongation Factor Thermounstable (EF-Tu). We present crystal structures of ancestral and consensus EF-Tus, accompanied by molecular dynamics simulations aimed at probing the strategies employed to enhance thermostability. All proteins adopt crystal structures similar to extant EF-Tus, revealing no difference in average structure between the methods. MD reveals that ASR-generated sequences retain dynamic properties similar to extant, thermostable EF-Tu from T. aquaticus, while consensus EF-Tu dynamics differ from evolution-based sequences. This work highlights the advantage of ASR for engineering thermostability while preserving natural motions in multidomain proteins.
eTOC Blurb
Ancestral sequence reconstruction (ASR) and the consensus approach are compared in the generation of thermostable EF-Tu homologs. Using a combination of X-ray crystallography and molecular dynamics simulations, Okafor et al. show that while both methods yield thermostable proteins, ASR, unlike consensus, preserves the natural protein motions in EF-Tu.
INTRODUCTION
Understanding the factors that contribute to protein thermostability is desirable for gaining insight into the physical and chemical mechanisms underlying protein folding and stability, eventually leading to the ability to design proteins that are functional at high temperatures. However, a thorough comprehension of the link between protein thermostability and other biophysical factors has been a long-standing challenge in protein research, limiting researchers’ abilities to rationally affect protein stability. Directed evolution (Asial et al., 2013), consensus sequence alignment (Lehmann and Wyss, 2001), rational design experiments (Renugopalakrishnan et al., 2005; Yang et al., 2015) and ancestral sequence resurrection (Cole and Gaucher, 2011) are a few of the methods that have been utilized to engineer protein stability. In spite of the emergence of these methods, there still exists a general lack of understanding of the mechanisms employed in proteins to improve stability. As these methods have been implemented in separate systems, it has not been possible to compare different stabilization methods to improve our knowledge of how thermostability is achieved in a given protein family. In this work, we present a structural and dynamical comparison, using the consensus and ancestral sequence reconstruction methods to investigate thermostability in the Elongation Factor Tu protein family.
To generate thermostable proteins, the consensus approach uses protein sequences from extant organisms in a sequence alignment. From the alignment, the most frequently observed amino acid at each position is incorporated into a consensus protein sequence. In addition, consensus variants can be generated by replacing individual protein residues with consensus amino acids most prevalent at individual sites in the extant sequences. Both consensus sequence and consensus variants approaches have been successfully used to engineer increased stability in proteins (Amin et al., 2004; Lehmann et al., 2000). Using this approach, thermophilic proteins can be generated, based solely on information derived from mesophilic sequences. Several limitations exist for this method, including its heavy dependence on the acquisition of homologous sequences and the quality of the sequence alignment (Lehmann et al., 2002; Porebski and Buckle, 2016). Additionally, a consensus sequence is not able to account for the context-dependent nature of amino acids in proteins (epistasis), nor has a consensus sequence been subjected to natural selection at any point in evolutionary history (Cole and Gaucher, 2011).
An emerging phylogenetic technique known as ancestral sequence reconstruction, ASR, has been used to interrogate protein evolution. Unexpectedly, multiple ASR studies have revealed that most resurrected ancestral proteins display increased thermostability over their extant homologs (Gaucher et al., 2008; Merkl and Sterner, 2016). ASR uses a multiple sequence alignment of extant proteins to build a phylogenetic tree, which is used to infer the sequences of ancestral proteins at different nodes in the phylogeny (Harms and Thornton, 2010; Pauling and Zuckerkandl, 1963). Introducing ancestral residues into modern sequences has resulted in more thermostable proteins, suggesting that ASR may be a useful approach to engineer thermostability while preserving function (Watanabe et al., 2006; Yamashiro et al., 2010).
Here, we compare both ancestral and consensus methods for increasing thermostability of Elongation Factor Tu, EF-Tu (bacteria)/Elongation Factor 1A (archaea and eukaryote). This gene family is a GTPase involved in the protein translation system. More specifically, EF-Tu binds to GTP, which in turn favors the binding of an aminoacyl-tRNA (Krab and Parmeggiani, 1998). This ternary complex binds to mRNA-programmed ribosomes, delivering an aminoacyl-tRNA to the ribosomal A site (Czworkowski and Moore, 1996). The biochemistry of EF-Tu has been studied for over three decades, giving rise to a clear understanding of the functional aspects of the protein. EF-Tu is highly conserved among all three domains of life and its thermostability correlates with the optimal growth temperature of its host organism. This implies that a strong selective constraint governs EF thermostability and this constraint is dictated by the host’s environment (Gaucher et al., 2003).
A previous study of the evolutionary history of 4-billion-year-old ancestral EF-Tu variants revealed a strong correlation between thermostability and the geothermal record of planetary temperature which showed a cooling trend from the emergence of life to present day (Gaucher et al., 2008; Hedges et al., 2015). To determine ancestral sequences, phylogenetic analysis of bacterial EF homologs was performed followed by maximum likelihood-based ancestral sequence reconstruction, as described in (Gaucher et al., 2008). The most ancient EFs were thermostable and the melting temperature (Tm) of each descendent decreased along the evolutionary tree en route to the extant bacterial EF-Tu. This provides a unique system in which to interrogate the mechanism by which a small defined number of historical amino acid replacements between each reconstructed protein variant altered thermostability. We hypothesized that this well-defined system, containing relatively few amino acid replacements between ASR variants with altered Tm, might offer insight into the forces that contribute to thermostability within this protein family. We compare this method with the more conventional consensus method whereby, a consensus EF-Tu sequence, which displayed increased thermostability (~ 20°) over an extant EF-Tu from E. coli (Cole and Gaucher, 2011), was generated and experimentally synthesized. The consensus sequence was generated from a sequence alignment (Notredame et al., 2000) of 161 sequences from a broad phylogenetic range (Table S1).
In this work, our goal is to understand and compare the mechanisms by which ASR and consensus approaches enhance thermostability in the EF-Tu family. We use a combination of x-ray crystallography and molecular dynamics (MD) simulations to investigate the structural mechanism by which each method generates a more stable EF-Tu variant. To enable these studies, we crystallized and determined the three-dimensional structures of the last common ancestor of bacterial EFs along with a more-derived ancestor within the bacterial lineage. We then employ all-atom MD simulations to investigate the mechanisms by which evolutionary sequence changes altered thermostability during the evolutionary history of EFs. We compare these atomic-level mechanisms with those employed to achieve thermostability in the consensus sequence, which we have also crystallized. These investigations offer insight into the evolution of atomic contacts and motions which permitted adaptation to altered temperatures while preserving protein function. MD simulations are also performed on the extant thermophilic and mesophilic EF-Tu homologs from T. aquaticus and E. coli, respectively. Higher temperature simulations are used to investigate protein unfolding in EF-Tus. Our key findings are as follows: we observe predictable behavior in ancestral sequences, where the trends in dynamics and stability are consistent with the protein Tm. Conversely, the consensus sequence displays unexpected instability in the simulations, inconsistent with its increased stability over E. coli EF-Tu. Ancestral EF-Tus show similarities with extant thermophilic EF-Tu from T. aquaticus, which highlights the important role of evolutionary selection in obtaining functional proteins, as well as previously observed limitations in the consensus method.
