

ABSTRACT
Cerebral ischemia-reperfusion injury triggers a deleterious process ending in neuronal death. This process has two components, a glutamate-dependent and a glutamate-independent mechanism. In the glutamate-independent mechanism, neurons undergo a slow depolarization eventually leading to neuronal death. However, little is known about the molecules that take part in this process. Here we show by using mice cortical neurons in culture and ischemia-reperfusion protocols that TRPM4 is fundamental for the glutamate-independent neuronal damage. Thus, by blocking excitotoxicity, we reveal a slow activating, glibenclamide- and 9-phenanthrol-sensitive current, which is activated within 5 min upon ischemia-reperfusion onset. TRPM4 shRNA-based silenced neurons show a reduced ischemia-reperfusion induced current and depolarization. Neurons were protected from neuronal death up to 3 hours after the ischemia-reperfusion challenge. The activation of TRPM4 during ischemia-reperfusion injury involves the increase in both, intracellular calcium and H2O2, which may act together to produce a sustained activation of the channel.
KEYWORDS: glutamate-independent neuronal death, ischemia-reperfusion, oxidative stress 9-phenanthrol, TRPM4
Introduction
Neuronal ischemia activates a mechanism of damage called excitotoxicity which consists in an uncontrolled glutamate release from synaptic terminals activating glutamatergic receptors and thus, initiating a persistent depolarization and tissue damage.1,2 This mechanism is self-limited and finishes after synaptic vesicles depletion.3 Despite the self-limited feature of excitotoxicity, it has been noted that neurons remain depolarized in a not well understood mechanism in which participates, among others, a Na+ inward current (NSCCa-ATP) that is activated by high [Ca2+]i and low [ATP]i4-7 and modulated by ROS.8 This mechanism is believed to be critical for the delayed neuronal death that occurs hours or even days after the initial injury.9
The interplay between high Ca2+i and high production of ROS are side effects of ischemia-reperfusion injury. Conversely, Ca2+i contributes to altered protein function such as increased calpain activity,10 increased ion permeability,11 activation of Ca2+-dependent phospholipases, and activation of ROS-generating pathways like NADPH oxidases (NOX)12 and xanthine oxidase conversion.13 Oxidants contribute to protein impairment, DNA breakdown, mitochondrial damage, increased membrane permeability and neuronal depolarization,1,14 altogether, perpetuating the damage and accounting for neuronal death.
TRPM4 is a non-selective cation channel permeable to monovalent cations and activated by intracellular Ca2+,15 blocked by ATP16 and modulated by ROS.8 Physiologically, TRPM4 is involved in the control of Ca2+i oscillations through the modulation of resting membrane potential,17,18 cell migration,19,20 rhythm generation in preBötzinger neurons,21 the afterdepolarization in cerebellar neurons,22 among other functions (for a review, see23). Interestingly, increased Ca2+i and ROS production during reperfusion favor the activation of TRPM4.16,24,25 Moreover, this channel has been previously involved in glutamate-dependent axonal degeneration by inducing large inward currents after pretreatment with glutamate.26 However, its role in the sustained depolarization during reperfusion is not well characterized. In this work by using chemical as well as oxygen and glucose deprivation ischemia-reperfusion models, we show that the continuous activation of TRPM4 during reperfusion leads neurons to a state of sustained depolarization leading to their death. The pharmacological inhibition or shRNA-based silencing of TRPM4 renders neurons resistant to reperfusion damage, increasing their survival rate. Thus, blockade of the sustained activation of TRPM4 might constitute a target for novel pharmacological tools addressed to reduce the damage during brain reperfusion.
Results
Persistent neuronal depolarization during ischemia-reperfusion
To figure out the potential role of TRPM4 in the glutamate-independent neuronal damage, we measured first the membrane potential of mouse cortical pyramidal neurons from primary cultures using the nystatin-perforated patch clamp technique. Neurons were exposed to 5 mM NaN3 in the absence (Fig. 1a) or presence (Fig. 1b) of an AET buffer to block excitotoxicity. We found that neurons depolarize after NaN3 exposure despite of AET presence, reaching a steady-state at 5 min (0 min = −69.5 ± 8.9 mV, 15 min = −9.2 ± 11.2 mV, n = 6, t-test, p < 0.0002). In the absence of AET, neurons undergo a continuous firing and depolarization (0 min = −72.2 ± 6.5 mV, 15 min −8.8 ± 8.2 mV, n = 6, t-test, p < 0.0001). In voltage-clamp experiments (Vh = −70 mV) we observed the development of a slow current that reached steady-state after 15 min (−359 ± 182.2 pA) upon NaN3 exposure in the absence (Fig. 1c) or presence (Fig. 1d) of the AET buffer. The current development correlated with the depolarization observed under current-clamp conditions (3.4 ± 1.2 min vs. 3.1 ± 1.6 min, respectively). During voltage-clamp experiments in the absence of the AET buffer, we observed an increased excitatory postsynaptic current (EPSC) frequency followed by a slow inward current (Vh = −70 mV, 0 min = −41.2 ± 19.8 pA, 15 min = −331.2 ± 143.8 pA, n = 6, t-test, p < 0.005). The firing rate and EPSC frequency after 10 min of NaN3 perfusion were completely suppressed at 20 min. These data suggest that excitotoxicity is a self-limited event and other conductances could be underlying the NaN3-induced depolarization.
Figure 1.

Excitotoxicity-independent neuronal depolarization after chemical ischemia-reperfusion. (a) Left panel shows a representative membrane potential recording in cortical pyramidal neurons (DIV 14–21) treated with 5 mM NaN3. Right panel depicts the membrane potential values at 0 and 15 min post NaN3 for each experiment (n = 6). (b) Left panel shows a representative membrane potential recording in cortical pyramidal neurons (DIV 14–21) treated with 5 mM NaN3 and AET. Right panel depicts the membrane potential values at 0 and 15 min post NaN3 and AET for each experiment (n = 6). (c) Left panel shows a representative current recording showing the effect of 5 mM NaN3. Right panel depicts the current values at 0 and 15 min post NaN3 for each experiment (n = 6). (d) Left panel shows a representative current recording showing the effect of 5 mM NaN3 and AET. Right panel depicts the current values at 0 and 15 min post NaN3 and AET for each experiment (n = 6). Black arrows indicate t = 0 and t = 15, respectively.
