

ABSTRACT
Rationale: Transient receptor potential channels of the ankyrin subtype-1 (TRPA1) are non-selective cation channels that show high permeability to calcium. Previous studies from our laboratory have demonstrated that TRPA1 ion channels are expressed in adult mouse ventricular cardiomyocytes (CMs) and are localized at the z-disk, costamere and intercalated disk. The functional significance of TRPA1 ion channels in the modulation of CM contractile function have not been explored.
Objective: To identify the extent to which TRPA1 ion channels are involved in modulating CM contractile function and elucidate the cellular mechanism of action.
Methods and Results: Freshly isolated CMs were obtained from murine heart and loaded with Fura-2 AM. Simultaneous measurement of intracellular free Ca2+ concentration ([Ca2+]i) and contractility was performed in individual CMs paced at 0.3 Hz. Our findings demonstrate that TRPA1 stimulation with AITC results in a dose-dependent increase in peak [Ca2+]i and a concomitant increase in CM fractional shortening. Further analysis revealed a dose-dependent acceleration in time to peak [Ca2+]i and velocity of shortening as well as an acceleration in [Ca2+]i decay and velocity of relengthening. These effects of TRPA1 stimulation were not observed in CMs pre-treated with the TRPA1 antagonist, HC-030031 (10 µmol/L) nor in CMs obtained from TRPA1−/− mice. Moreover, we observed no significant increase in cAMP levels or PKA activity in response to TRPA1 stimulation and the PKA inhibitor peptide (PKI 14–22; 100 nmol/L) failed to have any effect on the TRPA1-mediated increase in CM contractile function. However, TRPA1 stimulation resulted in a rapid phosphorylation of Ca2+/calmodulin-dependent kinase II (CaMKII) (1–5 min) that correlated with increases in CM [Ca2+]i and contractile function. Finally, all aspects of TRPA1-dependent increases in CM [Ca2+]i, contractile function and CaMKII phosphorylation were virtually abolished by the CaMKII inhibitors, KN-93 (10 µmol/L) and autocamtide-2-related peptide (AIP; 20 µmol/L).
Conclusions: These novel findings demonstrate that stimulation of TRPA1 ion channels in CMs results in activation of a CaMKII-dependent signaling pathway resulting in modulation of intracellular Ca2+ availability and handling leading to increases in CM contractile function. Cardiac TRPA1 ion channels may represent a novel therapeutic target for increasing the inotropic and lusitropic state of the heart.
KEYWORDS: CaMKII, cardiomyocytes, [Ca2+]i, contractility, TRPA1
Introduction
The transient receptor potential (TRP) ion channel of the ankyrin 1 (TRPA1) subtype is a member of the TRP superfamily of structurally related and is the only member of the ankyrin subfamily so far identified in mammals. Similar to other TRP ion channel superfamily members, TRPA1 acts as a non-selective cation channel that is highly permeable to calcium.1 The TRPA1 ion channel was first discovered in human fetal lung fibroblasts2 and later found to be primarily expressed in sensory neurons of the dorsal root, nodose and trigeminal ganglion3,4 where it is co-expressed with TRP ion channels of the vanilloid receptor subtype 1 (TRPV1) and together, both were proposed to serve a role in pain by acting as transducers of noxius stimuli.3 However, over the past decade it has become increasingly evident that TRPA1 ion channels are expressed in a variety of tissues and cell types including those of the cardiovascular system.5-7
We recently reported for the first time the expression of TRPA1 at the protein level in cardiac muscle and it's co-localization with TRPV1 ion channels at the costamere, z-disk and intercalated discs in murine cardiomyocytes (CM).8 However, the extent to which TRPA1 ion channel stimulation plays a role in the regulation and/or modulation of CM contractile function have not been previously reported. In the current study, we tested the hypothesis that stimulation of TRPA1 ion channels in electrically-stimulated mouse ventricular CMs results in increases in peak intracellular free Ca2+ concentration ([Ca2+]i) and a concomitant increase in CM contractile function. Furthermore, we hypothesized based on our initial findings that the TRPA1-induced increase in CM contractile function and [Ca2+]i occurs via a cAMP/PKA-dependent signal transduction pathway because the changes in CM [Ca2+]i dynamics and contractile function resemble those of classical β-adrenergic receptor activation of the heart. However, the major finding of the current study is that the observed increases in CM contractile function that occur in response to stimulation of TRPA1 ion channels are not dependent on cAMP production and/or PKA activation but instead result from activation of a CaMKII-dependent signaling pathway leading to an increase in the amplitude and an acceleration in the timing of the intracellular Ca2+ transient.
Materials and methods
Animal model
Four-month-old C57BL/6 WT and TRPA1−/− male mice (Jackson Labs, Bar Harbor, ME) were used and maintained in accordance with the Guide for the Care and Use of Laboratory Animals (NIH). All animals were housed at the Kent State University animal care facility (Kent, OH), which is accredited by the American Association for Accreditation of Laboratory Animal Care.
Isolation of CMs
Murine hearts were excised and transferred to a modified Langendorff apparatus and perfused with collagenase for the purpose of isolating CMs as described previously by our laboratory.8 In brief, mice were killed by cervical dislocation and hearts were collected into a bath of perfusion buffer. Hearts were cannulated via the aorta and blood was flushed with 0.2 mL perfusion buffer. The hearts were subjected to retrograde perfusion on a modified Langendorff apparatus at 37°C (pH 7.4) with a modified Krebs-Henseleit buffer (in mmol/L: 120.4 NaCl, 4.8 KCl, 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4–7HsO, 10 Na-HEPES, 4.6 NaHCO3, 30 taurine, 10 B.M. and 5.5 glucose). The perfusion buffer (calcium-free) was sterile-filtered and paced with a peristaltic pump (Masterflex) at a rate of 4 mL/min. Following perfusion for 4 minutes, the perfusion buffer containing collagenase type II (309 U/mg, Worthington Biochemical) perfused the heart for an additional 14 minutes. When the hearts became spongy, the left ventricles were removed, minced and triturated in Krebs-Henseleit buffer containing fetal bovine serum (FBS; 10% of final volume). The resulting cellular digest was washed and resuspended in HEPES-buffered saline (in mmol/L: 118 NaCl, 4.8 KCl, 0.6 KH2PO4, 4.6 NaHCO3, 0.6 NaH2PO4, 5.5 glucose, pH 7.4) at 23°C. CM yield was consistently ∼80–90%. CMs were then subjected slow calcium reintroduction to 1.23 mM and prepared for contractility assessment or Western blot.
Simultaneous measurement of [Ca2+]i and shortening
Simultaneous measurement of [Ca2+]i and contractile function was performed in individual freshly isolated CMs as described previously by our laboratory.9 Following isolation, freshly prepared CMs were incubated at room temperature for 40 min with fura-2 acetoxy methylester (fura-2/AM; 2 µmol/L) in HEPES-buffered saline (in mmol/L: 118 NaCl, 4.8 KCl, 1.23 CaCl2, 0.8 MgSO4–7H2O, 0.6 KH2PO4, 4.6 NaHCO3, 0.6 NaH2PO4, 5.5 glucose, pH 7.4). Fura-2-loaded CMs were loaded onto coverslips previously mounted on the stage of an Olympus IX-71 inverted fluorescence microscope (Olympus America). CMs were superfused continuously with HEPES-buffered saline at a flow rate of 1.5 mL/min and compounds (agonists, antagonists) were delivered for ∼5 minutes. In protocols where CMs received both agonists and antagonists, agonists were delivered initially (after a brief normalization period) and antagonists were subsequently introduced once agonist-induced effects plateaued (roughly 5 minutes). Healthy CMs (rod shaped) were selected for experimentation and paced at a frequency of 0.3 Hz (30V, 5 ms duration) applied using a Grass SD9 field stimulator (Grass). Felix GX software (with sarcomere length module) was used to select a region of the CM with discernable striations and excellent optical density to image the changes in sarcomere length with each contraction. [Ca2+]i measurements were simultaneously recorded on individual CMs. Real-time calcium tracing data were acquired via a D-104 microscope photometer and a PMT (Photon Technology International) using an alternating excitation wavelength protocol (340, 380 nm/20 Hz) and emission wavelength of 510 nm. Background fluorescence was automatically corrected for the experiments using Easy Ratio Pro. The ratio of the 2 intensities was used to measure changes in [Ca2+]i due to the fact that calibration of the system relies upon several assumptions. Hardware and software for data acquisition and analysis were generously provided by Horiba Scientific (Edison, NJ).