RESULTS
EF-Tu structure is conserved
Previously studied resurrected EF-Tus include ancestral proteins represented by nodes (N) 168, 170, and 253 from the phylogeny (Gaucher et al., 2008). EF-Tu N168 is dated at 3.4 – 4.3 billion years ago (Hedges et al., 2015) and represents the last common ancestor of all bacteria. N170 (3.1 – 3.3 billion years ago) represents the last common ancestor of T. aquaticus and E. coli and N253 (2.8 – 3.0 billion years ago) is a distant ancestor of proteobacterial EF-Tus (Figure 1A). Tm’s for N168, N170, N253 were determined as 73.3°, 66.2°, 55.7° respectively and are indicated on the cladogram (Figure 1A) (Gaucher et al., 2008). Consensus EF Tm was determined as 60.2° (Cole and Gaucher, 2011). High-quality crystals were obtained for ancestral proteins N168 and N253 that diffracted to 2.35 and 2.15Å resolution, respectively (Table 1). Crystals for consensus EF-Tu diffracted to 2.49Å. Each EF-Tu structure contains 387 amino acid residues and a single bound molecule of GDP and Mg2+. They all share the same canonical structure observed in the E. coli EF-Tu consisting of three distinct domains (Figure 1). Domain 1 contains the GDP/GTP binding site and consists of an α/β domain with a central parallel β-pleated sheet flanked by 7 α-helices on either side. Domains 2 and 3 both preserve the classic “greek key” antiparallel β-barrel structure common to many EFs. The expected conformation for GDP-bound EF-Tu is observed, with a large hole present in the center of the molecule, separating domains 1 and 2. Superposition of the ancestral crystal structures with the E. coli (PDB 1EFC) and T. aquaticus (PDB 1TUI) EF-Tus reveals a high degree of structural conservation between the ancestral proteins (~ 4 billion years old) and extant proteins, in spite of the presence of a 10-residue loop (i.e. the thermophile loop) in domain 1 of T. aquaticus (Figure 1B, red loop). Closer inspection of the secondary structure reveals slight differences. (Figure S1). As expected, the GDP binding site is highly conserved and the position of the bound GDP differs only slightly in each complex (Figure 1C). Sequence conservation in all EF-Tus is high with identity between 70% and 95% for all pairwise comparisons and sequence similarity between 81.6 – 97.5 % (Table S2).
Figure 1. EF-Tu crystal structures display a conserved fold and cofactor binding site.
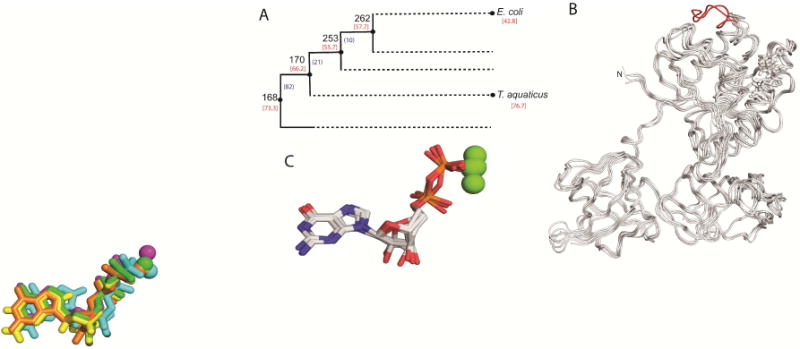
(A) Schematic cladogram showing the nodes targeted in this work. Number of amino acid residue replacements between consecutives nodes (black) and Tms (red) are indicated. This schematic represents a highly compressed version of the bacterial phylogeny used in the analyses. (B) Overlay of polypetide chain of structures for resurrected nodes targeted in this work. The thermophile-specific loop indel of T. aquaticus is colored red. (C) Overlay of GDP-Mg2+ complexes co-crystallized with EF-Tus. GDP is shown in stick representation and Mg2+ is shown as spheres.
Table 1.
Data Collection and Refinement Statistics
| Data Collection | Node 168 | Node 253 | ConcEF |
|---|---|---|---|
| Space group | P212121 | P212121 | P21 |
| Cell dimensions | |||
| a,b,c (Å) | 91.4, 173.4, 208.2 | 57.3, 73.4, 114.4 | 40.5, 104.1, 43.8 |
| α, β, γ(°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90, 111.3, 90 |
| No. Unique reflections | 145,229 | 23,450 | 10,877 |
| Resolution (Å) | 40-2.2.98 (2.380-2.298)* | 35.45-2,152 (2.23-2.152) | 26.5-2.494 (2.583-2.494) |
| Rsym (%) | 0.08 (0.47) | 0.08 (0.22) | 0.09 (0.43) |
| I/σI | 15.87 (4.02) | 23.2 | 18.8 (3.1) |
| Completeness (%) | 98.4 (93.5) | 87.3 (65.6) | 92.2 (82.4) |
| Redundancy | 5.9 (5.6) | 5.3 (4.0) | 4.3 (3.9) |
|
| |||
| Refinement | |||
| Resolution (Å) | 40-2.2.98 (2.380-2.298) | 35.45-2,152 (2.23-2.152) | 26.5-2.49 (2.58-2.49) |
| Rwork/Rfree (%) | 19.1/21.2 | 19.0/22.3 | 19.7/24.7 |
| No. atoms | |||
| Protein | 12060 | 2963 | 2852 |
| Ligand/ion | 313 | 58 | 29 |
| Water | 652 | 114 | 27 |
| B-factors (Å2) | |||
| Protein | 45.1 | 50.3 | 48.7 |
| Ligand/ion | 71.1 | 50.7 | 38.0 |
| Water | 50.2 | 48.9 | 34.4 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.008 | 0.004 | 0.003 |
| Bond angles (°) | 1.51 | 0.72 | 0.63 |
| Ramachandran favored (%) | 97.9 | 97 | 96 |
| Ramachandran outliers (%) | 0.08 | 0 | 0 |
| PDB accession code | 5W75 | 5W76 | 5W7Q |
Values in parentheses are for highest shell
Since most amino acid changes affect protein dynamics rather than protein structure, visualizing the impact of amino acid replacements is challenging when superimposing X-ray crystal structures. Therefore, we performed pairwise comparisons of EF-Tu structures using ProSMART, which quantifies the differences in local amino acid conformation between two structures (Nicholls et al., 2014). Thermostable EF-Tus (T. aquaticus, N168, N253 and consensus EF-Tus) are compared against mesophilic EF-Tu from E. coli to identify regions that might be implicated in thermostability. Overall, ProSMART analysis reveals differences in local structure distributed through all three EF-Tu domains (Figure 2). We observe that domain 1 shows higher conformational similarity than domains 2 and 3 for all comparisons. Conversely, domain 2 displays greater occurrences of either high structural dissimilarity or regions that were omitted from the analysis.
Figure 2. ProSMART Procrustes analysis of EF-Tu structures.
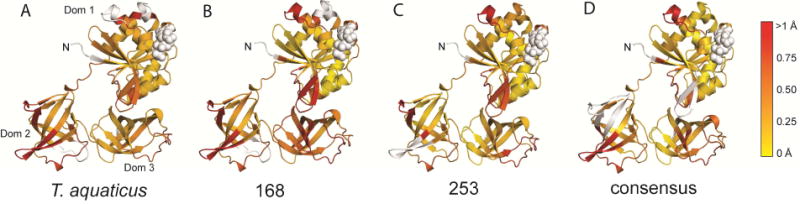
Pairwise ProSMART analyses of T. aquaticus, N168, N253 and consensus EF-Tus were performed with E. coli EF-Tu. Models were colored by the Procrustes score of the central residue of an aligned fragment pair. The Procrustes score is equivalent to the pairwise RMSD of atomic coordinates after superposition. Yellow implies local similarity, red implies local dissimilarity between compared regions. Areas colored white were not included in the analysis.
EF-Tu thermostability is correlated with domain 1 stability observed by unfolding in high temperature simulations
MD simulations were used to investigate the dynamics of thermostable EF-Tus generated by consensus or ancestral approaches. Root mean square deviations (RMSD) and fluctuations (RMSF) of individual EF-Tu domains are compared between proteins at both low and elevated temperatures. RMSDs and RMSFs are calculated in two ways, globally and by individual domains. Calculated by individual domains, RMSD and RMSF analyses provide specific information about observed changes in flexibility that may not be coupled to inter-domain interactions. Global analysis of RMSD and RMSF allow us to see how the combined domain effects change the flexibility of the overall EF-Tu structure compared to the starting conformation. RMSD analysis of individual domains reveals that domain 1 is most affected at increased temperatures. In 360K simulations, all EF-Tus have elevated RMSDs relative to 300K simulations, consistent with temperature-induced unfolding (Figure 3). The greatest increase is observed in E. coli, consistent with this having the lowest Tm. The smallest increases are observed in N168 and T. aquaticus EF-Tu, which are the most thermostable proteins. Overall, little to no RMSD increases are observed in domain 3 in the individual domain analysis for all EF-Tus. Analysis of RMSFs (Figure 4) obtained in simulations corroborate RMSD observations, including i) smallest temperature-induced increases in domain 3; ii) highest domain 1 unfolding in E.coli, reflected by high RMSF values in several regions. In general, high-mobility regions observed across all six simulated structures correspond to loops and beta turns.
