

ABSTRACT
Muscleblind-like (MBNL) proteins bind to hundreds of pre- and mature mRNAs to regulate their alternative splicing, alternative polyadenylation, stability and subcellular localization. Once MBNLs are withheld from transcript regulation, cellular machineries generate products inapt for precise embryonal/adult developmental tasks and myotonic dystrophy, a devastating multi-systemic genetic disorder, develops. We have recently demonstrated that all three MBNL paralogs are capable of fine-tuning cellular content of one of the three MBNL paralogs, MBNL1, by binding to the first coding exon (e1) of its pre-mRNA. Intriguingly, this autoregulatory feedback loop grounded on alternative splicing of e1 appears to play a crucial role in delaying the onset of myotonic dystrophy. Here, we describe this process in the context of other autoregulatory and regulatory loops that maintain the content and diverse functions of MBNL proteins at optimal level in health and disease, thus supporting the overall cellular homeostasis.
KEYWORDS: Alternative splicing, autoregulation, feedback loop, MBNL1, muscleblind-like, myotonic dystrophy, RBFOX, therapeutic strategies
Introduction
One could think of the whole cellular system in a simplistic manner as a fragile assembly built from a deck of cards with one lacking card as the prerequisite for the catastrophe. The alternate vision would be more complex, dynamic, evolved over many years, where each loss is monitored by an intricate web of feedback loops, bringing the cell back, despite the deficit, to its optimal status. In that case, one missing element would be sensed and replaced, often by upregulation of a homologous protein. The feedback loop mechanisms would be particularly important for major players, having a role in pushing the cell's fate towards a particular direction depending on the state of the organism. Of course, the range of such self-repair interventions would be limited, with some alterations overtaxing adaptive responses. Nevertheless, these reactions might constitute the first defenses and despite their limited efficiency, could help avoid cell death, for example, and in turn prolong the patient's life. Understanding this intricate web could allow us to design the best therapeutic intervention as not always it is restoring the missing element. Sometimes, the imperfect counterpart might be a better option due to, for instance, immune rejection of the normally best fitting protein1 or feasibility of using an alternative response path.2
In line of this view comes our recent discovery showing that one of the three members of the Muscleblind-like (MBNL) family, MBNL1, a developmental sensor that controls hundreds of alternative splicing events and alternative 3′UTRs, self-regulates its content and function, feeding back to its spliceable mRNA template, with other MBNLs contributing to this phenomenon3 (Figs. 1 and 2). This autoregulatory course is of particular importance as once overtaxed it may lead to MBNL1 functional deficiency, and in turn, myotonic dystrophy (DM), a heritable, incurable and devastating genetic disorder.4 Here, we look at this process from a broader perspective, including regulatory expression of other MBNL paralogs, showing the overall multilayered complexity of MBNL's overlapping and opposing roles in maintaining homeostasis. In all of these processes, RNA molecules serve as flexible templates for the building blocks, the proteins, that not only perform their functions but also respond back to the mother particles. In the end, we ponder whether we could benefit from this acquired knowledge in designing a therapy, having in mind an intervention allowing each cell in the body to find its optimal status independently, using feedback loop mechanisms, and thus reaching the overall health of the organism without causing toxicity or unwanted side-effects.
Figure 1.
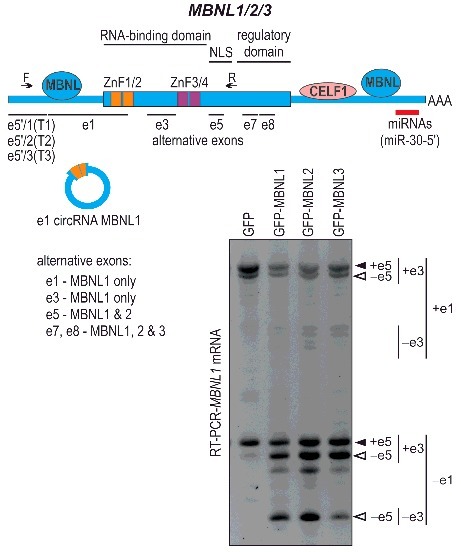
Autoregulatory alternative splicing of MBNLs. Upper panel shows schematic representation of MBNL1/2/3 transcripts (not to scale). Approximate binding sites of MBNL and CELF1 proteins as well as miRNAs within MBNLs mRNAs are indicated. Horizontal lines indicate 5′ exons (derived from transcription start site T1, T2 and T3) and alternatively spliced exons (e1, e3, e5, e7 and e8). Zinc finger motifs 1/2 and 3/4 are marked orange and purple, respectively. Schematic structure of MBNL1-dependent circRNA generated by circularization of e1 is also depicted underneath the mRNA schematic. Lower panel shows representative RT-PCR analysis of MBNL1 splicing in HeLa cells transiently transfected with GFP (control) or either of the expression constructs encoding for MBNL1, 2 or 3. Forward primer is located in e5′/2 (T2-derived) and reverse primer in e6 (marked with arrows in the upper panel schematic). Bands representing +e1 and –e1 MBNL1 mRNA splice isoforms differing also in e3 and e5 content are indicated. Note that the splicing pattern is similar regardless of the MBNL paralog used for transfection and all three alternative exons are negatively regulated by MBNLs. The data shown have not been previously published. Detailed preparation of the samples was described in3.