Analysis of [Ca2+]i and shortening data
The following variables were calculated for each individual contraction: fractional shortening (% of sarcomere length change during shortening), maximum velocity of cell shortening and relengthening (µm/sec), peak [Ca2+]i (340/380 ratio), time to peak [Ca2+]i (msec) and [Ca2+]i decay to baseline (msec). Variables from 10 contractions were averaged to obtain mean values at baseline and in response to the intervention. Averaging the variables over time minimizes beat-to-beat variation. Results were quantified and are expressed as mean ± SEM. Slavitzky-Golay Filter (x15 moving average) was used where indicated to increase the signal-to-noise ratio for the purpose of enhancing the clarity of the individual Ca2+ transient overlays to highlight changes in timing parameters.
cAMP production and PKA activity assay
Following isolation, CMs were pretreated with 3-isobutyl-1-methylxanthine (IBMX; 1 mmol/L) before treatment with allyl isothiocyanate (AITC; 1–300 µmol/L) or isoproterenol (ISO; 1–100 nmol/L) in the presence or absence of PKA inhibitor, PKI 14–22 (1–100 nmol/L). Samples were then subjected to protocols designed to analyze intracellular cyclic AMP levels or PKA activity. Intracellular cyclic AMP levels were analyzed by ELISA as per the manufacturer's instructions (Enzo Life Sciences). Each treatment was performed in duplicate and repeated 4 times.
For the PKA activity assay, protein was extracted from pelleted CMs (∼5 × 107 cells/set) using modified RIPA buffer (in mmol/L: 50 Tris-HCl, 1 EDTA, 150 NaCl, 0.25% deoxycholic acid, 1% NP-40, pH 7.4) supplemented with 10 mmol/L benzamidine chloride, 0.1% β-mercaptoethanol 1 mmol/L PMSF, 1 mmol/L sodium orthovanadate, 1 µmol/L Calyculin A to inhibit protease/phosphatase activities as described previously with minor modifications.10 After 15 min of incubation on ice, the lysed cells were centrifuged at 13,000 × g for 15 min at 4°C. The supernatant was used as a source of PKA for the activity assays. Cellular protein extract was incubated for 15 min at 30°C with the assay buffer containing 1.6 µCi p32-ATP, 0.2 mmol/L ATP, 10 mmol/L MgCl2, 20 mmol/L Tris-HCl/pH 7.6, 500 µmol/L IBMX, 10 mmol/L DTT, 5 mmol/L NaF, 200 µmol/L Kemptide (WRTDQEIEUYKAKV-PXILYFGCSA-N); 50 µmol/L H89 was added for each set of sample as control. The reaction was stopped using 1N HCl. The assay was done in duplicate and from each set 20 µl of sample was spotted on 1cm x1 cm phospho-cellulose paper; subsequently it was air-dried and washed with 1% phosphoric acid twice before putting them in scintillation vials with water for counting. The kinase activity units were counted as per the following equation:
Kinase activity = ((sample cpm – blank cpm) / (Specific activity × reaction time × mg protein)) X ((reaction volume/spot volume))
cpm: count per min; Specific activity = (cpm value of 5 µl of p32-ATP mix) / (pmoles of ATP)
Preparation of cell lysates and immunoblot analysis
Immunoblot analysis was performed as described previously.8 In brief, CMs obtained from WT or TRPA1−/− mice were treated with AITC (100 µmol/L) in the presence or absence of CaMKII inhibitors, KN-93 (10 µmol/L) or AIP (20 µmol/L). CMs were homogenized in a lysis buffer (in mM: 25 Tris-HCl, 150 NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, pH 7.6) containing protease and phosphatase inhibitors at varying time points (0–5 minutes) and protein concentration was assessed using the Bradford method (Bradford, 1976). Samples containing equal amount of protein lysates (50 µg) were heated at 95ºC then subjected to SDS-PAGE on a 4–15% precast polyacrylamide gels (Bio-Rad) through the use of a minigel apparatus and subsequently transferred to nitrocellulose membranes. Nonspecific binding was blocked with 1% BSA in Tris-buffered saline solution (0.2% [vol/vol] Tween-20 in 20 mM Tris base, 137 mM NaCl, pH 7.6) for 30 minutes at room temperature. Antibodies generated to recognize phosphorylated CaMKII (Cell Signaling) and GAPDH (Millipore) were diluted 1:1000 in Tris-buffered saline containing 1% BSA and incubated at 4°C overnight. After washing in Tris-buffered saline, membranes were incubated for 1 h at room temperature with horseradish-peroxidase linked secondary antibody (goat anti-mouse and goat anti-rabbit) diluted 1:5000 in Tris-buffered saline with 1% BSA. Enhanced chemiluminescence was performed to detect antibody binding using an ImageQuant LAS 4000 Mini (General Electric). Immunoreactivity was quantified by scanning densitometry and analyzed using ImageJ software (NIH)
Statistical analysis
Dose response curves to AITC and isoproterenol (ISO) were repeated in CMs obtained from 6 different wild-type mouse hearts. The effects of AITC on TRPA1−/− CMs were conducted in 4 different hearts. Results obtained from each heart were averaged so that all hearts were weighted equally. Within group comparisons were made using one-way analysis of variance with repeated measures and the Bonferroni post hoc test. Differences were considered statistically significant at p < 0.05. All results are expressed as means ± SEM.
Results
AITC stimulates dose-dependent increases in peak [Ca2+]i and contractile function in CMs
Figure 1 depicts representative traces demonstrating a marked increase in shortening (panel A, change in sarcomere length) and peak [Ca2+]i (panel B, change in 340/380 ratio) in an individual CM following stimulation of TRPA1 with AITC (100 μmol/L). Exploded views illustrating dose-dependent changes in sarcomere length and [Ca2+]i following exposure to AITC (1–300 µmol/L) are depicted in panels C and D, respectively. Summarized data demonstrating dose-dependent changes in [Ca2+]i dynamics and contractile function following exposure to AITC are shown in Table 1. The AITC-induced (100 µmol/L) increase in [Ca2+]i and contractile function were completely blocked by pre-treatment with the TRPA1 antagonist, HC-030031 (10 µmol/L), resulting in a peak [Ca2+]i and fractional shortening that were 98 ± 3.1% and 101 ± 2.2% of steady-state baseline control value, respectively.
Figure 1.
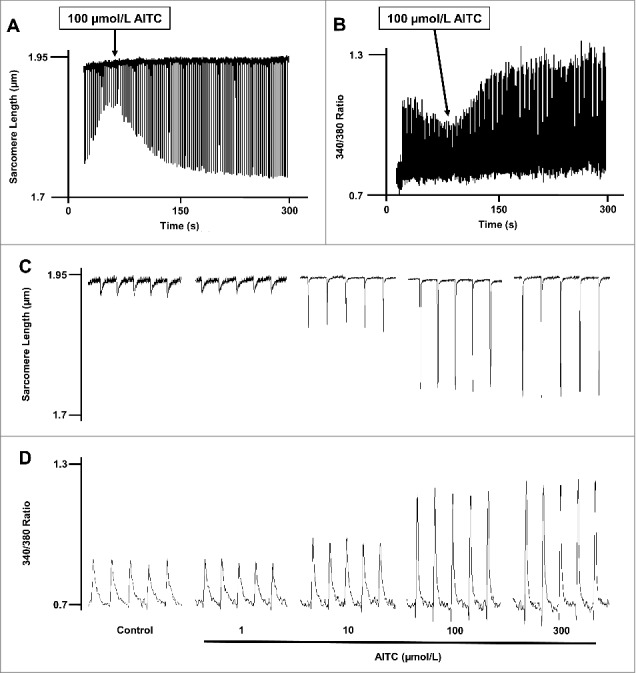
Allyl isothiocyanate (AITC) increases [Ca2±]I and shortening in CMs. Original traces demonstrating the effect of AITC (100 μmol/L) on steady-state sarcomere length (µm; panel A) and [Ca2+]i (340/380 ratio; panel B) in an individual mouse ventricular myocyte. AITC was added where indicated on the figure. Exploded views depicting the dose-dependent changes in sarcomere length and [Ca2+]i (panels C and D, respectively) before (control) and after addition of AITC (1–300 µmol/L) to single ventricular myocyte. AITC was added to the bath in a cumulative fashion. n = 20 cells from 6 hearts.
Table 1.