Figure 3. Root mean square deviation (RMSD) analyses of EF-Tu domain 1 correlate with thermostability.
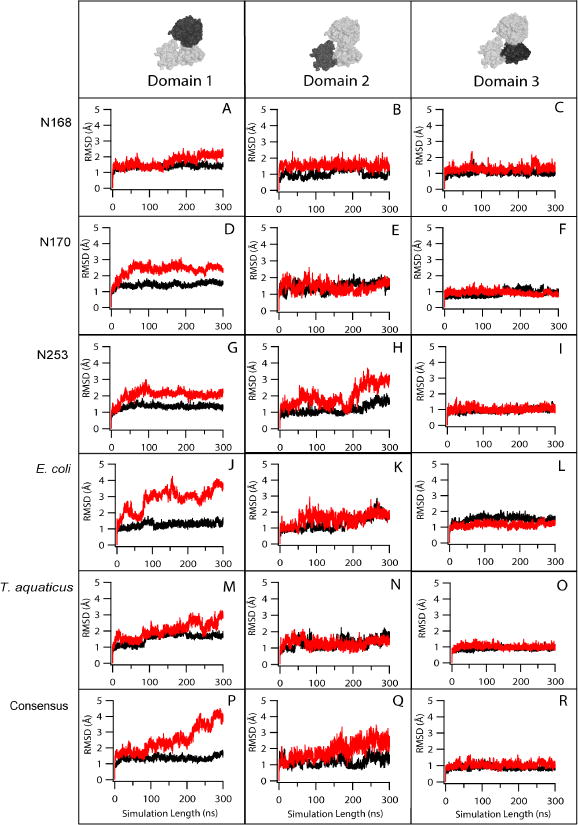
RMSD of protein backbone atoms (C, Cα, N, O atoms) for EF-Tus from MD simulations at 300 K (black) and 360 K (red).
Figure 4. Root mean square fluctuations (RMSF) correlate with thermostability.
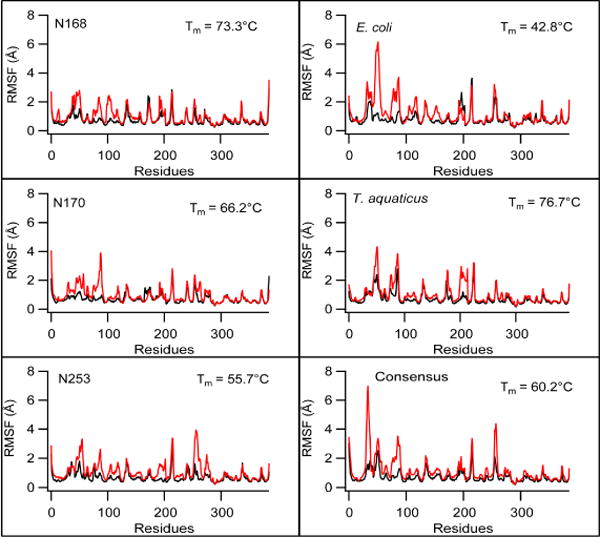
RMSF of Cα atoms for each residue in EF-Tu complexes from MD simulations at 300 K (black) and 360 K (red). RMSFs are shown by domain; residues in loops connecting adjacent domains are not included in the analysis.
Sequence changes explain stability differences in ancestral sequences
While increases in domain 1 RMSD correlate with thermostability of ancestral EF-Tus, an additional notable difference in dynamics occurs in domain 2 of N253 (Figure 3H). Higher temperature simulations result in increased domain 2 RMSD that is only observed in N253 and not N168 or N170 (Figure 3B, 3E). RMSF analysis also reveals this increased unfolding (i.e. higher RMSF values) in domain 2. (Figure 3E, residues 208 – 298, red trace). Included in this flexible region is a loop containing acidic residues D267 and E268 (Figure 5, orange). A comparison of the domain 2 sequence of N253 with those of N168 and N170 reveals residue replacements that could be implicated in the increased flexibility observed in that region (Figure 5). Domain 2 contains ionic interactions between D267 and E268 on one strand and positively charged K238/R238 (Figure 5, cyan) on the adjacent strand. The strand containing K238/R238 is in turn stabilized by a hydrophobic contact with a short helical turn that is contained in the hinge peptide between domains 1 and 2 (Figure 5, green). The helical turn is comprised of residues DVD, DID and AID in N168, N170 and N253 respectively. The hydrophobic contacts last throughout the simulation for all three structures. However, while the ionic interactions persist for the majority of the simulation in N168 and N170, they are reduced to 49% and < 30% in N253 for segments E268-K238 and D267-K238, respectively. This weakening and eventual loss of ionic interactions occurs in the second half of the simulation and is accompanied by the formation of new contacts between the flexible loop (i.e. containing D267, E268) with domain 1 residues. By the end of the simulation, N253 transitioned from the open GDP-bound conformation to the closed but active EF-Tu conformation that is associated with GTP binding (Figure 5E). This conformational change appears to be responsible for the increased flexibility observed in domain 2. N170 and N253 are the most similar of all the sequences studied here (94.7% identical, table S2), so this observation illustrates how the small differences in protein sequence can drive dramatic changes in conformational dynamics.
Figure 5. Lowered stability is observed in domain 2 of N253 EF-Tu.
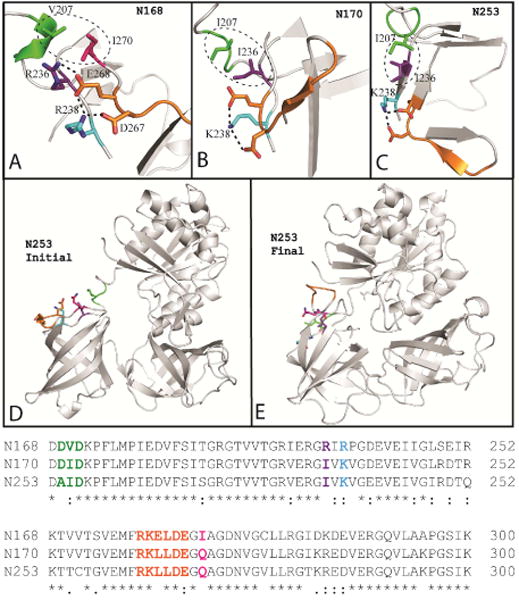
Network of ionic and hydrophobic interactions responsible for stabilizing domain 2 loop containing residues 263 to 268 (orange loop). These interactions are present in A) N168, B) N170, and C) N253. D) At the beginning of the 360 K simulation, the ionic interactions are intact in N253 and the protein is in the canonical GDP-bound conformation. E) At the end of the simulation, ionic interactions in the loop are disrupted as loop residues (orange) form new contacts with domain 1. N253 EF-Tu undergoes a conformational transition becoming more similar to the closed, GTP-bound conformation.
Consensus protein displays distinct dynamic behavior from ancestral or extant sequences
Consensus EF-Tu displays high mobility in comparison to all other EF-Tus in the simulations. Unusually high global RMSD (up to 6Å) values are observed in consensus EF-Tu for over half of the 300 K simulation suggesting that regions of consensus EF-Tu may be more dynamic than ancestral or extant EF-Tus (Figure 6A, black trace). To identify the major conformations sampled in the simulation, particularly those with high RMSD, we performed a cluster analysis. Three clusters were identified, all differing by the extent of unwinding and subsequent rotation in the hinge peptide between domains 1 and 2 (Figure S4). To identify significant residue motions in the EF-Tus, a principal component analysis (PCA) was performed on the 300 K simulations. In PCA, the correlation matrix is determined for the simulation, then diagonalized to obtain eigenvectors and eigenvalues of the system. Each eigenvector describes a dimension of positional fluctuations in the system and is weighted by its corresponding eigenvalue. A projection of the trajectory onto each eigenvector is a principal component. Typically, the top (~ 3-5) principal components are sufficient to describe up to 70% of the motions observed over a simulation. PCA reveals that the most dominant motion (PC1) in consensus EF-Tu is the unwinding of the functionally important hinge region, resulting in rotation of domains 2+3 relative to domain 1. Thus, the structure transitions from a compact state to an open/extended state (Figure 6B, C). This motion is responsible for 80% of the fluctuations in the simulation. The major PCs identified in simulations of ancestral and extant EF-Tus show motions distinct from the dominating hinging motion of consensus EF-Tu (Figure S5). In addition, the first PC for ancestral and extant EFs, at the most, accounts for 58% of total fluctuations.
