

ABSTRACT
Site-directed RNA editing (SDRE) is a general strategy for making targeted base changes in RNA molecules. Although the approach is relatively new, several groups, including our own, have been working on its development. The basic strategy has been to couple the catalytic domain of an adenosine (A) to inosine (I) RNA editing enzyme to a guide RNA that is used for targeting. Although highly efficient on-target editing has been reported, off-target events have not been rigorously quantified. In this report we target premature termination codons (PTCs) in messages encoding both a fluorescent reporter protein and the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) protein transiently transfected into human epithelial cells. We demonstrate that while on-target editing is efficient, off-target editing is extensive, both within the targeted message and across the entire transcriptome of the transfected cells. By redirecting the editing enzymes from the cytoplasm to the nucleus, off-target editing is reduced without compromising the on-target editing efficiency. The addition of the E488Q mutation to the editing enzymes, a common strategy for increasing on-target editing efficiency, causes a tremendous increase in off-target editing. These results underscore the need to reduce promiscuity in current approaches to SDRE.
KEYWORDS: RNA editing, site-directed RNA editing, ADAR, off-target RNA editing
Introduction
The conversion of A to I is one of the most common forms of base modifications in the messenger RNAs of multicellular metazoans. The process is catalyzed by a family of enzymes called Adenosine Deaminases that Act on RNA (ADARs).1–6 Biochemically, ADARs convert A's to I's by a simple hydrolytic deamination reaction.1,2 Because ribosomes read I's as guanosine (G),7 ADARs can alter protein function by recoding codons. From the standpoint of therapeutics, this activity could be useful. For example, mutations caused by G to A transitions can be reverted. PTCs (UGA, UAG, and UAA) can be recoded to tryptophan (UGG), allowing translation of full-length proteins. This process could also be used to engineer protein function. Given the broad range of therapeutic possibilities, strategies to redirect ADAR function are receiving increased attention.
The idea of hijacking ADAR activity for therapeutic purposes was first introduced 22 years ago.8 The strategy was to use an antisense RNA oligonucleotide to create a double-stranded RNA around a specific A that endogenous ADAR would recognize.1 Others have made variations on this strategy.9 More recently, new SDRE strategies have been developed.10,11 ADARs are modular enzymes, composed of a deaminase domain (DD) for catalysis and two or more N-terminal double-stranded RNA binding motifs (dsRBMs) for targeting. New methods for SDRE use an engineered ADAR where an antisense RNA oligonucleotide is substituted for the dsRBMs and guides the catalytic domain to a specific A.9,10,12–17 SDRE has successfully been employed in vitro, in mammalian cells, and in a simple animal model.9–17 It has been used to target a variety of genetic mutations that lead to diseases, including Cystic Fibrosis,10 Duchenne muscular dystrophy8 and Factor V Leiden thrombophilia.13 Targeting ADAR activity has also been proven useful for basic research. For instance, by tethering RNA binding proteins to ADAR's DD, their endogenous binding partners can be identified using a novel technique called TRIBE (targets of RNA-binding proteins identified by editing).18 Undoubtedly, there are many more applications.
To achieve selective editing, the Rosenthal lab has developed a genetically encoded, two component system for SDRE (Fig. 1A).10,16 The first is a recombinant editing enzyme which uses the DD from human ADAR2. The second is an antisense RNA guide, complementary to an A of interest within an mRNA, which serves two functions: it acts as a targeting domain and it provides the double-stranded structure required for editing.19 The interaction between the bacteriophage λN peptide and boxB RNA hairpin is used to link the guide and the DD:20 a λN peptide is fused to the N-terminus of the DD (λN-DD) and a boxB hairpin is linked to the RNA guide (Fig. 1A). Although this configuration worked well in vitro, its effectiveness was limited in transfected cells.10 To improve editing in the cellular environment, additional λN peptides and boxB hairpins were added to the system, and a specific mutation known to increase catalytic activity (E488Q)21 was added to the DD.16 Although these modifications greatly increased editing efficiency, they also led to the production of numerous unintended edits within the targeted message.16
Figure 1.
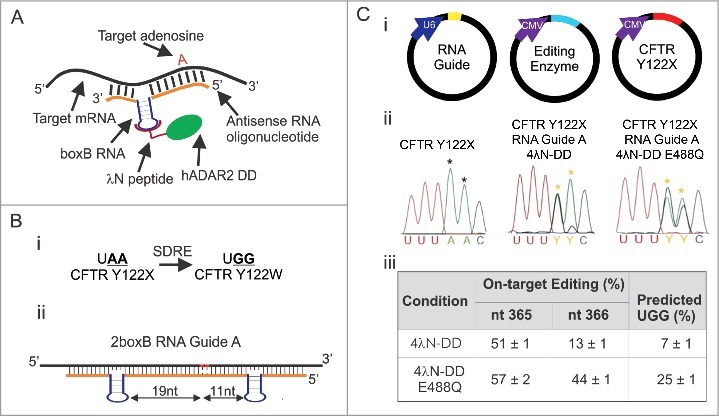
SDRE corrects a UAA PTC in CFTR Y122X. (A) Schematic showing the basic components of SDRE. hADAR2′s dsRBMs were removed and replaced with the RNA binding protein, λN (magenta). The DD is shown in green. An RNA guide (orange) complementary to the target mRNA (black) is fused to the boxB RNA hairpin (purple). The target A is shown in red. (B) Cartoon depicting overall strategy for correcting CFTR Y122X (i). Note that two A's need to be deaminated for conversion to W. (ii) Schematic of 2boxB RNA Guide A for targeting CFTR Y122X in human cells. The RNA guide is complementary to the target mRNA except at the boxB's and the target A's (shown in red), each having a C mismatch. One boxB is inserted 19 nt 3′ and another 11 nt 5′ from the first A in the UAA PTC. (C) Plasmid delivery of the components of SDRE for transient transfections in HEK293T cells (I). Electropherograms of directly sequenced RT-PCR products of corrected HEK293T cells and cells transfected with CFTR Y122X alone (ii). Yellow asterisks point to the edited A's and black asterisks refer to unedited A's. (iii) Quantification of correction at each target A. Editing percentages are based on direct sequencing results from RT-PCR. The estimated percent conversion to UGG was the product of the fractional conversion for each A; n = 3, mean ± s.e.m.
Off-target edits have never been rigorously quantified for any system of SDRE. For the approach to move towards therapeutic consideration, off-targets must be carefully assessed. If they are widespread, then approaches to reduce them must be employed. Others have reported few or no off-target edits, yet they have only looked for them in the targeted message.9,11–15,17 Our group uncovered numerous off-target events within the targeted message, and they could be mostly, but not completely, eliminated by reducing the amount of RNA guide transfected.16 We also explored off-target edits in five endogenous mRNAs expressed in HEK293T cells that were not targeted by our RNA guide and saw no editing. However, we did see increased editing, in two well-known substrates for endogenous ADAR (GLI1 and NEIL1).22,23 These results were ambiguous and suggested that SDRE could edit non-targeted mRNAs, but the extent of the phenomenon was unknown. In this study, we systematically evaluate off-target editing, both within the targeted message, and across the entire HEK293T cell transcriptome.