TRPM4 expression in cortical pyramidal neurons
To explore whether TRPM4 is expressed in cortical pyramidal neurons, we performed imaging studies using 5 weeks-old C57BL/6 mice brain slices. First, we evaluated the efficacy of the TRPM4shRNA. To that end, cortical pyramidal neuron cultures were transfected with a shRNA against TRPM4 (TRPM4shRNA) expressing EGFP as an expression marker. As depicted in Fig. 2a-d, neurons expressing the TRPM4shRNA have a ∼ 55% reduction in TRPM4 expression. Next, we observed the expression of TRPM4 in slice of medial prefrontal cortex. As shown, we observed TRPM4 labeling in several neurons across the cortical region (Fig. 2e, h). A zoomed area showed that most of the labeling is somatic with few neurons presenting labeling in neurites (Fig. 2h). MAP2 labeling showed the expected somatodendritic pattern (Fig. 2f, i). Superposition of images showed several overlapping sites (Fig. 2g, j) suggesting the presence of TRPM4 in the neurons.
Figure 2.
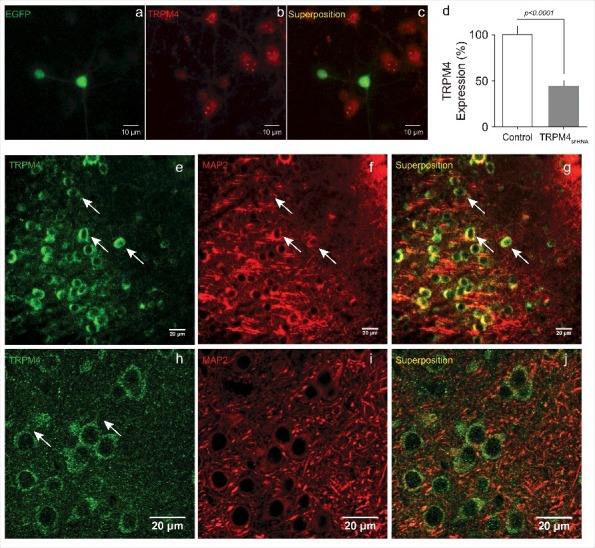
TRPM4 expression in cortical pyramidal neurons. (a) illustrates a representative experiment showing two neurons transfected with the TRPM4shRNA positive for EGFP expression. (b) shows the immunofluorescence for TRPM4. (c) shows superposition of images. (d) depicts the percentage of TRPM4 reduction of expression (55.3 ± 5.5%; n = 4 independent experiments, 55 cells in total). Representative images of the cerebral cortex showing the somatic immunoreactivity for TRPM4 (e), MAP2 (f) and the superposed signal (g) at 40x magnification and (h), (i), (j) at 63x magnification. Arrows indicate somata and neurite labeling for TRPM4.
TRPM4 involvement in the reperfusion current
Previous studies indicate the involvement of TRPM4 in glutamate-induced neuronal death and axonal degeneration,26,27 however, its role in glutamate-independent neuronal death is still unclear. To assess whether TRPM4 participates in the depolarization observed in this process, we used an shRNA-based silencing strategy. In these experiments, we found that cortical neurons transduced with TRPM4shRNA exhibit a small depolarization after NaN3 + AET exposure (0 min = −76.4 ± 5.9 mV, 15 min = −55.8 ± 21 mV, n = 5, t-test, p = 0.0638, Fig. 3a), as compared to TRPM4scrambled-transduced neurons (0 min = −72.6 ± 3.14 mV, 15 min = −18.6 ± 5.6 mV, n = 5, Fig. 3b). In voltage-clamp experiments, TRPM4shRNA-transduced neurons expressed a small inward current (Vh = −70 mV, 0 min = −44.5 ± 26.9 pA, 15 min = −190.2 ± 52.6 pA, n = 5, t-test, p < 0.0017, Fig. 3c), as compared to TRPM4scrambled-transduced neurons (0 min = −76.7 ± 11 pA, 15 min = −497.5 ± 40.8 pA, n = 6, Fig. 3d).
Figure 3.

TRPM4shRNA and pharmacological blockade protects neurons from ischemia and reperfusion-induced depolarization. (a) and (b) Representative membrane potential recordings in cortical pyramidal neurons (DIV 14–21) treated with 5 mM NaN3 and AET expressing TRPM4shRNA or TRPM4scramble. Right panels depict the membrane potential values at 0 and 15 min post NaN3 and AET for each experiment (n = 5). (c) and (d) Representative current recordings showing the effect of 5 mM NaN3, and AET expressing TRPM4shRNA (n = 5) or TRPM4scramble. Right panels depict the current values at 0 and 15 min post NaN3 and AET for each experiment (n = 6). Black arrows indicate t = 0 and t = 15, respectively. (e) Representative current recordings in cortical pyramidal neurons treated with NaN3 and AET (blue trace), NMDG+ replacement (red trace) and NMDG+ in the presence of 10 µM 9 Ph (green trace). (f) Representative current recordings of neurons expressing TRPM4shRNA (black trace) or TRPM4scramble (blue trace) exposed to NaN3 and AET. (g) Representative current recordings of neurons exposed to NaN3 + AET; control trace (black trace), TRPM4shRNA (grey trace) and TRPM4shRNA + 9 Ph (red trace). (h) Graph depicting the current reduction with TRPM4shRNA and TRPM4shRNA + 9 Ph (n = 4 independent experiments, mean ± S.D., one-way ANOVA).