Figure 6. Consensus EF-Tu demonstrates highly dynamic behavior.
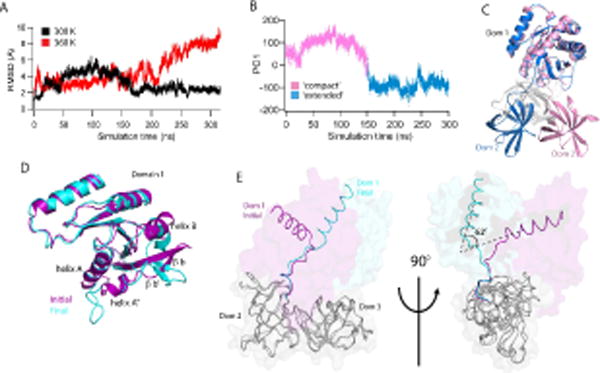
(A) Global RMSDs of the consensus EF-Tu over the simulation, 300 K (black) and 360 K (red). (B) Principal component analysis of Consensus 300 K simulation. First principle component mode (PC1) is the unwinding of the Domain1-Domain2 hinge peptide from a ‘compact’ state to an ‘extended’ state where the domains (1 and 2) are rotated away from each other. Two distinct conformations are observed. (C) Representative structures from each major subspace of PC1. Domain 1 of each conformation is superposed to show unwinding in the hinge peptide from the initial, compact conformation (pink) to the open, extended conformation (blue). Domain 3 is colored in gray for clarity. (D) Local structural changes in consensus 360 K simulation lead to increased RMSD of domain 1. Initial (purple) and final (cyan) structures of the simulation are superposed. Changes include rotation of helix B and unraveling of helices A, A″ and beta strands b′, b. (E) Initial and final conformations of consensus EF-Tu from 360 K simulation are overlaid with domains 2 and 3 (grey) superposed to highlight rotation of domains 2+3 relative to domain 1 (~ 62° rotation). Observed 360 K global RMSD changes result largely from this rotation.
Increased mobility is also apparent in the 360 K simulation of consensus EF-Tu, in both global and intra-domain RMSD analyses. A steady RMSD increase is observed in domain 1 (Figure 3), which results from unraveling in structure of residues 24 to 63 (helices A, A″, beta strands b′, b), as well as rotation of helix B, leading to enhanced interactions of this region with domain 3 (Figure 6D). Global RMSD changes at 360 K corresponds to unwinding of the hinge region, to an even greater extent compared to 300 K simulations of consensus EF-Tu (Figure 6C). We observe that in the simulations, domains 2 and 3 maintain a consistent orientation relative to each other. Observed movements change the positions of domains 2+3 relative to domain 1. A global overlay of initial and final structures obtained in the 360 K simulation shows severe unwinding in the hinge peptide, which is highlighted with the alignment of domains 2 and 3 (Figure 6E). This is reflected in the 360 K RMSD plot (Figure 6A, red trace), showing a RMSD of about 10 angstroms by the end of the simulation. It is also seen in the global RMSF analysis (Figure S3) where increased mobility is noticeably observed in domains 2 and 3 (residues 201 to 387) in the 360 K simulation compared to the 300 K simulation. Secondary structure analysis of 360 K simulations show that while unfolding is observed as expected at higher temperatures in various regions throughout the protein, there is no significant loss of structure over the course of the simulation. Therefore, we attribute the large changes seen in global RMSD and RMSF to a greater propensity for hinge unwinding in consensus EF-Tu.
Hydrophobicity is important for EF-Tu stability
Many intramolecular interactions (e.g. hydrogen bonds, ionic interactions, salt bridges, hydrophobic contacts and solvent accessible surface) have been implicated in promoting thermostability. We quantified these factors over the course of our simulations (Table S3) and find that only the buried area, which is a measure of the average area that is buried by each residue in a protein upon folding, shows high linear correlation with Tm in thermophilic EF-Tus (Figure 7). This measure, described previously by Rose et al (Rose et al., 1985), is proportional to the hydrophobic contribution of each residue to the overall conformational free energy (Richards, 1977). However, hydrophobic packing cannot account for the adoption of defined tertiary structures, suggesting that EF-Tu proteins are likely stabilized by a combination of different intermolecular forces in a non-obvious and complex manner. E. coli EF-Tu, the only mesophilic protein in our study, is an outlier on this plot (Figure 7, inset). Given a lack of experimentally determined Tms for other mesophilic bacterial EF-Tus, it is not possible to ascertain whether loss of the correlation between hydrophobicity and melting temperature is a general feature of mesophilic proteins or of this particular extant protein sequence.
Figure 7. Buried area correlates with Tm in thermophilic EF-Tu proteins.
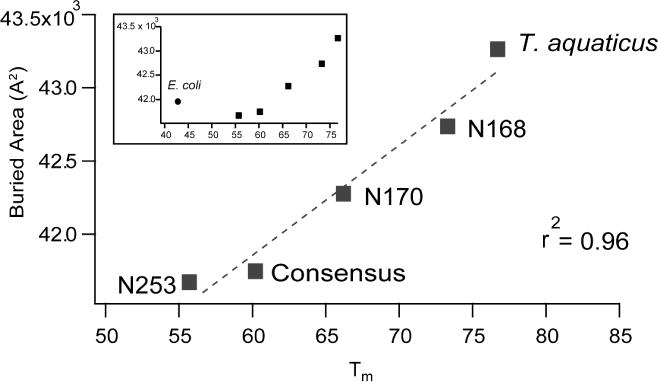
Mesophilic EF-Tu is an outlier (inset).
Community analysis reveals similarities between extant and ancestral thermostable sequences
To compare communication in EF-Tu between homologs and investigate potential links between interdomain communication and thermostability, we performed a community network analysis. The community analysis identifies groups of atoms undergoing correlated motion during the simulation. Community analyses are linked with information transfer in the protein, providing information about direct and allosteric communication between different regions in the molecule (Sethi et al., 2009). For analyses shown for each EF-Tu homolog in Figure 8, each separate community is represented by a differently colored circle. The size of the circle is proportional to the community size (i.e. the number of residues in each community) while the width of the line connecting the circles is proportional to the strength of communication between the pair of communities. In all EF-Tus, we observed that domains 2 and 3 partitioned into individual communities (red and green circles, respectively). In every case, domain 1 contained multiple communities.
Figure 8. Community analysis of EF-Tu proteins show that communities are built by domains.
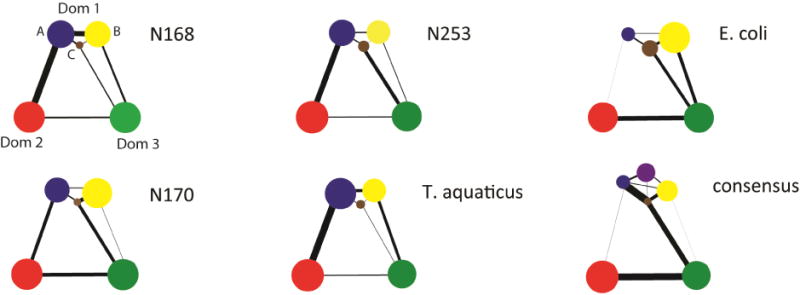
Domain 1 contains multiple communities in each EF-Tu studied while domains 2 and 3 each form individual communities. All ASR homologs and T. aquaticus contain three domain 1 communities with nearly identical size distributions. E. coli also has three communities in domain 1 with size distributions different than those in the thermostable homologs. Consensus EF-Tu is unique with four domain 1 communities. Consensus and E. coli both show diminished communication between domains 1 and 2.