Figure 2.

Switching MBNL1 on and off: the e1 loop. Upper panel (a) shows schematic representation of e1 splicing in MBNL1 pre-mRNA depending on transcription initiation start site T1 (left), T2 (middle) and T3 (right). T1- and T3-derived MBNL1 transcripts exhibit always skipped or included e1, respectively, independent of MBNL1. In contrast, T2-derived MBNL1 transcripts are subject to MBNL-dependent e1-splicing and feedback loop regulation outlined in lower panel (b). T2-derived MBNL1 transcripts are subject to MBNL-dependent e1-splicing. Fully functional MBNL1 protein translated from e1-containing MBNL1 mRNAs switches off its own expression by prompting e1 exclusion from MBNL1 pre-mRNA (exOFF). This results in MBNL1 proteins with compromised splicing activity and stability due to the absence of ZnF1/2. Truncated MBNL1 is no longer able to hinder e1 inclusion into mature mRNA, thus switching back the expression of the fully functional e1-containing MBNL1 (exON). Similarly, functional depletion of MBNL in DM, caused by sequestration on expanded C/CUG repeats, hinders MBNL1 splicing activity resulting in e1 inclusion and increased production of a fully functional MBNL1 (CNBP/DMPK C/CUG-expanded transcripts folding into toxic hairpin structures are indicated). Note, however, that in DM, the e1 feedback loop based on activation of e1 inclusion is overtaxed with time, depending on the length of sequestering C/CUG repeats and their somatic expansion. Orange and blue rectangles represent MBNL1 exons (numbered), and translation start sites (AUG) in specific splice-isoforms (+e1 or –e1) are indicated. MBNL1 proteins are depicted as large blue circles with ZnF1/2 and ZnF3/4 shown as smaller orange and purple circles, respectively. Truncated MBNL1 with compromised stability is shown as fading blue circles. Arrows point to direction of the e1-feedback loop. The model shown in a and b is based on our recently published data3.
DM as a disease of overtaxed autoregulatory processes
DM is an autosomal dominant multi-systemic disorder, which primarily affects musculature.5-7 According to the current state of knowledge, the disease is caused mainly by MBNL sequestration by expanded CUG or CCUG repeats harbored within the DMPK or CNBP transcripts, and respectively, distinguished as one of the two types, DM1 or DM2. The inherited mutation somatically expands with time, which further worsens the phenotype of affected patients and adds to the progressive character of DM. Entrapped MBNL cannot fulfill its normal function which, among others, is the regulation of hundreds of alternative splicing events that adapts mRNA transcripts to the given developmental state.8,9 This leads to a paradox – while the patients' body ages, the splicing patterns of many mRNAs reverse to the early developmental stage, and the translated proteins are unfit to perform the adult functions. In all, this results in a number of pathologies such as myotonia, muscle wasting, weakness, cardiac conduction defects, cataracts, breathing problems, and cognitive deficits, among others.
Switching MBNL1 on and off: the e1 loop
While searching for MBNL1 targets we found, among many other mRNAs, MBNL1's own transcript and, to advance the comprehension of our finding in a broader context, we expanded the previously reported autoregulation of MBNL110 by describing a new feedback loop that switches MBNL1 expression on and off3 (Fig. 2). With cells having too much MBNL, the protein binds to the first coding exon (e1) of MBNL1 mRNA, which spliceosome then omits in the assembly process to produce the e1-depleted mature transcript giving rise to a very unstable MBNL1 protein isoform. Moreover, once a GFP tag is placed in front of the coding sequence, the truncated MBNL1 is stabilized, however, its splicing function is heavily compromised. The reason for this is that the lack of e1 shifts the translation start to e2 and the resulting protein lacks two of the four zinc fingers (ZnFs) that are normally used to attach to mRNA targets. Altogether, this feedback loop keeps the level of MBNL1 in check. During development, the MBNL1 RNA and protein level rise and, with time, mRNAs without e1 start to dominate while only a fraction of total MBNL1 transcript gives rise to the highly functional isoform containing e1. One could envision this additionally generated mRNA as a backup for more demanding times. While the content of MBNL1 drops, repression of e1 is lifted and the cell starts to produce the highly functional MBNL1 to reach the appropriate protein level. On the other hand, this mechanism might also prevent cells from producing too much MBNL1, which in higher concentrations could be toxic. In DM, this feedback loop based on activation of e1 inclusion is obviously overtaxed with time, depending on the length of sequestering C/CUG repeats and their somatic expansion, which results in incongruent splicing of RNA transcripts and generation of proteins inapt for adult tasks.