Comparison of AITC- and isoproterenol (ISO)-induced changes in CM [Ca2±]i and contractile function. Data are expressed as mean ± SEM. * P < 0.05 compared with untreated control (ctrl).
| AITC (µmol/L) n = 20 | 0 (Ctrl) | 1 | 10 | 100 | 300 |
|---|---|---|---|---|---|
| Fractional Shortening (% sarcomere length) | 4.5 ± 0.1 | 5.0 ± 0.2 | 6.3 ± 0.2* | 8.7 ± 0.3* | 10.8 ±0.3* |
| Maximum Velocity of Cell Shortening (μm/sec) | 4.7 ± 0.1 | 5.4 ± 0.2 | 7.1± 0.3* | 9.9 ± 0.3* | 11.2 ± 0.3* |
| Maximum Velocity of Cell Relaxation (μm/sec) | 3.6 ± 0.1 | 4.1 ± 0.1 | 5.1 ± 0.3* | 7.1 ± 0.3* | 8.3 ± 0.3* |
| Peak [Ca2+]i (change in 340/380 ratio) | 0.13 ± 0.02 | 0.15 ± 0.04 | 0.19 ± 0.04* | 0.22 ± 0.06* | 0.26 ± 0.04* |
| Tp [Ca2+]I (msec) | 158 ± 9.4 | 144 ± 8.4 | 112 ± 9.0* | 58 ± 6.1* | 46 ± 3.3 |
| [Ca2+]i Decay (msec) | 610 ± 19 | 563 ± 18 | 458 ± 15* | 326 ± 12* | 235 ± 9* |
| ISO (nmol/L) n = 12 | 0 (Ctrl) | 1 | 5 | 10 | 100 |
| Fractional Shortening (% sarcomere length) | 4.6 ± 0.2 | 5.5 ± 0.3 | 7.1 ± 0.3* | 10.7 ± 0.6* | 11.5 ±0.5* |
| Maximum Velocity of Cell Shortening (μm/sec) | 4.7 ± 0.1 | 5.1 ± 0.1 | 7.4 ± 0.2* | 11.1 ± 0.4* | 12.7 ± 0.4* |
| Maximum Velocity of Cell Relaxation (μm/sec) | 3.6 ± 0.1 | 4.0 ± 0.1 | 5.6 ± 0.2* | 8.6 ± 0.3* | 10.0 ± 0.2* |
| Peak [Ca2+]i (change in 340/380 ratio) | 0.11 ± 0.02 | 0.12 ± 0.03 | 0.17 ± 0.05* | 0.22 ± 0.05* | 0.23 ± 0.05* |
| Tp [Ca2+]I (msec) | 140 ± 17 | 124 ± 16 | 104 ± 9 | 56 ± 7* | 52 ± 6* |
| [Ca2+]i Decay (msec) | 615 ± 16 | 586 ± 15 | 381 ± 16* | 231 ± 21* | 174 ± 16* |
AITC increases fractional shortening, maximum velocity of shortening and maximum velocity of relengthening in CMs
Figure 2 represents overlays depicting the dose-dependent increases in CM fractional shortening (panel A), maximum velocity of shortening (panel C) and maximum velocity of relaxation (panel E) following TRPA1 stimulation with AITC (1–300 μmol/L). AITC (100 µmol/L) increased fractional shortening, maximum velocity of shortening and maximum velocity of relaxation to 195 ± 6.8%, 213 ± 7.9% and 196 ± 8.7% of control, respectively. The summarized dose response data for panels A, C and E are depicted in panels B, D and F, respectively and are expressed as a percent of control. The summarized raw data for these parameters are listed in Table 1 and are expressed as % change in sarcomere length (fractional shortening) and µm/sec (velocity of shortening/relengthening).
Figure 2.
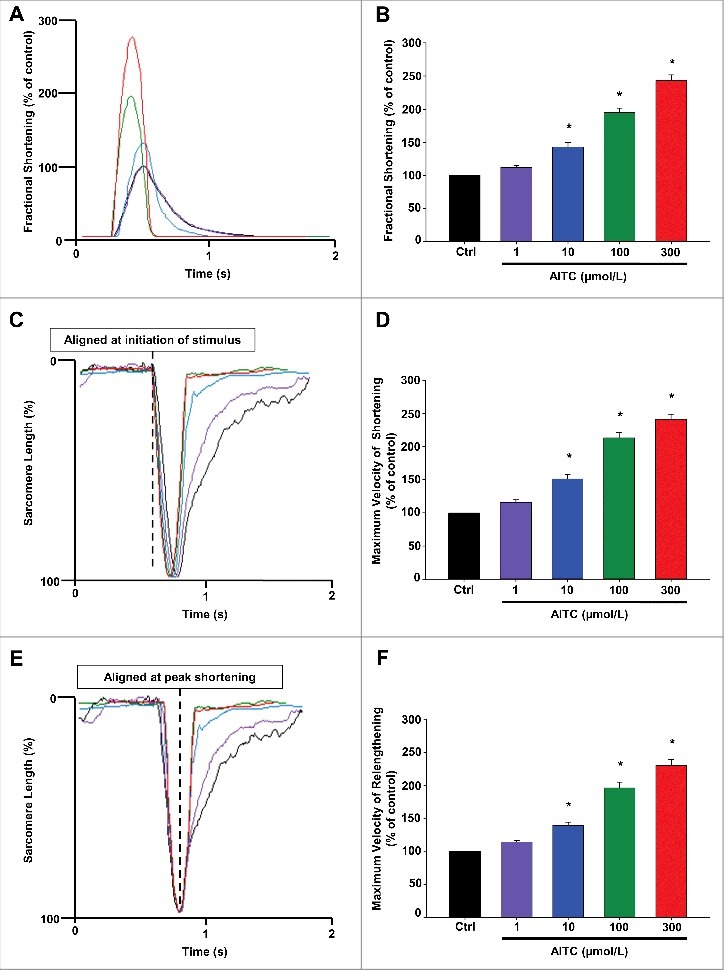
AITC increases fractional shortening, maximum velocity of shortening and maximum velocity of relengthening in CMs. Overlays of individual cell shortening and relengthening events illustrating the dose-dependent effects of AITC (1–300 μmol/L) on fractional shortening are depicted in panel A. Representative overlays assessing changes in sarcomere length normalized to peak height (set at 100%) and aligned at initiation of stimulus or at peak shortening to illustrate dose-dependent changes in timing of shortening and relengthening are depicted in panels C and E, respectively. Summarized data for panels A (fractional shortening), C (maximal velocity of shortening) and E (maximal velocity of relengthening) are depicted in panels B, D and F, respectively. Results are expressed as percent of steady-state baseline control (Ctrl) value set at 100%. Changes in fractional shortening were measured as percent of sarcomere length. Changes in velocity were measured in micrometers/sec. * P < 0.05 compared with Ctrl. n = 20 cells from 6 hearts.
AITC increases peak [Ca2+]i and accelerates time to peak [Ca2+]i and the rate of [Ca2+]i decay in CMs
Figure 3 represents overlays depicting the dose-dependent increases in peak [Ca2+]i (panel A) as well as an acceleration in time to peak [Ca2+]i (panel C) and in [Ca2+]i decay (panel E) following TRPA1 stimulation with AITC (1–300 μmol/L). AITC (100 µmol/L) increased peak [Ca2+]i to 180 ± 14.0% of control. Time to peak [Ca2+]i and the Ca2+ decay were accelerated by 63 ± 3.1% and 45 ± 2.9%, respectively, compared with control. The summarized data for panels A, C and E are depicted in panels B, D and F, respectively and are expressed as a percent of control. The summarized raw data for these parameters are listed in Table 1 and are expressed in msec. Individual Ca2+ transient traces were smoothed using the Savitzky-Golay filter to increase the signal-to-noise ratio and enhance the clarity of the figure to highlight changes in timing parameters.
Figure 3.
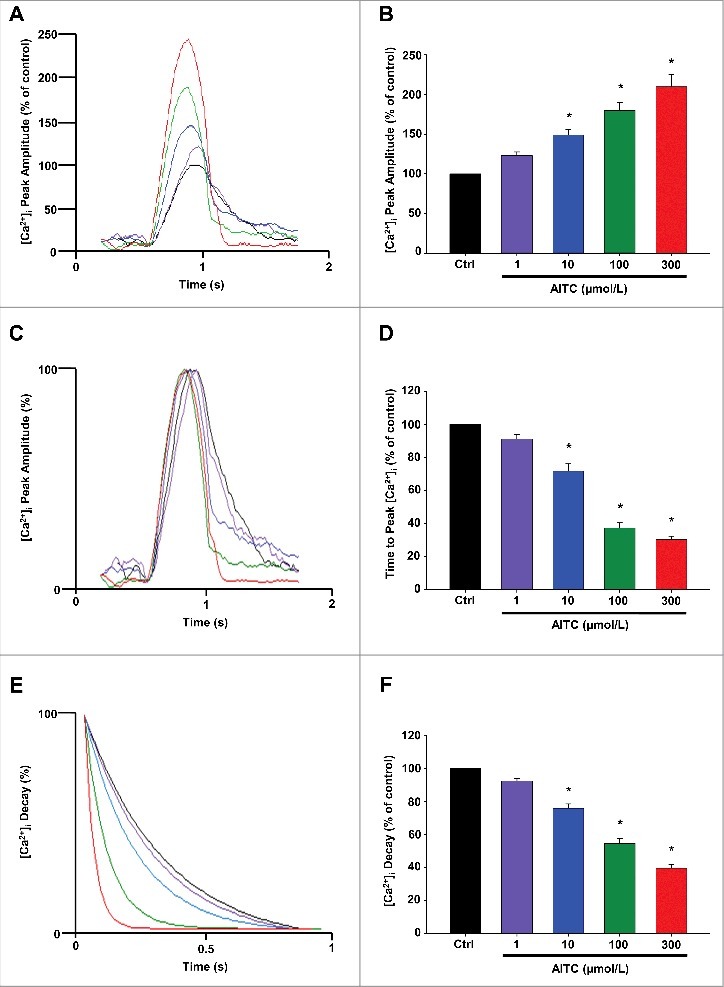
AITC increases peak [Ca2±]I and accelerates time to peak [Ca2±]I and the rate of [Ca2±]I decay in CMs. Overlays illustrating the dose-dependent effects of AITC (1–300 μmol/L) on peak [Ca2+]i are depicted in panel A. Representative overlays assessing dose-dependent changes in [Ca2+]i peak amplitude normalized to peak height (set at 100%) to illustrate changes in time to peak [Ca2+]i and the time of [Ca2+]i decay are depicted in panels C and E, respectively. Summarized data for panels A ([Ca2+]i peak amplitude), C (time to peak [Ca2+]i) and E ([Ca2+]i decay) are depicted in panels B, D and F, respectively. Results are expressed as percent of steady-state baseline control (Ctrl) value set at 100%. Changes in peak [Ca2+]i are measured as the change in the 340/380 ratio. Changes in timing are measured in milliseconds. Individual traces were smoothed using the Savitzky-Golay filter to increase the signal-to-noise ratio. * P < 0.05 compared with Ctrl. n = 20 cells from 6 hearts.