Results
Determining the efficiency of SDRE to target CFTR Y122X
The goal of this study was to determine whether we could efficiently edit a therapeutic target and also rigorously quantify off-target edits produced by SDRE. In our past study, we focused on editing a fluorescent reporter.16 Here, we test our system on a target with potential therapeutic value: the Y122X mutation within the CFTR mRNA. This mutation, as with many others within CFTR, leads to a severe form of Cystic Fibrosis, the most common inherited fatal disorder in Caucasians.24,25 Targeting Y122X presents a challenge because two A's need to be edited to convert the PTC to a tryptophan for read-through (UAA>UGG; Fig. 1Bi). As a first step, we set out to determine the efficiency at which our basic strategy can edit CFTR Y122X in transiently transfected HEK293T cells. In general, ADARs have specific preferences for residues that neighbor a targeted A (5′ U>A>C>G and 3′ G>C>U = A).26,27 Based on these rules, we reasoned that both A's in codon 122 (UAAC) would be practical candidates for editing.
For this experiment, we designed a suitable RNA guide specific for Y122X using the 2boxB strategy outlined in our previous report.16 In brief, this guide (i.e. RNA guide A) was designed with one boxB inserted 19 nt 5′, and a second boxB 11 nt 3′, from the first A in the UAA PTC (Fig. 1Bii). Besides the boxB's, all sequences were complementary to CFTR except for two C mismatches under the targeted A's which, according to previous studies, increase editing efficiency.10,14,16,28 CFTR Y122X, RNA Guide A, and the recombinant editing enzymes (4λN-DD and 4λN-DD E488Q) were delivered in separate plasmids (Fig. 1Ci) using transient transfections (CFTR Y122X and the editing enzymes were driven by the CMV promoter and the guide was driven by a U6 promoter). RNA was then extracted, and the extent of editing was estimated by RT-PCR followed by direct sequencing. Examples of the resulting electropherograms are shown in Fig. 1Cii. There was no editing at either A in cells transfected with CFTR Y122X alone. Transfections that included 4λN-DD and the RNA guide resulted in 50% editing at the first A and 13% at the second. For 4λN-DD carrying the E488Q mutation, editing was 56% and 44% at the first and second A's, respectively. Quantification of editing at both A's from three independent experiments with either 4λN-DD or 4λN-DD E488Q is shown in Fig. 1Ciii. Because both A's must be edited to convert this codon to tryptophan (single edits still result in a PTC), we estimated our success as the fractional conversion for both, assuming the editing events are independent (later we show that this assumption is not accurate). For 4λN-DD the correction of the PTC was 7 ± 1% and for 4λN-DD E488Q, 25 ± 1% (Fig. 1Ciii). Overall, these results suggest that SDRE can be used to target CFTR Y122X in human cells, however the efficiency is low.
Off-targets in CFTR Y122X mRNA
Another important concern associated with editing Y122X was the production of off-target edits. In our previous report, we showed that SDRE often generates off-targets, particularly with 4λNDD carrying the E488Q mutation.16 While we also showed that most off-targets within the fluorescent reporter mRNA that was targeted (mCherry-eGFP) could be controlled by limiting the amount of RNA guide and using 4λN-DD WT instead of 4λN-DD E488Q, some off-targets persisted. For the present study, we worried that off-targets could be more extensive, given that CFTR mRNA is significantly larger than the fluorescent reporter used before (4,440 nt versus 1,512 nt). Accordingly, we determined the extent of off-target editing in the complete CFTR mRNA when targeting the Y122X mutation. For these experiments, we transfected HEK293T cells as before and quantified off-target edits by RT-PCR and direct sequencing. Using 4λN-DD, there were six off-target edits, three of which resulted in silent mutations (Table 1A). For 4λN-DD E488Q, there were seventeen off-target edits, all 6 produced by 4λN-DD WT plus 11 additional ones. For all off-targets that were shared by both enzymes, editing was higher for the E488Q version. All off-targets but three (I199V, I121V, and L123L) occurred in areas not base-paired with the RNA guide. These results show that while SDRE can correct Y122X in HEK293T cells, it also generates multiple off-target editing events, particularly when using 4λN-DD E488Q.
Table 1.
Off-target editing events in CFTR Y122X produced by SDRE. Table indicating off-target editing sites found within the full-length CFTR mRNA after transient transfections in HEK293T cells using the non-NLS (A) or NLS-tagged (B) editing enzymes. Off-target edits that lead to a silent mutation are shown in bold. Off-targets that occur in areas complementary to the RNA guide are underlined. Editing percentages are based on direct sequencing results from RT-PCR products. NE = no editing detected. Values are reported as mean ± s.e.m; n = 2.
| A | |||
|---|---|---|---|
| nt Position | Codon Change | 4λN-DD | 4λN-DD E488Q |
| 355 | I199V | NE | 13 ± 2 |
| 361 | I121V | NE | 10 ± 1 |
| 369 | L123L | 39 ± 4 | 46 ± 3 |
| 761 | K254R | NE | 12 ± 5 |
| 1378 | T460A | NE | 16 ± 5 |
| 1458 | G486G | NE | 11 ± 1 |
| 2378 | K793R | NE | 29 ± 1 |
| 2379 | K793K | NE | 16 ± 5 |
| 3493 | K1165E | NE | 12 ± 2 |
| 3703 | S1235G | NE | 18 ± 1 |
| 3733 | R1245G | NE | 15 ± 1 |
| 3924 | E1308E | 18 ± 2 | 55 ± 1 |
| 3926 | Q1309R | 20 ± 5 | 46 ± 1 |
| 3973 | R1325G | NE | 16 ± 1 |
| 4191 | V1397V | 14 ± 1 | 14 ± 2 |
| 4376 | K1459R | 13 ± 1 | 19 ± 1 |
| 4381 | K1461E | 12 ± 1 | 36 ± 1 |
| B | |||
| nt Position | Codon Change | NLS 4λN-DD | NLS 4λN-DD E488Q |
| 355 | I199V | NE | NE |
| 361 | I121V | NE | NE |
| 369 | L123L | 27 ± 2 | 35 ± 1 |
| 761 | K254R | NE | NE |
| 1378 | T460A | NE | 10 ± 3 |
| 1458 | G486G | NE | NE |
| 2378 | K793R | NE | NE |
| 2379 | K793K | NE | NE |
| 3493 | K1165E | NE | NE |
| 3703 | S1235G | NE | NE |
| 3733 | R1245G | NE | NE |
| 3924 | E1308E | NE | 38 ± 2 |
| 3926 | Q1309R | NE | 27 ± 1 |
| 3973 | R1325G | NE | NE |
| 4191 | V1397V | NE | NE |
| 4376 | K1459R | NE | 15 ± 2 |
| 4381 | K1461E | NE | 30 ± 1 |
Adding an NLS to the editing enzymes efficiently transports SDRE to the nucleus
Consistent with our previous data, we have shown that our current system of SDRE generates off-target events. When devising strategies to reduce them, we hypothesized that the subcellular localization of the editing enzymes could play an important role. In contrast to WT human ADAR2, which is localized to the nucleus,29–31 4λN-DD and 4λN-DD E488Q would be expected to be expressed in the cytoplasm because they lack a nuclear localization signal (NLS; in their construction, the putative NLS of human ADAR2 was removed along with the dsRBMs).10,16 We hypothesized that a cytoplasmic localization for 4λN-DD and 4λN-DD E488Q would extend the duration of their access to RNAs, and this could result in increased off-target editing. In addition, in the cytoplasm the enzymes would need to compete with ribosomes for binding. Thus, we decided to transport our enzymes to the nucleus by adding three NLS's from the SV40 Large T Antigen to the N-terminus of 4λN-DD WT and 4λN-DD E488Q (Fig. 2A).32,33 For these new constructs we then asked whether they were indeed expressed in the nucleus, whether they produced adequate on-target editing, and whether they generated fewer off-target edits.
Figure 2.