All three ancestral proteins show similar community features, including i) three primary communities present in domain 1, labeled here as A, B and C (Figure 8) and ii) stronger communication is seen between domain 1 and domain 2. In ancestral homologs, the phosphate binding loop (P-loop, i.e. sequence responsible for coordinating the phosphate group of the nucleotide and binding GDP/GTP) lies on the interface of the two major domain 1 communities, and the peptide hinge between domains 1 and 2 is divided midway between domain 2 and domain 1 communities (Figure 9). We observe that communities from ancestral EF-Tus are nearly identical to the community distribution in T. aquaticus (Figure 8).
Figure 9. Community analysis is mapped on to selected EF-Tu proteins.
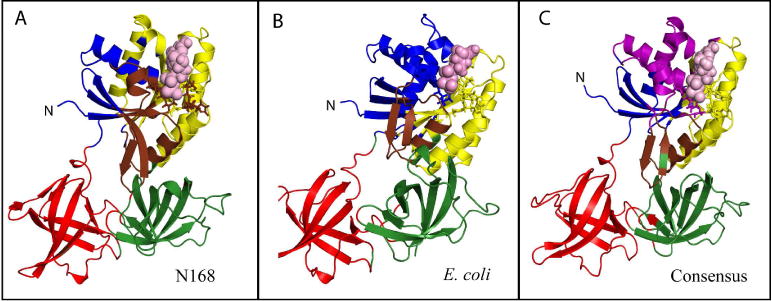
A) N168 B) E. Coli C) Consensus. Restudies are colored by community and bound GDP is shown in space-filling representation (pink) and the p-loop (i.e. GDP-binding loop) is shown in stick representation.
In contrast, consensus EF-Tu reveals dramatically different community features, with domain 1 partitioning into 4 communities instead of 3 (Figure 8, 9C). There is also weakened communication between domain 1 and 2, which may be because the connected domain 1 community is much smaller than those in the ancestral and T. aquaticus EF-Tu. These comparative analyses suggest that information transfer is similar between ancestral resurrected EF-Tus and extant T. aquaticus EF-Tu. Communication observed in consensus EF-Tu is unique, differing both from ancestral and extant EF-Tus. Communication between domains 1 and 2 is weakened drastically but enhanced between domains 2 and 3. This observation is consistent with the large motions and apparent instability observed in the hinge region. E. coli EF-Tu also shows novel community features, with 3 different communities in domain 1 but with different size distributions compared to other EF-Tus (Figure 8, Figure 9B). This observation could be a general indicator of different communication mechanisms employed in mesophilic EF-Tus compared to thermostable orthologs.
DISCUSSION
Comparison of mesophilic and thermophilic proteins (Razvi and Scholtz, 2006) have revealed differences between the two that might relate to thermostability, including increased hydrophobic interactions (Kumar and Nussinov, 2001; Zhou et al., 2007), increased hydrogen bonding (Sadeghi et al., 2006), electrostatic interactions (Kumar et al., 2000), specific amino acid substitutions (Ebrahimie and Ebrahimi, 2010), increased polar surface area (Haney et al., 1997), better packing (Scandurra et al., 1998), increased frequency of certain classes of amino acids (Panja et al., 2015), alpha helix stabilization (Li et al., 2005) and presence of aromatic clusters (Kannan and Vishveshwara, 2000). However, there remains no general consensus on engineering strategies to guide rational protein stability design. Sequence-based methods such as ASR and consensus approaches have shown success and were used to generate thermostable EF-Tu variants. Here, ancestral and consensus EF-Tu sequences are compared to probe the dynamic differences between thermostable structures obtained by either method. Crystal structures of ancestral and consensus proteins reveal high conservation in EF-Tu structure enabling an analysis of extant mesophilic and thermophilic EF-Tus. MD simulations reveal biophysical properties and dynamics that correlate with thermostability within this protein family. Analysis of elevated temperature simulations show that fluctuations in domain 1 are a predictor of overall thermostability. This result is consistent with previous work showing that the EF-Tu G-domain is important for thermostability when compared between mesophilic and thermophilic proteins (Šanderová et al., 2004). ASR and consensus sequences both recapitulate this domain 1-dependent stability observed in extant EFs.
Several other features which have been previously implicated in protein stability were also investigated, including hydrogen bonds, salt bridges, solvent accessible surface area and hydrophobic contacts. No large differences emerged among these features, so thermostability in EF-Tu cannot be attributed to any individual property. A near-linear correlation between EF-Tu thermostability and buried area might be an indicator of the importance of hydrophobicity in EF-Tu., which was previously identified as a determinant of thermostability within this protein family (Gromiha et al., 2013).
We used MD simulations to assess the dynamics of consensus and ancestral EF-Tus to link sequence-driven architectural changes to changes in protein stability. By identifying high-fluctuating regions in N253, we provide a specific illustration of how small sequence changes between ASR EF-Tu sequences modulate stability. Several aspects of the simulations revealed structural instability in consensus EF-Tu, identifying the sequence as a clear outlier both in magnitude and direction of conformational fluctuations. The overall highest fluctuations and RMSDs are observed for this protein. Unwinding of the peptide linker region between domains 1 and 2 was observed both in 300K and 360K simulations, which had the effect of rotating domains 2 and 3 relative to domain 1. This motion was identified by principal component analysis to be the most dominant movement in the simulation of the consensus protein. The instability observed in consensus domain 1, the catalytic domain of EF-Tu that is critical for function, may explain why the consensus protein has lower activity compared to wild-type EF in both E. coli and T. Thermophilus-based in vitro protein translation assays (data not shown).
In contrast, we observed similarities between ancestral resurrected EF-Tus and extant, thermophilic EF-Tu from T. aquaticus, suggesting that these ancestral proteins could indeed be active thermostable homologs. The three ancient proteins analyzed by MD (N168, N170 and N253) have all been tested in an in vitro reconstituted protein translation assay and all three displayed the ability to facilitate protein synthesis (Zhou et al., 2012). Of the similarities observed, community analysis shows that communication between groups of residues is the same between ancestral proteins and T. aquaticus EF-Tu. However, communication is different compared to the consensus and E. coli EF-Tu indicating that E. coli accumulated epistatic changes that altered the ancient communication network present in the ancestors and extant T. aquaticus EF-Tu. The inability of the consensus method to reproduce epistatic features of evolutionary processes is a known limitation (Trudeau et al., 2016).
While both ancestral reconstruction and consensus methods are able to successfully generate EF-Tu proteins that display improved thermostability and share a conserved fold, only ASR recapitulates the evolutionary conserved dynamical networks that appear to be a hallmark of the protein family. Experimental validation of protein function (i.e. translation) for these sequences at optimal temperatures is currently not possible, as this temperature varies for each EF-Tu and is closely correlated with their individual thermostabilities. All translation assay components would have to be optimized at corresponding temperatures for each EF-Tu. While it is not a straightforward task to use data from MD simulations to make inferences about functional properties of proteins, our analyses suggest that ASR EF-Tu proteins will function similarly to T. aquaticus EF-Tu. MD reveals significant divergence of the consensus protein from the extant and ancestral in its dynamics that would likely impact its function given that it requires productive association and dissociation with multiple components of the translational machinery. A previous study that compared enzymatic function of ASR and consensus-generated Precambrian β-lactamases showed consistent activity in ASR sequences but inconsistent, varying levels of activity in consensus sequences (Risso et al., 2014). Therefore, the results observed here appear consistent with previously determined limitations of the consensus method for generating proteins with predictable function and behavior. Based on our work, we provide two considerations for the design of functional, thermostable proteins.