AITC-induced increases in [Ca2+]i and contractile function are absent in CMs obtained from TRPA1−/− mice
Figure 4 depicts a representative trace demonstrating that AITC (100 µmol/L) has no effect on [Ca2+]i (panel A) or contractile function (panel B) in CMs obtained from TRPA1−/− mice. Fig. 4 (panels C-F) represent exploded views of changes in sarcomere length (C and D) and individual Ca2+ transients (E and F) before and after stimulation of TRPA1 with AITC in CMs obtained from TRPA1−/− mice (taken from panels A and B, respectively). In CMs obtained from TRPA1−/− mice, peak [Ca2+]i and fractional shortening were 102 ± 4.1% and 101 ± 5.2% of steady-state baseline control, respectively, following exposure to AITC.
Figure 4.
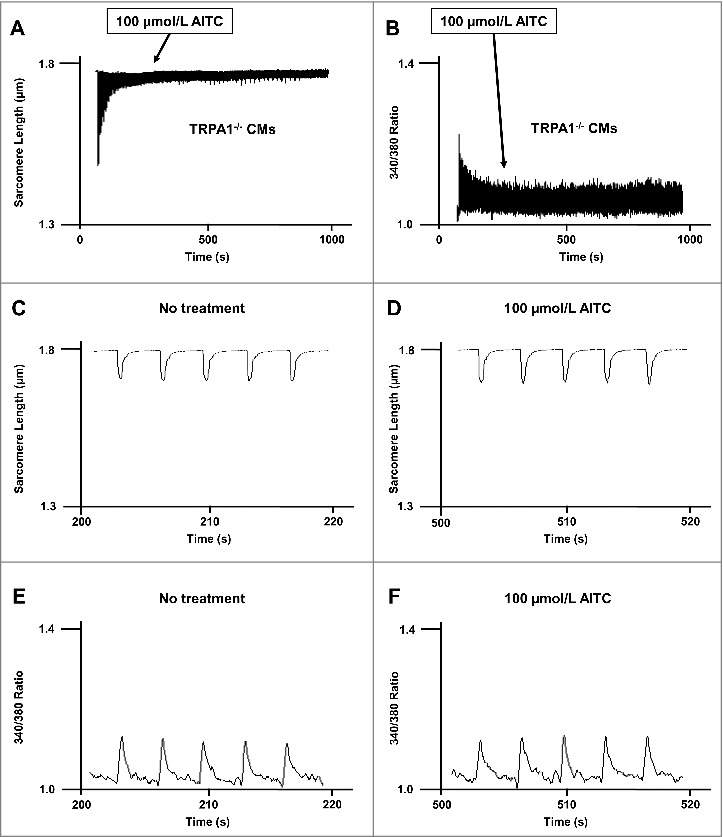
AITC has no effect on [Ca2±]I and shortening in CMs obtained from TRPA1 null mice. Original traces demonstrating the lack of effect of AITC (100 μmol/L) on steady-state sarcomere length (µm; panel A) and [Ca2+]i (340/380 ratio; panel B) in an individual mouse ventricular myocyte obtained from TRPA1 null mice (TRPA1−/−). AITC was added where indicated on the figure. Exploded views of changes in sarcomere length and [Ca2+]i before and after addition of AITC are depicted in panels C-F. n = 8 cells from 4 hearts.
AITC increases in CM [Ca2+]i and contractile function are similar to those observed following β-Adrenergic receptor (β-AR) stimulation with Isoproterenol (ISO)
For comparison, we also assessed the dose-dependent effects of β-AR stimulation with ISO (1–100 nmol/L) on [Ca2+]i and contractile function in CMs. The summarized data are depicted in Table 1. The dose-dependent effects of AITC (1–300 µmol/L) on CM [Ca2+]i and contractile function were qualitatively and quantitatively very similar to those observed with ISO (1–100 nmol/L). Representative traces and overlays and summarized data expressed as a percent of control can be found in the Supplemental Data.
TRPA1-induced increases in [Ca2+]i and contractile function occur independently of cyclic AMP (cAMP) and Protein kinase a (PKA) in CMs
Figure 5 depicts summarized data quantifying the extent to which AITC (1–300 µmol/L) or ISO (1–100 nmol/L) stimulate activity of the cAMP/PKA pathway in CMs. ISO induced a dose-dependent elevation in cAMP production (panel A) whereas AITC had no effect in CMs obtained from WT mice except at the highest concentration tested (panel B). Furthermore, ISO (10 nmol/L) induced a significant increase in PKA activity (3.22 × 104 ± 1.98 × 103 PKA activity units/107 CMs) which was dose-dependently eliminated in the presence of PKA inhibitor, PKI 14–22 (1–100 nmol/L; panel C). CMs treated with AITC (100 µmol/L) demonstrated no significant alterations in basal PKA activity when compared with the untreated control (1.12 × 103 ± 1.11 × 103 vs. 1.09 × 103 ± 0.45 × 103, respectively). The efficacy of PKI 14–22 (1–100 nmol/L) in reversing the ISO-induced increase in CM fractional shortening are depicted in the inset of panel C.
Figure 5.
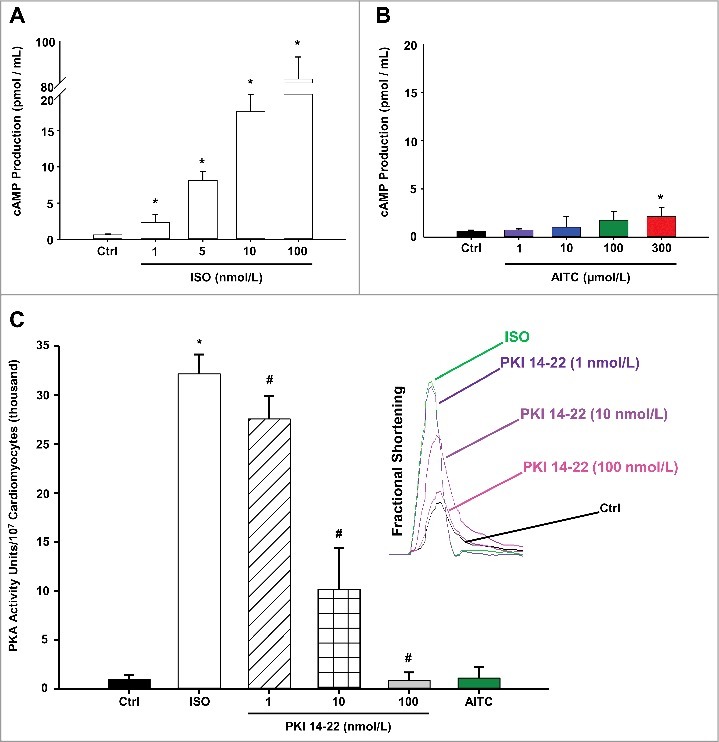
TRPA1 stimulation does not signal through the cAMP/Protein Kinase A (PKA) pathway in CMs. Summarized data demonstrating the effects of ISO (1–100 nmol/L) and AITC (1–300 µmol/L) on cAMP production in CMs obtained from WT mice are depicted in panels A and B, respectively. Results are expressed as total cAMP production (pmol/mL; n = CMs isolated from 5 separate hearts repeated in triplicate). Summarized data demonstrating the effects of AITC (100 µmol/L) or ISO (10 nmol/L) in the presence or absence of PKA inhibitor, PKI 14–22 (1–100 nmol/L), on PKA activity is shown in panel C (n = CMs isolated from 3 separate hearts repeated in triplicate). The dose-dependent effects of PKI 14–22 treatment following isoproterenol-induced increases in contractile function are shown in the subfigure on the right side of panel C (n = 6 CMs from 3 hearts). * P < 0.05 compared with untreated control value. # P < 0.05 compared with isoproterenol-treated CMs.