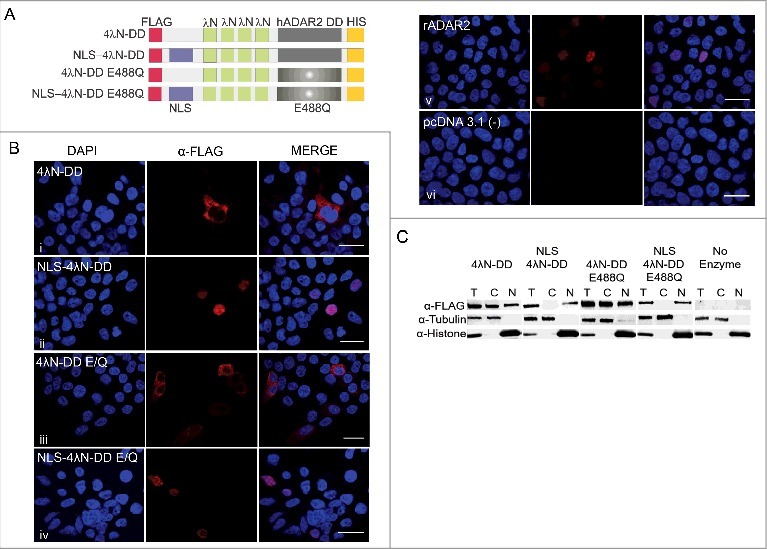
NLS-4λN-DD and 4λN-DD E488Q are localized in the nucleus. (A) Schematic of our editing enzymes (4λN-DD WT and E488Q) with and without the NLS fused to the N-terminus. (B) Immunostaining of both NLS and non-NLS versions of our editing enzymes transiently transfected in HEK293T cells visualized with a Nikon A1R Confocal Laser Microscope.100X images are shown and scale bars = 19μm; n = 2. (C) Western blot analysis of expression of 4λN-DD WT and E488Q with and without NLS in total, cytoplasmic and nuclear fractions extracted from HEK293T cells. 4λN-DD WT and E488Q are 60 kDa while both enzymes with the appended NLS are 64 kDa. Tubulin is 50 kDa and histone is 18 kDa; n = 2.
To examine the cellular localization of the editing enzymes, their expression patterns were determined by immunofluorescence. All enzymes contained an N-terminal FLAG epitope tag (Fig. 2A). HEK293T cells were transfected with plasmids encoding all versions of the editing enzymes and two days post-transfection cells were fixed and probed with an α-FLAG antibody. Results show that enzymes with an appended NLS are expressed exclusively in the nucleus, with a similar expression pattern to rat ADAR2, a well-known nucleolar-localized enzyme (Fig. 2B; panels ii, iv, and v).29,34 In contrast, enzymes without the NLS are mainly, but not exclusively, expressed in the cytoplasm (Fig. 2B; panels i and iii). Specificity of the staining was confirmed by the fact that cells transfected with the empty vector (pcDNA 3.1 (−)) showed no signal (Fig. 2B; panel vi). The extent of the enzyme's subcellular localization was also quantified because some individual cells showed staining in both the cytoplasm and nucleus. All cells transfected with the non-NLS enzymes showed cytoplasmic expression. Of these, 27% of cells transfected with WT 4λN-DD also showed nuclear staining, as did 33% of those transfected with 4λN-DD E488Q. By contrast, all cells transfected with either the NLS-tagged editing enzymes or rADAR2 showed exclusive nuclear staining. Western blot analyses of cells transfected by the same constructs were used to corroborate these results (Fig. 2C). Expression of tubulin and histone H3 were used to verify cytoplasmic and nuclear fractions, respectively. Interestingly, there was some expression of the non-NLS versions of the enzymes in both cytoplasmic and nuclear compartments. However, the overall expression pattern confirmed that the addition of three NLS's to our recombinant enzymes led to effective nuclear localization. Additionally, quantification of the relative intensity for all transfections showed that there was no difference in the total expression between the NLS and non-NLS tagged editing enzymes (4λN-DD = 1.4 ± 0.01, NLS 4λN-DD = 1.1 ± 0.1, 4λN-DD E488Q = 1.3 ± 0.28, and NLS 4λN-DD E488Q = 1.1 ± 0.05 relative to tubulin measured from the total protein fractions).
The nuclear-localized enzymes can correct a UAG PTC within eGFP
Once we determined that our enzymes were targeted to the nucleus, we next tested whether they could efficiently edit a target. For these experiments we used an mCherry-eGFP dual fluorescence reporter as described previously.16 Our reporter consists of a single message that encodes mCherry and eGFP W58X, separated by a 2A self-cleaving peptide (Fig. 3A). In this construct, green fluorescence has been abolished by adding a UAG PTC in eGFP at codon 58 (Fig. 3A, lower panel). We also made a reference construct that lacks the PTC as a tool for calibrating the ratio of green to red fluorescence (mCherry-eGFP WT, Fig. 3A, upper panel). When this construct was transfected into HEK293T cells, we could measure a consistent ratio of green to red fluorescence in individual cells. Using this number, we could then estimate the expected maximum green fluorescence of an experimental cell, based solely on its red fluorescence. Using a 2boxB RNA guide for eGFP W58X,16 we tested the efficiency of our nuclear localized enzymes. HEK293T cells were transfected with mCherry-eGFP W58X, a 2boxB RNA guide, and either 4λN-DD WT or 4λN-DD E488Q with the appended NLS. For comparison, the non-NLS versions were also tested. After transfection, we estimated eGFP correction by quantifying fluorescence and also by directly sequencing RT-PCR products. Examples of electropherograms are shown in Fig. 3Bi. For the NLS versions, editing efficiency based on sequencing was estimated at 39% and 52% for 4λN-DD WT and 4λN-DD E488Q, respectively. For the non-NLS versions it was 44% and 57%. Editing efficiency based on fluorescence was estimated to be 41 ± 2% for NLS-4λN-DD and 49 ± 2% for NLS-4λN-DD E488Q (Fig. 3Bii; dark grey bars). For the non-NLS versions, it was 39 ± 4% and 56 ± 2%. There was no significant difference between the NLS and non-NLS versions based on either quantification method. These data show that the NLS versions of the enzymes edit this target at levels equivalent to the original versions.
Figure 3.
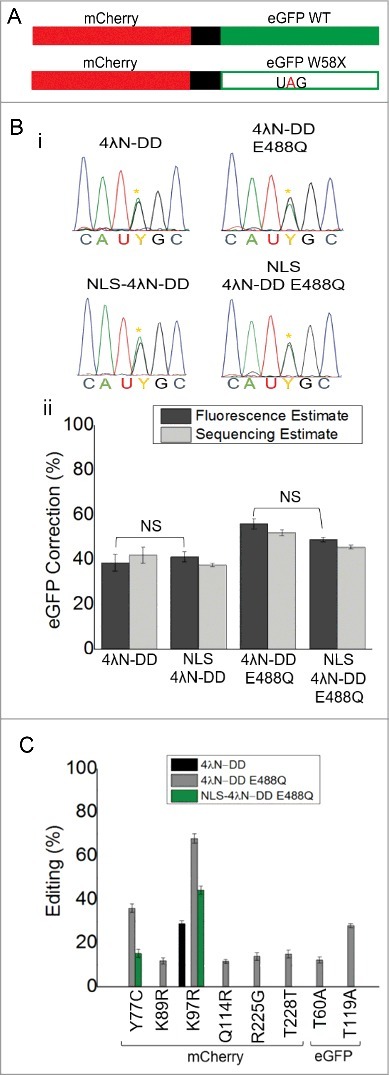
Nuclear localization reduces off-target editing while maintaining on-target editing. (A) Cartoon of fluorescent reporters used for calibration and experimental conditions. Full-length mCherry and eGFP WT were fused together and separated by a 2A self-cleaving peptide shown in black. Another construct was made that had a UAG PTC in eGFP that disrupted green but not red fluorescence (lower panel). (B) Electropherograms from directly sequenced RT-PCR products of experimental cells transfected with all the components of SDRE. Yellow asterisks represent the edited A. (C) Quantification of correction in transfections by both fluorescence (dark grey) and direct sequencing (light grey); Fluorescence estimates were based on n = 200-400 cells and sequencing estimates were taken from the same samples; n = 3, mean ± s.e.m. NS = not significantly different by ANOVA and Tukey's test where p>0.05. (D) Off-target editing events in the complete mCherry-eGFP mRNA transcript. All percentages are based on direct sequencing of RT-PCR products. No off-targets were seen in cells transfected with NLS-4λN-DD and it is not included in the legend; n = 3, mean ± s.e.m.