To explore the ionic nature of this current, we performed experiments replacing extracellular Na+ with the non-permeable cation NMDG+ after 10 min of perfusion with 5 mM NaN3 + AET and measured the current using a voltage ramp protocol (−80 to 80 mV, dV/dt 0.4 V/s). We found that replacing Na+ with NMDG+ decreases the inward current in 92 ± 10.3% (Fig. 3e, red trace) compared to control (blue trace), showing that Na+ is the main inward cation carried by this current. The exposure of neurons to 10 µM 9-phenantrol (9Ph, a non-specific TRPM4 inhibitor) in the presence of NaN3 + AET and NMDG+ as the main cation reduces the outward current (Fig. 3e, green trace), as expected for a cation-selective current carried by TRPM4. To confirm whether this current is driven by TRPM4, we performed experiments in neurons transduced with TRPM4shRNA (black trace in Fig. 3f) and TRPM4scramble (blue trace in Fig. 3f). We found that in neurons subjected to TRPM4 silencing the current induced NaN3 + AET exposure was significantly reduced. To explore whether TRPM4 silencing and 9 Ph present an additive effect, we performed experiments in neurons transduced with TRPM4shRNA (grey trace, Fig. 3g) and in neurons transduced with TRPM4shRNA + 9 Ph (red trace, Fig. 3g), both compared to control (black trace, Fig. 3g). Fig. 3h shows a summary graph of the current recorded under these conditions. As seen, there is no additive effect.
Increased H2O2 and Ca2+ levels during ischemia-reperfusion
In a previous work, we demonstrated that TRPM4 desensitization is removed by H2O2, leading to cell death.8 Because an important feature of the ischemia-reperfusion injury is ROS generation, we monitored chemically-induced ischemia-reperfusion H2O2 increase. Using the genetically encoded H2O2 probe HyPer-Cyto,28 we found an increase of H2O2 levels at 3 min reaching steady-state at 7 min post NaN3 (Fig. 4a, black circles). Preincubation for 1 h with 1 mM N-acetyl cysteine (NAC) abolished the H2O2 increase induced by NaN3 + AET (Fig. 4a, blue circles). As a control, to rule out an effect of the TRPM4 inhibitor 9 Ph on the H2O2 increase neurons were exposed to 10 µM 9 Ph (Fig. 4a, red circles).
Figure 4.

Reperfusion increases ROS and [Ca2+]i in cortical pyramidal neurons. (a) H2O2 quantification using HyPer-Cyto in cortical pyramidal neurons treated with NaN3+AET (black circles, n = 7), NaN3+AET+9 Ph (red circles, n = 7) and NaN3+AET+1 mM NAC (blue circles, n = 7). (b) Intracellular Ca2+ measurements in cortical pyramidal neurons treated with 5 mM NaN3+AET (n = 8). (c) Effect on intracellular Ca2+ of NaN3+AET+10 µM 9 Ph (red circles, n = 8) and NaN3+AET +1 mM NAC (blue circles, n = 8). Data are expressed as mean ± SEM.
Because Ca2+i increase is one of the landmarks of ischemia-reperfusion injury and is crucial for TRPM4 activation, we explored whether NAC affected [Ca2+]i during ischemia-reperfusion. As expected, NaN3 + AET increases Ca2+ (Fig. 4b). This increase was partially abolished by 5 min preincubation with 1 mM NAC (Fig. 4c, blue circles). TRPM4 inhibition by 10 µM 9 Ph did not affect Ca2+i levels (Fig. 4c, red circles) showing that, at least in the experimental protocol used for chemical ischemia-reperfusion, Ca2+i increase is partially dependent on ROS increase.
TRPM4 activation depends on ROS and a Ca2+ influx
One prediction from the above described results is that suppression of Ca2+i and/or ROS levels should hamper the depolarization and TRPM4 current activation induced by ischemia-reperfusion injury. We tested this prediction first by performing chemically induced ischemia-reperfusion experiments in the absence of extracellular Ca2+. We found that extracellular Ca2+ exclusion protected neurons from depolarization (0 min = −71.2 ± 9.9 mV, 15 min = −68 ± 9.6 mV, n = 5 t-test, p = 0.5, Fig. 5a), and current activation (0 min = −24.6 ± 12.1 pA, 15 min = −56.5 ± 24.85 pA, n = 5, t-test, p = 0.96, Fig. 5c). Then, we evaluated the effect of NAC, a small depolarization was recorded (0 min = −75 ± 5.7 mV, 15 min = −48.4 ± 8.5 mV, n = 5, t-test, p = 0.0117, Fig. 5b), which is related to a small current activation (0 min = −40.8 ± 26.6 pA, 15 min = −83.3 ± 22 pA, n = 6, t-test, p = 0.1205, Fig. 5d).
Figure 5.

Ca2+ and reducing agents modulate ischemia and reperfusion-induced depolarization. (a) Representative membrane potential recording in cortical pyramidal neurons (DIV 14–21) treated with 5 mM NaN3 +AET in the absence of extracellular Ca2+. Right panel shows the membrane potential values at 0 and 15 min post NaN3+AET for each experiment (n = 5). (b) Representative membrane potential recording in cortical pyramidal neurons (DIV 14–21) treated with 5 mM NaN3+AET and 1 mM NAC. Right panel shows the membrane potential values at 0 and 15 min post NaN3+AET and 1 mm NAC for each experiment (n = 5). (c) Representative current recording showing the effect of 5 mM NaN3+AET in the absence of extracellular Ca2+. Right panel shows the current values at 0 and 15 min post NaN3+AET for each experiment (n = 5). (d) Representative current trace showing the effect of 5 mM NaN3+AET and 1 mM NAC. Right panel shows the current values at 0 and 15 min post NaN3+AET and 1 mm NAC for each experiment (n = 6). Black arrows indicate t = 0 and t = 15, respectively. (e) Current time course measured at 100 mV from a voltage ramp as described in Methods in cortical pyramidal neurons exposed to 30 min OGD in the presence of AET (OGD+AET) and during reperfusion (Reperfusion+AET). (f) Mean current values during Reperfusion+AET and upon exposure to 100 µM glibenclamide (Glib+AET, n = 5).