Figure 6 demonstrates that PKI 14–22 does not significantly alter AITC-induced increases in contractile function or [Ca2+]i in electrically-paced CMs. Representative overlays illustrating the lack of effect of PKI 14–22 (100 nmol/L) following AITC (100 µmol/L) treatment on fractional shortening (panel A) and [Ca2+]i peak amplitude (panel B) are shown. The summarized data depicting the lack of effect of PKI 14–22 on AITC-induced changes in fractional shortening, maximum velocity of shortening and maximum velocity of relengthening are depicted in panels C, E, and G, respectively. Similarly, the summarized data for PKI 14–22 on [Ca2+]i indices including [Ca2+]i peak amplitude, time to peak [Ca2+]i and [Ca2+]i decay are shown in panels D, F and H, respectively. Individual Ca2+ transient traces were smoothed using the Savitzky-Golay filter to increase the signal-to-noise ratio and enhance the clarity of the figure to highlight changes in timing parameters.
Figure 6.
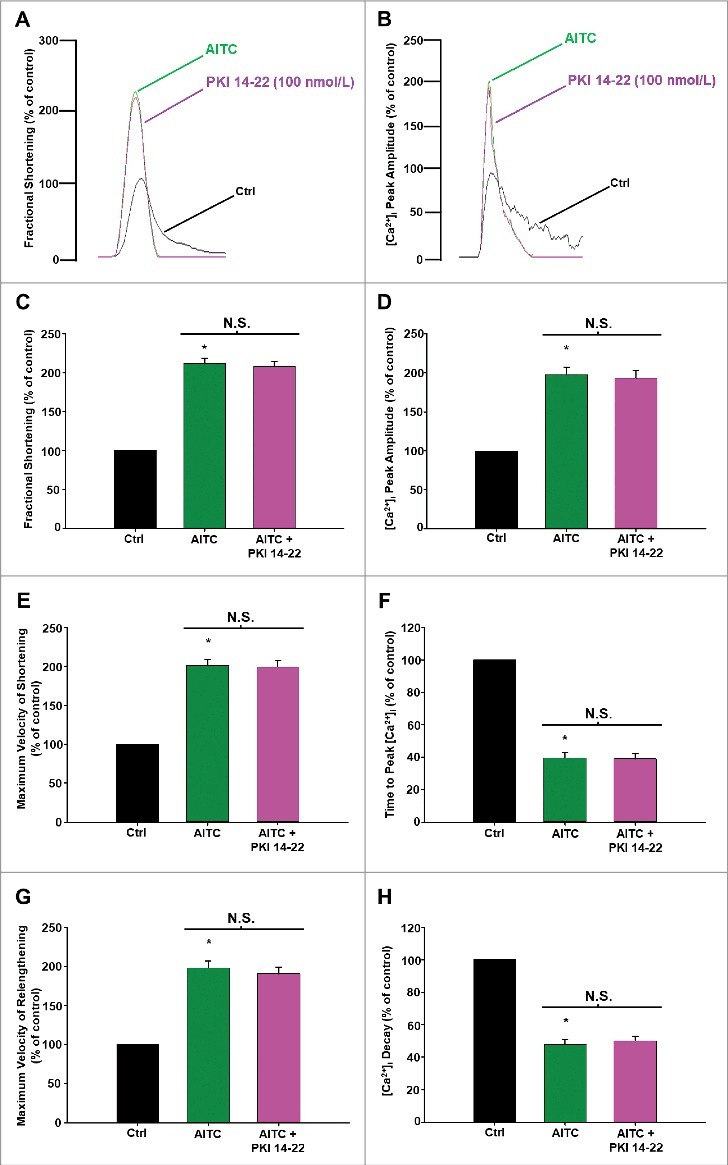
AITC-induced increases in contractile function and [Ca2±]i occur independently of PKA in CMs. Overlays of fractional shortening and [Ca2+]i peak amplitudes in electrically-paced CMs illustrating the effects of AITC (100 µmol/L) in the presence and absence of PKI 14–22 (100 nmol/L) are depicted in panels A and B, respectively. Summarized data for parameters measuring contractile function are depicted in panels C (fractional shortening), E (maximum velocity of shortening) and G (maximum velocity of relengthening). Summarized data analyzing [Ca2+]i dynamics are depicted in panels D ([Ca2+]i peak amplitude), F (time to peak [Ca2+]i) and H ([Ca2+]i decay). Results are expressed as percent of steady-state baseline control (Ctrl) value set at 100%. Individual [Ca2+]i traces were smoothed using the Savitzky-Golay filter to increase the signal-to-noise ratio. N.S. = not statistically significant. * P < 0.05 compared with Ctrl. n = 18 cells from 6 hearts.
TRPA1 stimulation elicits CaMKII phosphorylation at threonine 286 in a time-dependent manner in CMs
Figure 7 (panel A) depicts a representative immunoblot illustrating the time-dependent manner by which AITC elicits CaMKII phosphorylation at threonine 286 (pCaMKII-t286) in CMs. AITC-induced increases in pCaMKII-t286 at 1, 2 and 5 minutes in quiescent CMs obtained from WT mice. The summarized data illustrating the time-dependency of AITC-induced pCaMKII-t286 is shown in panel B.
Figure 7.
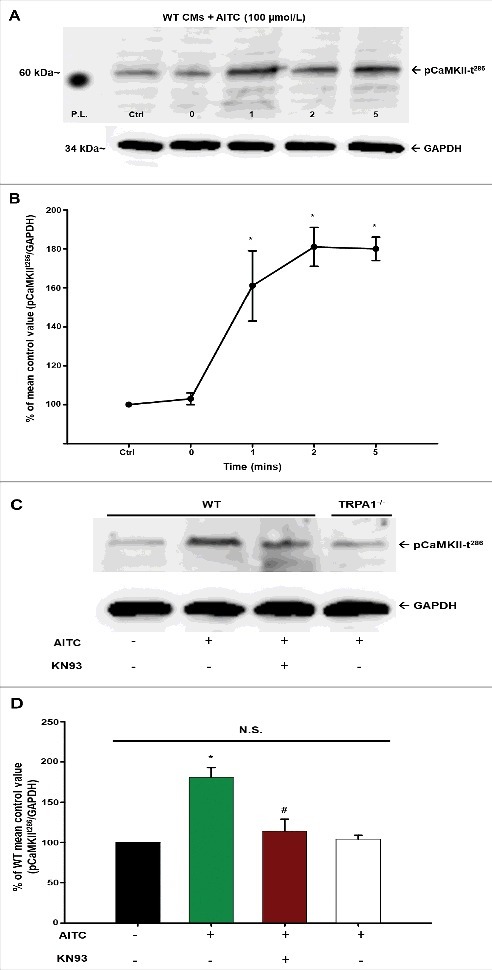
AITC-induced phosphorylation of CaMKII at threonine 286 occurs in a time- and TRPA1-dependent manner in CMs. Representative immunoblots demonstrating the effect of AITC (100 µmol/L) on CaMKII phosphorylation at threonine 286 (pCaMKII-t286) at 0–5 minutes in CMs obtained from WT mice are depicted in panel A. All samples were lysed at the indicated time points (mins). Summarized data are shown as the % of the untreated mean control value (pCaMKII-t286/total protein) in panel B (n = CMs obtained from 5 separate hearts). Representative immunoblots demonstrating the effects of AITC (100 µmol/L) on CaMKII phosphorylation in the presence (+) and absence (−) of KN93 (10 µmol/L) is shown in panel C. Samples from WT and TRPA1−/− were either treated with AITC or pretreated with KN93 for 10 minutes before AITC administration. All samples were lysed 5 minutes following treatment of AITC. Summarized data are expressed as the percent of the untreated WT mean control value in panel D (n = CMs isolated from 5 separate hearts). GAPDH was probed as the loading control. N.S. = not statistically significant. P.L. = protein ladder. * P < 0.05 compared with the WT mean control value. # P < 0.05 compared with the AITC-treated WT CMs.