First, our results validate the use of ASR as a protein design strategy for multidomain proteins revealing that ASR proteins employ mechanisms similar to T. aquaticus EF-Tu for achieving thermostability. Domain-specific effects that were common to both ASR and T. aquaticus sequences include i) stabilization of domain 1, mediated by intermolecular contacts such as hydrophobic interactions; and ii) enhanced interdomain communication between domains 1 and 2. This observation reinforces the importance of the use of evolutionary history as a guide for protein design. In addition to improved thermostability, ASR conserves the natural protein motions necessary for function, as demonstrated here in EF-Tu, a multidomain protein containing a GTPase domain (domain 1) and two oligonucleotide-binding domains (domains 2,3). Given the vast diversity in protein folds, it is unclear whether ASR would service as a tool to engineer stability in all protein families or in a finite subset. However, increased thermostability has been observed for ASR variants in other bacterial protein families such as β-lactamases (Risso et al., 2014), adenylate kinases (Nguyen et al., 2017) and nucleotide diphosphate kinases (Akanuma et al., 2013). While MD simulations were not performed to evaluate protein motions in these previous studies, retention of function in the ASR variants suggests that functional motions are conserved. Our work therefore complements previous work, demonstrating that ASR can be used to robustly design variants (single and multidomain) that are both thermostable and conserve natural protein motions across several diverse protein folds.
Second, an alternative design strategy for thermostability may be to emulate evolution by sampling amino acid sequence space in MD simulations while monitoring conformational fluctuations to identify thermostable scaffolds. A potential limitation is that the method would optimize thermostability but could also produce non-functional proteins. To mitigate this possibility, all simulations would have to be performed in a functional state of the protein complex (e.g. in complex with ligand or with an appropriate partner protein).
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric Ortlund (eortlun@emory.edu).
Experimental Model and Subject Details
Escherichia coli strain BL21(DE3) (Invitrogen, Carlsbad, CA) was transformed with pET-21a vector containing the N-terminal Hisx6-tagged ancestral and variant EF-Tus (Gaucher et al., 2008), and expressed by overnight auto-induction using Overnight Express Media (Novagen, Madison, WI) at 37 °C (Studier, 2005).
Method Details
Protein expression and purification
Cells were removed from media by centrifugation at 4550 × g and 4 °C for 15 minutes, resuspended in lysis buffer (50 mM potassium phosphate pH 7.5, 300 mM NaCl, 5 % (v/v) glycerol, 25 mM imidazole, 100 μM GDP, 10 μg/ml DNase, 176 μM PMSF and 5 mM BME), and lysed by sonication. Lysate was cleared by centrifugation at 31,000 × g and 4 °C for 1 hour and the supernatant purified using a 5 mL HisTrap FF column (GE Healthcare, Piscataway, NJ) according to manufacturer’s protocol except base buffer (50 mM potassium phosphate pH 7.5, 300 mM NaCl, 5 % (v/v) glycerol 100 μM GDP) containing either 25 or 500 mM imidazole using a 2% wash/30% elution/100% wash method. Protein fractions were pooled and dialyzed twice against 1000× buffer volume (20 mM Tris HCl, 100 mM KCl, 10 mM MgCl2 and 5 μM GDP) and stored at 4 °C.
Crystallography and structure determination
Ancestral EF-Tu Node 168 (N168) crystals were grown in 1.35 M ammonium sulfate and 0.1 M HEPES pH 7.5. Crystals were harvested by briefly soaking in cryoprotectant (2.0 M ammonium sulfate, 25 % sucrose) and flash-freezing in liquid nitrogen. N253 was crystallized in 40% PEG 200 and 0.1 M HEPES pH 7.0. Data were collected at the APS 22-BM without further cryoprotection. Consensus EF-Tu was crystallized in 20% PEG 10000 and 0.1 M HEPES, pH 7. And crystal were cryoprotected with 15% glycerol added to the crystallant. Data for all three complexes was conducted at the SER-CAT (Southeast Regional Collaborative Access Team) bending magnet (22-BM) beamline at the Advanced Proton Source (APS) at Argonne National Laboratory (Argonne, IL) and the data were processed and scaled with HKL2000 (Table)1 (Otwinowski et al., 1997). For N128, an earlier 3.2 Å model of N168 was used as a molecular replacement search model in Phaser (McCoy et al., 2007). Phases N253 and the Consensus EF were determined by molecular replacement using final N168 structure for initial phasing. Structure refinement and validation for all complexes was performed using PHENIX (Phenix, Berkeley, CA) (version 1.11.1), and model building was performed in COOT (MRC Laboratory of Molecular Biology, Cambridge, UK) (Adams et al., 2010; Emsley et al., 2010). PyMOL (version 1.8.2; Schrödinger, New York, NY) was used to visualize structures and generate figures. The Molprobity server was also used to assist in the refinement (Davis et al., 2007). A homology model of N170 was produced and energy minimized using best practices in MODELLER based on N168’s refined structure (Eswar et al., 2006). The percentage of residues in favored Ramachandran space is greater than 96% for all final models. Secondary structure assignment was made using x-ray structures (PDB IDs 5W75, 5W76, 1EFC, 1TUI, 5W7Q) with STRIDE (Heinig and Frishman, 2004) implemented in VMD.
ProSMART analysis
The ProSMART analysis tool (Nicholls et al., 2014) was used to perform a structural comparison of protein structure pairs. Pairwise comparisons were conducted between crystal structures of E. coli and i) T. aquaticus; ii) N168; iii) N253 and iv) Consensus. Comparisons generated a Procrustes score which is the RMSD of the central residue of two corresponding structural fragments of length n, where n is an odd number of amino acids.
Molecular dynamics
Six systems were prepared for MD simulations: ancestral EF-Tu nodes 168 (N168), 170 (N170), and 253 (N253), E. coli EF-Tu (PDB 1EFC), T. aquaticus EF-Tu (PDB 1TUI) and consensus EF-Tu. Each protein was in complex with a GDP and Mg2+ ion. Crystal structures reported here were used for N168 (PDB 5W75), N253 (PDB 5W76) and consensus EF-Tu (PDB 5W7Q). A structure for N170 was modeled from N168. The complexes were solvated in an octahedral box of TIP3P water with a 10 Å buffer around the protein complex. Na+ and Cl− ions were added to neutralize the protein and achieve 150 mM NaCl, mimicking physiological conditions. The xleap module of AmberTools (Case and Kollman, 2012) was used to set up the systems with the parm99-bsc0 forcefield (Pérez et al., 2007). Parameters for GDP were obtained using Antechamber (Wang et al., 2001) in AmberTools. All minimizations and simulations were performed with Amber14 (Case et al., 2014). All systems were subjected to 5000 steps of steepest descent minimization followed by 5000 steps of conjugate gradient minimization with 500 kcal/mol. Å2 restraints on all atoms. Restraints were removed from atoms (except for GDP, Mg2+ and surrounding residues) and the previously described minimization was repeated. Heating from 0 to 300 K was performed in a 100-ps run using constant volume periodic boundaries with 5 kcal/mol. Å2 harmonic restraints on all protein atoms. 12 ns of MD equilibration was performed with 10 kcal/mol. Å2 restraints on GDP, Mg2+ and surrounding residues using the NPT ensemble. All restraints were removed and 300 ns production simulations were performed for each system at both 300 K and 360 K in the NPT ensemble. A 2-fs timestep was used in all simulations and all bonds between hydrogen and heavy atoms were fixed with the SHAKE algorithm (Ryckaert et al., 1977). A cut-off distance 10 Å was used to evaluate long-range electrostatics with Particle Mesh Ewald (PME) and for van der Waals forces. For all analyses, 15000 evenly spaced frames were taken from each simulation.