Off-target events in mCherry-eGFP are reduced with the nuclear SDRE
After determining that on-target editing was similar with the nuclear-localized enzymes, we wanted to assess whether off-target edits were reduced. For these experiments, the complete mCherry-eGFP transcript was sequenced from the experiments outlined in the previous paragraph (Fig. 3). Consistent with our previous results,16 there was one off-target produced with 4λN-DD (K97R) and eight with 4λN-DD E488Q (Y77C, K89R, K97R, Q114R, R225G, T228T, T60A, and T119A; Fig. 3C). Upon addition of the NLS, all off-target edits were abolished for 4λN-DD and all but two (Y77C and K97R) were eliminated for the E488Q mutant. Additionally, editing efficiency at the remaining off-targets was lower for NLS-4λN-DD E488Q (15 ± 1% for Y77C and 44 ± 2% for K97R) compared to the non-NLS version (36 ± 2% for Y77C and 68 ± 2% for K97R). Taken together, these results suggest that transporting SDRE to the nuclear compartment significantly reduced off-target editing while maintaining on-target editing in the mCherry-eGFP target. However, it must be noted that off-target edits at lower than 10% efficiency could not be detected by this method.
Nuclear-localized enzymes can target CFTR Y122X
We next determined whether the nuclear localized enzymes could efficiently correct CFTR Y122X. To do so, we transfected CFTR Y122X, RNA guide A, and plasmids encoding our NLS-tagged editing enzymes in HEK293T cells. Editing efficiency was then determined by RT-PCR and also by direct sequencing. As shown in Fig. 4A, transfections with NLS-4λN-DD resulted in 52 ± 2% editing at the first A and 12 ± 1% correction at the second A. For NLS-4λNDD E488Q, editing at the first A was 55 ± 3% and 40 ± 1% at the second. We also estimated the efficiency of conversion to tryptophan (UGG) at 6 ± 1% and 22 ± 1% for NLS-4λN-DD and NLS-4λN-DD E488Q, respectively. There was no difference between the NLS and non-NLS versions (compare Figs. 1Ciii and 4A). These data suggest that a conversion of CFTR Y122X to tryptophan is possible, but the overall efficiency is fairly low.
Figure 4.
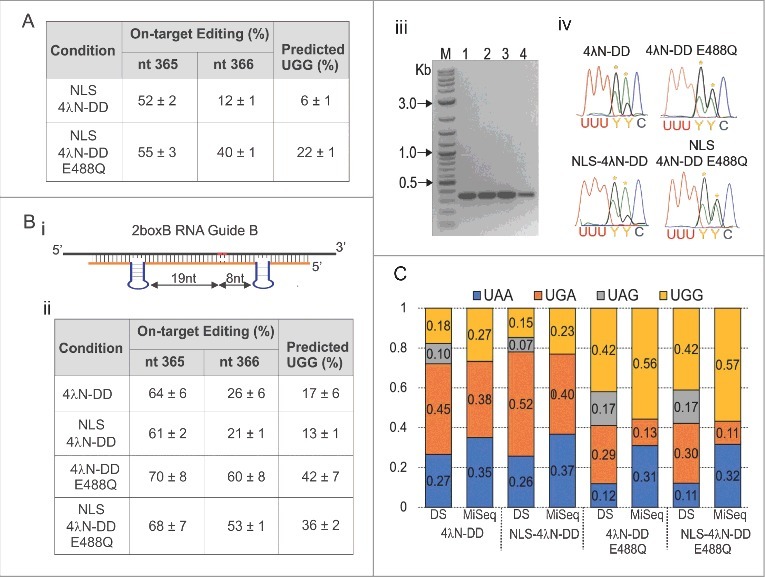
SDRE efficiently corrects a UAA PTC in CFTR Y122X at the level of RNA. (A) Quantification of editing percent of the two A's in the UAA PTC in CFTR Y122X after transfection in HEK293T cells using the NLS-tagged enzymes. Editing estimates were based on RT-PCR products from experimental cells. Percent conversion to UGG for each condition is also shown; n = 3, mean ± s.e.m. (B) An optimized RNA Guide was designed for increasing editing efficiency at the UAA PTC in CFTR Y122X. The optimized RNA guide (2boxB RNA Guide B) was designed with one boxB inserted 19 nt 3′ and another 8 nt 5′ from the second A (i). HEK293T cells were transfected as before followed by RT-PCR and direct sequencing. Quantification of editing estimates at each A and the extent of UGG conversion is shown in (ii). Estimates were based on directly sequenced RT-PCR products from experimental cells. n = 4, mean ± s.e.m. (iii) Image of the 380 bp CFTR amplicon for each condition tested (M = 2-log DNA ladder, 1 = 4λN-DD WT, 2 = NLS-4λN-DD, 3 = 4λN-DD E488Q, and 4 = NLS-4λN-DD E488Q). Bands were extracted and sent for direct sequencing. Examples of electropherograms from the RT-PCR products are shown in (iv). Yellow asterisks indicate the target A's. (C) Conversion to UAG, UGA, UGG, and UAA based on both direct sequencing (DS) and Miseq from the same samples. For the Miseq data, a total of 111,753 transcripts were analyzed in cells transfected with 4λN-DD. For NLS-4λN-DD = 104,011, 4λN-DD E488Q = 92,479 and NLS-4λN-DD E488Q = 113,997.
Optimized guide for increasing editing efficiency of Y122X mutation
To better edit CFTR Y122X, we focused on improving the RNA guide. In our previous report, we showed that as long as the target A was in a good neighboring context for editing, its precise position with respect to the boxB loops could vary.16 For example, placing a boxB hairpin 8 nt 3′ from an A in a favorable context for editing (AAG) resulted in efficient editing.16 Even though the second A in CFTR Y122X is in a AAC context, we hypothesized efficient editing could occur based on the nearest neighbor rules.26,27 Accordingly, we designed an RNA guide (i.e. RNA boxB Guide B) with this spacing where the first boxB is 19 nt 5′ from the first A and the second boxB is 8 nt 3′ from the second A, and determined its ability to edit Y122X (UAA; Fig. 4Bi). Using this guide, the editing efficiency at the second A was increased to 21 ± 1% with NLS-4λN-DD and to 53 ± 1% with NLS-4λN-DD E488Q (Fig. 4Bii). In addition, the editing efficiency of the first A was increased to 61 ± 2% and 68 ± 7% with NLS-4λN-DD and NLS-4λN-DD E488Q, respectively. As before, there was no difference between the NLS and non-NLS versions. We also estimated the frequency of full conversion to UGG; for NLS-4λN-DD it increased from 6 ± 1 to 13 ± 1% and for NLS-4λN-DD E488Q it increased from 22 ± 1 to 36 ± 2% (Fig. 4Bii). Thus, by manipulating the geometry of the RNA guide, we were able to increase editing efficiency.