Figure 7 (panel C) depicts a representative immunoblot illustrating the effects of AITC (100 µmol/L; 5 min) on pCaMKII-t286 in CMs obtained from WT and TRPA1−/− mice. AITC-induced pCaMKII-t286 elevations in CMs obtained from WT mice but not in CMs isolated from TRPA1−/− mice (panel A; 105 ± 2.1% of unstimulated control). Moreover, the TRPA1-induced increases in pCaMKII-t286 were markedly attenuated in WT CMs pretreated with CaMKII inhibitor, KN-93 (10 µmol/L; 118 ± 18.0% of unstimulated control) as well as the structurally and mechanistically different inhibitor AIP (20 µmol/L; 107 ± 14.0% of unstimulated control). Summarized data are depicted in panel D.
TRPA1 stimulation modulates contractile function and [Ca2+]i via a CaMKII-dependent mechanism in CMs
Figure 8 depicts the extent to which CaMKII inhibition reverses AITC-induced increase in [Ca2+]i and contractile function. Representative overlays demonstrate that the effects of TRPA1 stimulation with AITC (100 µmol/L) on fractional shortening (panel A) and [Ca2+]i peak amplitude (panel B) are dose-dependently reversed following treatment with KN-93 (1–10 µmol/L) in electrically-paced CMs. Further analysis showed that the effects of AITC on fractional shortening (panel C), maximum velocity of shortening (panel E) and maximum velocity of relengthening (panel G) were similarly reversed by treatment with KN-93. As expected, the effects of AITC on [Ca2+]i peak amplitude (panel D), time to peak [Ca2+]i (panel F) and [Ca2+]i decay (panel H) were also reversed following treatment with KN-93. Parallel experiments were also performed to assess the effects of KN-93 on steady-state contractile parameters. Addition of KN-93 (10 µmol/L) reduced [Ca2+]i peak amplitude by 12 ± 6% and fractional shortening by 15 ± 8% (4 out of 12 cells, n = 3 hearts). Control experiments using KN-92 (inactive analog of KN-93) resulted in no significant alterations in baseline contractility or AITC-induced increases in CM [Ca2+]i or contractile function (data not shown). Furthermore, additional experiments were conducted using the CaMKII inhibitory peptide AIP, (20 µmol/L) following treatment of CMs with AITC. Addition of AIP following AITC reduced [Ca2+]i peak amplitude and fractional shortening to 106 ± 9% and 105 ± 8% of control (n = 18 cells obtained from 5 hearts). Individual Ca2+ transient traces were smoothed using the Savitzky-Golay filter to increase the signal-to-noise ratio and enhance the clarity of the figure to highlight changes in timing parameters.
Figure 8.
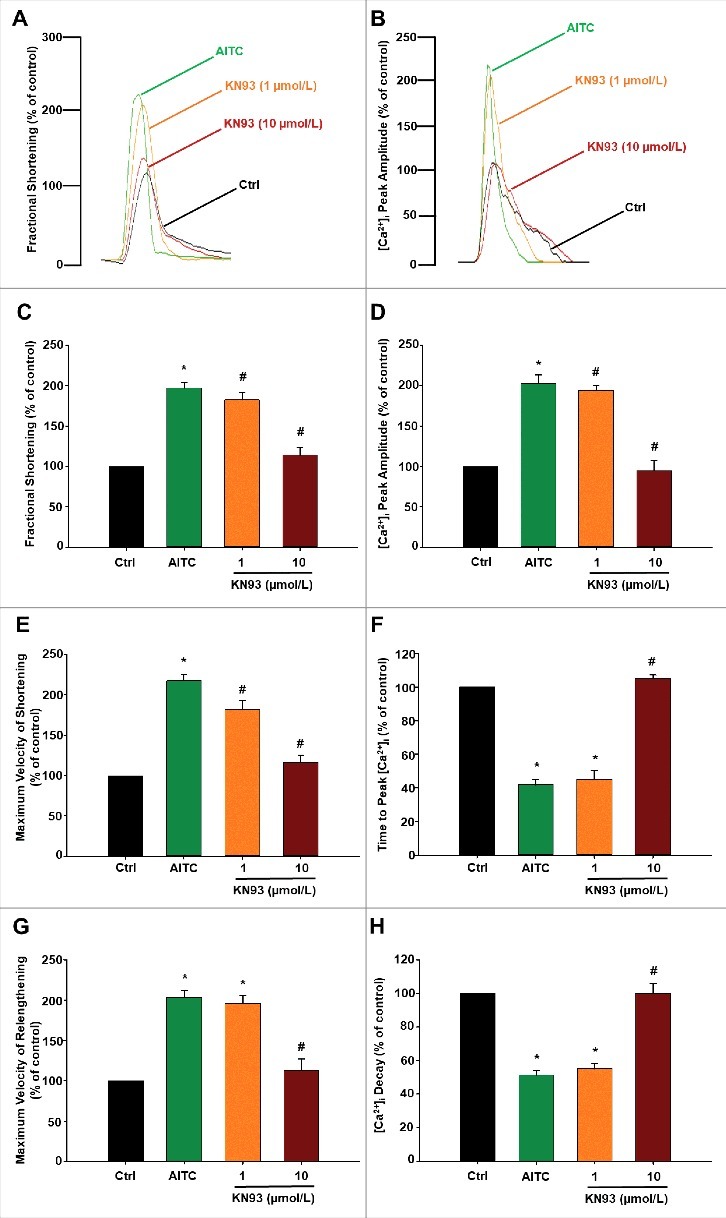
TRPA1 stimulation modulates contractile function and [Ca2±]i through a CaMKII-dependent mechanism in CMs. Overlays of fractional shortening and [Ca2+]i peak amplitudes in electrically-paced CMs illustrating the effects of AITC (100 µmol/L) in the presence and absence of KN93 (1–10 µmol/L) are depicted in panels A and B, respectively. Summarized data for parameters measuring contractile function are depicted in panels C (fractional shortening), E (maximum velocity of shortening) and G (maximum velocity of relengthening). Summarized data analyzing [Ca2+]i dynamics are depicted in panels D ([Ca2+]i peak amplitude), F (time to peak [Ca2+]i) and H ([Ca2+]i decay). Results are expressed as percent of steady-state baseline control (Ctrl) value set at 100%. Individual [Ca2+]i traces were smoothed using the Savitzky-Golay filter to increase the signal-to-noise ratio. * P < 0.05 compared with Ctrl. # P < 0.05 compared with AITC-treated cells. n = 15 cells from 5 hearts.
Discussion
We recently were the first laboratory to demonstrate the functional expression of TRPA1 in the adult mouse heart and it's localization in costameres, z-discs and intercalated discs of mouse CMs.8 To our knowledge, the current study is the first to thoroughly characterize the effects of TRPA1 stimulation on [Ca2+]i and contractile function in electrically-stimulated adult mouse CMs. The major finding of the current study is that TRPA1 agonist, AITC, stimulates dose-dependent increases in peak [Ca2+]i and contractile function in freshly isolated CM's. Moreover, a dose-dependent acceleration in the timing parameters associated with the rise and fall of the Ca2+ transient as well as shortening and relengthening of the CM were also observed. These effects were not observed in the presence of the TRPA1 antagonist, HC-030031 nor in CMs obtained from TRPA1−/− mice indicating the effects of AITC on [Ca2+]i and contractile function were TRPA1 dependent. Although the effects of TRPA1 stimulation on [Ca2+]i and contractile function were qualitatively and quantitatively similar to the classical CM responses observed following β-AR stimulation with ISO, we did not observe any increase in CM cAMP production or PKA activity following TRPA1 activation with AITC, and the PKA inhibitor peptide was without effect on AITC induced modifications in CM [Ca2+]i and contractile function. However, we did observe an increase in CaMKII phosphorylation following TRPA1 stimulation and inhibition of CaMKII with KN-93 completely reversed the effect of TRPA1 stimulation with AITC on CM [Ca2+]i and contractile indices. Therefore, TRPA1 activation increases CM contractile function via a CaMKII-dependent pathway.