To verify our observations with a different protocol of ischemia-reperfusion injury we performed an oxygen-glucose deprivation-reperfusion protocol (OGD). As depicted in Fig. 5e, during 30 min of exposure to an OGD protocol we observed no current activation, however, 10 min after switching to normal ACSF we observed a current increase which was inhibited by 100 µM glibenclamide, a known TRPM4 inhibitor (Fig. 5f).
TRPM4 inhibition increases neuronal survival
We sought to explore whether TRPM4 inhibition increases neuronal survival after ischemia-reperfusion injury. As depicted in Fig. 6a neuronal death measured with the trypan-blue exclusion method was reduced in neurons exposed to NaN3 + AET plus 9 Ph and in neurons transduced with TRPM4shRNA (63 ± 12%, n = 6, one-way ANOVA, p < 0.0055).
Figure 6.

TRPM4 inhibition protects from the reperfusion-induced neuronal death. (a) Quantification of neuronal death after 2 h treatment with 5 mM NaN3 under 10 µM 9Ph, TRPM4shRNA and TRPM4scramble (n = 6). (b) LDH release time course up to 24 h treatment with 5 mM NaN3 under 9Ph, TRPM4shRNA and TRPM4scramble (n = 6). *, control vs 9Ph, 1 h, p < 0.05; &&, control vs TRPM4shRNA, 1 h, p < 0.001; ***, control vs 9Ph, 2 h, p < 0.0001; &&&, control vs TRPM4shRNA, 2 h, p < 0.0001; *, control vs 9Ph, 3 h, p < 0.05; &&&, control vs TRPM4shRNA, 3 h, p < 0.0001; &, control vs TRPM4shRNA, 24 h, p < 0.05.
To follow the temporal course of cell death, cortical pyramidal neurons transduced with TRPM4shRNA or TRPM4scramble were exposed to NaN3 + AET for 0, 15, 30, 60, 120, 180 min and 24 h, and then tested for cell death with the LDH release method (Fig. 6b). The data show that neurons begin to die after 15 min. TRPM4 silencing or TRPM4 inhibition by 9 Ph decreases cell death up to three hours with no further protective effect after that time (Control = 77 ± 2, 9 Ph = 58.2 ± 10, TRPM4shRNA = 71.3 ± 4.5, TRPM4scramble = 78.3 ± 4, n = 6, Fig. 6b).
Discussion
Here, we report a glutamate-independent mechanism of neuronal reperfusion damage dependent on TRPM4 activation. First, we demonstrate that TRPM4 is expressed in cortical pyramidal neurons. We found that high levels of intracellular ROS and Ca2+ during ischemia-reperfusion act to produce a sustained activation of TRPM4, which depolarizes neurons eventually triggering cell death. Pharmacological inhibition with 9-phenanthrol and glibenclamide or shRNA-based silencing of TRPM4 protects neurons from depolarization and cell death up to 3 hours after reperfusion. Additionally, TRPM4shRNA and 9-phenanthrol did not show and additive effect on the currents. Furthermore, we found that neuronal protection induced by TRPM4 inhibition becomes evident once the glutamate-induced damage i.e. excitotoxicity is blocked, suggesting that TRPM4 participates in the glutamate-independent neuronal damage observed under ischemia-reperfusion injury.3,29
Several reports show that TRPM4 activation strictly depends on intracellular Ca2+ ions.15,30-32 Our Ca2+ imaging experiments showing that chemically-induced reperfusion increases intracellular Ca2+ with a similar time course as the development of the TRPM4-dependent inward current. Moreover, in the absence of extracellular Ca2+ ions neuronal depolarization as well as current activation was abolished.
We have previously described a sustained activation of TRPM4 in HeLa and HEK293 cells treated with H2O2 by a mechanism that involves the oxidation of the cysteine 1093, which removes channel desensitization, allowing TRPM4 to be permanently activated as long as intracellular Ca2+ remains elevated. Also, we demonstrated that cysteine 1093 plays a key role in the increased vulnerability to necrotic cell death.8 Here we expanded our previous study by measuring endogenous H2O2 production induced upon chemical ischemia and reperfusion in a neuronal model. In the presence of NaN3 + AET, H2O2 increases with a time course that parallels Ca2+i increase and current development that was partially blocked by NAC. Reperfusion induces mitochondrial and cellular depolarization which is tightly coupled to ROS production.13,24,33 Neurons accumulate high amounts of unpaired electrons, due to the opening of the mitochondrial transition pore.13,34 Upon reperfusion, these unpaired electrons combine with faster rate with O2 producing large amounts of O2−, which reacts with several proteins in the electron transport chain, impairing its activity and decreasing its capability to produce the ATP necessary to power the Na+/K+ ATPase and perpetuating neuronal depolarization.
During reperfusion, initial Na+ influx plays a key role in neuronal swelling and oncotic death. Several routes for Na+ influx have been demonstrated or suggested.6,35,36 Recently, Weilinger et al. showed a non-conducting function of NMDA receptor (NMDAR metabotropic) which activates Src and pannexin-1, which in turn increases Ca2+i producing an increase in the dendritic volume (blebbing) and dendritic destruction.37 Although a pannexin-1 effect on Na+ influx was not addressed in our experimental model, we observed that extracellular N-Methyl-D-glucamine (NMDG+), an organic pannexin-permeating cation,38 reduces almost completely the reperfusion-activated current, suggesting that pannexin-1 may not have a significant role, at least in our model, in the reperfusion-induced depolarization.