A better estimation of the frequency of correcting Y122X to W by MiSeq
We have shown that we can edit both A's in CFTR Y122X, but the estimated efficiency is low. These predictions, however, were based on the idea that the two editing events are independent, an assumption that may not be accurate. Studies have shown that editing sites are often clustered and that specific editing events can rely on the prior editing of another site;35 if so, our predictions could be either over or under-representations. To better estimate the true extent of the double edit, we sequenced RT-PCR products, which contained the Y122X codon, by MiSeq. For these experiments, transfections were performed in HEK293T cells as before using the optimized 2boxB RNA guide B, CFTR Y122X, and either 4λN-DD WT or 4λN-DD E488Q, with and without the NLS. The 380 bp CFTR PCR products are shown in Fig. 4Biii. The same samples were also sent for Sanger sequencing. Sanger sequencing results confirmed our previous data and examples of electropherograms are shown in Fig. 4Biv. From these same samples we predicted the extent of UGG conversion by direct sequencing (DS) to be 15% for NLS-4λN-DD and 42% with NLS-4λN-DD E488Q (Fig. 4C). For the non-NLS editing enzymes, the percent conversion to UGG was 18% for 4λN-DD and 42% for 4λN-DD E488Q (Fig. 4C). Miseq data from the same experiments, however, showed that the conversion to UGG was much higher in all cases (23% for NLS-4λN-DD, 57% for NLS-4λN-DD E488Q, 27% for 4λN-DD and 56% for 4λN-DD E488Q). The reason for the discrepancy was clear: the MiSeq data showed that the editing of the second A does not occur in the absence of editing the first A (i.e. UAA > UAG does not happen). Therefore, estimates neglecting the correlation between the two editing sites are inaccurate (Fig. 4C). As a result, far more codons are fully converted to tryptophan than originally estimated.
The nuclear-localized SDRE decreases off-target events in CFTR mRNA
Once we determined that the nuclear-localized enzymes function with equivalent efficiency, we wanted to assess whether they produced fewer off-target events. Sanger sequencing of full-length CFTR Y122X cDNA from our editing reactions was performed. In general, for both editing enzymes, appending the NLS reduced off-target edits (Table 1B). When using NLS 4λN-DD, all off-targets were abolished except for one which produced a silent mutation (L123L). For NLS 4λN-DD E488Q, six off-targets persisted. Of these, two were silent (L123L and E1308E). Thus, the nuclear localized enzymes reduced off-targets in the targeted CFTR mRNA. However, some remained and this led to questions regarding the extent of off-target edits in mRNAs that were not targeted by our RNA guide.
Transcriptome-wide quantification of off-target editing
No previous studies have systematically explored the extent of transcriptome-wide off-target editing produced by systems that couple the catalytic domain of ADAR to an antisense RNA oligonucleotide. To quantify transcriptome-wide off-target events, matched RNA and DNA samples from HEK293T cells transfected with different combinations of editing enzymes and guide RNAs were sequenced using the Illumina platform. Sequence data was analyzed using the REDITools package36 in order to locate RNA-DNA differences residing in the protein-coding region of the transcriptome (see Materials and Methods). Fig. 5 presents the number of mismatches observed, clearly showing abundant A>G off-target editing in all samples containing the guide RNA and an editing enzyme. As expected, other types of mismatches were far less frequent and probably represent the inherent noise in the system. In all, there are from ∼250K-870K off-target editing sites within coding sequences, depending on the specific enzyme-guide combination (Fig. 5A). Many more off-targets are observed in non-coding regions (Figure S1 and Table S1). In general, enzymes containing the E488Q mutation generate many more off-target events and the presence of an NLS reduces the off-targets by ∼50%. Considering only high confidence (at least 3 supporting reads) and highly edited (>10%) sites, there are far fewer off-target events, but the number is still substantial (Fig. 5B). At ∼77,000 sites, 4λN-DD E488Q produced the most. NLS 4λN-DD produced the least (∼8,800), about an order of magnitude less. In these data, the E488Q mutation produced an even more pronounced increase in off-targets, and the addition of an NLS reduced them by about a half. It is notable that the enzymes with E488Q produce many off targets even in the absence of a guide RNA. A histogram of editing sites binned by their editing levels clearly shows that the vast majority of sites are edited at low levels (<5%; Fig. 5C).
Figure 5.
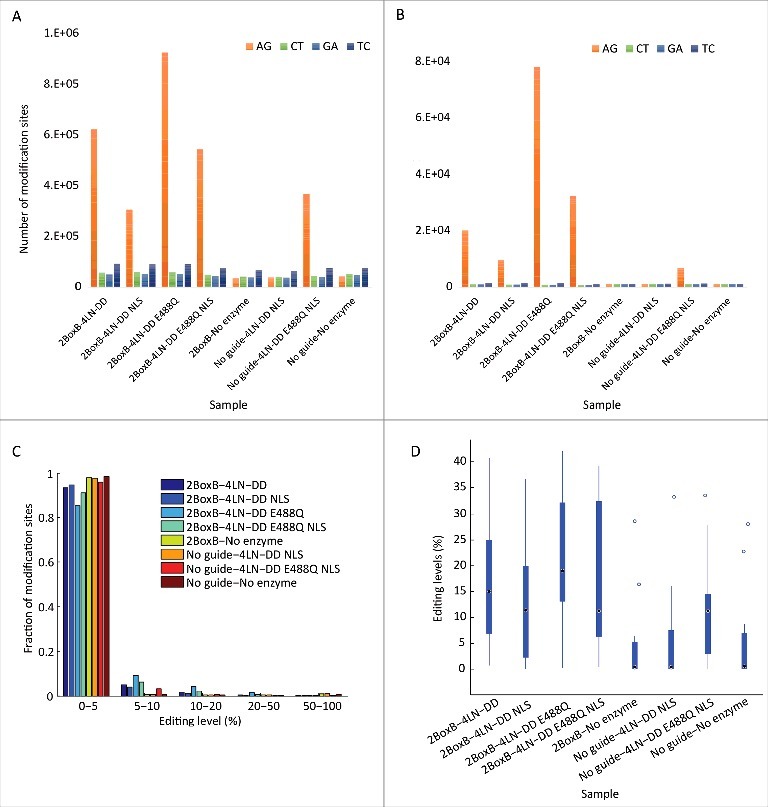
Transcriptome-wide off-targets produced by SDRE. (A) Number of RNA-DNA mismatches identified within coding sequences in each of the transfected cells and controls. All samples transfected with an enzyme and a guide RNA exhibit a sizable over-representation of A-to-G mismatches, attesting for many thousands of editing sites. The number of G-to-A and C-to-T mismatches may be used as an estimate for the background detection noise (false positives). (B) The same pattern is observed when considering only high-confidence, highly edited (>10%) mismatches. (C) The majority of editing sites found in all samples are edited at low levels. (D) Treatment with the various SDRE approaches increases editing levels at 14 well covered, well documented conserved mammalian editing sites (see Table S2).
Next we asked whether SDRE can affect editing at endogenous sites. As a reference, we looked at the 38 human RNA editing sites that are conserved in mammals (Table S2).37 Of these, 14 sites had sufficient coverage in our samples for analysis. Fig. 5D presents a box plot of the distribution of editing levels at these sites. Editing was low or undetectable in samples that lacked the enzyme and guide RNA, consistent with the idea that HEK293T cells exhibit little editing activity on their own. However, all samples that contained both enzyme and guide showed elevated editing. Data for individual sites is given in Table S2. These data suggest that SDRE promotes higher editing at sites that are recognized by endogenous ADAR.