The e1 feedback loop is particularly active in skeletal and cardiac muscles, which correlates with the overall high MBNL expression level in these tissues11 and the fact that several muscle-specific mRNAs controlled by MBNL, such as ABLIM1, BIN1, CACNA1S, CLCN1, INSR, once translated to the developmentally incorrect isoform, markedly contribute to the DM phenotype.12–18 The reason for the tissue-dependent difference in the activity of this loop is that MBNL1 transcription may start from one of the three sites (T1-T3; Figs. 1 & 2a), with each giving rise to a different 5′UTR exon, preceding e1, and with each operated autonomously by a distinct promoter. Rare T1 and the second most frequent in striated muscles T3 transcripts exhibited always skipped and included e1, respectively. However, in contrast to T2 mRNAs, the former were never regulated by the content of MBNL, but rather their splicing pattern seemed to be solely based on the length of the intron, that is the distance between the 5′UTR exon and e1, which varies enormously, ranging from 55 kb to only 0.2 kb. On the contrary, the in-between starting T2-derived transcripts had either included or excluded e1 depending on the MBNL content. Consequently, we conclude that tight regulation of MBNL cellular level in striated muscles prevents rapid changes in its cellular content, signifying important functions of MBNL1 in these tissues, with presumably less significant roles of the e1 loop in tissues, in which T3-derived transcripts are relatively more abundant.
Shuttling MBNL1/2 from the cytoplasm to the nucleus: the e5 loop
While the e1-dependent loop is an on/off switch for MBNL1 expression, the second, aforementioned loop based on autoregulatory splicing of the 54 nucleotide-long exon (e5), shifts the protein content from predominantly cytoplasmic to exclusively nuclear (Fig. 3).10,19-22 Here, MBNL1 binds to several sites in intron 4, which presumably alters the RNA structure and hinders formation of a functional spliceosome.23 As a consequence, e5 is omitted from the mature mRNA and the protein lacks a half of its bipartite nuclear localization signal (NLS). The resulting alteration leaves a portion of the protein in the cytoplasm, turning it away from its splicing function. Consistently, our comparative analysis of force expressed MBNL1 without and with e5, both devoid of a GFP tag, revealed higher splicing activity of the latter in the splicing of four endogenous targets.3 At first, this regulatory exclusion of e5 appears to have the same negative effect on MBNL1 as skipping of e1. However, cytoplasmic isoforms also participate in controlling RNA metabolism, as stabilizers of mRNA transcripts and as their transporters to various sites in the cell, such as the membrane, polysomes, or neurites in neurons8,24, and the resultant affected stability of mRNA targets in MBNL functional deficiency might contribute to the DM development.25 Nevertheless, it could be that these roles are just secondary to regulation of alternative splicing because in MBNL1 functional deficiency, like in DM, alterations based on the function of both autoregulatory feedback loops, are aimed, above all, to increase the nuclear, splicing function of MBNL1. This subcellular shift of MBNL1 might also pertain to the order of time, that is, the cargo has to be generated first to be then moved to various destination places. On the other hand, in a healthy cell, there could be a cross-talk between the e1 and e5 pathways, possibly involving other RNA binding proteins or solely based on binding stoichiometry of MBNL to different sites of the transcript in order to maintain the total protein level as well as its cytoplasmic/nuclear ratio optimal.
Figure 3.
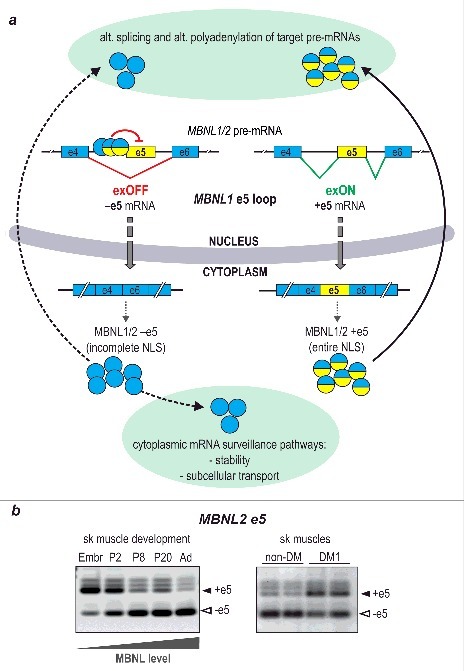
Shuttling MBNL1 and MBNL2 (MBNL1/2) from the cytoplasm to the nucleus: the e5 loop. In schematic diagram depicting e5 loop (a), MBNL1/2 proteins bind to several sites in intron 4 leading to e5 skipping from mature mRNA. MBNL1/2 proteins derived from –e5 mRNAs (blue circles) contain incomplete nuclear localization signal (NLS), which leaves a portion of the protein in the cytoplasm turning it away from its nuclear splicing function (only part of the protein derived from –e5 mRNA is transported back to the nucleus; depicted as curved dotted line arrow). In the absence of MBNLs, or when the protein level is low, e5 is included into mature mRNA translating into a protein with entire NLS (+e5 MBNL proteins marked as blue/yellow circles). This leads to MBNL1/2 transport into the nucleus (curved line arrow), where it fulfills its regulatory alternative splicing function and also loops back by shutting off e5 inclusion into its own mRNA. Blue color indicates constitutive exons, yellow indicates alternatively spliced e5. Panel (b) shows representative RT-PCR analyses of MBNL2 e5 splicing in developing murine skeletal muscles (left) as well as adult skeletal muscles of non-DM1 (control) and DM1-patients (right). Similar analyses have been previously performed for MBNL1 e5 in humans, mice and chickens22,57. Splice isoforms are indicated with black (+e5) and white (–e5) arrowheads. Increasing level of MBNL proteins throughout the course of murine skeletal muscles development is indicated with triangle below left panel. Abbreviations: sk muscle, skeletal muscle; Embr, embryonic day 18; P2, P8 and P20, postnatal day 2, 8 and 20, respectively; Ad, adult. The model shown in a is based on the previously published data10,21-23,57.