It has been also shown that extracellular acidification occurring during ischemia-reperfusion injury triggers ASIC-1 activation.39,40 We ruled out this possibility by the continuous perfusion (2-3 mL/min) with a buffer at constant pH. On the other hand, TRPM7 has been described as cation influx pathway during delayed neuronal death.3,41,42 To rule out TRPM7 contribution, our experimental protocol included Gd3+.3,43
TRPM4 activation is involved in hemorrhagic bleeding observed during ischemia.5,44 Additionally, several studies suggest that glibenclamide and 9 Ph protect animals from ischemia-reperfusion injury.26,45,46 Moreover, in multiple sclerosis, the activation of TRPM4 triggers neuronal degeneration.26 In parallel, the post-transcriptional silencing of TRPM4 protects cardiomyocytes from ischemia reperfusion injury,46-48 increasing its capability to recover.
A possible mechanism for TRPM4 activation under ischemia-reperfusion injury may comprise first an uncontrolled glutamate release and the activation of glutamatergic and calcium channels and thus, increasing Ca2+i and mitochondrial depolarization, which generates the accumulation of unpaired electrons. During reperfusion, O2 reacts with unpaired electrons at a faster rate than the electron transport capability producing substantial amounts of O2−, which impairs further ATP production and thus, perpetuating the damage. ROS produced in the mitochondria would remove TRPM4 desensitization and in combination with high Ca2+i would trigger a sustained activation of the channel, leading to a permanent depolarized state and neuronal death.
In this study, we show the involvement of TRPM4 in the sustained depolarization observed during reperfusion, and that its inhibition (pharmacological or post-transcriptional silencing) improves the survival of cortical pyramidal neurons in culture, suggesting that TRPM4 is the relevant monovalent cation-selective inward current activated during reperfusion. Therefore, blockade of this channel could increase the survival of neurons during ischemia-reperfusion injury and TRPM4 might constitute a therapeutic target under this condition.
Materials and methods
Cortical neuron culture
Dissociated cortical neuron cultures, glia free, were prepared from embryonic day 18 C57/BL6 mice of either sex as described previously.49 Cells were seeded (5 × 104 cells/mL) on 12 mm round coverslips coated with poly-L-Lysine (100 µg/mL) and cultured for 14–21 days in Neurobasal Medium-B27 (Invitrogen).
Neuronal transfection
Neurons were transfected in DIV0 after its dissociation. Briefly, neurons were incubated with 1 µg of DNA of a shRNA against TRPM4 (TRPM4shRNA) expressing EGFP as an expression marker in 2 µL Lipofectamine 2000 (Invitrogen) diluted in 500 µL Minimum Essential Media supplemented (MEM) (Sigma) with 10% horse serum for 5 h. Then, media was replaced with fresh Neurobasal-B27 media and incubated at 37°C in 5% CO2 until DIV14.
Cell immunofluorescence
Neurons grown in 12 mm coverslip were fixed in 4% w/v paraformaldehyde (10 min exposure) dissolved in 0.01 M PBS pH 7.4 and then washed 3 times in PBS. Cells were permeabilized with 0.01% Triton X-100 (Sigma) (10 min exposure), then blocked in normal goat serum 10% in PBS 1 h at RT (Sigma). Cells were incubated overnight at 4°C with the primary antibody (anti-TRPM4, Alomone, Catalogue Number ACC-044, RRDI AB_2040250) diluted in the same blocking solution. After incubation, cells were washed 3 times with PBS before incubation with the secondary antibody (Alexa Fluor 546, Thermofisher Catalogue Number A21123, RRDI AB_2535765) for 1.5 h at RT. After 3 five min washes with PBS, cells were mounted with Prolong Gold mounting media (Thermofisher). Fluorescence was detected in an IX70 DSU spinning disk microscope (Olympus) using 40 × 1.4 N.A. magnification objective.
Neuronal transduction
Neuronal cultures at 6 DIV (days in vitro) were transduced with engineered lentiviruses expressing a shRNA against TRPM4 (TRPM4shRNA) or the scrambled sequence (TRPM4scramble); both expressing EGFP as an expression marker kindly provided by D.J. Linden, John Hopkins University.22
Chemical ischemia and oxygen-glucose deprivation
For chemically induced ischemia and reperfusion, neurons were perfused with 5 mM NaN3 added to the ACSF equilibrated in O2. Oxygen and glucose deprivation was induced by perfusing neurons with an anoxic ACSF containing 10 mM 2-D-glucose instead of glucose and equilibrated with 95% N2, 5% CO2 for 1 h previous to the experiments. Neurons were bathed with this buffer for 30 min and then, switched to normal ACSF equilibrated with O2 (95% O2, 5% CO2).
Immunofluorescence
For immunofluorescence experiments, we used 5 weeks old C57/BL6 mice. Briefly, after approval by the local ethics committee, animals were anesthetized with isoflurane and heart was perfused with PBS 0.1 M, pH 7.4. The brain was extracted and fixed overnight in 4% w/v paraformaldehyde diluted in PBS 0.1 M, pH 7.4. Brains were sectioned at 50 µm thick using a Vibratome VT1000 (Vibratome). Floating sections were permeabilized in PBS 0.01% Triton, then blocked for unspecific binding with goat serum. Sections were incubated with anti-TRPM4 (1:100, Alomone) primary antibodies for 48 h at 4°C in agitation, and then washed and incubated with the respective secondary antibody (1:1000). We used MAP2 as neuronal marker (1:100, Abcam). Cells were mounted on glass slides with VectaShield and observed on a confocal microscope (Zeiss LSM 510, Zeiss).