Discussion
Clearly, off-target editing events present a significant challenge for SDRE. Even when considering only high frequency events (>10%), and using the least promiscuous enzyme (NLS-4λN-DD), there were close to 8,800 events. In general, nuclear localization improves, but does not abolish, off-target events. In addition, one must proceed with caution when using the E488Q mutation to increase on-target editing efficiency: this mutation alone generates many off-target events. It is difficult to estimate the detrimental effects of off-target editing events on cell physiology. When expressing the editing enzymes and RNA guides used in this study, HEK293T cells showed no obvious effects (i.e. they survived post-transfection with normal cell densities), however this should be examined in greater detail. Unlike mutations in DNA, changes in RNA are transient and would not be expected to lead to malignant transformation. Regardless, with the extent of off-target RNA editing, it would be reasonable to hypothesize that SDRE can lead to a general proteotoxic stress or more acute stress should vital transcripts be edited in important codons. In addition, it can augment the editing levels at endogenous sites, disrupting their regulation of physiology.
Looking forward, strategies to decrease or eliminate off-target editing should be a priority for SDRE. Nuclear localization of the editing enzyme was a useful first step. When compared to the cytoplasmic editing enzymes, all NLS versions maintained similar on-target editing efficiencies while exhibiting reduced off-target editing. The reasons underlying this difference are not entirely clear. In the cytoplasm, they would have prolonged access to the RNA, perhaps promoting greater editing at lower affinity sites. Higher affinity on-target sites, without any ribosomal interference, could be rapidly edited in the nucleus. Other approaches to controlling off-target editing certainly exist. In a recent publication,18 we showed that the extent of off-target editing in the targeted message correlated strongly with the amount of guide RNA expressed. Accordingly, RNA guides that create higher affinity structures for on-target editing can be used in lower quantities, thereby reducing off-target editing. In addition, catalytic domains from different ADARs, or a re-engineered version of the DD from human ADAR2, might be more specific. These avenues should be explored before employing SDRE for directed correction.
In this work we explored using SDRE to correct the Y122X mutation in CFTR. This mutation is most prevalent in the population of Reunion Island, where it displays a frequency of about 24%38,39 as compared to 0.07% in all of Europe.40 It was challenging because two A's had to be edited to erase the PTC. However, the resulting codon was a tryptophan, not the WT tyrosine. It will be important to determine whether a tryptophan at this position will be tolerated, and will not significantly alter CFTR function. In addition, the general method should be applicable to other PTCs in CFTR. After the ΔF508 mutation, which is the most frequent mutation and not currently correctable by our approach, mutations such as W1282X and G542X are among the most common.41 A nuclear localized version of SDRE gives the added bonus of correcting transcripts that harbor PTCs before they can be cleared by nonsense-mediated decay. Besides PTCs, many potentially correctable (G>A) missense mutations exist in CFTR, and some are among the most common mutations.
SDRE holds much promise outside of CFTR as well. Any genetic mutation that can be corrected by a site-specific A>G change is a potential candidate for correction. More importantly, SDRE can be used to directly modify protein function for therapeutic ends. The lifetimes of mRNAs, and the proteins that they encode, make SDRE particularly appealing for transient applications. Perhaps a similar approach can be used for modifications other than adenosine deamination. As a first step, the system's specificity needs to be improved.
Materials and methods
Molecular biology
Production of the 4λN-DD and 4λN-DD E488Q constructs in pcDNA 3.1 (+) has been described previously.16 The SV40 Large T Antigen NLS (PKKKRKV) repeated 3X and separated by a GSTSGV linker was added to the N-terminus of each editing enzyme by synthesizing a geneblock and cloning it into the original vector by the Gibson Assembly Method (New England Biolabs, Cat. #E5510). The NLS-linker unit is PKKKRKVDPKKKRKVDPKKKRKVDGSTSGV. This particular NLS was chosen because it is a very robust signal and widely used for this purpose.32,33 FLAG-tagged rat ADAR2 in pcDNA 3.1 was kindly provided by Dr. Marie Öhman from Stockholm University.
CFTR Y122X and mCherry-eGFP were the target mRNAs used in this study. The CFTR Y122X in pcDNA 5.1 was provided by Dr. Martin Mense from the Cystic Fibrosis Foundation Therapeutics laboratory in Lexington, MA. The production of mCherry-eGFP and mCherry-eGFP W58X in pcDNA 3.1(−) has been described previously.16 Both constructs were used for editing assays in HEK293T cells.
The production of the mCherry-eGFP 2boxB RNA guide has been described previously.16 For the production of the 2boxB RNA guides targeting CFTR Y122X, dsDNA oligonucleotides were designed and cloned into the BLOCK-iT U6 RNAi Entry Vector (Invitrogen, Cat. #K4945-00).
All DNA oligonucleotides and gene blocks were purchased from Integrated DNA Technologies, Inc. For the mCherry-eGFP experiments, the oligonucleotides used have been reported.16 The sequences for all other oligonucleotides and synthetic gene blocks are listed in Table S3.
Cell culture
HEK293T cells were maintained in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin solution, 1mM sodium pyruvate, and 2mM glutamine. For each transfection, 300 × 103 cells were grown on 35mm dishes. All transfections were performed using the Effectene® Transfection Reagent kit (QIAGEN, Cat. #301425) according to protocol.
Immunofluorescence
For immunofluorescence experiments, dishes were coated with 0.1% poly-D-lysine and seeded with HEK293T cells. Two days later, 0.1 μg of plasmid DNA encoding for either 4λN-DD, 4λN-DD E488Q, 4λN-DD NLS, or 4λN-DD E488Q NLS, all in pcDNA 3.1(+), were transfected. As negative control, 0.1 μg of pcDNA 3.1 (+) empty plasmid was transfected and as nuclear control, 0.1 μg rADAR2 in pcDNA 3 was used. Forty-eight hours later, cells were washed with 1X PBS and fixed with 4% paraformaldehyde-PBS for 15 min on ice, permeabilized in 0.1% Triton X-100 in PBS for 15 minutes, and blocked with 10% normal donkey serum / 1% BSA solution for 10 minutes at room temperature. Cells were then incubated with anti-FLAG (1:500; Sigma, Cat. #F3165) primary antibody overnight at 4°C. Cells were then probed with Alexa Fluor 546 donkey anti-mouse (1:800; Thermo Fisher Scientific, Cat. #A11126) for 1 hour at room temperature. Hoestch 33342 (1:10000; Thermo Fisher Scientific, Cat. #H3570) in PBS was added to the cells for nuclear visualization. All images were acquired using a Nikon A1R confocal system. For analysis, a total of 100 cells or each experiment were visually counted using the Fiji ImageJ software. Experiments were performed in duplicate.
Cell fractionation
For corroborating nuclear expression of the NLS-tagged editing enzymes, HEK293T cells were transfected with 0.1 μg of either 4λN-DD, 4λN-DD E488Q, 4λN-DD NLS, or 4λN-DD E488Q NLS, all in pcDNA 3.1(+). For negative controls, pcDNA 3.1(−) empty vector was used instead of the editing enzymes. Forty-eight hours post-transfection, cells were washed with 1ml 1X cold PBS and centrifuged at 200 rcf for 5 min. at 4°C. Cells were then fractionated into cytoplasmic and nuclear compartments with the NE-PER extraction kit (Thermo Fisher Scientific, Cat. #78833) according to protocol. For the total fraction, cells were lysed with the CEB I and CEB II buffers provided with the kit and samples were taken before centrifugation.