Other autoregulatory loops
Besides e1 and e5, some of the other MBNL1 alternatively spliced exons are also sensitive to MBNL content, including e3, e7 and e8 (201, 36, and 95 nucleotides, respectively; Fig. 1).11,21,22 In our experiments, exclusion of e1 in HeLa cells was paralleled by skipping of e3 from the mature mRNA, once MBNL1 content reached a relatively high threshold level (Fig. 1).3 As e3 encodes a linker joining two pairs of ZnFs, its absence further incapacitates MBNL1-related splicing19,26. Consequently, the e3-based loop might be an auxiliary preventive measure protecting the cell from MBNL excess, given that we detected low activity level of MBNL1 lacking e1. Similarly, e7 and e8 analogously respond to MBNL content and their inclusion into mature mRNA is significantly higher in DM.21 While not much is known about the function of e8, e7 has been implicated in increasing MBNL1 affinity to RNA and the protein self-dimerization.19,27 As a result, MBNL1 protein generated from mRNAs containing e7 as well as e5 had the highest processing capacity.19 In a more recent study, however, MBNL1 isoforms with/without e7 and e8 did not differ much in the splicing of the majority of the tested endogenous targets, while for other exons a splicing pattern dependent on the mRNA sequence and the binding position relative to the alternative exon was noted.21 Nevertheless, it is important to mention that most of the MBNL1 isoform comparative studies use GFP fusion proteins and do not take into the account that the GFP tag affects the protein properties, such as the stability3, thus further studies elucidating their exact functions should follow. Altogether, we hypothesize that all MBNL1 alternative exons sensitive to MBNL protein content are in one way or another negatively regulated in terms of feedback loops. Furthermore, we conclude that the control of e1 and e5 splicing constitute central autoregulatory pathways for MBNL1 cellular activity.
Complementary functions of MBNL paralogs
For the most part, the DM phenotype results from sequestration of the three MBNL paralogs, which bind to vastly analogous sequences in RNA transcripts, including expanded C/CUG sequences, owing to the conserved structure of ZnFs.8,21,28-30 Sequential analyses of MBNL knock-out (KO) mice revealed their discernible roles, grounded mostly on their differential, time- and tissue-restricted localizations, as well as highly complementary functions.
MBNL1 partially compensates for the lack of MBNL2 and vice versa
MBNL1 KO mice show generalized muscular spliceopathy, with approximately 80–90% alterations akin to the ones observed in the transgenic mouse model of DM9 expressing over 200 CTG repeats driven by human skeletal actin promoter (HSALR)31, yet again underscoring the role of MBNL1 as the main splicing factor contributing to the skeletal muscle pathology in DM. In comparison, MBNL1 KO mice exhibit only modest splicing alterations in the brain32, with more severe phenotype observed in MBNL2 KOs29, consistent with the previous reports demonstrating that the latter is the major paralog in the central nervous system.11,33 Notably, deletion of only one paralog resulted in the increasing concentration of the other, which was especially evident for the high molecular isoform derived from mRNA containing exons normally skipped from the transcript upon high overall content of MBNLs.8,21,29,34 The importance of this modulation is further emphasized by the fact that double KO mice are embryonic lethal and that conditional deletion of MBNL2 on the MBNL1 KO background causes very robust splicing shifts, exceeding the ones seen in either of the single KO mouse models34 but resembling the level of spliceopathy observed in highly affected DM patients.
These studies show that once the adult pool of MBNL, that consists almost exclusively of MBNL1 and MBNL211,33, is diminished, an increase in the expression of the highly potent, nuclear isoform of either of the paralog containing e5 is triggered. However, while MBNL2 contains alternatively spliced e5 that is regulated by both proteins, the e1 loop is virtually non-active in MBNL23. Furthermore, an increase in the MBNL2 protein content is presumably preceded by the rise in transcript amount, while such pattern might be secondary to the e1 inclusion in MBNL1 (Fig. 4;3). In summary, an increase of MBNL1 can be solely explained by inclusion of e1 into the mature mRNA, a process that is regulated by all MBNLs, and higher stability of the translated product; while the upregulation of MBNL2 seems to be controlled by other mechanisms (see also below).
Figure 4.