Electrophysiology
Coverslips were transferred to a temperature controlled (30–35°C) submerged recording chamber continuously perfused at 2–3 mL/min ACSF containing (in mM): 124 NaCl, 25 Na2HCO3, 11 glucose, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.25 NaH2PO4, equilibrated with 95% O2, 5% CO2. Multiclamp 700 A and Axopatch 200 B amplifiers equipped with Digidata 1322A or 1440 data acquisition boards and pCLAMP10 software (all from Molecular Devices) were used. Bridge balance and access resistance were monitored during recordings and experiments with >20% changes were discarded. All experiments were performed with nystatin-perforated patch clamp (300 µg/mL nystatin were daily made and added to the intracellular buffer) using patch pipettes with an open tip resistance of 4–6 MΩ. Intracellular solution contained (in mM) 130 KCl, 5 NaCl, 10 HEPES, 0.1 BAPTA (pH 7.2, 285 mOsm/kg). The anti-excitotoxic cocktail (AET cocktail) contained: CNQX (10 µM), AP5 (25 µM), picrotoxin (100 µM), nifedipine (100 µM), TTx (1 µM), TEA (10 mM), 4-AP (1 mM), and GdCl3 (100 µM) to block TRPM7.3
H2O2 and Ca2+ imaging
Changes in [Ca2+]i were monitored as described before.8,50 Briefly, neurons grown in 12 mm round coverslip were incubated with 1 µM Fura 2-AM (Invitrogen) diluted in 0.01% pluronic acid for 1 hour at 37°C in ACSF, then neurons were washed and kept protected from light for 30 min before the measurements. Coverslips were mounted on the stage of an Olympus IX70 microscope, attached to a Sutter Lambda DG-4 high-speed filter unit (Sutter Instruments). Changes in [Ca2+.i were observed using a 40x objective during exposure to 340 and 380 nm, and the intensity of the fluorescence emission 520 nm was recorded using a Hamamatsu Orca-ER CCD camera unit (Hamamatsu) controlled with Micro-manager 1.4. The ratio of fluorescence intensity values at 340/380 reflects the changes in the [Ca2+.i.
Total H2O2 was measured using the genetic encoded probe HyPer-Cyto (Evrogen).28 Briefly, neurons grown in 12 mm round coverslip were transfected at DIV 6 and then measured at DIV 14–21. Changes in intracellular fluorescence were measured by 420 nm excitation and 520 nm emission wavelengths. The vector encoding the protein probe was transfected using Lipofectamine 2000 (Invitrogen). The changes (ΔF) in H2O2 levels were quantified using the formula:
Were Fc is the fluorescence intensity of the cell and Fb is the fluorescence intensity of the background. Values were normalized to a percentage scale against Ca2+i at starting time.
Cell death assay
Neurons superfused with ACFS were treated with 5 mM NaN3 for 1 h and viability was determined up to 6 h later, neurons were kept at 37°C in a controlled atmosphere (95% O2. 5% CO2) during the whole experiment. To quantify neuronal death, we used two methods, trypan blue incorporation and LDH release. Briefly, neurons were incubated with trypan blue (0.4% for 5 min at room temperature). Dishes were mounted on an optical microscope and stained neurons were counted. LDH activity was measured in the neuronal supernatant by a colorimetric end-point kit (Roche) following the manufacturer's instructions and using a calibration curve to ensure linearity. Data were expressed as the fraction of maximum release in the presence of 1% Triton.
Reagents
Unless otherwise stated, all chemicals and reagents were purchased from Merck KgaA-Chemicals and Sigma.
Statistics and graphic software
Unless stated otherwise, all data are presented as a mean ± SEM; statistical differences were tested by two-way ANOVA or Student´s t-test. Graphics were generated using Clampfit 10 (Molecular Devices), GraphPad Prism 6.0 (GraphPad) and Adobe Illustrator CS6 (Adobe) for image composition.
All experiments involving animals were in accordance with the animal protocol approved by the ethical committee of the Universidad de Chile following the rules and guidelines from the Chilean Council of Science and Technology (CONICYT).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias (FONDAP) 15010006 grant to A.S., Fondo de Desarrollo Científico y Tecnológico (FONDECYT) 1160518 grant to O.C. and FONDECYT 11140731 and Programa de Atracción e Inserción Capital Humano (PAI) 79140059 grants to E.L.-S.
Author contributions
E.L.-S. and A.S. designed research; D.R., O.C., and E.L.-S. performed research; D.R., O.C., E.L.-S., and A.S. analyzed data; E.L.-S. and A.S. wrote the paper.
References
- [1].Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79(4):1431-568. PMID:10508238 [DOI] [PubMed] [Google Scholar]
- [2].Won SJ, Kim DY, Gwag BJ. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol. 2002;35(1):67-86. PMID:16248972 [DOI] [PubMed] [Google Scholar]
- [3].Aarts M, Iihara K, Wei W-L, Xiong Z-G, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115(7):863-77. doi: 10.1016/S0092-8674(03)01017-1. PMID:14697204 [DOI] [PubMed] [Google Scholar]