Western blot
After cellular fractionation, samples were run on a 4-20% gradient gel based on relative volume. Samples were then transferred onto PVDF membranes followed by blocking with the Odyssey® TBS Blocking Buffer (LI-COR Biosciences, Cat. #92750000). Membranes were probed with either α-FLAG at 1:3000 (Sigma, Cat. #F3165), α-Tubulin at 1:4000 (Sigma, Cat. #SAB3501071), or α-HIS3 at 1:4000 (Sigma, Cat. #H1029) primary antibody overnight at 4°C. Membranes were then probed with either IRDye 680RD goat anti-rabbit (1:20000) or IRDye 800CW goat anti-mouse (1:20000) secondary antibody (LI-COR Biosciences, Cat. #925-68071 and Cat. #925-32210, respectively). Images were acquired using the Odyssey® Fc imaging system (LI-COR Biosciences) according to manufacturers instructions. For quantification, total expression of each recombinant enzyme was normalized to tubulin and the relative intensity was analyzed using the LI-COR Image Studio Software. Experiments were performed in duplicate.
Editing assays in HEK293T cells
Editing assays in HEK293T were performed as described previously.16 In short, all SDRE components were co-transfected in the following amounts: pcDNA 3.1 (−) mCherry-eGFP, mCherry-eGFP W58X, or CFTR Y122X in pcDNA 5 (25 ng), 4λN-DD, NLS-4λN-DD, 4λN-DD E488Q, and NLS-4λN-DD E488Q, all in pcDNA 3.1(+); 100 ng, and U6 pENTR guide RNA vector (1.5 μg). In our previous report, we showed that transfecting 1.5 μg of 2boxB RNA significantly reduced off-target edits while still maintaining robust on-target correction.16 Accordingly, we decided to transfect this amount of 2boxB RNA guide in all our editing assays. For either the no RNA guide or no editing enzyme controls, the pcDNA 3.1(−) empty vector was used for transfection in equal proportions. Cells were analyzed 4 days post-transfection.
Quantification of editing efficiency
For the mCherry-eGFP editing assays, cells were imaged with the Cellometer® Vision imaging system (Nexcelom Bioscience) using the dual fluorescence mode to measure correction at the level of fluorescence.16 The extent of eGFP correction at the RNA level was also determined by RT-PCR and direct sequencing. For editing assays with CFTR Y122X, correction was only measured at the level of RNA by RT-PCR and direct sequencing. For determining RNA level correction by RT-PCR, RNA samples were extracted from HEK293T cells with the RNAqeous kit (ThermoFisher Scientific, Cat. #AM1912) according to protocol. After RNA extraction, cDNAs were synthesized using gene-specific RT-primers and amplified by PCR. The final products were directly sequenced and quantified based on C/T peak heights of the anti-sense strand.42 For clarity, however, electropherograms in all Figures are shown as the reverse complement. Off-target edits were also identified by RT-PCR and quantified as described by analyzing the full-length mRNA targets. For quantification of the complete CFTR sequence, products were also quantified based on A/G peak heights, depending on the primer used.
Quantification of editing at Y122X using MiSeq
A 380 nt amplicon surrounding codon 122 in CFTR was amplified from cDNA made from transient transfections of HEK293T cells as previously described. The first round of PCR used the primers Y122XFwdMiseq and Y122XRevMiseq (see Table S3). First round PCR products were then reamplified using a low cycle number with the Illumina sequencing primers (see Table S3). 2nd round amplicons were purified using Qiagen MinElute columns (Qiagen, Cat. #28004) and their quality was assessed using a Bioanalyzer (Agilent) on a DNA1000 chip. Samples were pooled and quantified by qPCR using a KAPA Illumina Library Quantification Kit (Kapa Biosystems, Cat. #KK4828-07960166001). The pool of 8 samples was included on a Miseq run with 88 other samples in equal proportions using the 2 × 300 (v3) format. Sequences were immediately trimmed to 250 nt. Different editing patterns were counted using the “grep” command on total sequence files.
Transcriptome-wide off-target editing
To determine and quantify transcriptome-wide off-targets, RNA extracts and genomic DNA from transfected HEK293T cells were sent for Illumina sequencing to the University of Chicago Genomic Facility. The RNA-Seq libraries for all the samples were prepared using the TruSeq Stranded mRNA Sample Prep Kit, as described by the manufacturer (Illumina, Cat. #RS-122-2103), and were sequenced using one lane for samples 1-5 and one lane for samples 6-8 of Illumina HiSeq 4000 instrument, generating 45-141M 100nt, paired-end reads (Table S4). The genomic DNA sequencing library was prepared from the same cells using the TruSeq DNA Sample Prep kit, as described by the manufacturer (Illumina, Cat. #FC-121-2003), and sequenced using one lane of the Illumina HiSeq 4000 instrument, generating 374,246,311 100 nt, paired-end reads. All reads will be submitted to the NCBI SRA upon manuscript acceptance.
In order to detect off-target editing sites, The RNA and DNA reads were aligned separately to the human genome (hg19) using Bowtie2 with local alignment configuration and default parameters.43 Then, REDITools package (V-1.0.3) was used to locate RNA-DNA differences.36 To improve detection accuracy, we kept only uniquely aligned reads, discarded the first and last six bases of each read, as well as bases with a quality score ≤ 30. We focused on sites residing in coding regions only (according to RefSeq CDS table, downloaded from UCSC on April 2017), and removed all genomic locations identified as common SNPs (dbSNP147 Common SNPs table, downloaded from UCSC table browser on August 2017).44
The number of G-to-A and C-to-T mismatches may be used as an estimate for the background detection noise (false positives). Other mismatch types were always less abundant than these two types. Note that some of the T-to-C mismatches could be due to A-to-I editing on the complementary strand.
Statistical analysis
One-way ANOVA and the Tukey's test were used for mean comparisons using p<0.05.
Supplementary Material
Funding Statement
This study was supported by the National Institutes of Health under Grant 1R01NS087726 (JJCR); the United States-Israel Binational Science Foundation under Grant No. 2013094 (JJCR and EE), and The Cystic Fibrosis Foundation Therapeutics under Grant Rosent14XXO (JJCR).
Disclosure of potential conflicts of interest
The authors disclose no conflicts of interest.
Acknowledgements
We would like to thank Dr. Hilary Morrison for help with Miseq amplicon preparation and sequencing, Dr. Martin Mense for useful discussions on CFTR mutants, Dr. Gail Mandel for discussions regarding appropriate nuclear localization signals, and Drs. Garrett Seal and Amelia Merced for help with confocal microscopy. We would also like to thank Mrs. Sonia Soto for technical support. Finally, NLB was supported by a post-doctoral scholarship from the Center for Nanoscience and Nanotechnology at Tel Aviv University and we thank them for their support.