Overlapping and compensatory roles of MBNL1 and MBNL2. Total mRNA (upper panel) and e5-containing mRNA (lower panel) expression level of MBNL1 (left) and MBNL2 (right) transcripts in DM1 fibroblasts (∼1000 CUGs) with siRNA-mediated knock-down of MBNL1 or MBNL2 or both. AllStars negative control siRNA (Qiagen) was used as a control (siCtrl). Note that siRNA-mediated MBNL1 knock-down results in MBNL2 mRNA increase, but not vice-versa (upper panel). Also, combined knock-down of MBNL1 and MBNL2 augments e5 inclusion when compared to knock-down of either of the paralogs alone (lower panel). Also note that 1000 CUGs DM1 fibroblasts have already partially depleted basal level of functional MBNLs due to their sequestration by expanded CUG repeats. In non-DM1 fibroblasts the following values for e5 splicing were obtained, MBNL1: Mock 3.50% (±1.49), siCtrl 5.03% (± 0.74), siMBNL1 12.32% (±3.81); MBNL2: Mock 2.52% (±0.59), siCtrl 2.92% (±0.13), siMBNL1 18.45% (±1.06) (data not included in bar graphs). The data shown have not been previously published. Detailed preparation of the samples was described in3.
MBNL3 and its own autoregulatory loop
While MBNL1 and MBNL2 are ubiquitously expressed and have highly complementary roles in postnatal tissues, MBNL3 expression is rather restricted to the embryonic life and the adult muscle differentiation, where it drives efficient myotube reconstitution following injury30. This paralog also differs most from the others in terms of the sequence and protein structure, as highlighted by the fact that neither of the major feedback loops (e1 and e5) operate in MBNL3 in the way they work for other paralogs. First, opposing the results with MBNL1, isoforms lacking e1 can be readily detected for MBNL3 and one could reason that MBNL3-specific sequences, not present in MBNL1, are responsible for this enhanced isoform stability. The excision and inclusion of MBNL3 e1 did not depend on MBNL1 administration in HeLa cells (our unpublished data, not shown), although an increase in the content of the low molecular isoform is observed in mice devoid of the high molecular isoform.30 Interestingly, this truncated isoform also relocates to the nucleus, where it can partially compensate for the full-length protein as shown by increased number of associated truncated MBNL3 to mRNA targets upon deletion of its full-length counterpart. Intriguingly, this nuclear relocalization is not based on the e5 loop, since the corresponding exon and the necessary sequence encoding the first half of the bipartite NLS, present in MBNL1 and MBNL2, is non-existent in MBNL3. Since MBNL3 protein levels do not change in MBNL1 KO mice, as opposed to MBNL234, and because MBNL1 content is unaffected by the absence of the high molecular MBNL330, we hypothesize that MBNL3 expression is regulated independently from other paralogs. One form of such regulation could be transcription of the full-length isoform and its truncated counterpart from different transcription start sites.
Other regulatory pathways of MBNL expression
An additional negative feedback loop mechanism based on circularization of e1, at the expense of the full-length transcript, has been described in Drosophila and also indicated for humans.35 The overall model points to the role of MBNL1 as the factor that halts production of its own mRNA by binding to introns flanking e1, bridging them and initiating spliceosome-dependent formation of e1 circular RNA (e1 circRNA; Fig. 1). In that case, the MBNL-specific consensus binding sites contained in e1 circRNA would also act as a sponge absorbing the excess of the protein. However, while circRNA biogenesis reacted to modulation of Mbl, a Drosophila ortholog of MBNL1, we detected comparable levels of e1 circRNA in control, DM and HSALR muscle samples, despite significant increase in MBNL1 transcript levels following MBNL sequestration.3 It is not known if/to what extent e1 is alternatively spliced in Drosophila, however, it could be that the biogenesis of mbl is rather dependent on generation of circRNA, while the similar effect in mammals is achieved by the e1 loop grounded on alternative splicing. In all, this indicates the importance of precise regulation of MBNL expression during evolution.
Micro RNAs and RNA binding proteins can also modulate MBNL levels by binding to the 3′UTRs of their mRNAs (Fig. 1). Particularly, a set of miR30-5p miRNAs distinctively repressed MBNL paralogs36 while knock-down of CELF1, the MBNL functional antagonist, decreased the decay rate of MBNL1 mRNA.37 Intriguingly, marked stabilization was observed for MBNL2 mRNA following administration of siRNA against either CELF1 or MBNL1 in C2C12 cells37 (see also Fig. 4). Apart from affecting the half-life of MBNL transcripts by binding to its 3′UTR, CELF proteins antagonistically regulate alternative splicing of over 200 exons regulated by MBNL, among them the 95 nucleotide-long MBNL2 exon.38,39 The referred data indicate that MBNL binding to the 3′UTR of their own mRNAs supplements the loops based on alternative splicing in the autoregulatory expression of MBNLs.