- [4].Chen M, Simard JM. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J Neurosci. 2001;21(17):6512-21. PMID:11517240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gerzanich V, Woo SK, Vennekens R, Tsymbalyuk O, Ivanova S, Ivanov A, Geng Z, Chen Z, Nilius B, Flockerzi V, et al.. De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat Med. 2009;15(2):185-91. doi: 10.1038/nm.1899. PMID:19169264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sheldon C, Diarra A, Cheng YM, Church J. Sodium influx pathways during and after anoxia in rat hippocampal neurons. J Neurosci. 2004;24(49):11057-69. doi: 10.1523/JNEUROSCI.2829-04.2004. PMID:15590922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Simard JM, Kilbourne M, Tsymbalyuk O, Tosun C, Caridi J, Ivanova S, Keledjian K, Bochicchio G, Gerzanich V. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J Neurotrauma. 2009;26(12):2257-67. doi: 10.1089/neu.2009.1021. PMID:19604096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Simon F, Leiva-Salcedo E, Armisén R, Riveros A, Cerda O, Varela D, Eguiguren AL, Olivero P, Stutzin A, Armisen R, et al.. Hydrogen peroxide removes TRPM4 current desensitization conferring increased vulnerability to necrotic cell death. J Biol Chem. 2010;285(48):37150-8. doi: 10.1074/jbc.M110.155390. PMID:20884614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meloni BP, Meade AJ, Kitikomolsuk D, Knuckey NW. Characterisation of neuronal cell death in acute and delayed in vitro ischemia (oxygen-glucose deprivation) models. J Neurosci Methods. 2011;195(1):67-74. doi: 10.1016/j.jneumeth.2010.11.023. PMID:21134398 [DOI] [PubMed] [Google Scholar]
- [10].Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277(32):29181-6. doi: 10.1074/jbc.M204951200. PMID:12042324 [DOI] [PubMed] [Google Scholar]
- [11].Lehotský J, Kaplán P, Matejovicová M, Murín R, Racay P, Raeymaekers L. Ion transport systems as targets of free radicals during ischemia reperfusion injury. Gen Physiol Biophys. 2002;21(1):31-7. PMID:12168723 [PubMed] [Google Scholar]
- [12].Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29(8):2545-52. doi: 10.1523/JNEUROSCI.0133-09.2009. PMID:19244529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27(5):1129-38. doi: 10.1523/JNEUROSCI.4468-06.2007. PMID:17267568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pundik S, Xu K, Sundararajan S. Reperfusion brain injury: focus on cellular bioenergetics. Neurology. 2012;79(13 Suppl 1):S44-51. doi: 10.1212/WNL.0b013e3182695a14. PMID:23008411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109(3):397-407. doi: 10.1016/S0092-8674(02)00719-5. PMID:12015988 [DOI] [PubMed] [Google Scholar]
- [16].Nilius B, Prenen J, Voets T, Droogmans G. Intracellular nucleotides and polyamines inhibit the Ca2+- activated cation channel TRPM4b. Pflügers Arch Eur J Physiol. 2004;448(1):70-5. doi: 10.1007/s00424-003-1221-x. [DOI] [PubMed] [Google Scholar]
- [17].Cheng H, Beck A, Launay P, Gross SA, Stokes AJ, Kinet J-P, Fleig A, Penner R. TRPM4 controls insulin secretion in pancreatic beta-cells. Cell Calcium. 2007;41(1):51-61. doi: 10.1016/j.ceca.2006.04.032. PMID:16806463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Launay P, Cheng H, Srivatsan S, Penner R, Fleig A, Kinet JP. TRPM4 regulates calcium oscillations after T cell activation. Science. 2004;306(5700):1374-7. doi: 10.1126/science.1098845. PMID:15550671 [DOI] [PubMed] [Google Scholar]
- [19].Cáceres M, Ortiz L, Recabarren T, Romero A, Colombo A, Leiva-Salcedo E, Varela D, Rivas J, Silva I, Morales D, et al.. TRPM4 is a novel component of the adhesome required for focal adhesion disassembly, migration and contractility. PLoS One. 2015;10(6):e0130540. doi: 10.1371/journal.pone.0130540. PMID:26110647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shimizu T, Owsianik G, Freichel M, Flockerzi V, Nilius B, Vennekens R. TRPM4 regulates migration of mast cells in mice. Cell Calcium. 2009;45(3):226-32. doi: 10.1016/j.ceca.2008.10.005. PMID:19046767 [DOI] [PubMed] [Google Scholar]
- [21].Crowder EA, Saha MS, Pace RW, Zhang H, Prestwich GD, Del Negro CA. Phosphatidylinositol 4,5-bisphosphate regulates inspiratory burst activity in the neonatal mouse preBötzinger complex. J Physiol. 2007;582(Pt 3):1047-58. doi: 10.1113/jphysiol.2007.134577. PMID:17599963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim YS, Kang E, Makino Y, Park S, Shin JH, Song H, Launay P, Linden DJ. Characterizing the conductance underlying depolarization-induced slow current in cerebellar Purkinje cells. J Neurophysiol. 2013;109(November 2012):1174-81. doi: 10.1152/jn.01168.2011. PMID:23197456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vennekens R, Nilius B. Insights into TRPM4 Function, regulation and physiological role [Internet] In: Transient Receptor Potential (TRP) Channels. Berlin, Heidelberg: Springer Berlin Heidelberg; 2007. page 269-85 [DOI] [PubMed] [Google Scholar]
- [24].Brady NR, Hamacher-Brady A, Westerhoff H V, Gottlieb RA. A wave of reactive oxygen species (ROS)-induced ROS release in a sea of excitable mitochondria. Antioxid Redox Signal. 2006;8(9–10):1651-65. doi: 10.1089/ars.2006.8.1651. PMID:16987019 [DOI] [PubMed] [Google Scholar]
- [25].Nilius B, Prenen J, Tang J, Wang C, Owsianik G, Janssens A, Voets T, Zhu MX. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J Biol Chem. 2005;280(8):6423-33. doi: 10.1074/jbc.M411089200. PMID:15590641 [DOI] [PubMed] [Google Scholar]
- [26].Schattling B, Steinbach K, Thies E, Kruse M, Menigoz A, Ufer F, Flockerzi V, Brück W, Pongs O, Vennekens R, et al.. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2012;18(12):1805-11. doi: 10.1038/nm.3015. PMID:23160238 [DOI] [PubMed] [Google Scholar]
- [27].Simard C, Sallé L, Rouet R, Guinamard R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br J Pharmacol. 2012;165(7):2354-64. doi: 10.1111/j.1476-5381.2011.01715.x. PMID:22014185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Belousov VV, Fradkov AF, Lukyanov KA, Staroverov DB, Shakhbazov KS, Terskikh AV, Lukyanov S. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nat Methods. 2006;3(4):281-6. doi: 10.1038/nmeth866. PMID:16554833 [DOI] [PubMed] [Google Scholar]