MD trajectories were analyzed with various tools. Root mean square deviation (RMSD) and root mean square fluctuations of residues (RMSF) were performed with the CPPTRAJ module of AmberTools(Roe and Cheatham III, 2013). RMSD and RMSF analysis were both performed in two ways: i) Locally, by domain, where calculations took only atoms of individual domains into account, and ii) Globally, where all atoms (all domains) are used in calculations. DSSP (Kabsch and Sander, 1983) was used to assign protein secondary structure as well as to calculate accessible surface area in the folded state of the protein (Rose et al., 1985). HBPLUS (McDonald and Thornton, 1994)was used to calculate the number of hydrogen bonds in the protein along the trajectory. A hydrogen bond is defined by a maximum distance of 3.9 Å between a donor (non-hydrogen) and acceptor atoms with a minimum DHA angle of 90°. The Salt Bridges plugin in VMD (Humphrey et al., 1996) was used to identify pairs of amino acids engaged in ionic interactions. Ionic interactions are defined between residue pairs (R,K,H): (D,E) that are within 6 A for at least 75% of the simulation. Surrounding hydrophobicity was calculated as previously described (Gromiha et al., 2013) using the hydrophobic indices of surrounding residues with a 6.5 Å cutoff. CPPTRAJ was used for principal component analysis over backbone atoms. Network theory, implemented in the NetworkView (Eargle and Luthey-Schulten, 2012; Sethi et al., 2009) plugin in VMD and the Carma program (Glykos, 2006) were used to construct protein structure networks from each trajectory. Briefly, networks are constructed by defining all protein C-α atoms as nodes, using Cartesian covariance to measure communication within the network. Edges are drawn between pairs of nodes that reside within a 4.5 Å cutoff for > 75% of the simulation. Networks were divided into communities, subnetworks which are defined by dynamics of the proteins. Communities are generated using the Girvan-Newman algorithm to find groups of nodes with correlated motions. The minimum number of communities possible was generated while maintaining at least 98% maximum modularity (Newman, 2006). The MMTSB toolset was used to perform a cluster analysis with a 2 Å RMSD cutoff (Feig et al., 2004).
Quantification and Statistical Analysis
N/A.
Data and Software Availability
Coordinates have been deposited into the Protein Data Bank under accession codes 5W75, 5W76, and 5W7Q.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| BL21 (DE3) | Invitrogen, Carlsbad, CA | C602003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Deoxyribonuclease (DNase) | Spectrum chemicals | CAS: 9003-98-9 |
| Phenylmethylsulfonyl fluoride (PMSF) | Roche | CAS: 329-98-6 |
| Beta mercaptoethanol (BME) | Acros Organics | CAS: 60-24-2 |
| Guanosine diphosphate (GDP) | Sigma | CAS: 43139-22-6 |
| Deposited Data | ||
| Consensus EF-Tu-GDP-Mg | This study | PDB 5W7Q |
| Node 168 EF-Tu-GDP-Mg | This study | PDB 5W75 |
| Node 253 EF-Tu-GDP-Mg | This study | PDB 5W76 |
| Recombinant DNA | ||
| pET-21a vectors containing EF-Tu sequences | Gaucher et al., 2008 | N/A |
| Software and Algorithms | ||
| PHENIX (version 1.11.1) | Adams et al., 2010 | RRID:SCR_014224 |
| COOT | Emsley et al., 2010 | RRID:SCR_014222 |
| Phaser | (McCoy et al., 2007) | RRID:SCR_014219 |
| PyMOL (version 1.8.2) | Schrodinger, New York, NY | RRID:SCR_000305 |
| MODELLER | Eswar et al., 2006 | RRID:SCR_008395 |
| Molprobity server | Davis et al., 2007 | RRID:SCR_014226 |
| ProSMART | Nicholls et al., 2014 | http://smb.slac.stanford.edu/facilities/software/ccp4/html/prosmart.html |
| Amber Molecular Dynamics Package (Amber14, AmberTools 15) | Case et al., 2014 | http://ambermd.org/ |
| HBPlus | McDonald and Thornton, 1994 | https://www.ebi.ac.uk/thornton-srv/software/HBPLUS/ |
| Visual Molecular Dynamics | Humphrey et al., 1996 | RRID:SCR_001820 |
| Carma | Glykos, 2006 | https://utopia.duth.gr/glykos/carma.html |
| MMTSB | Feig et al., 2004 | http://blue11.bch.msu.edu/mmtsb/Main_Page |
| Other | ||
| HisTrap FF Column | GE Healthcare, Piscataway, NJ | Cat#: 17-5255-01 |
Supplementary Material
HIGHLIGHTS.
Ancestral and consensus methods generate thermostable EF-Tus and conserve structure
Ancestral sequences reveal stable dynamics similar to extant, thermostable EF-Tu
Consensus sequence displays distinct dynamics and instability in simulations
Study highlights benefit of ASR and role of evolution in achieving thermostability
Acknowledgments
This work was supported in part by the National Institutes of Health (K12-GM000680 to C.D.O.); W.M. Keck Foundation Medical Research Grant (to E.A.O.). Crystallographic data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, C.D.O., E.A.G., E.A.O.; Methodology, C.D.O., M.F.C., E.A.O.; Investigation, C.D.O., M.C.P. C.E.F., N.C.B.; Writing – Original Draft, C.D.O., M.C.P., C.E.F., N.C.B., E.A.G., and E.A.O.; Writing – Review & Editing, C.D.O., E.A.G., and E.A.O.; Funding Acquisition, E.A.G. and E.A.O.; Resources, E.A.G. and E.A.O; Supervision, E.A.O.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akanuma S, Nakajima Y, Yokobori S-i, Kimura M, Nemoto N, Mase T, Miyazono K-i, Tanokura M, Yamagishi A. Experimental evidence for the thermophilicity of ancestral life. Proceedings of the National Academy of Sciences. 2013;110:11067–11072. doi: 10.1073/pnas.1308215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin N, Liu AD, Ramer S, Aehle W, Meijer D, Metin M, Wong S, Gualfetti P, Schellenberger V. Construction of stabilized proteins by combinatorial consensus mutagenesis. Protein Engineering, Design and Selection. 2004;17:787–793. doi: 10.1093/protein/gzh091. [DOI] [PubMed] [Google Scholar]
- Asial I, Cheng YX, Engman H, Dollhopf M, Wu B, Nordlund P, Cornvik T. Engineering protein thermostability using a generic activity-independent biophysical screen inside the cell. Nat Commun. 2013;4 doi: 10.1038/ncomms3901. [DOI] [PubMed] [Google Scholar]
- Case D, Babin V, Berryman J, Betz R, Cai Q, Cerutti D, Cheatham T, III, Darden T, Duke R, Gohlke H. Amber. 2014;14 [Google Scholar]
- Case D, Kollman P. AmberTools. University of California, San Francisco; 2012. p. 12. [Google Scholar]
- Cole MF, Gaucher EA. Utilizing Natural Diversity to Evolve Protein Function: Applications Towards Thermostability. Current opinion in chemical biology. 2011;15:399–406. doi: 10.1016/j.cbpa.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czworkowski J, Moore PB. The elongation phase of protein synthesis. Progress in nucleic acid research and molecular biology. 1996;54:293–332. doi: 10.1016/s0079-6603(08)60366-9. [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eargle J, Luthey-Schulten Z. NetworkView: 3D display and analysis of protein·RNA interaction networks. Bioinformatics. 2012;28:3000–3001. doi: 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimie E, Ebrahimi M. Searching for patterns of thermostability in proteins and defining the main features contributing to enzyme thermostability through screening, clustering, and decision tree algorithms 2010 [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006 doi: 10.1002/0471250953.bi0506s15. Chapter 5 Unit 5 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig M, Karanicolas J, Brooks CL. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. Journal of Molecular Graphics and Modelling. 2004;22:377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Gaucher EA, Govindarajan S, Ganesh OK. Palaeotemperature trend for Precambrian life inferred from resurrected proteins. Nature. 2008;451:704–707. doi: 10.1038/nature06510. [DOI] [PubMed] [Google Scholar]
- Gaucher EA, Thomson JM, Burgan MF, Benner SA. Inferring the palaeoenvironment of ancient bacteria on the basis of resurrected proteins. Nature. 2003;425:285–288. doi: 10.1038/nature01977. [DOI] [PubMed] [Google Scholar]
- Glykos NM. Carma: a molecular dynamics analysis program. J Comput Chem. 2006;27:1765–1768. doi: 10.1002/jcc.20482. [DOI] [PubMed] [Google Scholar]
- Gromiha MM, Pathak MC, Saraboji K, Ortlund EA, Gaucher EA. Hydrophobic environment is a key factor for the stability of thermophilic proteins. Proteins: Structure, Function, and Bioinformatics. 2013;81:715–721. doi: 10.1002/prot.24232. [DOI] [PubMed] [Google Scholar]