MBNLs as a part of cellular splicing factor machinery
Alternative exons are often regulated by more than one splicing factor, which indicates that MBNL loops are a part of a larger splicing factor system that adjust transcript message in a cell for specific needs in a given time. For instance, the final message encoded in RNA may be an outcome of cooperative or antagonistic regulation of MBNLs and RBFOXs, PTBPs, STAUFEN, or previously mentioned CELF proteins.38,40-42 Autoregulatory feedback loops have been reported for many splicing paralogs, including RBFOX1/2, PTBP1/2 or hnRNP L/LL43–46. These loops, similarly to the ones described for MBNLs, act in two distinct but complementary ways: (1) an excess of a splicing regulator is paralleled by exclusion of an exon in its own (and often its paralog) mRNA and the resultant transcript is either subject to nonsense-mediated decay, as in the case of PTBP and hnRNP L, or generates a truncated protein that is unstable or antagonizes the full-length counterpart as reported for MBNL1 and RBFOX, respectively; and (2) a loss of one paralog is accompanied by a compensatory increase of the other (for instance, RBFOX1 deletion in the brain results in 60% increase of RBFOX247). In general, one could conclude that these autoregulatory pathways act to prevent loss of a splicing regulator due to cancelling mutations and to keep the factor functional homeostasis depending on the cellular demand. Until now, MBNL1 expression has been indicated to be directly coupled to RBFOX loops44,47,48, where, similarly to RBFOX itself, it binds and excludes an alternative exon in RBFOX that is necessary for generation of a fully functional isoform9,41.
Therapies based on increasing the pool of functional MBNL
One could envision two distinct ways to increase functional content of MBNL in DM tissues, that is, by: (1) releasing MBNL molecules from pathogenic sequestration by expanded C/CUG repeats, and (2) boosting the content of MBNL to oversaturate the mutant DMPK and CNBP mRNAs with MBNL molecules, so that the excess protein could perform its normal tasks. In the first scenario, chemically modified antisense oligonucleotides (AONs) or small compounds with relatively high affinity to the RNA repeats, but not to other MBNL binding sites contained in pre-mRNA targets, can be designed and effectively administered to the cells.11,49 In addition to blocking C/CUG repeats within DMPK and CNBP mRNAs, AON analogs with specific chemical modifications and short interfering RNAs can be employed to degrade the mutated transcripts by triggering Ribonuclease H (RNase H) and RNA interference mechanisms, respectively.4,49,50 Conversely, delivery of recombinant AAV vectors carrying MBNL1 corrected the splicing and reversed myotonia in HSALR mice, showing that the overall expansion of the MBNL pool is a viable, alternative therapeutic option51. Such increases and the resultant correction of the DM-like phenotype have also been obtained with: miRNA sponge constructs specific for dme-miR-277 and dme-miR-304 targeting 3′UTR of mbl mRNAs in a Drosophila model of DM52; histone deacetylase inhibitors in DM1 patient-derived cells53; and following delivery of phenylbutazone, a non-steroidal anti-inflammatory drug, into HSALR mice.54 Intriguingly, phenylbutazone elevated transcription by suppressing methylation of a specific region in intron 1, which most presumably coincided with activation of T3 transcription start site3, and generation of mRNAs that on one hand would translate into highly functional MBNL1 but on the other, would not be regulated by the feedback loop grounded on alternative splicing of e1.
Conclusions
Restoration of MBNL function in DM cells and tissues corrects the alternative splicing and revokes pathology such as myotonia. Therefore, the therapy of choice should aim at increasing functional pool of MBNL, either by releasing it from expanded C/CUG repeats or increasing its overall quantity. At the same time, side effects caused by overloading of the MBNL pool could be limited by taking into the account the feedback loop mechanisms that maintain the cellular homeostasis as well as other processes that might be involved in development of myotonic dystrophy, such as higher activity of hyper-phosphorylated CELF155, non-canonical repeat-associated non-AUG (RAN) translation from the expanded repeats,56 or DMPK/CNBP loss.4 This is of particular importance when considering systemic and life-time application of therapeutic drugs. For exogenous activation of MBNL transcription, MBNL1 transcription start site T2 seems to be the most suitable target, as the ensuing pre-mRNA is susceptible to both major loops, e1 and e5, and hence, theoretically, following induction each cell in the body could reach the optimal MBNL content.
Funding Statement
This work was supported by the Foundation for Polish Science-TEAM program cofinanced by the European Union within the European Regional Development Fund (to K.S.); the National Centre for Research and Development under Grant ERA-NET-E-Rare-2/III/DRUG_FXSPREMUT/01/2016 (to K.S.); the Polish National Science Centre Grants 2014/15/B/NZ5/00142 (to E.S.-K.) and 2014/15/B/NZ2/02453 (to K.S); and the Ministry of Science and Higher Education of the Republic of Poland, from the quality-promoting subsidy, under the Leading National Research Centre (KNOW) program for the years 2012–2017 [KNOW RNA Research Centre in Poznan (No. 01/KNOW2/2014)].
Conflict of interest
The authors declare no conflict of interest.
Author contribution
PK wrote the manuscript, with contributions from ESK and KS. All co-authors designed and prepared the figures.