- [29].Annunziato L, Cataldi M, Pignataro G, Secondo A, Molinaro P. Glutamate-independent calcium toxicity: Introduction. Stroke. 2007;38(2 PART 2):661-4. doi: 10.1161/01.STR.0000247942.42349.37. PMID:17261710 [DOI] [PubMed] [Google Scholar]
- [30].Li M, Inoue K, Si H, Xiong Z. Calcium-permeable ion channels involved in glutamate receptor-independent ischemic brain injury. Acta Pharmacol Sin. 2011;32(6):734-40. doi: 10.1038/aps.2011.47. PMID:21552295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nilius B, Mahieu F, Prenen J, Janssens A, Owsianik G, Vennekens R, Voets T. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 2006;25(3):467-78. doi: 10.1038/sj.emboj.7600963. PMID:16424899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nilius B, Prenen J, Janssens A, Voets T, Droogmans G. Decavanadate modulates gating of TRPM4 cation channels. J Physiol. 2004;560(Pt 3):753-65. doi: 10.1113/jphysiol.2004.070839. PMID:15331675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Petrosillo G, Ruggiero FM, Pistolese M, Paradies G. Ca2+-induced reactive oxygen species production promotes cytochrome c release from rat liver mitochondria via mitochondrial permeability transition (MPT)-dependent and MPT-independent mechanisms: role of cardiolipin. J Biol Chem. 2004;279(51):53103-8. doi: 10.1074/jbc.M407500200. PMID:15475362 [DOI] [PubMed] [Google Scholar]
- [34].Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47(1):9-23. doi: 10.1007/s12035-012-8344-z. PMID:23011809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen QX, Perkins KL, Choi DW, Wong RK. Secondary activation of a cation conductance is responsible for NMDA toxicity in acutely isolated hippocampal neurons. J Neurosci. 1997;17(11):4032-6. PMID:9151719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kiedrowski L. Critical role of sodium in cytosolic [Ca2+] elevations in cultured hippocampal CA1 neurons during anoxic depolarization. J Neurochem. 2007;100(4):915-23. doi: 10.1111/j.1471-4159.2006.04308.x. PMID:17241128 [DOI] [PubMed] [Google Scholar]
- [37].Weilinger NL, Lohman AW, Rakai BD, Ma EMM, Bialecki J, Maslieieva V, Rilea T, Bandet MV, Ikuta NT, Scott L, et al.. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat Neurosci. 2016;19(3):432-42. doi: 10.1038/nn.4236. PMID:26854804 [DOI] [PubMed] [Google Scholar]
- [38].Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. 2006;25(21):5071-82. doi: 10.1038/sj.emboj.7601378. PMID:17036048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mari Y, Katnik C, Cuevas J. ASIC1a channels are activated by endogenous protons during ischemia and contribute to synergistic potentiation of intracellular Ca2+ overload during ischemia and acidosis. Cell Calcium. 2010;48(1):70-82. doi: 10.1016/j.ceca.2010.07.002. PMID:20678793 [DOI] [PubMed] [Google Scholar]
- [40].Simon F, Varela D, Eguiguren AL, Díaz LF, Sala F, Stutzin A. Hydroxyl radical activation of a Ca2+-sensitive nonselective cation channel involved in epithelial cell necrosis. Am J Physiol Cell Physiol. 2004;287(4):C963-70. doi: 10.1152/ajpcell.00041.2004. PMID:15163619 [DOI] [PubMed] [Google Scholar]
- [41].Alim I, Teves L, Li R, Mori Y, Tymianski M. Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci. 2013;33(44):17264-77. doi: 10.1523/JNEUROSCI.1729-13.2013. PMID:24174660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sun H, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, Kiyonaka S, Mori Y, Jones M, Forder JP, et al.. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci. 2009;12(10):1300-7. doi: 10.1038/nn.2395. PMID:19734892 [DOI] [PubMed] [Google Scholar]
- [43].Bouron A, Kiselyov K, Oberwinkler J. Permeation, regulation and control of expression of TRP channels by trace metal ions. Pflügers Arch Eur J Physiol. 2014;467(6):1143-64. doi: 10.1007/s00424-014-1590-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Simard JM, Chen M, Tarasov K V, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V. Newly expressed SUR1-regulated NC(Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med. 2006;12(4):433-40. doi: 10.1038/nm1390. PMID:16550187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kunte H, Schmidt S, Eliasziw M, del Zoppo GJ, Simard JM, Masuhr F, Weih M, Dirnagl U. Sulfonylureas improve outcome in patients with type 2 diabetes and acute ischemic stroke. Stroke. 2007;38(9):2526-30. doi: 10.1161/STROKEAHA.107.482216. PMID:17673715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Simard JM, Kahle KT, Gerzanich V. Molecular mechanisms of microvascular failure in central nervous system injury–synergistic roles of NKCC1 and SUR1/TRPM4. J Neurosurg. 2010;113(3):622-9. doi: 10.3171/2009.11.JNS081052. PMID:20035575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Guinamard R, Chatelier A, Demion M, Potreau D, Patri S, Rahmati M, Bois P. Functional characterization of a Ca2+-activated non-selective cation channel in human atrial cardiomyocytes. J Physiol. 2004;558(Pt 1):75-83. doi: 10.1113/jphysiol.2004.063974. PMID:15121803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang J, Takahashi K, Piao H, Qu P, Naruse K. 9-Phenanthrol, a TRPM4 inhibitor, protects isolated rat hearts from ischemia-reperfusion injury. PLoS One. 2013;8(7):e70587. doi: 10.1371/journal.pone.0070587. PMID:23936231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42(5):674-83. doi: 10.1002/jnr.490420510. PMID:8600300 [DOI] [PubMed] [Google Scholar]
- [50].He M-L, Zemkova H, Koshimizu T, Tomić M, Stojilkovic SS. Intracellular calcium measurements as a method in studies on activity of purinergic P2X receptor channels. Am J Physiol Cell Physiol. 2003;285(2):C467-79. doi: 10.1152/ajpcell.00042.2003. PMID:12711592 [DOI] [PubMed] [Google Scholar]