AITC stimulates TRPA1-dependent increases in [Ca2+]i and contractile function in CMs
Modulation of CM [Ca2+]i throughout the excitation contraction coupling process, as well as the timing parameters associated with intracellular Ca2+ handling is a critical determinant of the inotropic and lusitropic state of the heart under physiologic and/or pathophysiological conditions. The discovery of novel ligand gated ion channels capable of activating signaling pathways which modulate Ca2+ regulatory pathways and/or myofilament Ca2+ sensitivity may pave the way for the design of novel therapies capable of increasing the inotropic and or lusitropic state of the heart. Our findings indicate that TRPA1 stimulation results in the activation of a signaling pathway(s) that modulate cellular mechanisms leading to (1) an increase in the amount of cytosolic Ca2+ available to interact with the cardiac myofilaments; (2) an acceleration in the time to peak [Ca2+]i and (3) an acceleration in the removal of Ca2+ from the cytosol. These changes in intracellular Ca2+ availability and handling are also reflected as changes in CM contractile function including (1) an increase in fractional shortening; (2) an increase in maximum velocity of shortening and (3) an increase in maximum velocity of relengthening. Our current findings are consistent with a recent study indicating a role for other TRP channel family members (TRPV2) in the regulation and/or modulation of inotropic and lusitropic events in cardiac muscle.11
Cellular mechanisms of TRPA1-induced increases in CM [Ca2+]i and contractile function
Based on the current findings, we initially hypothesized that stimulation of TRPA1 channels may be coupled to the protein kinase A (PKA) signaling pathway in CMs. Our hypothesis is based on the fact that the current study demonstrates remarkable qualitative and quantitative similarities when comparing the effects of TRPA1 stimulation on CM [Ca2+]i and contractile function to those effects observed in response to β-AR stimulation.12,13 The β-AR signaling pathway has been well established in heart for decades and is known to be coupled to the cAMP/PKA signaling pathway resulting in phosphorylation and activation of sarcolemmal Ca2+ channels14,15 and the sarcoplasmic reticulum (SR) pump regulatory protein, phospholamban.16-18 Moreover, it is also well established that β-AR stimulation of intact hearts or isolated CMs results in phosphorylation of troponin I (TnI) on the cardiac myofilaments.18-20 The current studies indicate that TRPA1 stimulation had no effect on cAMP production at concentrations that markedly increased CM contractile function (1–100 µmol/L) or PKA activity (100 µmol/L) when compared with β-AR activation with ISO. Furthermore, electrically-paced CMs treated with PKI 14–22 following AITC yielded no quantifiable alterations in [Ca2+]i or contractile function. This data suggests that TRPA1-induced increases in [Ca2+]i and contractile function occur via a mechanism independent of cAMP and PKA.
Based upon several recent studies indicating the notable potential of CaMKII in the regulation and/or modulation of cardiac contractility,21-23 we aimed to investigate whether TRPA1 stimulation elicits increases in [Ca2+]i and contractile function through a CaMKII-mediated process. Initially, we examined the extent to which AITC elicited phosphorylation of CaMKII at threonine 286 (pCaMKII-t286), a known indicator of CaMKII activity.24 The current data suggests TRPA1 stimulation with AITC elicits an increase in pCaMKII-t286 within 1 minute following treatment and remains elevated thereafter (through 5 min). To our knowledge, this is the first data to elucidate a potential downstream signaling mediator of TRPA1 activation in CMs that is capable of modulating Ca2+ regulatory proteins such as the L-type Ca2+ channels,25 ryanodine receptors26 or phospholamban,27 as well as myofilament Ca2+ responsiveness mediated by TnI.28 We subsequently investigated the extent to which TRPA1-induced increases in [Ca2+]i and contractile function occur through a CaMKII-dependent mechanism in electrically-paced CMs. Our findings indicate that KN-93 dose-dependently reversed the TRPA1-induced increases in [Ca2+]i and contractile function. Similarly, a structurally and mechanistically different CamKII inhibitor (AIP) also reversed the TRPA1-induced increases in [Ca2+]i and contractile function further supporting the findings obtained using the KN compounds and confirming a role for CaMKII as a mediator of the TRPA1 stimulated effects on CM contractile function.
A theory proposed by Maier and Bers suggests that calmodulin activation may occur in a cyclical manner whereby incoming Ca2+ binds to and activates calmodulin during contraction and dissociates during relaxation.23 Therefore, one could base their hypothesis on the ‘Michaelis-Menten’ theory whereby stimulation of ion channels leading to Ca2+ influx would increase the ratio of active to inactive calmodulin by making more Ca2+ available to bind calmodulin and subsequently activate the downstream CaMKII. If true, this could partially explain why activation of TRPA1, a Ca2+-permeable ion channel, elicits increases in beat-to-beat [Ca2+]i and the corresponding contractile function. However, the current results are complicated by the pharmacological nature of ‘KN’ compounds. ‘KN’ compounds are allosteric inhibitors of CaMKII where they competitively bind the calmodulin binding site.29 KN-93, as well as the structurally-similar predecessor KN-62, are unable to inhibit the kinase when it is autonomously activated. The current results indicate that KN-93 treatment following AITC administration reverses the increases observed in [Ca2+]i and contractile function by TRPA1 stimulation. Therefore, we propose that CaMKII activation in CMs may not exhibit the autonomous phenomena observed in different cell types (including those in the nervous system30,31) but rather, that calmodulin and CaMKII activity is cyclical in nature, allowing the KN-93 compounds to take effect after Ca2+ dissociates from calmodulin rendering CaMKII inactive between each cardiac cycle. This would further support the previously noted theory proposed by Maier and Bers,23 as well as the findings of the current study.
As noted above, experiments in quiescent CMs revealed that KN-93 markedly attenuated AITC-induced pCaMKII-t286, and reversed the concomitant AITC-induced increases in [Ca2+]i and contractile function in paced CMs. We propose that pretreatment of CMs with CaMKII inhibitors (before AITC administration) essentially “lock” the kinase in its inactive conformation rendering CaMKII unable to expose its calmodulin- or ATP-binding sites and thereby limiting its ability to autophosphorylate at the threonine 286 residue. The current data indicate that CaMKII inhibition reverses TRPA1-induced increases in [Ca2+]i and contractile function.
The current conclusions rely on several assumptions and need to be addressed as such. First, as do most pharmacological activators and inhibitors, KN compounds have certain limitations. KN-93 and KN-62 have a documented propensity to modulate off-target mediators including ion channels such as L-type Ca2+ channels.32,33 However, control experiments using the inactive analog, KN-92, yielded no statistically significant alterations in TRPA1-induced increases in [Ca2+]i and contractile function suggesting that KN-93 is working primarily through CaMKII inhibition. Moreover, the use of a structurally and mechanistically different CaMKII inhibitor (AIP) supported the findings obtained with the active KN compound. We also noted on some occasions that, CMs pretreated with KN-93 (10 µmol/L) resulted in subtle and inconsistent declines in steady-state [Ca2+]i and contractile function in a manner similar to what was observed in intact ferret CMs34; however, this effect only took place in ∼33% of the CMs exposed to KN-93. The inconsistences may be related to anatomic location from which the CMs were derived as well as species variability.34
We propose that TRPA1 stimulation likely involves an intracellular signaling pathway(s) that trigger post-translational modifications (phosphorylation) of the L-type Ca2+ channel as well as phospholamban resulting increases in transsarcolemmal Ca2+ influx SR uptake and Ca2+ loading. Together these would account for, at least in part, (1) an increase in the amount of Ca2+ available to interact with the cardiac myofilaments and (2) the accelerated rate of decay of the Ca2+ transient due to the removal of inhibition of the SR Ca2+ pump by phosphorylated phospholamban. Parallel changes in contractility could also be explained by the aforementioned mechanisms and include an increase in (1) fractional shortening, (2) maximum velocity of shortening and (3) maximum velocity of relaxation. Alternatively, there could also be a role for TRPA1-dependent phosphorylation of TnI in mediating the observed changes in both contraction and relaxation that are independent of changes in Ca2+ handling.12 We observed a TRPA1-induced increase in maximum unloaded shortening velocity which could be explained by TnI phosphorylation. Moreover, an increased shortening velocity could contribute to the increased fractional shortening and a positive inotropic effect since, in theory, the power output of muscle is determined by the product force of velocity.35,36 Finally, TnI phosphorylation is also known to reduce myofilament Ca2+ sensitivity and contribute to an accelerated rate of relaxation also contributing to a positive lusitropic effect.12 Further studies are required to clarify the precise cellular and molecular mechanisms by which TRPA1 stimulation augments CM contractile function.
Summary and conclusions
TRPA1 stimulation of electrically-stimulated mouse CMs results in increases in [Ca2+]i and contractile function indicative of positive inotropic and positive lusitropic effects observed in cardiac muscle and intact hearts. The findings mirror those observed with β-AR stimulation of the cAMP/PKA signaling pathway although TRPA1-induced alterations of [Ca2+]i and contractile function occur via a CaMKII-dependent mechanism. Further studies will be required to identify the precise downstream effectors by which TRPA1 stimulation results in increases in CM [Ca2+]i and contractile function and whether these findings may be extrapolated to intact cardiac muscle and/or the human heart.
Supplementary Material
Abbreviations
- AITC
allyl isothiocyanate
- CaMKII
Ca2+/calmodulin dependent kinase II
- cAMP
cyclic AMP.
- CM
adult mouse ventricular cardiomyocyte
- [Ca2+]i
intracellular free calcium concentration
- PKA
protein kinase A
- TRPA1
transient receptor potential ankyrin channel subtype-1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Horiba Scientific for supplying our laboratory with the Felix GX software with accompanying sarcomere length module and Dr. Srinivasan Vijayaraghavan for his collaborations with our PKA activity assay.
Funding
This work was supported by a grant from NIH HL65701 to D.D.