References
- 1.Bass BL, Weintraub H. A developmentally regulated activity that unwinds RNA duplexes. Cell. 1987;48:607–13. doi: 10.1016/0092-8674(87)90239-X. [DOI] [PubMed] [Google Scholar]
- 2.Bass BL, Weintraub H. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell. 1988;55:1089–98. doi: 10.1016/0092-8674(88)90253-X. [DOI] [PubMed] [Google Scholar]
- 3.Melcher T, Maas S, a Herb, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–4. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 4.Bass BL. RNA editing and hypermutation by adenosine deamination. Trends Biochem Sci. 1997;22:157–62. doi: 10.1016/S0968-0004(97)01035-9. [DOI] [PubMed] [Google Scholar]
- 5.O'Connell MA, Gerber A, Keegan LP. Purification of native and recombinant double-stranded RNA-specific adenosine deaminases. Methods. 1998;15:51–62. doi: 10.1006/meth.1998.0605. [DOI] [PubMed] [Google Scholar]
- 6.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc Natl Acad Sci U S A. 1994;91:11457–61. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basilio C, Wahba AJ, Lengyel P, Speyer JF. Synthetic Polynucleotides and the Amino Acid Code, V*. Biochemistry. 1962;48:613–6. PMCID:PMC221128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woolf TM, Chase JM, Stinchcomb DT. Toward the therapeutic editing of mutated RNA sequences. Proc Natl Acad Sci U S A. 1995;92:8298–302. doi: 10.1073/pnas.92.18.8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stafforst T, Schneider MF. An RNA-deaminase conjugate selectively repairs point mutations. Angew Chem Int Ed Engl. 2012;51:11166–9. doi: 10.1002/anie.201206489. [DOI] [PubMed] [Google Scholar]
- 10.Montiel-Gonzalez MF, Vallecillo-Viejo I, Yudowski GA, Rosenthal JJC. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc Natl Acad Sci U S A. 2013;110:18285–90. doi: 10.1073/pnas.1306243110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuda M, Umeno H, Nose K, Nishitarumizu A, Noguchi R, Nakagawa H. Construction of a guide-RNA for site-directed RNA mutagenesis utilising intracellular A-to-I RNA editing. Sci Rep. 2017;7:41478. doi: 10.1038/srep41478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogel P, Stafforst T. Site-directed RNA editing with antagomir deaminases: a tool to study protein and RNA function. ChemMedChem. 2014;9:2021–5. doi: 10.1002/cmdc.201402139. [DOI] [PubMed] [Google Scholar]
- 13.Vogel P, Schneider MF, Wettengel J, Stafforst T. Improving site-directed RNA editing in vitro and in cell culture by chemical modification of the guideRNA. Angew Chemie – Int Ed. 2014;53:6267–71. doi: 10.1002/anie.201402634. [DOI] [PubMed] [Google Scholar]
- 14.Schneider MF, Wettengel J, Hoffmann PC, Stafforst T. Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res. 2014;42:e87. doi: 10.1093/nar/gku272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanswillemenke A, Kuzdere T, Vogel P, Jekely G, Stafforst T. Site-directed RNA editing in vivo can be triggered by the light-driven assembly of an artificial riboprotein. J Am Chem Soc 2015;jacs.5b10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montiel-González MF, Vallecillo-Viejo IC, Rosenthal JJC. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 2016;gkw738. doi: 10.1093/nar/gkw738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heep M, Mach P, Reautschnig P, Wettengel J, Stafforst T. Applying human ADAR1p110 and ADAR1p150 for Site-Directed RNA editing—G/C substitution stabilizes guideRNAs against editing. Genes (Basel). 2017;8. doi: 10.3390/genes8010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon AC, Rahman R, Jin H, Shen JL, Fieldsend A, Luo W, Rosbash M. TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell. 2016;165:742–53. doi: 10.1016/j.cell.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macbeth MR, Schubert HL, VanDemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–9. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su L, Radek JT, Hallenga K, Hermanto P, Chan G, Labeots LA, Weiss MA. RNA recognition by a bent alpha-helix regulates transcriptional antitermination in phage lambda. Biochemistry. 1997;36:12722-32. doi: 10.1021/bi971408k. [DOI] [PubMed] [Google Scholar]
- 21.Kuttan A, Bass BL. Mechanistic insights into editing-site specificity of ADARs. Proc Natl Acad Sci U S A. 2012;109:E3295–304. doi: 10.1073/pnas.1212548109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeo J, a Goodman R, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc Natl Acad Sci U S A. 2010;107:20715–9. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimokawa T, Rahman MF-U, Tostar U, Sonkoly E, Ståhle M, Pivarcsi A, Palaniswamy R, Zaphiropoulos PG. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013;10:321–33. doi: 10.4161/rna.23343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–73. Available from: http://www.ncbi.nlm.nih.gov/pubmed/2475911 [DOI] [PubMed] [Google Scholar]
- 25.Rommens J, Iannuzzi M, Kerem B, Drumm M, Melmer G, Dean M, Rozmahel R, Cole J, Kennedy D, Hidaka N, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science (80−). 1989;245:1059–65. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 26.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–84. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 27.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun [Internet]. 2011;2:319 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3113232&tool=pmcentrez&rendertype=abstract doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong SK, Sato S, Lazinski DW. Substrate recognition by ADAR1 and ADAR2. RNA. 2001;7:846–58. doi: 10.1017/S135583820101007X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desterro JMP, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M. Dynamic association of RNA-editing enzymes with the nucleolus. J Cell Sci. 2003;116:1805–18. doi: 10.1242/jcs.00371. [DOI] [PubMed] [Google Scholar]
- 30.Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci U S A. 2003;100:14018–23. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maas S, Gommans WM. Identification of a selective nuclear import signal in adenosine deaminases acting on RNA. Nucleic Acids Res. 2009;37:5822–9. doi: 10.1093/nar/gkp599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arenal A, Pimentel R, García C, Pimentel E, Aleström P. The SV40 T antigen nuclear localization sequence enhances nuclear import of vector DNA in embryos of a crustacean (Litopenaeus schmitti). Gene. 2004;337:71–7. doi: 10.1016/j.gene.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 33.Brandén LJ, Mohamed a J, Smith CI. A peptide nucleic acid-nuclear localization signal fusion that mediates nuclear transport of DNA. Nat Biotechnol. 1999;17:784–7. doi: 10.1038/11726. [DOI] [PubMed] [Google Scholar]
- 34.Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc Natl Acad Sci U S A. 2003;100:14018–23. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ensterö M, Daniel C, Wahlstedt H, Major F, Öhman M. Recognition and coupling of A-to-I edited sites are determined by the tertiary structure of the RNA. Nucleic Acids Res. 2009;37:6916–26. doi: 10.1093/nar/gkp731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picardi E, Pesole G. REDItools: High-throughput RNA editing detection made easy. Bioinformatics. 2013;29:1813–4. doi: 10.1093/bioinformatics/btt287. [DOI] [PubMed] [Google Scholar]
- 37.Pinto Y, Cohen HY, Levanon EY. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 2014;15:R5. doi: 10.1186/gb-2014-15-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duguépéroux I, Bellis G, Lesure JF, Renouil M, Flodrops H, De Braekeleer M. Cystic fibrosis at the Reunion Island (France): Spectrum of mutations and genotype-phenotype for the Y122X mutation. J Cyst Fibros. 2004;3:185–8. doi: 10.1016/j.jcf.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Cartault F, Steffan J, Vidaud D, Bousquet S, Lesure F, Renouil M, McDonell N, Feingold J, Beldjord C, Bienvenu T. Detection of more than 91% cystic fibrosis mutations in a sample of the population from Reunion Island and identification of two novel mutations (A309G, S1255L) and one novel polymorphism (L49L). Clin Genet. 1998;54:437–9. [PubMed] [Google Scholar]
- 40.Estivill X, Bancclls C, Ramos C. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. Hum Mutat. 1997;10:135–54. doi: 10.1002/(SICI)1098-1004(1997)10:2%3c135::AID-HUMU6%3e3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 41.Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations–correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 42.Rinkevich FD, Schweitzer PA, Scott JG. Antisense sequencing improves the accuracy and precision of A-to-I editing measurements using the peak height ratio method. BMC Res. Notes. 2012;5:63. doi: 10.1186/1756-0500-5-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hinrichs A, Karolchik D, Baertsch R, Barber G, Bejerano G, Clawson H. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res. 2006;34:D590–8. doi: 10.1093/nar/gkj144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.