- Haney P, Konisky J, Koretke K, Luthey-Schulten Z, Wolynes P. Structural basis for thermostability and identification of potential active site residues for adenylate kinases from the archaeal genus Methanococcus. Proteins Structure Function and Genetics. 1997;28:117–130. doi: 10.1002/(sici)1097-0134(199705)28:1<117::aid-prot12>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Harms MJ, Thornton JW. Analyzing protein structure and function using ancestral gene reconstruction. Current opinion in structural biology. 2010;20:360–366. doi: 10.1016/j.sbi.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Marin J, Suleski M, Paymer M, Kumar S. Tree of life reveals clock-like speciation and diversification. Molecular biology and evolution. 2015;32:835–845. doi: 10.1093/molbev/msv037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Research. 2004;32:W500–W502. doi: 10.1093/nar/gkh429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of molecular graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kannan N, Vishveshwara S. Aromatic clusters: a determinant of thermal stability of thermophilic proteins. Protein engineering. 2000;13:753–761. doi: 10.1093/protein/13.11.753. [DOI] [PubMed] [Google Scholar]
- Krab IM, Parmeggiani A. EF-Tu, a GTPase odyssey. Biochimica et Biophysica Acta (BBA)-Gene Structure and Expression. 1998;1443:1–22. doi: 10.1016/s0167-4781(98)00169-9. [DOI] [PubMed] [Google Scholar]
- Kumar S, Nussinov R. How do thermophilic proteins deal with heat? Cellular and Molecular Life Sciences CMLS. 2001;58:1216–1233. doi: 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tsai CJ, Nussinov R. Factors enhancing protein thermostability. Protein engineering. 2000;13:179–191. doi: 10.1093/protein/13.3.179. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Kostrewa D, Wyss M, Brugger R, D’Arcy A, Pasamontes L, van Loon AP. From DNA sequence to improved functionality: using protein sequence comparisons to rapidly design a thermostable consensus phytase. Protein Engineering. 2000;13:49–57. doi: 10.1093/protein/13.1.49. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Loch C, Middendorf A, Studer D, Lassen SF, Pasamontes L, van Loon AP, Wyss M. The consensus concept for thermostability engineering of proteins: further proof of concept. Protein engineering. 2002;15:403–411. doi: 10.1093/protein/15.5.403. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Wyss M. Engineering proteins for thermostability: the use of sequence alignments versus rational design and directed evolution. Current Opinion in Biotechnology. 2001;12:371–375. doi: 10.1016/s0958-1669(00)00229-9. [DOI] [PubMed] [Google Scholar]
- Li WF, Zhou XX, Lu P. Structural features of thermozymes. Biotechnology Advances. 2005;23:271–281. doi: 10.1016/j.biotechadv.2005.01.002. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. Journal of Applied Crystallography. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. Journal of molecular biology. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- Merkl R, Sterner R. Reconstruction of ancestral enzymes. Perspectives in Science. 2016;9:17–23. [Google Scholar]
- Newman ME. Modularity and community structure in networks. Proceedings of the national academy of sciences. 2006;103:8577–8582. doi: 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V, Wilson C, Hoemberger M, Stiller JB, Agafonov RV, Kutter S, English J, Theobald DL, Kern D. Evolutionary drivers of thermoadaptation in enzyme catalysis. Science. 2017;355:289–294. doi: 10.1126/science.aah3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls RA, Fischer M, McNicholas S, Murshudov GN. Conformation-independent structural comparison of macromolecules with ProSMART. Acta Crystallographica Section D: Biological Crystallography. 2014;70:2487–2499. doi: 10.1107/S1399004714016241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment 11. In: Thornton J, editor. Journal of Molecular Biology. Vol. 302. 2000. pp. 205–217. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W, Charles W, Carter Jr. Methods in Enzymology. Academic Press; 1997. [20] Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Panja AS, Bandopadhyay B, Maiti S. Protein Thermostability Is Owing to Their Preferences to Non-Polar Smaller Volume Amino Acids, Variations in Residual Physico-Chemical Properties and More Salt-Bridges. PloS one. 2015;10:e0131495. doi: 10.1371/journal.pone.0131495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L, Zuckerkandl E. Chemical paleogenetics. Acta chem scand. 1963;17:S9–S16. [Google Scholar]
- Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE, Laughton CA, Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophysical journal. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porebski BT, Buckle AM. Consensus protein design. Protein Engineering, Design and Selection. 2016;29:245–251. doi: 10.1093/protein/gzw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razvi A, Scholtz JM. Lessons in stability from thermophilic proteins. Protein Science : A Publication of the Protein Society. 2006;15:1569–1578. doi: 10.1110/ps.062130306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renugopalakrishnan V, Garduno-Juarez R, Narasimhan G, Verma C, Wei X, Li P. Rational design of thermally stable proteins: relevance to bionanotechnology. Journal of nanoscience and nanotechnology. 2005;5:1759–1767. doi: 10.1166/jnn.2005.441. [DOI] [PubMed] [Google Scholar]
- Richards FM. Areas, Volumes, Packing, and Protein Structure. Annual Review of Biophysics and Bioengineering. 1977;6:151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- Risso VA, Gavira JA, Gaucher EA, Sanchez-Ruiz JM. Phenotypic comparisons of consensus variants versus laboratory resurrections of Precambrian proteins. Proteins: Structure, Function, and Bioinformatics. 2014;82:887–896. doi: 10.1002/prot.24575. [DOI] [PubMed] [Google Scholar]
- Roe DR, Cheatham TE., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. Journal of Computational Physics. 1977;23:327–341. [Google Scholar]
- Sadeghi M, Naderi-Manesh H, Zarrabi M, Ranjbar B. Effective factors in thermostability of thermophilic proteins. Biophysical Chemistry. 2006;119:256–270. doi: 10.1016/j.bpc.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Šanderová H, Hůlková M, Maloň P, Kepková M, Jonák J. Thermostability of multidomain proteins: Elongation factors EF-Tu from Escherichia coli and Bacillus stearothermophilus and their chimeric forms. Protein science. 2004;13:89–99. doi: 10.1110/ps.03272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scandurra R, Consalvi V, Chiaraluce R, Politi L, Engel PC. Protein thermostability in extremophiles. Biochimie. 1998;80:933–941. doi: 10.1016/s0300-9084(00)88890-2. [DOI] [PubMed] [Google Scholar]
- Sethi A, Eargle J, Black AA, Luthey-Schulten Z. Dynamical networks in tRNA: protein complexes. Proceedings of the National Academy of Sciences. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Trudeau DL, Kaltenbach M, Tawfik DS. On the Potential Origins of the High Stability of Reconstructed Ancestral Proteins. Molecular Biology and Evolution. 2016;33:2633–2641. doi: 10.1093/molbev/msw138. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang W, Kollman PA, Case DA. Antechamber: an accessory software package for molecular mechanical calculations. J Am Chem Soc. 2001;222:U403. [Google Scholar]
- Watanabe K, Ohkuri T, Yokobori S-i, Yamagishi A. Designing Thermostable Proteins: Ancestral Mutants of 3-Isopropylmalate Dehydrogenase Designed by using a Phylogenetic Tree. Journal of Molecular Biology. 2006;355:664–674. doi: 10.1016/j.jmb.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Yamashiro K, Yokobori SI, Koikeda S, Yamagishi A. Improvement of Bacillus circulans β-amylase activity attained using the ancestral mutation method. Protein Engineering Design and Selection. 2010:gzq021. doi: 10.1093/protein/gzq021. [DOI] [PubMed] [Google Scholar]
- Yang H, Liu L, Li J, Chen J, Du G. Rational Design to Improve Protein Thermostability: Recent Advances and Prospects. ChemBioEng Reviews. 2015;2:87–94. [Google Scholar]
- Zhou XX, Wang YB, Pan YJ, Li WF. Differences in amino acids composition and coupling patterns between mesophilic and thermophilic proteins. Amino Acids. 2007;34:25–33. doi: 10.1007/s00726-007-0589-x. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Asahara H, Gaucher EA, Chong S. Reconstitution of translation from Thermus thermophilus reveals a minimal set of components sufficient for protein synthesis at high temperatures and functional conservation of modern and ancient translation components. Nucleic Acids Research. 2012;40:7932–7945. doi: 10.1093/nar/gks568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.