Author contributions
SA, IB and DD participated in research design and analysis. SA, PS, MG and SD conducted the experiments. SA and DD wrote or contributed to the writing of the manuscript.
References
- [1].Fernandes ES, Fernandes MA, Keeble JE. The functions of TRPA1 and TRPV1: moving away from sensory nerves. Br J Pharmacol. 2011;166:510-21. doi: 10.1111/j.1476-5381.2012.01851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jaquemar D, Schenker T, Trueb B. An ankyrin-like protein with transmembrane domains is specifically lost after oncogenic transformation of human fibroblasts. J Biol Chem. 1999;274:7325-33. doi: 10.1074/jbc.274.11.7325. PMID:10066796 [DOI] [PubMed] [Google Scholar]
- [3].Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, et al.. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperature. Cell. 2003;112:819-29. doi: 10.1016/S0092-8674(03)00158-2. PMID:12654248 [DOI] [PubMed] [Google Scholar]
- [4].Nagata K, Duggan A, Kumar G, Garcia-Anoveros J. Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci. 2005;25:4052-61. doi: 10.1523/JNEUROSCI.0013-05.2005. PMID:15843607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yue Z, Zie J, Yu AS, Stock J, Du J, Yue L. Role of TRP channels in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2015;308(3):H157−82. doi: 10.1152/ajpheart.00457.2014. PMID:25416190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vennekens R. Emerging concepts for the role of TRP channels in the cardiovascular system. J Physiol. 2011;589:1527-34. doi: 10.1113/jphysiol.2010.202077. PMID:21173080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nilius B. TRP channels in disease. Biochim Biophys Acta. 2007;1772:805-12. doi: 10.1016/j.bbadis.2007.02.002. PMID:17368864 [DOI] [PubMed] [Google Scholar]
- [8].Andrei SR, Sinharoy P, Bratz IN, Damron DS. TRPA1 is functionally co-expressed with TRPV1 in cardiac muscle: Co-localization at z-discs, costameres and intercalated discs. Channels. 2016; 10(5): 395-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kurokawa H, Murray PA, Damron DS. Propofol attenuates b-adrenoreceptor-mediated signal transduction via a protein kinase C-dependent pathway in cardiomyocytes. Anesthesiology. 2002;96:688-98. doi: 10.1097/00000542-200203000-00027. PMID:11873046 [DOI] [PubMed] [Google Scholar]
- [10].Vijayaraghavan S, Goueli SA, Davey MP, Carr DW. Protein kinase A-anchoring inhibitor peptides arrest mammalian sperm motility. J Biol Chem. 1997;272:4747-52. doi: 10.1074/jbc.272.8.4747. PMID:9030527 [DOI] [PubMed] [Google Scholar]
- [11].Rubinstein J, Lasko VM, Koch SE, Singh VP, Carreira V, Robbins N, Patel AR, Jiang M, Bidwell P, Kranias EG, et al.. Novel role of transient receptor potential vanilloid 2 in the regulation of cardiac performance. Am J Physiol Heart Circ Physiol. 2014;306:H574−84. doi: 10.1152/ajpheart.00854.2013. PMID:24322617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Solaro RJ. Modulation of Cardiac Myofilament Activity by Protein Phosphorylation: Comprehensive Physiology; The Cardiovascular System, The Heart. 2002. Online Supplement 6, 2011. Jan. doi: 10.1002/cphy.cp020107 [DOI] [Google Scholar]
- [13].Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht:Kluwer Academic Publishers; 2001. [Google Scholar]
- [14].Hess P, Lansman JB, Nilius B, Tsien RW. Calcium channel types in cardiac myocytes: modulation by dihydropyridines and beta-adrenergic stimulation. J Cardiovasc Pharmacol. 1986;8 Suppl 9:S11−21. doi: 10.1097/00005344-198611001-00002. PMID:2433538 [DOI] [PubMed] [Google Scholar]
- [15].Catterall WA. Molecular properties of voltage-sensitive sodium and calcium channels. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas. 1988;21:1129-44. PMID:2855029 [PubMed] [Google Scholar]
- [16].Katz AM, Tada M, Kirchberger MA. Control of calcium transport in the myocardium by the cyclic AMP-Protein kinase system. Advances in cyclic nucleotide research. 1975;5:453-72. PMID:165680 [PubMed] [Google Scholar]
- [17].Tada M, Inui M. Regulation of calcium transport by the ATPase-phospholamban system. J Mol Cell Cardiol. 1983;15:565-75. doi: 10.1016/0022-2828(83)90267-5. PMID:6313949 [DOI] [PubMed] [Google Scholar]
- [18].Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in beta-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol Heart Circ Physiol. 2000;278:H769−79. PMID:10710345 [DOI] [PubMed] [Google Scholar]
- [19].Onorato JJ, Rudolph SA. Regulation of protein phosphorylation by inotropic agents in isolated rat myocardial cells. J Biol Chem. 1981;256:10697-703. PMID:6169722 [PubMed] [Google Scholar]
- [20].Kranias EG, Garvey JL, Srivastava RD, Solaro RJ. Phosphorylation and functional modifications of sarcoplasmic reticulum and myofibrils in isolated rabbit hearts stimulated with isoprenaline. Biochem J. 1985;226:113-21. doi: 10.1042/bj2260113. PMID:3156585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Erickson JR, Patel R, Ferguson A, Bossuyt J, Bers DM. Fluorescence resonance energy transfer-based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ Res. 2011;109:729-38. doi: 10.1161/CIRCRESAHA.111.247148. PMID:21835909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang W, Zhu W, Wang S, Yang D, Crow MT, Xiao RP, Cheng H. Sustained beta1-adrenergic stimulation modulates cardiac contractility by Ca2+/calmodulin kinase signaling pathway. Circ Res. 2004;95:798-806. doi: 10.1161/01.RES.0000145361.50017.aa. PMID:15375008 [DOI] [PubMed] [Google Scholar]
- [23].Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34:919-39. doi: 10.1006/jmcc.2002.2038. PMID:12234763 [DOI] [PubMed] [Google Scholar]
- [24].Erickson JR. Mechanisms of CaMKII Activation in the Heart. Frontiers in pharmacology. 2014;5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kamp TJ, Hell JW. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ Res. 2000;87:1095-102. doi: 10.1161/01.RES.87.12.1095. PMID:11110765 [DOI] [PubMed] [Google Scholar]
- [26].Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80-5. doi: 10.1016/j.bbrc.2008.08.084. PMID:18755143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mattiazzi A, Mundina-Weilenmann C, Guoxiang C, Vittone L, Kranias E. Role of phospholamban phosphorylation on Thr17 in cardiac physiological and pathological conditions. Cardiovasc Res. 2005;68:366-75. doi: 10.1016/j.cardiores.2005.08.010. PMID:16226237 [DOI] [PubMed] [Google Scholar]
- [28].Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12-21. doi: 10.1016/j.cardiores.2004.12.022. PMID:15769444 [DOI] [PubMed] [Google Scholar]
- [29].Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Frontiers in pharmacology. 2014;5:21. doi: 10.3389/fphar.2014.00021. PMID:24600394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J Neurochem. 1997;68:2119-28. doi: 10.1046/j.1471-4159.1997.68052119.x. PMID:9109540 [DOI] [PubMed] [Google Scholar]
- [31].Coultrap SJ, Buard I, Kulbe JR, Dell'Acqua ML, Bayer KU. CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J Biol Chem. 2010;285:17930-7. doi: 10.1074/jbc.M109.069351. PMID:20353941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li G, Hidaka H, Wollheim CB. Inhibition of voltage-gated Ca2+ channels and insulin secretion in HIT cells by the Ca2+/calmodulin-dependent protein kinase II inhibitor KN-62: comparison with antagonists of calmodulin and L-type Ca2+ channels. Mol Pharmacol. 1992;42:489-8. PMID:1328847 [PubMed] [Google Scholar]
- [33].Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180-7. doi: 10.1097/FJC.0b013e3181a25078. PMID:19333131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Li L, Satoh H, Ginsburg KS, Bers DM. The effect of Ca(2+)-calmodulin-dependent protein kinase II on cardiac excitation-contraction coupling in ferret ventricular myocytes. J Physiol. 1997;501 (Pt 1):17-31. doi: 10.1111/j.1469-7793.1997.017bo.x. PMID:9174990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Layland J, Grieve DJ, Cave AC, Sparks E, Solaro RJ, Shah AM. Essential role of troponin I in the positive inotropic response to isoprenaline in mouse hearts contracting auxotonically. J Physiol. 2004;556:835-47. doi: 10.1113/jphysiol.2004.061176. PMID:14966306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Herron TJ, Korte FS, McDonald KS. Power output is increased after phosphorylation of myofibrillar proteins in rat skinned cardiac myocytes. Circ Res. 2001;89:1184-90. doi: 10.1161/hh2401.101908. PMID:11739284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.