References
- 1.Bengtsson NE, Seto JT, Hall JK, Chamberlain JS, Odom GL. Progress and prospects of gene therapy clinical trials for the muscular dystrophies. Hum Mol Genet. 2016;25:R9–17. doi: 10.1093/hmg/ddv420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrar MA, Park SB, Vucic S, Carey KA, Turner BJ, Gillingwater TH, Swoboda KJ, Kiernan MC. Emerging therapies and challenges in spinal muscular atrophy. Annals of neurology. 2017;81:355–68. doi: 10.1002/ana.24864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konieczny P, Stepniak-Konieczna E, Taylor K, Sznajder LJ, Sobczak K. Autoregulation of MBNL1 function by exon 1 exclusion from MBNL1 transcript. Nucleic Acids Res. 2017;45:1760–75. doi: 10.1093/nar/gkw1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thornton CA, Wang E, Carrell EM. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev. 2017;44:135–40. doi: 10.1016/j.gde.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thornton CA. Myotonic dystrophy. Neurol Clin. 2014;32:705–19, viii. doi: 10.1016/j.ncl.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meola G, Cardani R. Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochimica et biophysica acta. 2015;1852:594–606. doi: 10.1016/j.bbadis.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 7.Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. The Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- 8.Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al.. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–24. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, et al.. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010;17:187–93. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kino Y, Washizu C, Kurosawa M, Oma Y, Hattori N, Ishiura S, Nukina N. Nuclear localization of MBNL1: splicing-mediated autoregulation and repression of repeat-derived aberrant proteins. Hum Mol Genet. 2015;24:740–56. doi: 10.1093/hmg/ddu492. [DOI] [PubMed] [Google Scholar]
- 11.Konieczny P, Stepniak-Konieczna E, Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014;42:10873–87. doi: 10.1093/nar/gku767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Z, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT, Thornton CA. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet. 2012;21:1312–24. doi: 10.1093/hmg/ddr568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savkur R, Philips A, Cooper T. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–7. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 14.Kino Y, Washizu C, Oma Y, Onishi H, Nezu Y, Sasagawa N, Nukina N, Ishiura S. MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Res. 2009;37:6477–90. doi: 10.1093/nar/gkp681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fugier C, Klein A, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, et al.. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17:720–5. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 16.Kanadia R, Johnstone K, Mankodi A, Lungu C, Thornton C, Esson D, Timmers A, Hauswirth W, Swanson M. A muscleblind knockout model for myotonic dystrophy. Science (New York, NY) 2003;302:1978–80. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 17.Ho T, Charlet-B N, Poulos M, Singh G, Swanson M, Cooper T. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–12. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohsawa N, Koebis M, Mitsuhashi H, Nishino I, Ishiura S. ABLIM1 splicing is abnormal in skeletal muscle of patients with DM1 and regulated by MBNL, CELF and PTBP1. Genes to cells: devoted to molecular & cellular mechanisms. 2015;20:121–34. doi: 10.1111/gtc.12201. [DOI] [PubMed] [Google Scholar]
- 19.Tran H, Gourrier N, Lemercier-Neuillet C, Dhaenens CM, Vautrin A, Fernandez-Gomez FJ, Arandel L, Carpentier C, Obriot H, Eddarkaoui S, et al.. Analysis of exonic regions involved in nuclear localization, splicing activity, and dimerization of Muscleblind-like-1 isoforms. J Biol Chem. 2011;286:16435–46. doi: 10.1074/jbc.M110.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez-Costa J, Artero R. A conserved motif controls nuclear localization of Drosophila Muscleblind. Mol Cell. 2010;30:65–70. doi: 10.1007/s10059-010-0089-9. [DOI] [PubMed] [Google Scholar]
- 21.Sznajder LJ, Michalak M, Taylor K, Cywoniuk P, Kabza M, Wojtkowiak-Szlachcic A, Matłoka M, Konieczny P, Sobczak K. Mechanistic determinants of MBNL activity. Nucleic Acids Res. 2016;44:10326–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terenzi F, Ladd A. Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA biology. 2010;7:43–55. doi: 10.4161/rna.7.1.10401. [DOI] [PubMed] [Google Scholar]
- 23.Gates D, Coonrod L, Berglund J. Autoregulated splicing of muscleblind-like 1 (MBNL1) Pre-mRNA. J Biol Chem. 2011;286:34224–33. doi: 10.1074/jbc.M111.236547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taliaferro JM, Vidaki M, Oliveira R, Olson S, Zhan L, Saxena T, Wang ET, Graveley BR, Gertler FB, Swanson MS, et al.. Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol Cell. 2016;61:821–33. doi: 10.1016/j.molcel.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osborne RJ, Lin X, Welle S, Sobczak K, O'Rourke JR, Swanson MS, Thornton CA. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet. 2009;18:1471–81. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum Mol Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 27.Yuan Y, Compton S, Sobczak K, Stenberg M, Thornton C, Griffith J, Swanson MS. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35:5474–86. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teplova M, Patel D. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat Struct Mol Biol. 2008;15:1343–51. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al.. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–50. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poulos MG, Batra R, Li M, Yuan Y, Zhang C, Darnell RB, Swanson MS. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum Mol Genet. 2013;22:3547–58. doi: 10.1093/hmg/ddt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton C. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science (New York, NY) 2000;289:1769–73. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 32.Suenaga K, Lee KY, Nakamori M, Tatsumi Y, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Du H, Ares M Jr, et al.. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PloS one. 2012;7:e33218. doi: 10.1371/journal.pone.0033218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanadia R, Urbinati C, Crusselle V, Luo D, Lee Y-J, Harrison J, Oh SP, Swanson MS. Developmental expression of mouse muscleblind genes Mbnl1, Mbnl2 and Mbnl3. Gene Expr Patterns. 2003;3:459–62. doi: 10.1016/S1567-133X(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 34.Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, Warren SA, Chamberlain CM, Finn D, Hong H, et al.. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med. 2013;5:1887–900. doi: 10.1002/emmm.201303275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56:55–66. doi: 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 36.Zhang BW, Cai HF, Wei XF, Sun JJ, Lan XY, Lei CZ, Lin FP, Qi XL, Plath M, Chen H. miR-30-5p Regulates Muscle Differentiation and Alternative Splicing of Muscle-Related Genes by Targeting MBNL. Int J Mol Sci. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masuda A, Andersen HS, Doktor TK, Okamoto T, Ito M, Andresen BS, Ohno K. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci Rep. 2012;2:209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, Lambert NJ, Freese P, Saxena T, Cooper TA, et al.. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015;25:858–71. doi: 10.1101/gr.184390.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci U S A. 2008;105:20333–8. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bondy-Chorney E, Crawford Parks TE, Ravel-Chapuis A, Klinck R, Rocheleau L, Pelchat M, Chabot B, Jasmin BJ, Côté J. Staufen1 Regulates Multiple Alternative Splicing Events either Positively or Negatively in DM1 Indicating Its Role as a Disease Modifier. PLoS Genet. 2016;12:e1005827. doi: 10.1371/journal.pgen.1005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klinck R, Fourrier A, Thibault P, Toutant J, Durand M, Lapointe E, Caillet-Boudin ML, Sergeant N, Gourdon G, Meola G, et al.. RBFOX1 cooperates with MBNL1 to control splicing in muscle, including events altered in myotonic dystrophy type 1. PloS One. 2014;9:e107324. doi: 10.1371/journal.pone.0107324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gooding C, Edge C, Lorenz M, Coelho MB, Winters M, Kaminski CF, Cherny D, Eperon IC, Smith CW. MBNL1 and PTB cooperate to repress splicing of Tpm1 exon 3. Nucleic Acids Res. 2013;41:4765–82. doi: 10.1093/nar/gkt168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keppetipola N, Sharma S, Li Q, Black DL. Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol. 2012;47:360–78. doi: 10.3109/10409238.2012.691456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Damianov A, Black DL. Autoregulation of Fox protein expression to produce dominant negative splicing factors. RNA. 2010;16:405–16. doi: 10.1261/rna.1838210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell. 2004;13:91–100. doi: 10.1016/S1097-2765(03)00502-1. [DOI] [PubMed] [Google Scholar]
- 46.Rossbach O, Hung LH, Schreiner S, Grishina I, Heiner M, Hui J, Bindereif A. Auto- and cross-regulation of the hnRNP L proteins by alternative splicing. Mol Cell Biol. 2009;29:1442–51. doi: 10.1128/MCB.01689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Jr Ares M, Mody I, Black DL. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet. 2011;43:706–11. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Jr Ares M, Otis TS, Black DL. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev. 2012;26:445–60. doi: 10.1101/gad.182477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Konieczny P, Stepniak-Konieczna E, Sobczak K. Modified Antisense Oligonucleotides and Their Analogs in Therapy of Neuromuscular Diseases. In: Jurga S, Erdmann VA, Barciszewski J, eds. Rna Technol. Cham: Springer International Publishing, 2016:243–71. doi: 10.1007/978-3-319-34175-0_11. [DOI] [Google Scholar]
- 50.Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Molecular therapy: the journal of the American Society of Gene Therapy. 2013;21:380–7. doi: 10.1038/mt.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci U S A. 2006;103:11748–53. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cerro-Herreros E, Fernandez-Costa JM, Sabater-Arcis M, Llamusi B, Artero R. Derepressing muscleblind expression by miRNA sponges ameliorates myotonic dystrophy-like phenotypes in Drosophila. Sci Rep. 2016;6:36230. doi: 10.1038/srep36230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang F, Bodycombe NE, Haskell KM, Sun YL, Wang ET, Morris CA, Jones LH, Wood LD, Pletcher MT. A flow cytometry-based screen identifies MBNL1 modulators that rescue splicing defects in myotonic dystrophy type I. Hum Mol Genet. 2017;26:3056–68. doi: 10.1093/hmg/ddx190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen G, Masuda A, Konishi H, Ohkawara B, Ito M, Kinoshita M, Kiyama H, Matsuura T, Ohno K. Phenylbutazone induces expression of MBNL1 and suppresses formation of MBNL1-CUG RNA foci in a mouse model of myotonic dystrophy. Sci Rep. 2016;6:25317. doi: 10.1038/srep25317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cleary JD, Ranum LP. Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev. 2014;26:6–15. doi: 10.1016/j.gde.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–97. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]